
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectivity control in thiol–yne click reactions via visible light induced associative electron upconversion†
Julia V.
Burykina‡
 a,
Nikita S.
Shlapakov‡
ab,
Evgeniy G.
Gordeev
a,
Burkhard
König
*b and
Valentine P.
Ananikov
*a
a,
Nikita S.
Shlapakov‡
ab,
Evgeniy G.
Gordeev
a,
Burkhard
König
*b and
Valentine P.
Ananikov
*a
aZelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, Moscow, 119991, Russia. E-mail: val@ioc.ac.ru
bInstitut für Organische Chemie, Universität Regensburg, Universitätstrasse 31, 93053 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de
First published on 23rd July 2020
Abstract
An associative electron upconversion is proposed as a key step determining the selectivity of thiol–yne coupling. The developed synthetic approach provided an efficient tool to access a comprehensive range of products – four types of vinyl sulfides were prepared in high yields and selectivity. We report practically important transition-metal-free regioselective thiol–yne addition and formation of the demanding Markovnikov-type product by a radical photoredox process. The photochemical process was directly monitored by mass-spectrometry in a specially designed ESI-MS device with green laser excitation in the spray chamber. The proposed reaction mechanism is supported by experiments and DFT calculations.
Introduction
Transition-metal-catalyzed atom-economic carbon–sulfur bond construction has received significant interest in the last few decades.1–5 Many homogeneous and heterogeneous catalytic systems were developed for the addition reaction of thiols to alkynes, but there is still demand for a selective and simple catalytic synthesis of Markovnikov and anti-Markovnikov vinyl sulfides.6–8 Reported methods for selective thiol–yne reactions require expensive metal complexes or special ligands and harsh conditions. Moreover, most catalytic systems are limited in scope and metal catalyzed reactions may contaminate the products with metal traces.9–11Visible light photoredox catalysis has evolved into an important method in organic chemistry and can provide superior reaction conditions.12–15 The first example of a photoredox thiol–yne click reaction under metal-free conditions with good yields and selectivity for β-vinylsulfides was reported in 2016.16 Later, Wang and co-workers reported a photoredox process driven by 1 mol% mesityl-10-methyl-acridinium tetrafluoroborate, providing a range of mono- and bis-substituted sulfide products.17 However, the described processes based on the photoinduced free radical chain exclusively yield the linear (anti-Markovnikov) isomers.
The situation changed after Lei and co-workers18 published a light-mediated regioselective radical synthesis of α-substituted vinyl sulfones. The possibility of applying this protocol for branched (Markovnikov type) vinylsulfide synthesis was also demonstrated. The only example (phenylacetylene with p-tolylthiol) gives a 60% 1H-NMR yield of α-vinylsulfide. On the other hand, convincing and detailed mechanistic investigations shedding light on such an uncommon change of selectivity are of much interest.
Our goal was to develop a universal metal-free approach to access different thiol–yne products with high selectivity. The change of the photochemical conditions allows the control of the reaction selectivity in a desired manner.
The concept behind the control of thiol addition reaction selectivity in the absence of metals relies on an associative electron upconversion consisting in highly reducing radical-anion formation via orbital crossing. The electron upconversion19 and orbital crossing20–23 concepts have been elaborated by Alabugin and coworkers but have not yet been applied to alkyne functionalization to the best of our knowledge (Scheme 1).
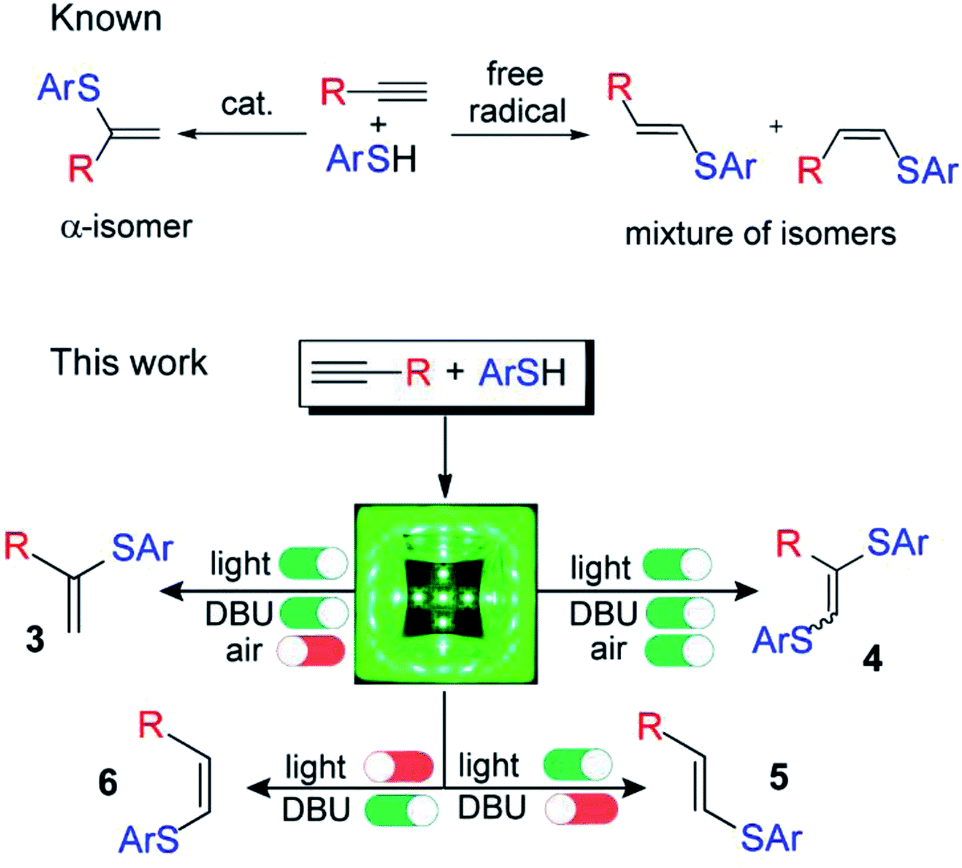 | ||
| Scheme 1 Controllable switching of the reaction selectivity; an argon atmosphere is not important for the formation of products 5 and 6; 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). | ||
In experimental and theoretical studies, we evaluated all plausible vinylsulfide/disulfide pathways of a photoredox thiol/yne reaction and determined the dependence of selective product formation on the reaction conditions. We report a photoredox system for the synthesis of four products (α-isomer (3), β(E)-isomer (5), β(Z)-isomer (6) and disulfide (4)) under simple conditions with high selectivity (Scheme 1).
Results and discussion
The reaction of phenylacetylene 1a with thiophenol 2a mediated by Eosin Y under light from green light-emitting diodes (LEDs) was chosen as a model thiol–alkyne coupling (Scheme 2), but the reaction turned out not to be selective leading to the formation of a broad range of products.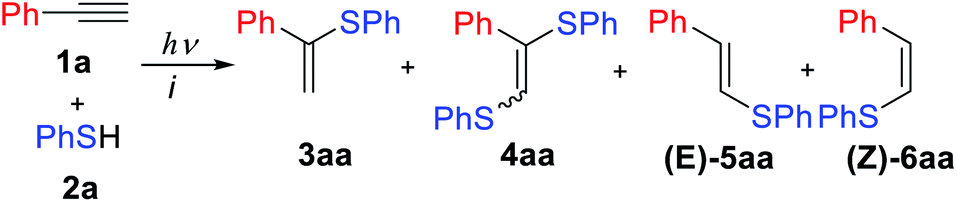 | ||
| Scheme 2 The model thiol–yne click reaction between 1a and 2a. | ||
Examination of typical bases revealed their crucial effect on the thiol–yne coupling. The previously reported photoredox system16 with pyridine as the base gave the anti-Markovnikov radical product 5aa in 90% yield (Table 1, entry 1). The use of NaHCO3 or KOAc as bases decreased the yield and selectivity (entries 2–3). However, the addition of a stronger base gave an acceptable yield for product 3aa (entry 4) which is consistent with Lei's work.18 Additional screening revealed that moderately strong bases are necessary for obtaining the Markovnikov product 3aa (entries 4–7). The best result gave a catalytic system of Eosin Y/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under green LED irradiation affording the α-isomer in 85% yield (entry 7).
