
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective aqueous C–H acylation of tyrosine-containing oligopeptides with aldehydes†
Marcos
San Segundo
 and
Arkaitz
Correa
*
and
Arkaitz
Correa
*
University of the Basque Country (UPV/EHU), Department of Organic Chemistry I, Joxe Mari Korta R&D Center, Avda. Tolosa 72, 20018 Donostia-San Sebastián, Spain. E-mail: arkaitz.correa@ehu.eus
First published on 6th October 2020
Abstract
The development of useful synthetic tools to label amino acids within a peptide framework for the ultimate modification of proteins in a late-stage fashion is a challenging task of utmost importance within chemical biology. Herein, we report the first Pd-catalyzed C–H acylation of a collection of Tyr-containing peptides with aldehydes. This water-compatible tagging technique is distinguished by its site-specificity, scalability and full tolerance of sensitive functional groups. Remarkably, it provides straightforward access to a high number of oligopeptides with altered side-chain topology including mimetics of endomorphin-2 and neuromedin N, thus illustrating its promising perspectives toward the diversification of structurally complex peptides and chemical ligation.
Introduction
Non-natural amino acids and peptides derived thereof are highly coveted compounds in proteomics and drug discovery due to their often enhanced biological activities and improved metabolic stability compared with their native analogues.1 As a result, there is an urgent demand to increase the available synthetic toolbox to perform chemical labelling processes of peptides and proteins in a late-stage fashion. Innovation is occurring at a rapid pace and a myriad of reliable methods have emerged within the last decade in the burgeoning area of bioconjugation.2 Metal catalysis has recently unlocked new tactics in the field,3 thereby enabling the development of a sheer number of metal-catalyzed modification techniques featuring the manipulation of otherwise unreactive C–H bonds embedded within the amino acid backbone4 and the corresponding side-chains.5 The latter have streamlined the straightforward assembly of biomolecules of paramount significance in a more sustainable manner by avoiding the use of pre-functionalized substrates. Despite the wealth of reports in the field, several challenges still remain: (a) most of the protocols entailed the diversification of a limited number of amino acid residues including tryptophan (Trp), glycine (Gly), alanine (Ala), cysteine (Cys) or phenylalanine (Phe), among others,6 and (b) toxic halide-counterparts and organic solvents are usually required. Accordingly, the selective tagging of other canonical amino acid residues while implementing atom-economical C–H coupling partners represents an unmet challenge of capital significance within peptide chemistry and protein engineering.Tyrosine (Tyr), the 4-hydroxylated congener of Phe, constitutes an abundant proteinogenic amino acid, which is a privileged core in a vast array of relevant compounds such as neurotransmitters and hormones, as well as a versatile precursor to numerous alkaloids with potent antibiotic activity such as vancomycin, among others (Fig. 1).7
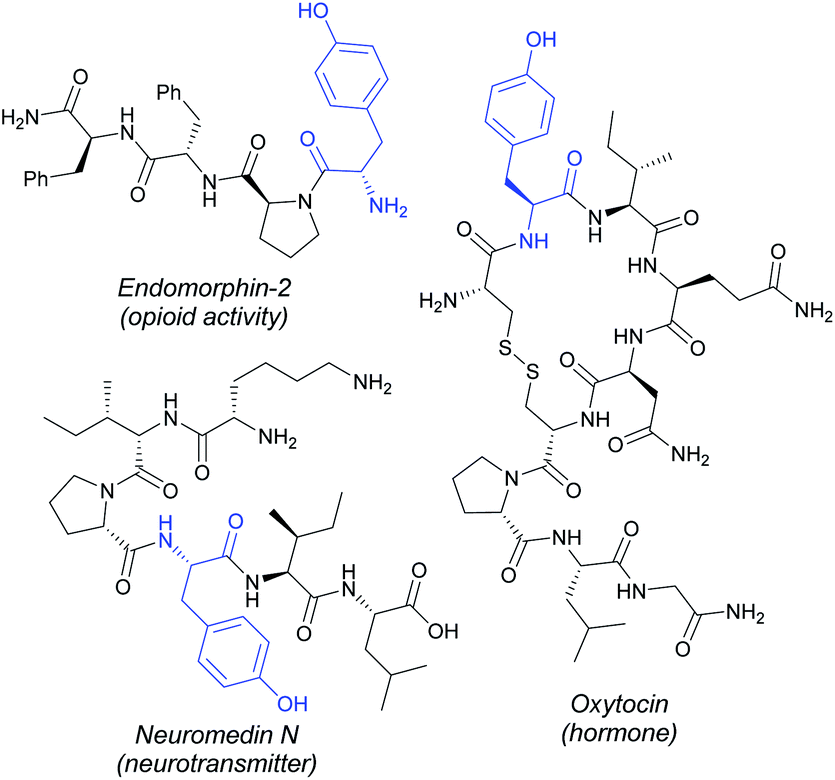 | ||
| Fig. 1 Illustrative Tyr-containing biologically relevant polypeptides. | ||
Likewise, Tyr-containing compounds are of widespread use as dietary supplements or food additives, and play a pivotal role in biological processes such as photosynthesis. Its inherent chemical reactivity is dictated by the pH-tunable and electron-rich phenol-containing side-chain. In this regard, several transformations performed in simple phenols have been translated into elegant tagging techniques of Tyr-containing peptides. Whereas O-functionalization reactions including arylation,8 alkylation9 and glycosylation10 reactions generally occur under basic conditions, C-targeted reactions at the ortho-C–H bond to the phenol motif usually proceed in acidic or neutral conditions. The introduction of highly reactive electrophiles upon Mannich-type reactions,11 diazonium couplings12 or ene-type reactions13 as well as N-centered radicals derived from phenothiazines14 has been achieved under metal-free reaction conditions. Conversely, metal catalysis has been crucial to install other coupling partners such as aryl halides,15 aryl trilfuoroborate salts,16 acrylates17 and the trifluoromethyl group18 (Scheme 1, route a). The latter have been rarely applied within a challenging peptide framework and hence the site-selective modification of tyrosine unit still remains elusive.