| Entry | Base | 3aa, % | 4aa, % | 5aa, % | 6aa, % |
|---|---|---|---|---|---|
| a Unless otherwise noted, the reactions were carried out using 1a (0.15 mmol), 2a (0.3 mmol), DMF (3 ml), base (0.33 mmol), Eosin Y (3 mol%), 40 °C, green LEDs (1.25 W), 24 h, and argon. b Reaction mixture was deeply colored. c Under air. d t = 80 °C, reaction time 72 h. e Reaction without Eosin Y and green light. f 0.125 ml of DMF. g Addition of TEMPO. h Addition of γ-terpinene. Several reaction mixtures' NMR spectra are shown in the ESI (Fig. S1–S4). | |||||
| 1 | Py | 0 | 0 | 90 | 10 |
| 2 | NaHCO3 | 29 | 2 | 62 | 7 |
| 3 | KOAc | 44 | 4 | 47 | 5 |
| 4 | K2CO3 | 70 | 13 | 7 | 10 |
| 5 | KF | 81 | 7 | 10 | 2 |
| 6b | t BuOK | 33 | 5 | 0 | 62 |
| 7 | DBU | 85 | 7 | 3 | 0 |
| 8c | DBU | 10 | 77 | 0 | 0 |
| 9c,d,e | DBU | 0 | 0 | 7 | 93 |
| 10f | DBU | 5 | 0 | 37 | 58 |
| 11e | DBU | 0 | 0 | 0 | 22 |
| 12e,c | DBU | 0 | 0 | 0 | 23 |
| 13g | DBU | 0 | 92 | 0 | 0 |
| 14h | DBU | 1 | 0 | 1 | 12 |
Further experiments revealed that air and argon have a significant influence on the selectivity of this transformation. Conducting the reaction under air and with DBU gave a good yield of disulfide 4aa (entry 8). Increasing the temperature and conducting the reaction under air without Eosin Y and light gave product 6aa (entry 9). Formation of Z-anti-Markovnikov products under basic conditions is discussed in Oshima's work on nucleophilic addition of thiols to alkynes.24 Thus, a selective synthesis of 3aa/4aa/5aa/6aa is possible by changing the base and the presence or absence of air and light.
The concentration of reaction reagents has a significant influence on the reaction selectivity. Increasing the reagent concentration (entry 10) leads to the formation of 5aa and 6aa as the main products and trace amounts of 3aa. Under standard reaction conditions (entry 7) the thiol dissociates heterolytically almost completely, which prevents it from participating in the radical process leading to product 5aa.
The control reactions without Eosin Y and light irradiation with DBU under argon (entry 11) or air (entry 12) gave the products of nucleophilic addition reactions. In the case of tBuOK addition the reaction mixture turns black under irradiation, and an ionic mechanism leading to by-product 6aa is dominant (entry 6).
To verify the radical pathway of the reaction we performed the transformation in the presence of radical traps: 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) (entry 13) and γ-terpinene (entry 14). The presence of γ-terpinene completely suppresses any radical processes, but the background Michael-type reactivity remains. TEMPO turned out to be an ineffective radical trap in our system. It acts as a one electron oxidant and can substitute air oxygen, as reflected by the excellent yield of product 4aa under these conditions.
Optimization of other reaction parameters such as the reagent ratio, solvent and photoredox system is summarized in Table S1† (ESI).
Next, we studied the application of this switchable photoredox system to the synthesis of specific thiol–yne products of various alkynes. The experiments confirmed the high selectivity and efficiency of the synthetic approach for the preparation of α-isomer (3), β(E)-isomer (5), β(Z)-isomer (6) and bis-sulfide (4). In all the cases, complete conversion of alkyne was achieved, and the corresponding product was obtained in good to very good yields (Scheme 3).
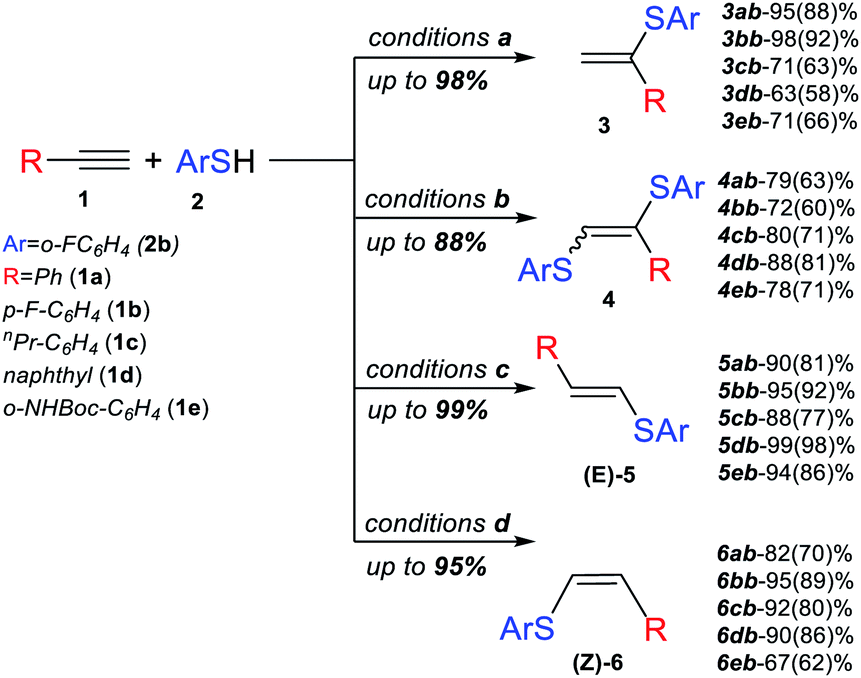 | ||
| Scheme 3 Conditions a: 1 (0.15 mmol), 2b (0.3 mmol), DMF (3 ml), DBU (0.33 mmol), Eosin Y (3 mol%), 40 °C, green LEDs (1.25 W), 24 h, and argon bubbling; conditions b: under the same conditions as for [a], but 2b (0.45 mmol) and without argon bubbling; Conditions c: 1 (1 mmol), 2b (1.1 mmol), DMF (0.5 ml), Py (0.3 mmol), Eosin Y (1 mol%), 40 °C, green LEDs (1.25 W), 6 h, and under air; Conditions d: 1 (1 mmol), 2b (1.5 mmol), DMF (0.5 ml), tBuOK (0.5 mmol), 70 °C, 24 h, and under air. Yields determined by 1H NMR and isolated yields are shown in parentheses. | ||
The substrate scope for the most challenging selective synthesis of the Markovnikov product (3) involving different types of alkynes and thiols was then investigated employing the optimized reaction conditions (Scheme 4). Due to the importance of fluorine containing compounds,25–27o-F-substituted thiol was chosen as the model thiol for alkyne scope investigation. A number of aromatic thiols, bearing various functional groups at the ortho-, meta- and para-position reacted smoothly with phenylacetylene to afford the α-isomers (3) in 74–95% yields. Even in the case of non-polar electron-donating thiols 2a, 2c and 2e, the corresponding products 3aa, 3ac and 3ae could be isolated in 70–78% yields.
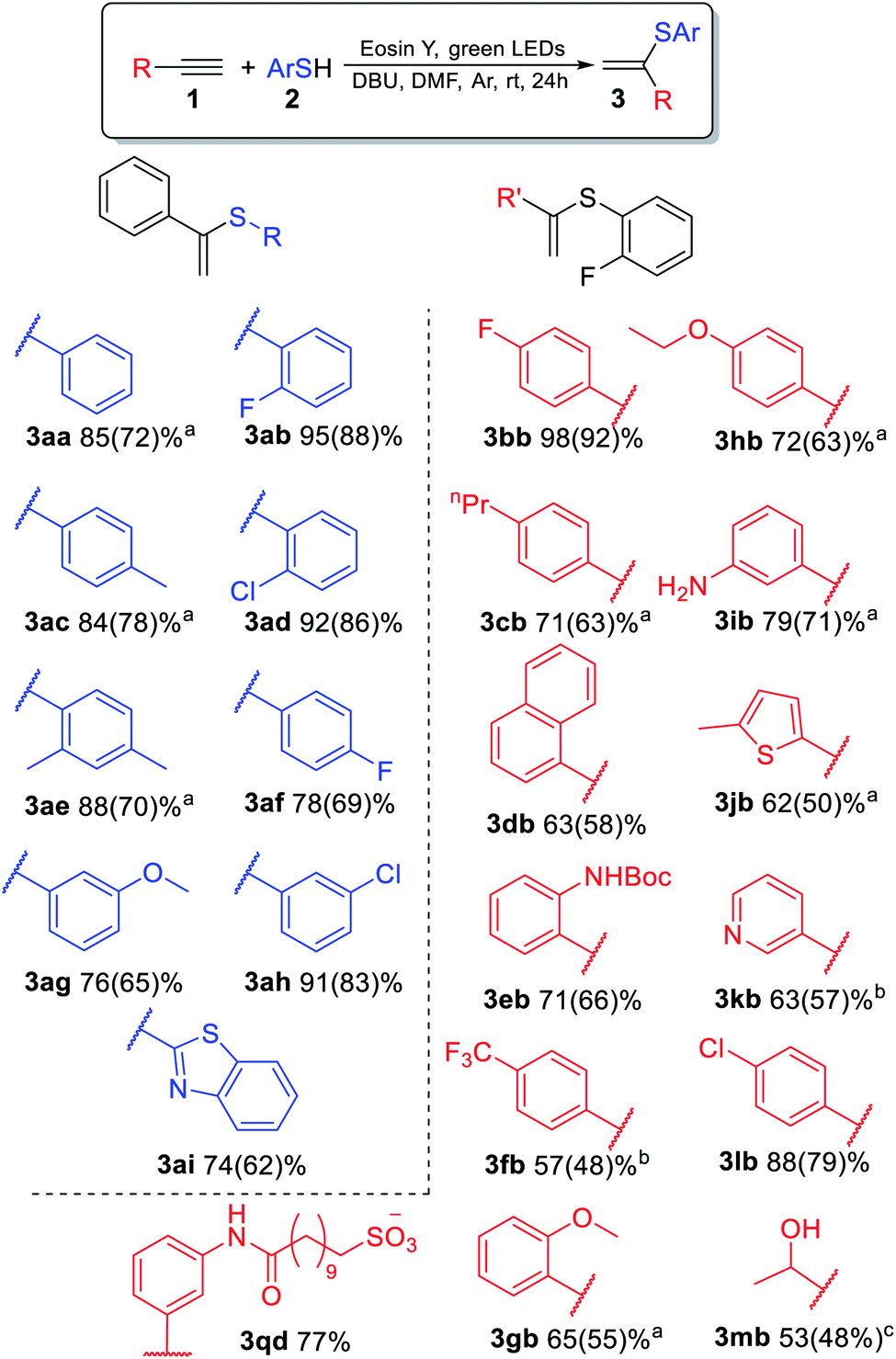 | ||
| Scheme 4 Conditions: 1 (0.15 mmol), 2 (0.3 mmol), DMF (3 ml), DBU (0.33 mmol), Eosin Y (3 mol%), 40 °C, green LEDs (1.25 W), 24 h, and argon bubbling. Yields determined by 1H NMR and isolated yields shown in parentheses. a48 h, b2 h, 30 W green LEDs, c24 h, 30 W. | ||
Similarly to phenylacetylene, terminal aromatic alkynes were selectively transformed into the expected adducts 3 in moderate to high yields. In the case of electron-withdrawing alkynes 1f and 1k, we use a more powerful 30 W green LED to speed up the synthesis of the Markovnikov product and outrun the competitive non photocatalytic process of β(Z)-isomer 6 product formation. In the case of electron-donating alkynes 1g–1j, the α-isomer was produced in moderate to good yields accompanied by by-products 4 and 7.
As a representative example of such a side process, we investigated the reaction between 1h and 2b and successfully isolated and characterized compound 7hb, the product of attack of radical R1 at the double bond of vinylsulfide 3hb. The electrophilic vinyl radicals get attacked by nucleophiles (3hb or SAr−), which explains why nucleophilic vinylsulfide 3hb reacts in the observed manner (Scheme 5).
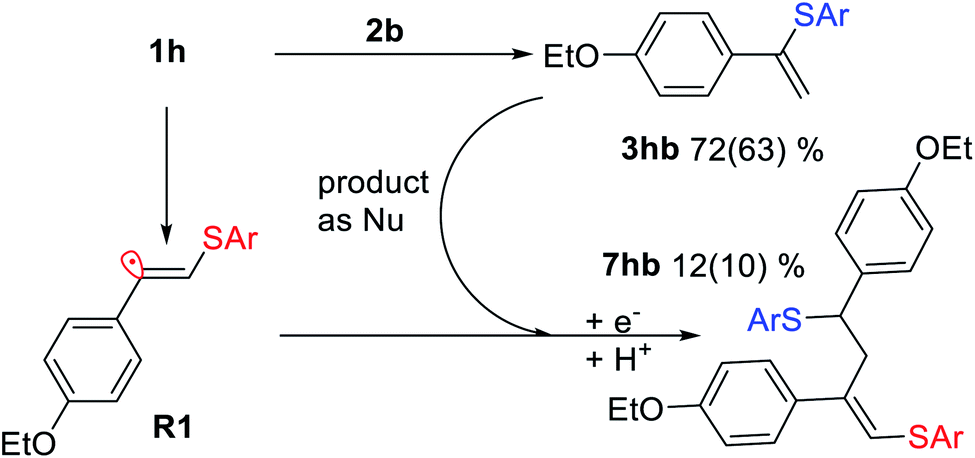 | ||
| Scheme 5 Interaction between vinyl radical R1 and vinylsulfide 3hb under the standard reaction conditions for 7hb formation. Yields determined by 1H NMR and isolated yields shown in parentheses. | ||
To determine the limitations of our method we have conducted experiments with aliphatic (1r) and internal (1s) alkynes (Scheme 6). Both alkynes react slowly under the developed conditions and full conversion was not reached after 2 days. The reaction of alkyne 1r with such an electron-deficient thiol as 2j gives only the oxidative product 4rj in the absence of air. This may be explained by the formation of a highly reductive intermediate not stabilized by the aromatic ring resulting in its oxidation by the solvent. On the other hand, alkyne 1s reacts with retention of the selectivity described above, and product 3sa and by-product 4sa have been isolated with an overall yield of 81%. However, replacing DBU with the weak base pyridine leads to formation of compounds 5sa and 6sa as expected for a free radical chain reaction.