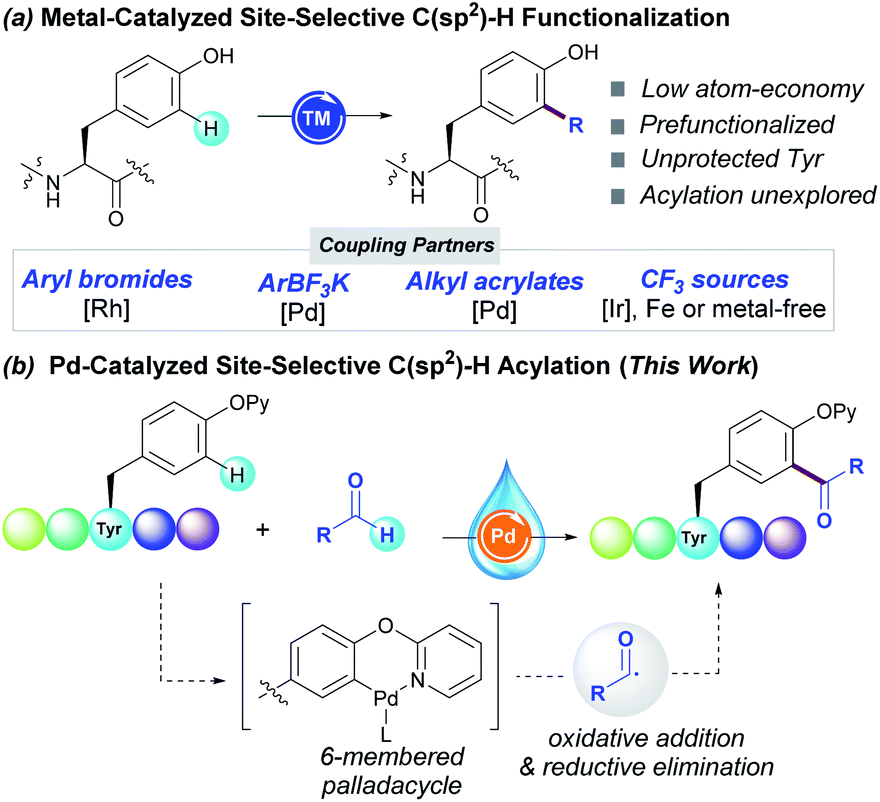 | ||
| Scheme 1 Ortho C–H functionalization of Tyr derivatives. | ||
As part of our interest in the radical modification of peptides,19 we have recently reported unprecedented Pd-catalyzed C–H acylation reactions for the efficient labelling of Phe-containing peptides.20 While conceptually innovative, the protocol suffered from certain downsides such as the requirement of stoichiometric amounts of silver salts to ensure high yields,21 the use of DMF as toxic organic solvent and it was found just applicable to Phe derivatives housing a picolinamide as directing group (DG) at the N-terminal position. Given that the use of aldehydes has been overlooked in the realm of bioconjugation, we sought to unveil their full synthetic potential in the virtually unexplored ortho-C–H acylation of Tyr residues within complex peptide settings. Building on precedents in C–H acylation reactions,22 we predicted that the use of a DG would be determinant for achieving site-selectivity23 through the formation of a 6-membered palladacycle prone to undergo further oxidative addition of the transient acyl radical species (Scheme 1, route b). In particular, we envisioned that the conversion of the native Tyr unit into the corresponding 2-pyridyl ether compound24 would be crucial to enable the site-selective modification of Tyr residues at any position within the peptide sequence. Herein we report on the first Pd-catalyzed C–H acylation of Tyr-containing peptides with aldehydes. This scalable method avoids the undesired use of toxic and expensive silver salts, and features the use of water as a non-flammable and environmentally-friendly solvent, thus resulting in a powerful diversification technique of paramount importance in peptide chemistry.
Results and discussion
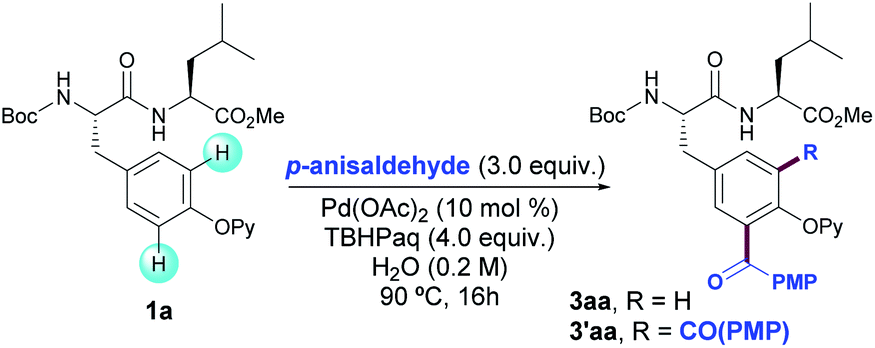
We commenced our studies by exploring the radical acylation of Boc-Tyr(OPy)-Leu-OMe (1a) with p-anisaldehyde (2a) as the model reaction. When submitting dipeptide 1a to the previously reported reaction conditions for the acylation of Phe-containing peptides20 just traces of 3aa were obtained, hence showing the subtleties of the modification of the Tyr scaffold. Initial exploratory solvent screening with tert-butyl hydroperoxide (TBHP) as oxidant showed the feasibility of our approach and moderate to good yields were obtained in solvents such as toluene, acetonitrile, chlorobenzene and even water (Table S1†).25 Driven by its clear benefits in the modification of biomolecules, we focused on the development of a practical acylation under an aqueous environment.26After considerable experimentation,25 we eventually found that the combination of Pd(OAc)2 (10 mol%), an aqueous solution of inexpensive TBHP (Luperox®) as oxidant in neat water as solvent at 90 °C provided 3aa in 78% yield as a mixture of mono- and diacylated products (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) (entry 1). Blank experiments underpinned the crucial role of both Pd catalyst and oxidant in the acylation reaction as not even traces of 3aa were detected in their absence (entries 2 and 3, respectively). The performance of the reaction under air resulted in lower