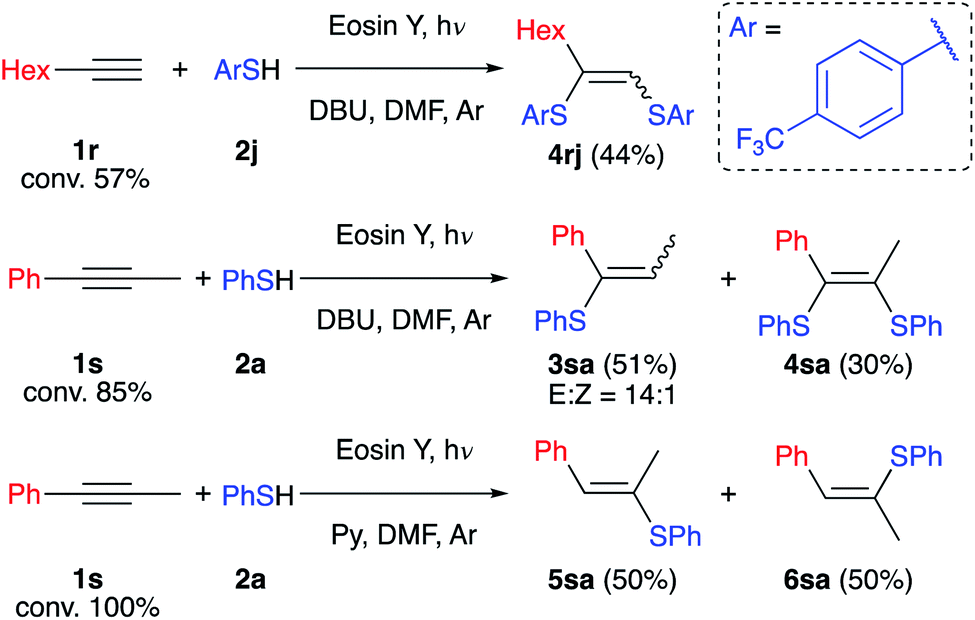 | ||
| Scheme 6 Limitations of the protocol for aliphatic (reaction conditions: 1r (0.15 mmol), 2j (0.3 mmol), DMF (2 ml), DBU (0.33 mmol), Eosin Y (3 mol%), 40 °C, green LEDs (1.25 W), 48 h, and argon flushing) and internal (reaction conditions: 1s (0.15 mmol), 2a (0.3 mmol), DMF (2 ml), DBU (0.33 mmol), Eosin Y (3 mol%), 40 °C, green LEDs (1.25 W), 48 h, and argon flushing) alkynes. The yields were determined by GC-FID. | ||
In the case of 1n which contains an o-Br substituent, the formation of an unexpected product 8na was observed in 62% yield (Scheme 7). Presumably, the nucleophilic attack of the thiolate anion to the electrophilic vinyl radical R1via orbital crossing leads to the formation of highly reducing anion-radical R2. A subsequent intramolecular ET process from the π*-orbital to the σ*(C–Br)-orbital with consequent bromine atom extrusion gave benzothiophene derivative 8na with loss of the Ar-radical (blue, Scheme 7), which gets trapped by a thiolate anion and oxidized.
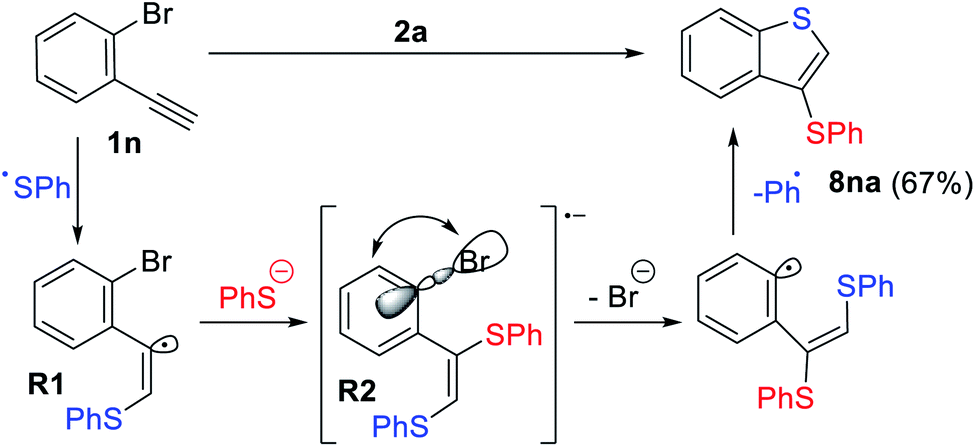 | ||
| Scheme 7 Observation of Intra ET between the π*-orbital of R2 and the σ*(C–Br)-orbital with consequent bromine atom extrusion leading to benzophenone 8nb; reaction conditions: 1 (0.15 mmol), 2 (0.45 mmol), DMF (3 ml), K2CO3 (0.5 mmol), Eosin Y (7 mol%), 40 °C, green LEDs (1.25 W), 24 h, and argon flushing. The yields were determined by 1H NMR. | ||
A similar idea to control the reactivity of substrates by intramolecular electron transfer in π-conjugated arene systems was demonstrated by Studer28 and is applicable to other reactions with arenes.29 Knowles30 described the intramolecular electron transfer from a hydroxy-group to a p-methoxyphenyl group using photoredox catalysis supporting our hypothesis.
Further investigation of the described process led to the assumption that Intra ET is only possible when the Br-containing benzene ring (purple, Scheme 8) is in π-conjugation with the alkyne fragment (blue, Scheme 8). In the case of electron transfer via the conjugated π-system onto the σ*(C–Br)-orbital, the C–Br bond becomes activated and a radical cyclization by-product 4ob′ forms along with the main products 3ob and 4ob. However, the Intra ET process depends on the spatial continuity of the upconverted anion-radical π-system. Factors that interfere with the π-conjugation between the alkyne fragment and the Br-atom containing aromatic ring (e.g. steric repulsion of peri-substituents in the biphenyl systems) may prevent the Intra ET process and lead to the superiority of Intermolecular ET processes, as indicated by the predominant formation of product 4pb.
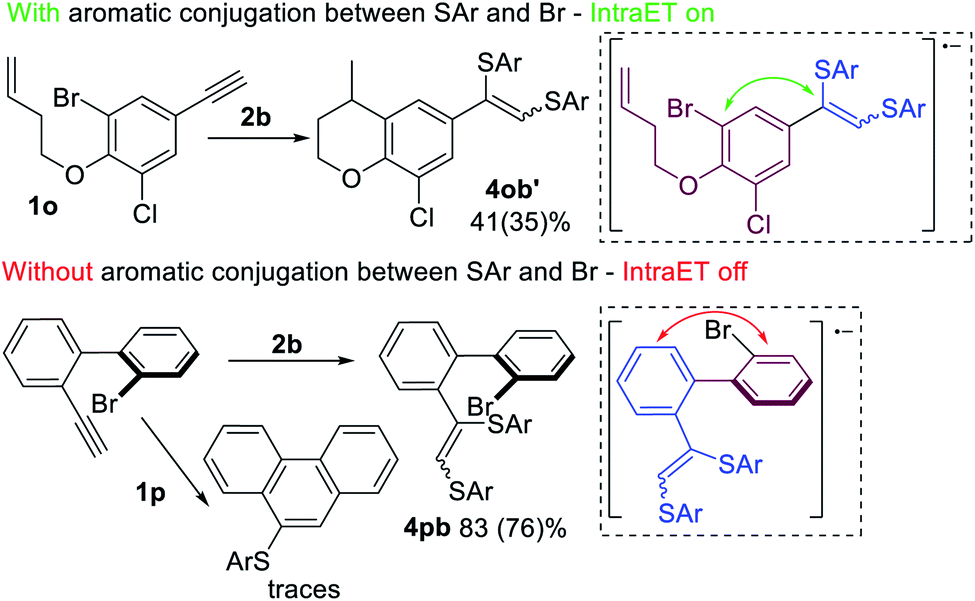 | ||
| Scheme 8 Structural limitations of the Intra ET process in the systems containing Br and a triple bond; reaction conditions: 1 (0.15 mmol), 2 (0.3 mmol), DMF (3 ml), K2CO3 (0.29 mmol), Eosin Y (3 mol%), 40 °C, green LEDs (30 W), 24 h, and argon flushing. The yields were determined by 1H NMR. | ||
DFT calculations of the C–Br bond dissociation energy for substrates 1n, 1o, and 1p do not explain the low activity of 1p. However, DFT molecular dynamics modeling has shown a significant drop of the dissociation rate of the R2 radical anion for 1p, because this method takes into consideration all conformations, but not the most stable one with the best conjugation (see the ESI† and computational details).
All the findings described above highlight the key role of the upconverted R2 radical-anion formation in the studied system.