yields of 3aa (entry 4) and the process was entirely inhibited under an oxygen atmosphere (entry 5). The use of variable amounts of oxidant and p-anisaldehyde led to an optimal balance between yield and mono-selectivity when using 4.0 and 3.0 equivalents, respectively. For example, when using 2.0 equivalents of TBHP, 3aa was obtained in a comparatively lower yield (entry 6) and when increasing the amount of aldehyde to 6.0 equivalents the higher yield was due to a loss in regioselectivity (entry 7). Other reaction parameters such as palladium source, supporting ligands and reaction temperature were analyzed but lower yields were obtained; indeed, higher selectivity toward the monoacylation product was only achieved at the expense of obtaining lower overall yields.25
:2 ratio) (entry 1). Blank experiments underpinned the crucial role of both Pd catalyst and oxidant in the acylation reaction as not even traces of 3aa were detected in their absence (entries 2 and 3, respectively). The performance of the reaction under air resulted in lower yields of 3aa (entry 4) and the process was entirely inhibited under an oxygen atmosphere (entry 5). The use of variable amounts of oxidant and p-anisaldehyde led to an optimal balance between yield and mono-selectivity when using 4.0 and 3.0 equivalents, respectively. For example, when using 2.0 equivalents of TBHP, 3aa was obtained in a comparatively lower yield (entry 6) and when increasing the amount of aldehyde to 6.0 equivalents the higher yield was due to a loss in regioselectivity (entry 7). Other reaction parameters such as palladium source, supporting ligands and reaction temperature were analyzed but lower yields were obtained; indeed, higher selectivity toward the monoacylation product was only achieved at the expense of obtaining lower overall yields.25
As depicted on Table 1, an aqueous solution of TBHP provided much better results than other related peroxides or persulfates (entries 8–12), which together with the use of water as solvent constitutes an additional bonus of the method in terms of economics and sustainability. Importantly, unlike other Pd-catalyzed C–H peptide modification reactions, silver additives were found to be unnecessary. Moreover, despite the highly oxidizing reaction system, undesired N-acylation of the peptide backbone with p-anisaldehyde was never observed.27
|
|
||
|---|---|---|
| Entry | Change from standard conditions | 3aa (%) |
|
a Reaction conditions: 1a (0.15 mmol), 2a (0.45 mmol), Pd(OAc)2 (10 mol%), TBHPaq (4.0 equiv.) in H2O (0.75 mL) at 90 °C for 16 h under Ar.
b Yield of isolated product after column chromatography.
c Ratio of mono- and diacylated product (3aa:3′aa). DTBP = di-tert-butyl peroxide; DCP = dicumyl peroxide; TBHPaq (Luperox®, 70 wt% in water).
|
||
| 1 | None | 78 (8:2)c |
| 2 | Without Pd(OA)2 | 0 |
| 3 | Without TBHPaq | 0 |
| 4 | Under air | 46 (93:7) |
| 5 | Under O2 | 0 |
| 6 | With 2.0 equiv. of TBHPaq | 65 (93:7)c |
| 7 | 6.0 equiv. of 2a | 88 (6:4)c |
| 8 | (NH4)2S2O8 instead of TBHPaq | Traces |
| 9 | DTBP instead of TBHPaq | Traces |
| 10 | H2O2 instead of TBHPaq | Traces |
| 11 | t BuOOBz instead of TBHPaq | 33 (7:3)c |
| 12 | DCP instead of TBHPaq | Traces |