To reveal the nature of the observed transformations we carried out the reaction inside the electrospray ionization chamber of a mass spectrometer in the neat state from the flask.31,32 Monitoring of photoredox-catalyzed reactions by coupling of spray-based ionization mass spectrometers with online laser irradiation has been established as a reliable method.33–35 For the thiol–yne click reaction, we coupled a green laser source with an electrospray ionization mass spectrometer (ESI-MS). The device used consisted of a green laser connected to the ESI chamber as shown in Fig. 1. The irradiation was directed to the tip of the nebulizer where small charged microdroplets of the reaction mixture are formed due to an applied potential between the electrode near the sprayer (the nebulizer is grounded) and the shield (Fig. 1A).36
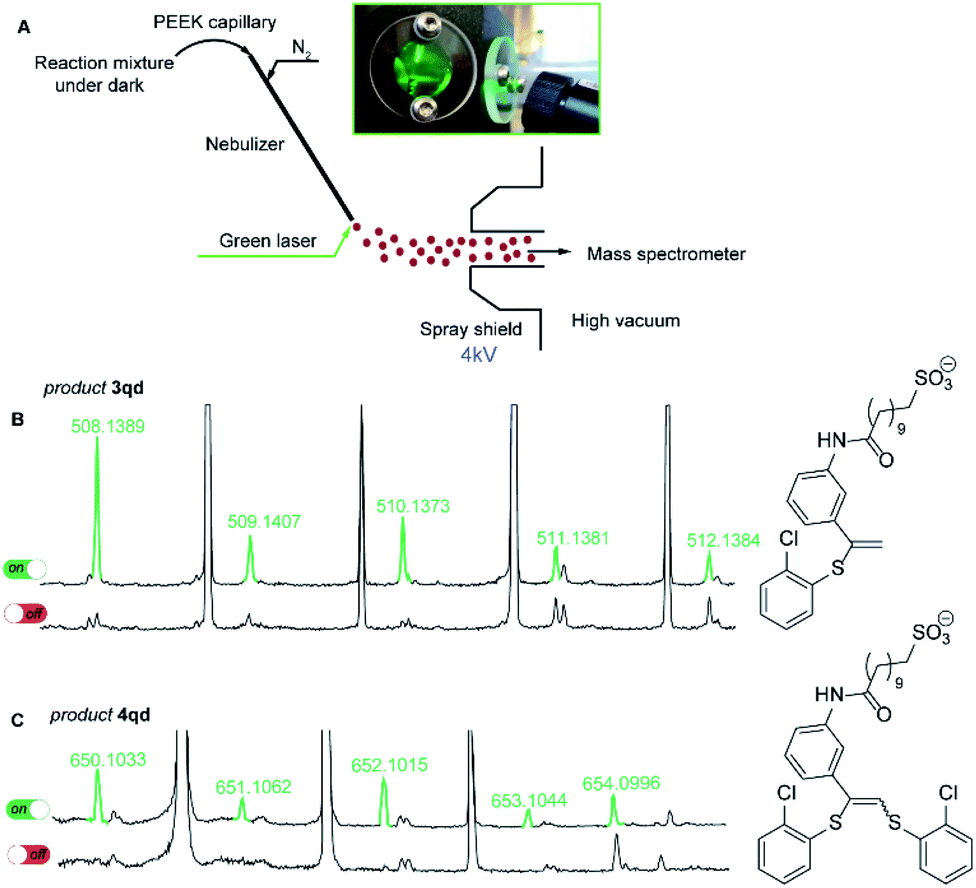 | ||
| Fig. 1 (A) Scheme of the coupled device for the online ESI-(−)MS experiment; the laser power is about 80 mW. A reaction mixture was introduced into the PEEK capillary and then photoexcited using the laser pointer inside the spray chamber and passed into the mass spectrometer; (B) the real-time spectra of the 3qd product for light/dark experiments in negative ion mode; (C) the real-time spectra of 4qd for light/dark experiments in negative ion mode. The light experiment results are shown in green and the dark experiment results are shown in black. | ||
For this experiment, we have synthesized alkyne 1q with an easily ionized sulfonate group and carried out the thiol–yne reaction between alkyne 1q and 2-chlorothiophenol 2d under standard reaction conditions (Scheme 4). In the negative ion mode in the absence of green light irradiation, the signals corresponding to alkyne 1q, thiol 2d, and Eosin Y species were dominant in the ESI mass spectra.
Under the green light irradiation of the nebulizing tip, the molecular ions corresponding to products 3qd (measured m/z 508.1389; calcd m/z 508.1389 for [C25H31NO4S2Cl]−) and 4qd (measured m/z 650.1033; calcd m/z 650.1032 for [C31H35NO4S3Cl2]−) began to appear a couple of milliseconds after the start of irradiation. However, when the LED light was switched off the signals of 3qd and 4qd dropped to zero in their intensities (Fig. 1C). Simultaneous appearance or disappearance of these peaks proved the key role of light in the formation of a highly reactive intermediate (conceivably R2), which is able to be transformed to either product 3 or 4.
Furthermore, a detailed investigation of the reaction pathway by continuous online ESI-(−)MS monitoring was conducted with irradiation of the Schlenk tube reaction vessel. This experiment allows real-time tracking of the starting material, photocatalyst degradation and product formation (see the ESI† for details).
The overall reaction mechanism is proposed on the basis of the collected experimental data and previous findings (Scheme 9). The catalytic cycle starts from Eosin Y affording the thiyl radical upon light irradiation. Next, ArS˙ is added to the alkyne 1 to produce R1, which is able to abstract a hydrogen atom yielding side product 5, which can be suppressed under optimized conditions. Nucleophilic addition of ArS− to R1via orbital crossing results in the formation of R2. The radical-anion R2 could be protonated, which leads to the formation of stabilized benzyl-type radical R3.
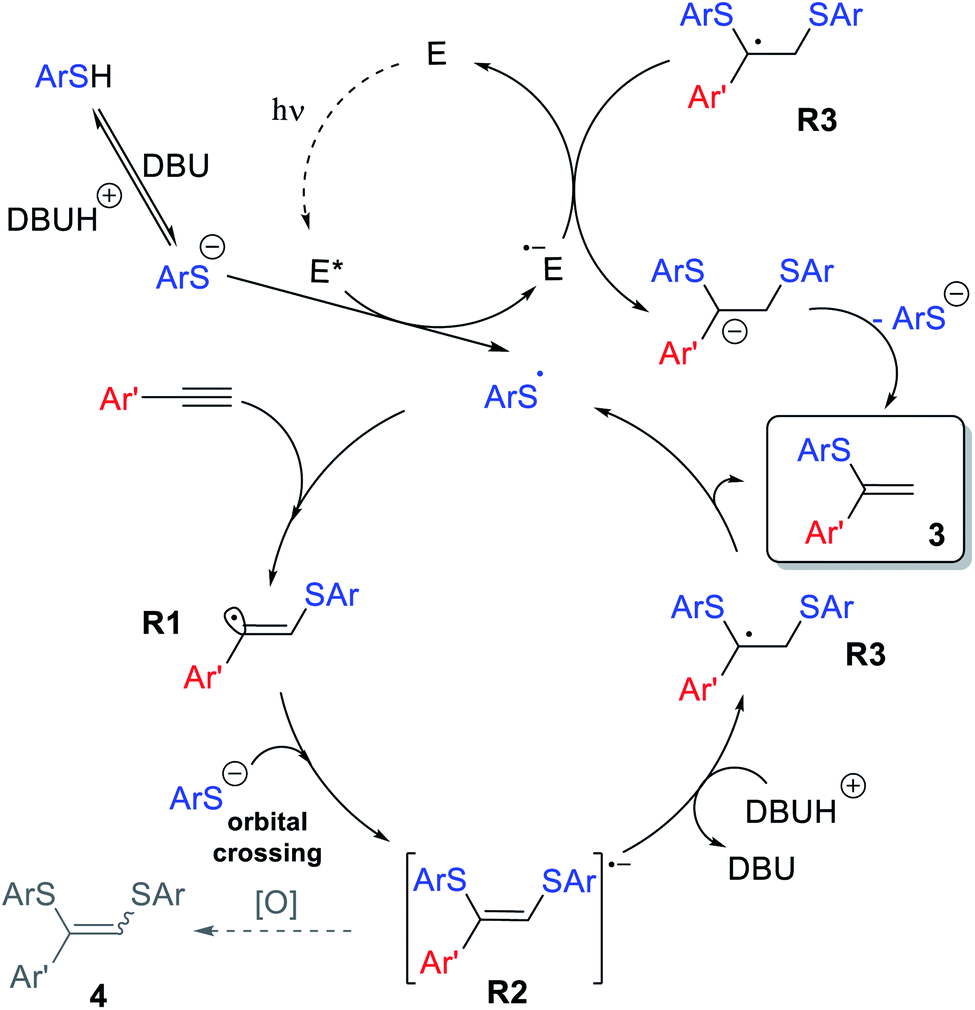 | ||
| Scheme 9 Plausible catalytic cycle of the photoredox thiol–yne click reaction. | ||
Subsequent elimination of a thiyl radical yields product 3, while the ArS˙ radical triggers the next ion-radical cycle. It should be noted that upconverted highly reducing intermediate R2 is extremely sensitive to oxidizing agents (oxygen impurities, excited forms of the photocatalyst or even the solvent in some cases), and therefore easily diverts towards the oxidative bis-addition product 4 during the single electron transfer stage. This Inter ET process strongly depends on the structure of the intermediate R2. The HOMO energy DFT-calculations for different R2 demonstrates that an aromatic substituent stabilizes the intermediate significantly better than an aliphatic one. For R = Ph, the HOMO energy is −3.40 eV, while for R = n-Hex, the HOMO energy is −3.00 eV, that is, in the case of an alkyl substituent, intermediate R2 has significantly more pronounced electron-donating properties and can more easily lose activity due to oxidation (see the ESI†). This explains the fact of lower yields in the case of 3mb and exclusive 4rj formation in the case of aliphatic alkyne 1j.