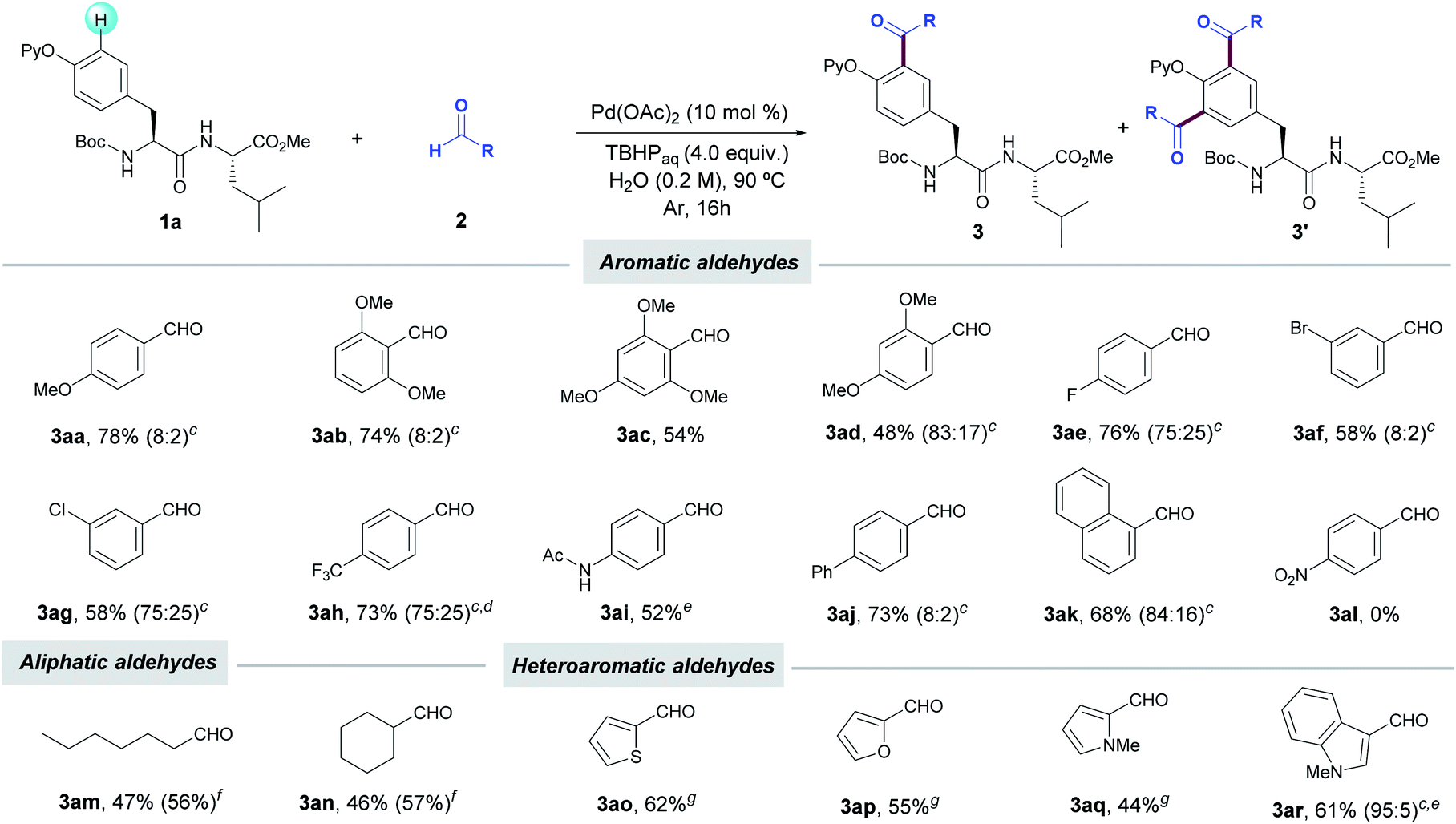
With the optimized conditions in hand, we next investigated the scope of the C(sp2)–H acylation protocol with Tyr-containing dipeptide 1a. Notably, a wide variety of electronically diverse aldehydes smoothly underwent the target oxidative coupling, thus enabling the rapid access to a variety of unknown acylated Tyr-containing dipeptides in a late-stage manner. In general, a variety of benzaldehydes regardless of their electronic nature provided the corresponding acylated products 3 in good yields as mixtures of mono- and diacylated compounds, with a preferential selectivity toward the monoacylated product (up to 8:2 ratio). The method was compatible with the presence of ethers (3aa–ad), halides (3ae–ah), acetamide (3ai) and naphthyl system (3ak). Conversely, the highly electron-withdrawing nitro group resulted in no conversion of dipeptide 1a. Interestingly, the lower tendency to oxidation of aliphatic aldehydes such as heptanal and cyclohexanecarboxaldehyde ushered in the exclusive formation of mono-acylated products 3am and 3an, respectively, in moderate yields under the standard reaction conditions featuring the use of water as the sole solvent. However, the performance of the process in chlorobenzene with a higher excess of the corresponding aldehydes resulted in slightly higher yields (up to 57%). Likewise, other challenging aldehydes housing pharmaceutically relevant heterocyclic scaffolds could be also employed as reaction partners, although the use of toluene as solvent at 100 °C was found determinant for the process to occur.25 Accordingly, 2-thiophene (2o), 2-furan (2p), N-methyl-2-pyrrole (2m) and N-methyl-3-indolyl carboxaldehydes (2n) selectively afforded the corresponding monoacetylated products 3ao–ar. It is noteworthy that a gram-scale acylation with p-(trifluoromethyl)benzaldehyde (2h) was successfully performed, which verified the robustness and synthetic utility of our peptide tagging manifold. Collectively, the aqueous acylation of Tyr derivatives reported herein underscores related protocols on simple 2-phenoxypyridine derivatives, which occurred at higher reaction temperatures (up to 140 °C), in chlorinated solvents and are restricted to the use of benzaldehydes (Table 2).24b,g
|
a As for Table 1, entry 1.
b Yield of isolated product after column chromatography, average of at least two independent runs.
c Ratio of mono- and diacylated product (3:3′).
d Gram scale experiment.
e Reaction performed in toluene.
f Reaction performed in PhCl with 5.0 equiv. of aldehyde.
g Reaction performed in toluene at 100 °C with 5.0 equiv. of aldehyde.
|
|---|
|
We next explored the synthetic scope in the challenging setting of short-to-medium size peptides (Table 3). Notably, peptides bearing Phe (1b), Val (1c), Gly (1d), Asn (1e), Ser (1f), Asp (1g), non-protected Tyr (1h), Pro (1i), Ala (1j), and Lys (1k) boded well with the reaction conditions and provided the corresponding acylated dipeptides (3b–k) in good yields. The success of the method did not rely on a specific situation of the Tyr along the peptide sequence, and was applicable to Tyr residues located both at the N- and C-terminal positions.