To support our hypothesis for the mechanism, we have measured the quantum yield at 528 nm (23%) and the quantum efficiency (29%). A high value of the quenching factor (0.8) indicates an efficient PET from the excited state of the photocatalyst to the ArS− anion (E(ArS˙/ArS−) = 0.1–0.4 V, E(Eosin*/Eosin−) = 0.8 V),37 but other deactivating pathways of the photocatalyst affecting the quenching factor were not considered. The back electron transfer from the reduced form of the photocatalyst to the ArS-radical is also thermodynamically feasible (E(Eosin/Eosin−) = −1.1 V).
Thus, we conclude that a short radical chain could be considered as a possible scenario for the reaction along with moderately efficient chain termination caused by reduction of the benzylic radical intermediate R3 (E(R3˙/R3−) should be less reductive then E(benzyl˙/benzyl−) = −1.4 V because of the SAr-group stabilization effect).
To gain insight into the radical formation and understand the reaction regioselectivity, we carried out DFT calculations for product 3 formation from the intermediate R2. The transformation of intermediate R2 to product 3 begins with protonation of one of the R2 double bond carbons by a DBUH+ cation (Fig. 2A). The protonation can occur either at the secondary carbon atom and proceed via the TS2 transition state or at the tertiary carbon atom and proceed via the TS1 transition state. The spin density corresponding to the unpaired electron of the R2 radical anion is predominantly localized on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C carbon atoms (see Fig. 3A).
C carbon atoms (see Fig. 3A).
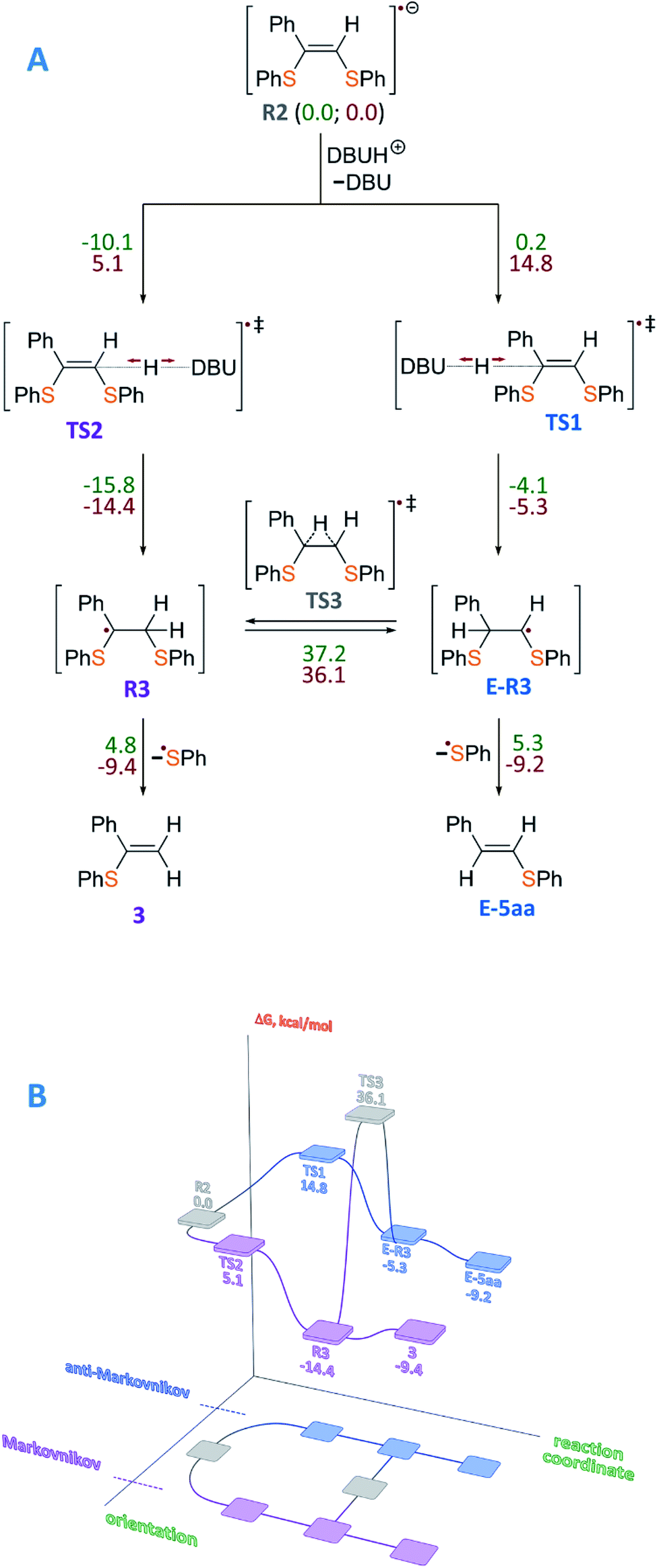 | ||
| Fig. 2 (A) Mechanism of product 3 formation from intermediate R2, which is taken as a reference point, with total energy values (ΔE, kcal mol−1) denoted by green color and free Gibbs energies (ΔG, kcal mol−1) denoted by red color. (B) Representation of the free energy profile for two possible reaction paths of product 3 formation (see conventional representation of the energy profiles and optimized molecular structures in the ESI†); UBMK/6-311+G(d,p) D3BJ & SMD(DMF). | ||
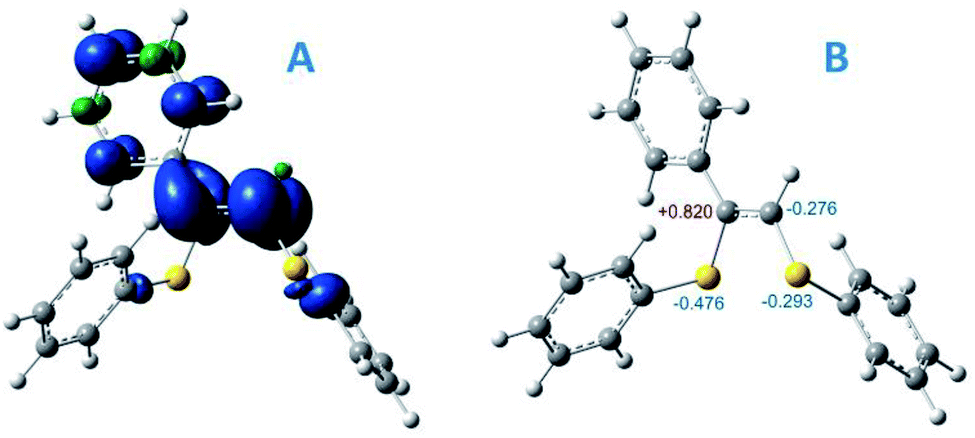 | ||
| Fig. 3 (A) spin density distribution for intermediate R2; (B) Mulliken atomic charges for some atoms of intermediate R2, UBMK/6-311+G(d,p) D3BJ & SMD(DMF). | ||
However, potential barriers of R2 protonation for the two reaction channels differ significantly: the protonation leading to the anti-Markovnikov product requires much higher activation energy (ΔG‡ = 14.8 kcal mol−1) compared with the protonation leading to the Markovnikov-type product (ΔG‡ = 5.1 kcal mol−1) (Fig. 2A and B). The lower activation energy for the TS2 transition state correlates with the charge distribution in the intermediate R2: the secondary carbon atom is negatively charged (−0.276), which ensures its increased nucleophilicity, while the tertiary carbon atom carries a significant positive charge (see Fig. 3B).