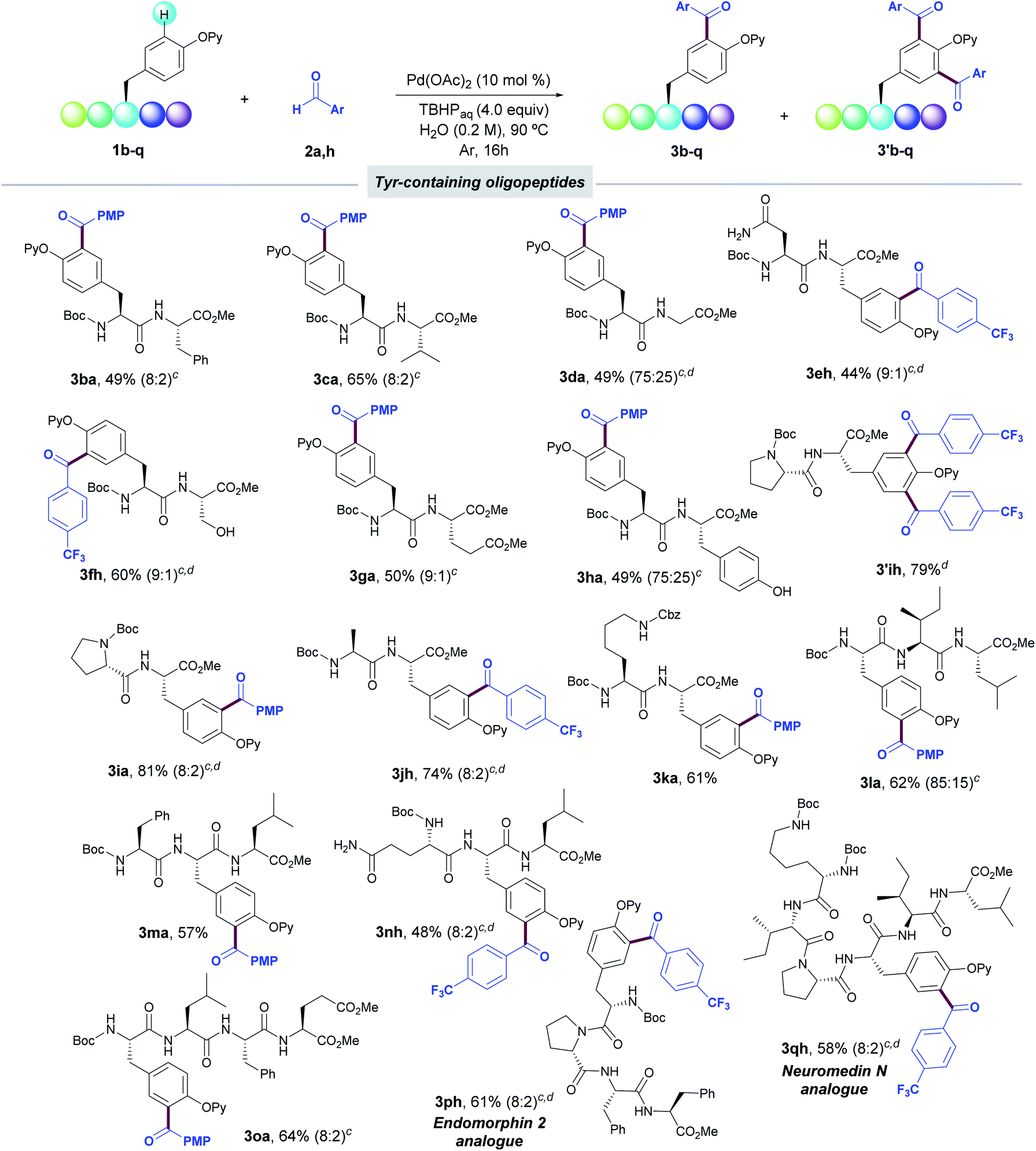
We next evaluated the viability of the Pd-catalyzed acylation for the late-stage diversification of more complex oligopeptides. In this respect, the process efficiently occurred in a selective manner in Tyr-containing tri- and tetrapeptides (1l–o), including even peptides housing the Tyr unit in inner positions (1m,n). Importantly, pentapeptide 1p and hexapeptide 1q bearing the amino acid sequence of biologically relevant Endomorphin-2 and Neuromedin N, respectively, were also acylated in a late-stage fashion. The latter examples illustrate the high synthetic potential of this acylation technique toward the site-selective labelling of biomolecules of high structural complexity. In all cases, the incorporation of the radical acyl species was biased by the 2-pyridyloxy group attached to the phenol ring within the Tyr residue and other sensitive functional groups such as carboxamides within Gln (3nh) and Asn (3eh) as well as oxidizable protic free-hydroxyl groups of Ser (3fh) and Tyr (3ha) remained intact under the reaction conditions. The free-amino group of Lys residue (3ka, 3qh) as well as the N-terminal residue of the peptide sequence was conveniently protected to avoid undesired oxidative aminations with the corresponding aldehyde.28 Peptides bearing other electron-rich aromatic residues such as His and Trp or guanidine-containing Arg unit were not tolerated.25 Likewise, thiol-containing Cys and Met residues were not accommodated, which could be due to competitive oxidative reactions with the corresponding aldehydes (Table S8, ESI†).29
With the aim to create molecular diversity, we extended the scope beyond the introduction of one single aldehyde into the Tyr unit, and found that the performance of two consecutive aroylation reactions with two distinct benzaldehydes enabled the assembly of fully decorated Tyr-containing dipeptide 3as in a selective manner (Scheme 2).
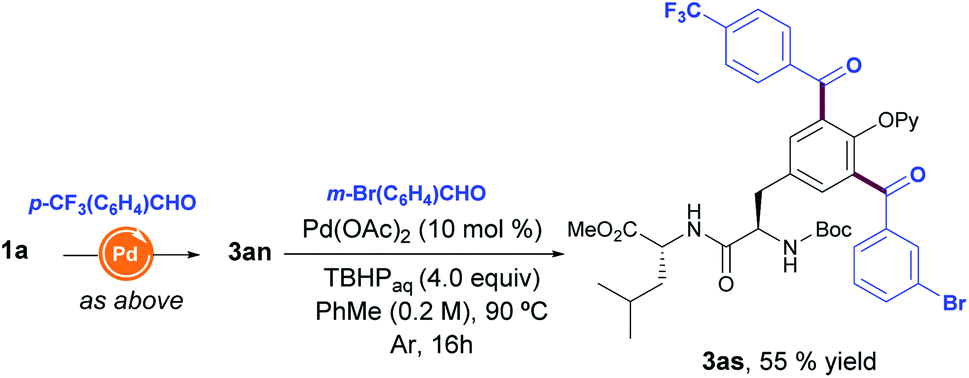 | ||
| Scheme 2 Consecutive C(sp2)–H aroylation reaction. | ||
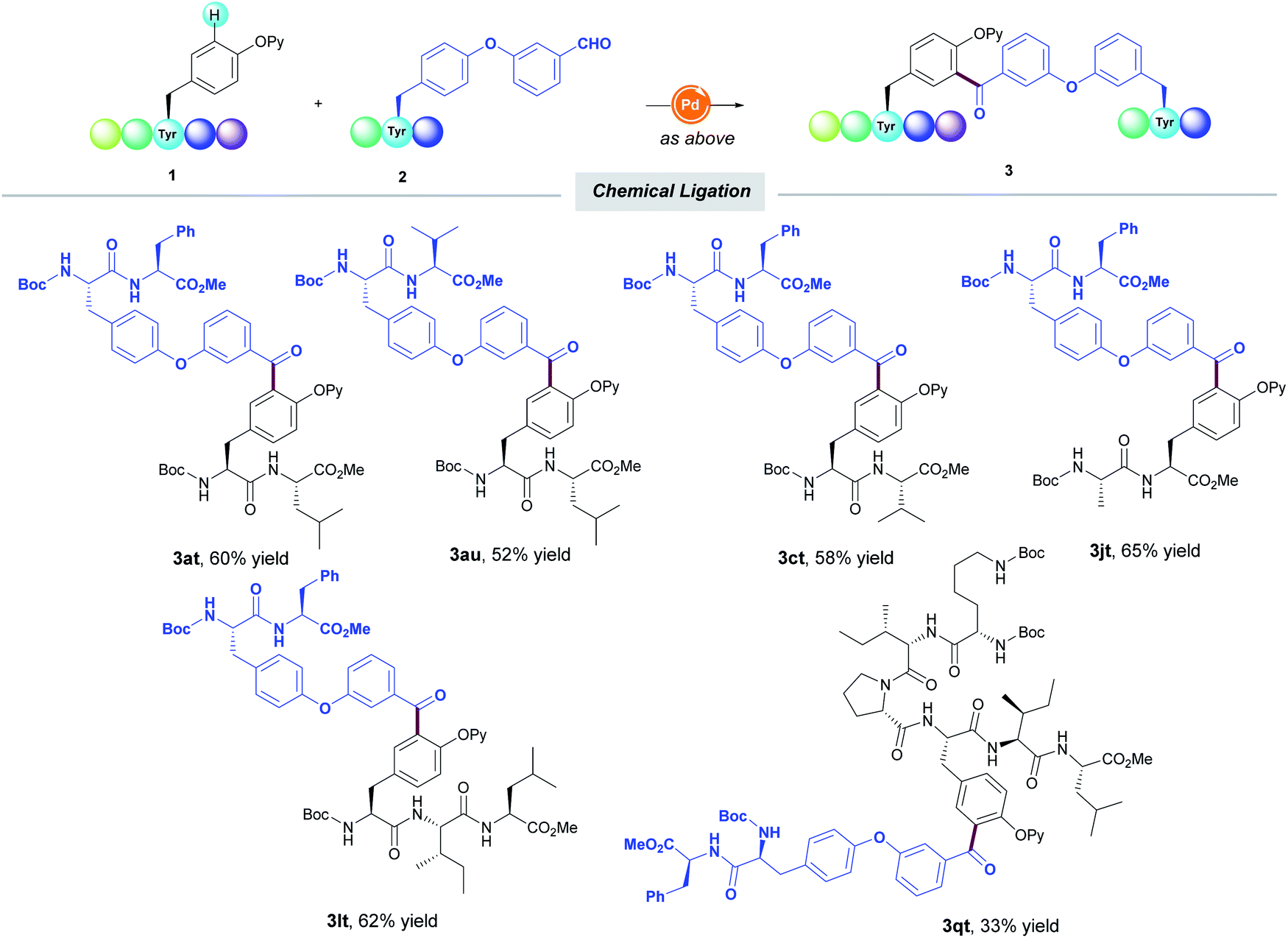
Likewise, the aldehyde unit could be tethered within a Tyr-containing peptide and efficiently coupled with another Tyr-containing short-to-medium peptide to deliver unprecedented oligopeptides featuring a unique diaryl ketone cross-linking (Table 4). Notably, the functionalization always occurred in a selective fashion toward the mono-acylated compounds. The practicality of the method within the realm of chemical ligation was verified by the selective mono-acylation of Neuromedin N analogue 1q, thereby enabling the assembly of octapeptide 3qt of high structural complexity. It must be noted that all the experiments were run at least twice with a variable yield by no more than 5% between runs, thus showing the reliability of the protocol.
Although the facile removal of the OPy group has been customarily described in simple aryl systems upon a two-step sequence entailing N-methylation with methyl triflate followed by the cleavage of the resulting C(pyridinium)–O bond by treatment with an alcoholic solution of sodium,24 its application in a peptide sequence resulted in mixtures of products and racemization of the existing chiral centers. Accordingly, we studied the use of modern metal-catalyzed borylation reactions for the ultimate, yet milder removal of the directing group.30 After careful evaluation of a number of Rh-, Ni- and Fe-catalyzed borylation reactions, the targeted borylative cleavage was never achieved within our Tyr-containing peptides and the removal of the OPy group occurred in moderate yield to produce the corresponding reduced product 4.30b Although it poses a limitation at first sight and remains an issue to be improved, further optimization of the process could provide a complementary avenue for the assembly of meta-aroylated Phe-containing peptides, thereby resulting in the direct conversion of a Tyr residue into the Phe analogue featuring the use of the OPy as a traceless directing group (Scheme 3). Likewise, the fully decorated Tyr(OPy)-containing peptides assembled herein could offer interesting possibilities within drug discovery.
 | ||
| Scheme 3 Reductive cleavage of the DG. | ||
Conclusions
In summary, we have developed a broadly applicable method for the site-selective tagging of Tyr-containing oligopeptides featuring a novel Pd-catalyzed C(sp2)–H acylation reaction with abundant aldehydes. This labelling platform represents a reliable, yet innovative, means for the radical diversification of a wide variety of Tyr-containing compounds, thus providing access to novel peptidomimetics of high structural complexity. Although one might anticipate that site-selectivity issues might come into play in the presence of multiple C–H bonds, our work meets this challenge, offering an unrecognized opportunity within the radical labelling of peptides. Salient features of our protocol are the compatibility with an aqueous environment, the widespread availability and low-cost of the aldehydes, the broad functional group tolerance, the preferential site-selectivity toward the functionalization of the Tyr unit and the facile installation and removal of the required 2-pyridyl ether group. Accordingly, we anticipate that this Pd-catalyzed oxidative acylation manifold could become a useful synthetic tool for the late-stage and rapid modification of a virtually unlimited set of Tyr-containing lead drug candidates.Conflicts of interest
A patent disclosing the Pd-catalyzed aqueous acylation of Tyr-containing oligopetides has been filed (P202030131, Spain).Acknowledgements
We are grateful to Ministerio de Ciencia e Innovación (RTI2018-093721-B-I00, MCI/AEI/FEDER, UE) and Basque Government (IT1033-16) for financial support. We thank for technical and human support provided by Central Service of Analysis-SGIker of UPV/EHU and European funding (ERDF and ESF).Notes and references
- (a) E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262 RSC; (b) R. J. Malonis, J. R. Lai and O. Vergnolle, Chem. Rev., 2020, 120, 3210 CrossRef CAS; (c) J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700 CrossRef CAS; (d) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382 CrossRef CAS.
- (a) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS; (b) N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 102 CrossRef; (c) O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495 RSC; (d) M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896 CrossRef CAS.
- (a) J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., Int. Ed., 2019, 58, 6176 CrossRef CAS; (b) P. G. Isenegger and B. G. Davis, J. Am. Chem. Soc., 2019, 141, 8005 CAS; (c) M. Jbara, S. K. Maity and A. Brik, Angew. Chem., Int. Ed., 2017, 56, 10644 CrossRef CAS.
- (a) M. San Segundo and A. Correa, Synthesis, 2018, 50, 2853 CrossRef CAS; (b) T. Brandhofer and O. García Mancheño, Eur. J. Org. Chem., 2018, 6050 CrossRef CAS.
- For recent reviews, see: (a) I. Guerrero and A. Correa, Asian J. Org. Chem., 2020, 9, 898 CrossRef CAS; (b) H. Gruß and N. Sewald, Chem.–Eur. J., 2020, 26, 5328 CrossRef; (c) D. G. Rivera, G. M. Ojeda-Carralero, L. Reguera and E. V. Van der Eycken, Chem. Soc. Rev., 2020, 49, 2039 RSC; (d) Z. Bai and H. Wang, Synlett, 2020, 31, 199 CrossRef CAS; (e) C. Bottecchia and T. Noël, Chem.–Eur. J., 2019, 25, 26 CrossRef CAS; (f) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700 CrossRef CAS; (g) L. R. Malins, Pept. Sci., 2018, 110, e24049 CrossRef; (h) X. Lu, S.-J. He, W.-M. Cheng and J. Shi, Chin. Chem. Lett., 2018, 29, 1001 CrossRef CAS; (i) S. Mondal and S. Chowdhury, Adv. Synth. Catal., 2018, 360, 1884 CrossRef CAS; (j) L. R. Malins, Curr. Opin. Chem. Biol., 2018, 46, 25 CrossRef CAS; (k) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS; (l) A. M. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1 CrossRef CAS; (m) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS.
- For a selection of recent examples, see: (a) L. Liu, Y.-H. Liu and B.-F. Shi, Chem. Sci., 2020, 11, 290 RSC; (b) J. Peng, C. Li, M. Khamrakulov, J. Wang and H. Liu, Org. Lett., 2020, 22, 1535 CrossRef CAS; (c) X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230 CrossRef CAS; (d) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633 CrossRef CAS; (e) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476 CrossRef CAS; (f) M. J. Terrey, C. C. Perry and W. B. Cross, Org. Lett., 2019, 21, 104 CrossRef; (g) B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858 CrossRef CAS; (h) W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10554 CrossRef CAS; (i) Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, J. Am. Chem. Soc., 2018, 140, 6797 CrossRef CAS; (j) M. Bauer, W. Wang, M. M. Lorion, C. Dong and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 203 CrossRef CAS; (k) T. Liu, J. X. Qiao, M. A. Poss and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10924 CrossRef CAS; (l) A. F. M. Noisier, J. García, I. A. Ionut and F. Albericio, Angew. Chem., Int. Ed., 2017, 56, 314 CrossRef CAS; (m) G. Liao, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, Nat. Commun., 2016, 7, 12901 CrossRef; (n) K. Li, Q. Wu, J. Lan and J. You, Nat. Commun., 2015, 6, 8404 CrossRef CAS; (o) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS.
- (a) J. Lee, M. Ju, O. H. Cho, Y. Kim and K. Tae, Adv. Sci., 2019, 6, 1801255 CrossRef; (b) C. A. Schenck and H. A. Maeda, Phytochemistry, 2018, 149, 82 CrossRef CAS; (c) F. Albericio and H. G. Kruger, Future Med. Chem., 2012, 4, 1527 CrossRef CAS.
- K. L. Seim, A. C. Obermeyer and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 16970 CrossRef CAS.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080 CrossRef CAS.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644 CrossRef CAS.
- (a) D. W. Romanini and M. B. Francis, Bioconjugate Chem., 2008, 19, 153 CrossRef CAS; (b) J. M: McFarland, N. S. Joshi and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 7639 CrossRef; (c) N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942 CrossRef CAS.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406 CrossRef CAS.
- H. Ban, J. Gavrilyuk and C. F. Barbas III, J. Am. Chem. Soc., 2010, 132, 1523 CrossRef CAS.
- C. Song, K. Liu, Z. Wang, B. Ding, S. Wang, Y. Weng, C.-W. Chiang and A. Lei, Chem. Sci., 2019, 10, 7982 RSC.
- (a) R. B. Bedford, M. F. Haddow, R. L. Webster and C. J. Mitchell, Org. Biomol. Chem., 2009, 7, 3119 RSC . For an oxidative cross-coupling between two tyrosine residues, see:; (b) M. Ben-Lulu, E. Gaster, A. Libman and D. Pappo, Angew. Chem., Int. Ed., 2020, 59, 4835 CrossRef CAS.
- (a) M. Vilaró, G. Arsequell, G. Valencia, A. Ballesteros and J. Barluenga, Org. Lett., 2008, 10, 3243 CrossRef; (b) N. Basse, S. Piguel, D. Papapostolou, A. Ferrier-Berthelot, N. Richy, M. Pagano, P. Sarthou, J. Sobczak-Thépot, M. Reboud-Ravaux and J. Vidal, J. Med. Chem., 2007, 50, 2842 CrossRef CAS.
- (a) Q.-L. Hu, K.-Q. Hou, J. Li, Y. Ge, Z.-D. Song, A. S. C. Chan and X.-F. Xiong, Chem. Sci., 2020, 11, 6070 RSC; (b) Y. Dou, L. J. Kenry, J. Jiang and Q. Zhu, Chem.–Eur. J., 2019, 25, 6896 CrossRef CAS.
- (a) C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180 CrossRef CAS; (b) N. Ichiishi, J. P. Caldwell, M. Lin, W. Zhong, X. Zhu, E. Streckfuss, H.-Y. Kim, C. A. Parish and S. W. Krska, Chem. Sci., 2018, 9, 4168 RSC; (c) K. L. Kirk, M. Nishida, S. Fujii and H. Kimoto, J. Fluorine Chem., 1992, 59, 197 CrossRef CAS.
- (a) I. Guerrero and A. Correa, Org. Lett., 2020, 22, 1754 CrossRef CAS; (b) M. San Segundo and A. Correa, ChemSusChem, 2018, 11, 3893 CrossRef CAS; (c) M. San Segundo, I. Guerrero and A. Correa, Org. Lett., 2017, 19, 5288 CrossRef CAS.
- M. San Segundo and A. Correa, Chem. Sci., 2019, 10, 8872 RSC.
- B. Bhaskararao, S. Singh, M. Anand, P. Verma, P. Prakash, A. C, S. Malakar, H. F. Schaefer and R. B. Sunoj, Chem. Sci., 2020, 11, 208 RSC.
- For selected reviews, see: (a) C. Santiago, N. Sotomayor and E. Lete, Molecules, 2020, 25, 3247 CrossRef CAS; (b) W.-C. Yang, J.-G. Feng, L. Wu and Y.-Q. Zhang, Adv. Synth. Catal., 2019, 361, 1700 CrossRef CAS; (c) A. Banerjee, Z. Lei and M.-Y. Nga, Synthesis, 2019, 51, 303 CrossRef CAS; (d) X.-F. Wu, Chem.–Eur. J., 2015, 21, 12252 CrossRef CAS; (e) A. Batra, P. Singh and K. N. Singh, Eur. J. Org. Chem., 2016, 4927 CrossRef CAS; (f) C. Pan, X. Jia and J. Cheng, Synthesis, 2012, 44, 677 CrossRef CAS.
- For selected reviews on the use of DGs in C–H functionalization, see: (a) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS; (b) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, T. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (c) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS.
- For selected C–H functionalization reactions directed by the 2-pyridyl ether unit on simple phenyl systems, see: (a) S.-J. Lou, Q. Chen, Y.-F. Wang, D.-Q. Xu, X.-H. Du, J.-Q. He, Y.-J. Mao and Z.-Y. Xu, ACS Catal., 2015, 5, 2846 CrossRef CAS; (b) J.-H. Chu, S.-T. Chen, M.-F. Chiang and M.-J. Wu, Organometallics, 2015, 34, 953 CrossRef CAS; (c) B. Liu, H.-Z. Jiang and B.-F. Shi, J. Org. Chem., 2014, 79, 1521 CrossRef CAS; (d) J. Yao, R. Feng, Z. Wu, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2013, 355, 1517 CrossRef CAS; (e) W. Ma and L. Ackermann, Chem.–Eur. J., 2013, 19, 13925 CrossRef CAS; (f) L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154 CrossRef CAS; (g) X. Jia, S. Zhang, W. Wang, F. Luo and J. Cheng, Org. Lett., 2009, 11, 3120 CrossRef CAS.
- For more details, see the ESI.†.
- (a) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC; (b) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS; (c) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS.
- J. Wang, C. Liu, J. Yuan and A. Lei, Chem. Commun., 2014, 50, 4736 RSC.
- (a) Y. Deng, J. Peng, F. Xiong, Y. Song, Y. Zhou, J. Zhang, F. S. Lam, C. Xie, W. Shen, Y. Huang, L. Meng and X. Li, Angew. Chem., Int. Ed., 2020, 59, 14965 CrossRef CAS; (b) W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064 CrossRef CAS.
- C.-L. Yi, Y.-T. Huang and C.-F. Lee, Green Chem., 2013, 15, 2476 RSC.
- (a) X. Zeng, Y. Zhang, Z. Liu, S. Geng, Y. He and Z. Geng, Org. Lett., 2020, 22, 2950 CrossRef CAS; (b) M. Tobisu, J. Zhao, H. Kinuta, T. Furukawa, T. Igarashi and N. Chatani, Adv. Synth. Catal., 2016, 358, 2417 CrossRef CAS; (c) H. Kinuta, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 1593 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, data for new compounds, 1H, 13C and 19F NMR spectra. See DOI: 10.1039/d0sc03791e |
| This journal is © The Royal Society of Chemistry 2020 |