Formation of product 3 is therefore promoted by kinetic and thermodynamic effects of the protonation stage: intermediate R3 is significantly stabilized as compared with intermediate E-R3. Transition between the intermediates R3 and E-R3 is unlikely, since the TS3 transition state is characterized by high energy, and if the protonation proceeds via the R2→R3 → 3 pathway, the transition to the R2→E-R3 → E-5aa pathway is unlikely. In the last step, regeneration of the ArS˙ radical occurs with the formation of the final product 3. Within the reaction mechanism, the regeneration of Eosin Y is mediated by R3. Similar results were obtained for alkyl-substituted intermediate R2 (see the ESI†). Thus, the performed calculations correlate well with our experimental findings.
Conclusions
We have developed the selective synthesis of Markovnikov-type thiol–yne products using a photoredox catalyzed reaction. The developed procedure affords Markovnikov-type vinylsulfides in up to 92% yield under mild reaction conditions. The key advantages of the developed system are simple and straightforward reaction conditions for selective synthesis of the desired products.Using standard ESI-MS spray-based ionization mass spectrometers with online laser irradiation, direct monitoring of the reaction was possible. Computational study at a UBMK/6-311+G(d,p) D3BJ & SMD(DMF) level of theory revealed a lower activation energy for the Markovnikov-type product formation under the presented conditions.
The associative electron upconversion process in the photoinduced reaction of thiols and alkynes is essential for the observed selectivities. The concept may find applications in the design of other transformations of alkynes with good leaving groups based on an intramolecular electron transfer (Intra ET) process.
Experimental
General procedures
The reagents were obtained from commercial sources and used as supplied (verified by NMR prior to use). The solvents were purified according to published methods. The solvents for NMR spectroscopy were obtained from Deutero GmbH. Acetonitrile (HPLC-grade) for the ESI-MS was obtained from Merck and used as supplied. Unless otherwise noted, the reactions were carried out in PTFE screw-capped tubes equipped with magnetic stirring bars. Magnetic stirring bars were cleaned with a boiling solution of alkali followed by a boiling solution of aqua regia and further rinsing with distilled water to ensure the removal of all absorbed metal traces. Column chromatography was performed using Merck 60 μm silica gel. All NMR measurements were performed with Bruker DRX 500, Bruker Avance III 400 and Bruker Fourier 300 spectrometers operating at 500.1, 400.1 and 300.1 MHz for 1H, 125, 100 and 75 MHz for 13C, and 470.5, 376.4 and 282.3 for 19F nuclei. 1H, 13C{1H} chemical shifts are given in ppm relative to the residual peak of the solvent DMSO-d6 (δ 2.5 ppm) for the proton spectra and relative to the solvent peak (δ 39.5 ppm) for the carbon spectra. All 19F NMR chemical shifts were referenced to internal CF3C6F5 (δ −55.85 ppm). The spectra were processed with the Bruker TopSpin 3.2 software package.High-resolution mass spectra were obtained on a Bruker maXis Q-TOF instrument (Bruker Daltonik GmbH, Germany) equipped with an electrospray ionization (ESI) ion source. The experiments were performed in positive (+) MS ion mode (HV Capillary: −4500 V; HV End Plate Offset: −500 V) or negative (−) MS ion mode (HV Capillary: +4000 V; HV End Plate Offset: −500 V) with a scan range of m/z 50–1500. External calibration of the mass spectrometer was performed using a low-concentration tuning mix solution (Agilent Technologies). Direct syringe injection was applied for the analysis of solutions in MeCN (flow rate: 3 μl min−1) for analytical characterization, and pressurized sample infusion was applied for reaction monitoring studies in DMF (additional experimental details provided below). Nitrogen was applied as the nebulizer gas (1 bar) and dry gas (4.0 l min−1, 200 °C). The spectra were processed using Bruker Data Analysis 4.0 software.
The GC-MS experiments were carried out with an Agilent 7890A GC system, equipped with an Agilent 5975C mass-selective detector (electron impact, 70 eV) and an HP-5MS column (30 m/0.25 mm/0.25 μm film) using helium as the carrier gas at a flow of 1.0 ml min−1.
The GC-FID measurements were performed with a SCION 436-GC gas chromatograph with a flame ionization detector and an HP-5MS (Agilent Technologies) column (30 m × 0.25 mm × 0.25 μm film) using helium as the carrier gas at a constant linear velocity of 30 cm s−1. The following temperature program was used in all GC-MS measurements: initial temperature: 60 °C, hold for 2 min, then 20 °C min−1 to 300 °C and hold for 6 min.
Computational details
Synthesis of product 3
Eosin Y (3 mg, 4.3 μmol), thiol (0.3 mmol), and DBU (50 μl, 0.33 mmol) were dissolved in 3 ml of DMF. The solution was flushed with argon for 15 minutes. After this, the alkyne (0.15 mmol) was added under an argon flow and the cap was closed. The reaction was irradiated using 1.25 W green LEDs for 24 h.Isolation protocol A: after completion of the reaction 10 ml of petroleum ether was added. The organic layer was washed with a 20% KOH solution in water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether/triethylamine, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.01).
:0.01).
Isolation protocol B: after completion of the reaction 10 ml of DCM was added. The organic layer was washed with 20% KOH solution in water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether/DCM/triethylamine, 10:1:0.01).
Synthesis of product 4
Alkyne (0.15 mmol), thiol (0.45 mmol), Eosin Y (3 mg, 4.3 μmol), DBU (75 μl, 0.48 mmol) and 3 ml of DMF were mixed in a reaction vessel under stirring and placed in a photoreactor equipped with green LEDs (λmax = 533 nm). The reaction was carried out in an open tube for 24 h. After completion of the reaction 10 ml of DCM was added. The organic layer was washed with water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether/ethyl acetate, 4:1).
Synthesis of product 5
Alkyne (1 mmol), thiol (1.1 mmol), Eosin Y (6.5 mg, 0.001 mmol), pyridine (25 μl, 0.3 mmol) and 0.5 ml of DMF were mixed in a reaction vessel under stirring and placed in a photoreactor equipped with green LEDs (λmax = 533 nm). The reaction was carried out in an open tube for 6 h. After completion of the reaction 10 ml of DCM was added. The organic layer was washed with water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether).Synthesis of product 6
Alkyne (1 mmol), thiol (1.5 mmol), tBuOK (92.5 mg, 0.5 mmol) and 0.5 ml of DMF were mixed in a reaction vessel under stirring. The reaction was carried out in a PTFE screw-capped tube at 70 °C for 24 h. After completion of the reaction 10 ml of DCM was added. The organic layer was washed with water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether).Product 7hb was obtained by column chromatography on Merck 60 μm silica gel (petroleum ether/DCM/triethylamine, 10:1:0.01) from the reaction mixture of 3hb.
Synthesis of product 4ob′
Thiol (2b) (0.3 mmol), Eosin Y (3 mg, 4.3 μmol), K2CO3 (40 mg) and 3 ml DMF were placed in a PTFE screw-capped tube under stirring. The solution was flushed under argon for 10 minutes. After this acetylene 1o (0.15 mmol) was added under the argon flow and the cap was closed hermetically. The reaction vessel was placed in a photoreactor equipped with green LEDs (W = 30 W, λmax = 533 nm) and stirring was conducted for 24 h. After completion of the reaction 10 ml of DCM was added. The organic layer was washed with water and brine and dried over MgSO4. The solvent layer was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether/ethyl acetate, 4:1).
Synthesis of product 4pb
Thiol (2b) (0.3 mmol), Eosin Y (3 mg, 4.3 μmol), K2CO3 (40 mg) and 3 ml DMF were placed in a PTFE screw-capped tube under stirring. The solution was flushed with argon for 10 minutes. After this o-(o-bromophenyl)phenylacetylene (1p) (0.15 mmol) was added under an argon flow and the cap was closed hermetically. The reaction was carried out in a photoreactor equipped with green LEDs (W = 30 W, λmax = 533 nm) and allowed to proceed for 24 h under stirring. After completion of the reaction 10 ml of DCM was added. The organic layer was washed with water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether/ethyl acetate, 5:1).
Synthesis of product 8na
Thiol (2a) (0.45 mmol), Eosin Y (6.5 mg, 0.001 mmol), K2CO3 (69 mg) and 3 ml of DMF were placed in a PTFE screw-capped tube under stirring. The solution was flushed with argon for 10 minutes. After this, o-bromophenylacetylene (1n) (0.15 mmol) was added under an argon flow and the cap was closed hermetically. The reaction was carried out in a photoreactor equipped with green LEDs (λmax = 533 nm) and allowed to proceed for 24 h under stirring. After completion of the reaction 10 ml of DCM was added. The organic layer was washed with water and brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on Merck 60 μm silica gel (petroleum ether).Monitoring of the photocatalytic thiol–yne click reaction by irradiation in the ionization chamber
Alkyne (1q) (11.6 mg, 30 μmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (10 μl, 66 μmol), 3 mg (4.3 μmol) of Eosin Y and 4 ml of DMF were mixed in a round-bottom flask, and arylthiol (2d) (7 μl, 62 μmol) was added. An aliquot of the reaction mixture was injected into the ESI ion source of a mass spectrometer. After 10 min the green laser (80 mW) was turned on and reaction monitoring was started after stabilization of the total ion current. The products were detected in negative ion mode as singly charged ions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Scientific Schools Development Program by Zelinsky Institute of organic chemistry is gratefully acknowledged. N. S. S. thanks BAYHOST for PhD scholarship, J. V. B. thanks Dr D. B. Eremin for helpful discussions.Notes and references
- E. S. Degtyareva, J. V. Burykina, A. N. Fakhrutdinov, E. G. Gordeev, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2015, 5(12), 7208–7213 CrossRef CAS
.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed
- A. Ogawa, T. Ikeda, K. Kimura and T. Hirao, J. Am. Chem. Soc., 1999, 121, 5108–5114 CrossRef CAS
-
A. Ishii and N. Nakata, in Hydrofunctionalization, ed. V. P. Ananikov and M. Tanaka, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 21–50 Search PubMed
- R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed
-
A. Ogawa, in Hydrofunctionalization, ed. V. P. Ananikov and M. Tanaka, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 325–360 Search PubMed
- N. V. Orlov, ChemistryOpen, 2015, 4, 682–697 CrossRef CAS PubMed
- R. Castarlenas, A. Di Giuseppe, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211–222 CrossRef CAS PubMed
- E. O. Pentsak, D. B. Eremin, E. G. Gordeev and V. P. Ananikov, ACS Catal., 2019, 9, 3070–3081 CrossRef CAS
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed
- A. S. Kashin and V. P. Ananikov, J. Org. Chem., 2013, 78, 11117–11125 CrossRef CAS PubMed
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC
- J. Zhu, W. C. Yang, X. D. Wang and L. Wu, Adv. Synth. Catal., 2018, 360, 386–400 CrossRef CAS
- S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Chem. Sci., 2016, 7, 6740–6745 RSC
- S. Kaur, G. Zhao, E. Busch and T. Wang, Org. Biomol. Chem., 2019, 17, 1955–1961 RSC
- H. Wang, Q. Lu, C. W. Chiang, Y. Luo, J. Zhou, G. Wang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 595–599 CrossRef CAS PubMed
- M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem., Int. Ed., 2019, 58, 5532–5550 CrossRef CAS PubMed
- C. J. Evoniuk, G. dos Passos Gomes, S. P. Hill, S. Fujita, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2017, 139, 16210–16221 CrossRef CAS PubMed
- I. V. Alabugin and M. Manoharan, J. Am. Chem. Soc., 2003, 125(15), 4495–4509 CrossRef CAS PubMed
- Q. Elliott, G. dos Passos Gomes, C. J. Evoniuk and I. V. Alabugin, Chem. Sci., 2020, 11, 6539–6555 RSC
- P. W. Peterson, N. Shevchenko, B. Breiner, M. Manoharan, F. Lufti, J. Delaune, M. Kingsley, K. Kovnir and I. V. Alabugin, J. Am. Chem. Soc., 2016, 138, 15617–15628 CrossRef CAS PubMed
- A. Kondoh, K. Takami, H. Yorimitsu and K. Oshima, J. Org. Chem., 2005, 70, 6468–6473 CrossRef CAS PubMed
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acenã, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS
- B. Janhsen, C. G. Daniliuc and A. Studer, Chem. Sci., 2017, 8, 3547–3553 RSC
- P. Camargo Solórzano, F. Brigante, A. B. Pierini and L. B. Jimenez, J. Org. Chem., 2018, 83, 7867–7877 CrossRef PubMed
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS PubMed
- A. S. Kashin, E. S. Degtyareva, D. B. Eremin and V. P. Ananikov, Nat. Commun., 2018, 9, 2936 CrossRef PubMed
- L. P. E. Yunker, R. L. Stoddard and J. S. McIndoe, J. Mass Spectrom., 2014, 49, 1–8 CrossRef CAS PubMed
- S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed., 2016, 55, 9345–9349 CrossRef CAS PubMed
- Y. Cai, J. Wang, Y. Zhang, Z. Li, D. Hu, N. Zheng and H. Chen, J. Am. Chem. Soc., 2017, 139, 12259–12266 CrossRef CAS PubMed
- J. Zelenka and J. Roithová, ChemBioChem, 2020, 21, 1–10 CrossRef
- J. S. McIndoe and K. L. Vikse, J. Mass Spectrom., 2019, 54, 466–479 CrossRef CAS PubMed
- A. Godsk Larsen, A. Hjarbæk Holm, M. Roberson and K. Daasbjerg, J. Am. Chem. Soc., 2001, 123, 1723–1729 CrossRef CAS PubMed
- A. D. Boese and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS
- T. Clark, J. Chandrasekhar and G. W. Spitznagel, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 115, 10291–10302 CrossRef CAS
- H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 114, 9758–9763 CrossRef CAS
- H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2002, 117, 8694–8704 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental data, copies of NMR spectra, DFT and ESI-MS data. See DOI: 10.1039/d0sc01939a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |