
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFungal siderophore biosynthesis catalysed by an iterative nonribosomal peptide synthetase†
Yang
Hai‡
 a,
Matthew
Jenner
*bc and
Yi
Tang
*ad
a,
Matthew
Jenner
*bc and
Yi
Tang
*ad
aDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, California 90095, USA. E-mail: yitang@ucla.edu
bDepartment of Chemistry, Warwick Integrative Synthetic Biology Center, University of Warwick, Coventry, UK. E-mail: M.Jenner@warwick.ac.uk
cWarwick Integrative Synthetic Biology (WISB) Centre, University of Warwick, Coventry, UK
dDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA
First published on 28th September 2020
Abstract
Siderophores play a vital role in the viability of fungi and are essential for the virulence of many pathogenic fungal species. Despite their importance in fungal physiology and pathogenesis, the programming rule of siderophore assembly by fungal nonribosomal peptide synthetases (NRPSs) remains unresolved. Here, we report the characterization of the bimodular fungal NRPS, SidD, responsible for construction of the extracellular siderophore fusarinine C. The use of intact protein mass spectrometry, together with in vitro biochemical assays of native and dissected enzymes, provided snapshots of individual biosynthetic steps during NPRS catalysis. The adenylation and condensation domain of SidD can iteratively load and condense the amino acid building block cis-AMHO, respectively, to synthesize fusarinine C. Our study showcases the iterative programming features of fungal siderophore-producing NRPSs.
Introduction
Iron is an essential element to life, but its bioavailability is very limited owing to the poor solubility of ferric iron in aerobic environments. Hence, virtually all living organisms have evolved sophisticated mechanisms for iron acquisition and storage.1,2 Siderophores are low-molecular-weight, high-affinity iron chelators and play a central role in maintaining iron homeostasis in bacteria, fungi, and plants.3 Two major hydroxamate-based peptidyl siderophores are employed by fungi: the depsipeptides, exemplified by fusarinine C (FSC, 1) and triacetylfusarinine C (TAFC, 2),4 and the coprogen family of siderophores (Fig. 1A).5 These compounds are excreted primarily to capture ferric iron, assisting iron uptake. In contrast, the intracellular siderophores, represented by the ferrichrome family, are primarily used for iron storage although some fungal species have been observed to secrete such siderophores (Fig. 1A).6 Both extra- and intracellular siderophores are indispensable for the virulence of many notorious pathogenic fungal species, such as rice blast fungus Magnaporthe oryzae and the human opportunistic pathogen Aspergillus fumigatus.7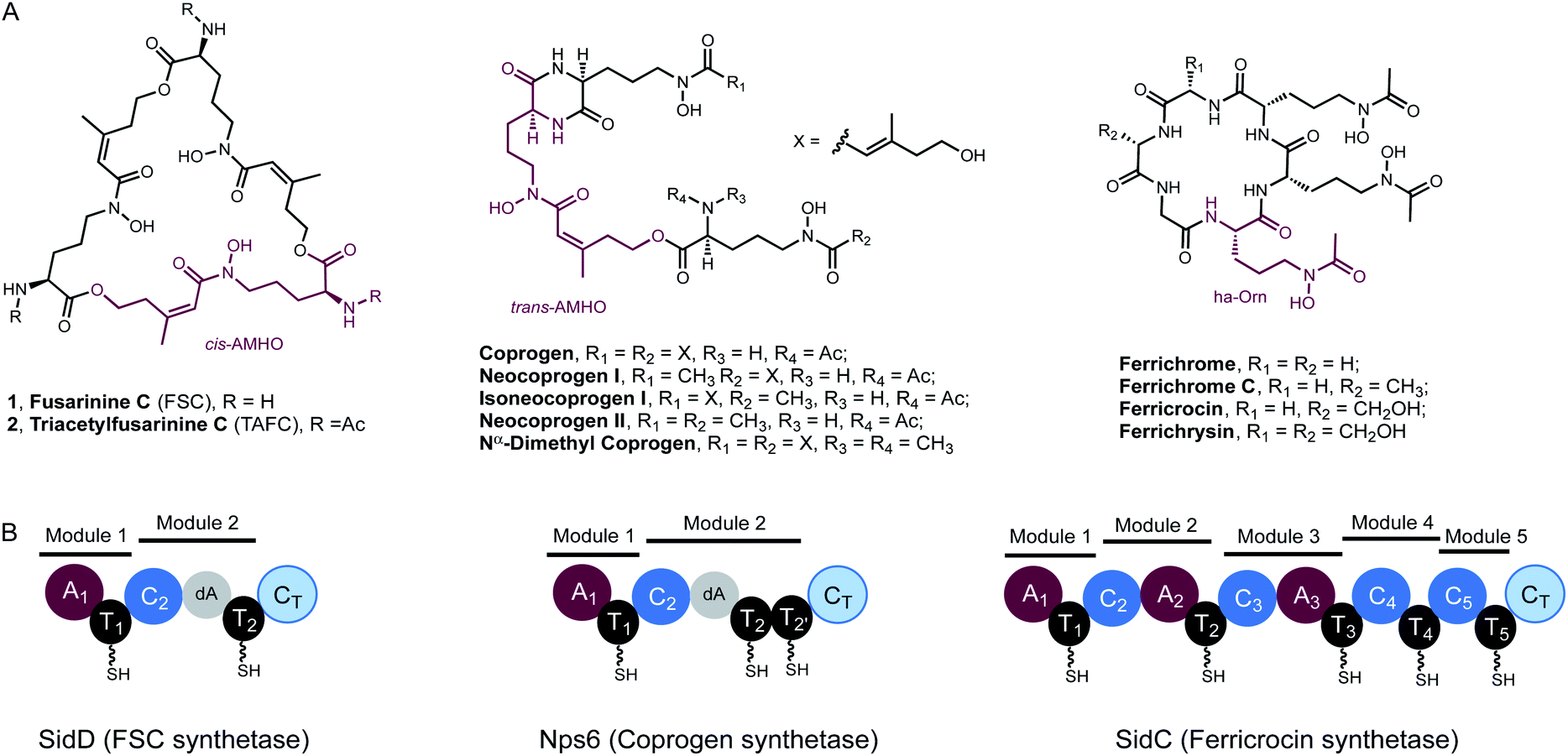 | ||
| Fig. 1 Fungal hydroxamate siderophores and the domain organisation of NRPSs responsible for their biosynthesis. (A) Structures of extra- and intra-cellular siderophores shown in the desferri-form. In each case, a repeating hydroxamate unit has been highlighted in purple. TAFC and ferricrocin possess Fe3+ binding constants of 1032 and 1027, respectively. (B) NRPS domain organisation of SidD, Nps6 and SidC (left to right). In SidD and Nps6, the dA notation represents a degenerated A domain. In SidC, module 4 and module 5 completely lack A domains. | ||
Despite the preponderance of siderophores in fungal physiology and pathogenesis, the enzymes involved in fungal siderophore biosynthesis remain largely uncharacterized, in particular the core modular nonribosomal peptide synthetases (NRPS) that are essential in assembling peptidyl siderophores.8,9 Representative examples of these NRPSs include the FSC synthetase, SidD, the coprogen synthetase, Nps6, and the ferricrocin synthetase, SidC (Fig. 1).10–12
Features unique to these NRPSs can be readily identified from their characteristic domain organizations. Firstly, they typically have incomplete C-terminal modules missing a functional adenylation (A) domain, which suggests the thiolation (T) domain in these modules must be aminoacylated by A domains from the upstream modules. Specifically, in both SidD and Nps6, module 2 harbours degenerate A (dA) domains which are truncated at their N-termini by nearly 290 amino acid residues and are predicted to be catalytically inactive.13 These dA domains are likely vestiges of evolution and only play a structural role within the NRPS. In comparison, module 4 and 5 in SidC have evolved to completely lack A domains. A second defining feature is the number and order of modules in these siderophore-producing NRPSs do not match the length and sequence of their peptide siderophore products, indicating a high degree of non-colinearity.13,14 These features distinguish them from the well-characterized fungal iterative cyclodepsipeptide (CDP) NRPSs,15,16 all of which harbour complete modules and assemble depsipeptide antibiotics (e.g. enniatin) consisting of repeated dipeptide units.
To understand the molecular mechanism of this unique family of NRPSs, we focused on the elucidation of SidD-catalysed biosynthesis of FSC. Here, employing a combination of in vitro biochemical assays and intact protein mass spectrometry (MS), we show how the bimodular NRPS, SidD, is precisely programmed to iteratively assemble the trimeric siderophore FSC.
Results and discussions
To decipher the mechanism of SidD, we purified recombinant SidD and its mutants (designed to dissect the domain function, vide infra) from Saccharomyces cerevisiae JHY686 strain (Fig. S1†).17 To ensure protein samples were all in the holo-form, purified SidD variants were enzymatically phosphopantetheinylated using the phosphopantetheinyl transferases NpgA (A. nidulans) or Sfp (B. subtilis), as described previously.18–21 The substrate, N5-cis-anhydromevalonyl-N5-hydroxy-L-ornithine (cis-AMHO) was prepared by alkaline hydrolysis of 1.22 We isolated 1 from an A. nidulans mutant in which the acetyltransferase gene sidG was knocked out to prevent acetylation of 1 to give 2in vivo (ESI methods†).12 In the presence of ATP and Mg2+, wild-type SidD could synthesize desferri-1 utilizing cis-AMHO as building blocks (Fig. 2A), while ferri-1 was observed when Fe3+ was present in the substrate solution (prepared from hydrolysis of ferri-FSC without removing Fe3+ ions). The product identity was confirmed by comparison to a chemical standard of ferri-1 on LC-MS (Fig. 2B). The reaction has an apparent KM of 120 μM for cis-AMHO and an overall kcat of 0.5 s−1 (Fig. 2C). Trace amounts of linear-FSC (3) were observed in both the authentic standard and the reaction mixture. This is likely the result of slow non-enzymatic hydrolysis of the ester bond in 1 under the assay conditions (phosphate buffer, pH 7.8), or in the reaction mixture, where additional linear-FSC (3) could arise from both ester hydrolysis or non-enzymatic thioester hydrolysis of the cis-AMHO trimer appended to the SidD protein.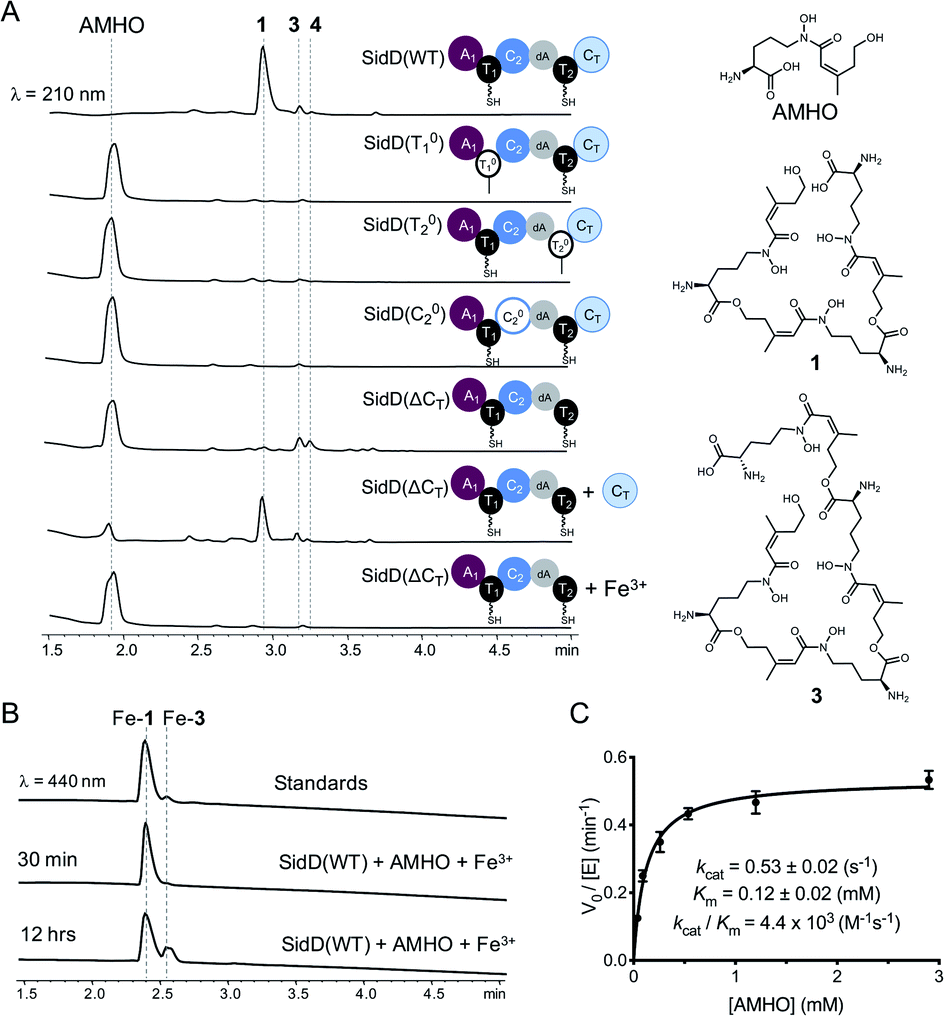 | ||
| Fig. 2 Comparison of in vitro enzymatic assays for wild-type SidD and mutational variants. (A) HPLC traces of the enzymatic reactions for different SidD variants. The domain architectures of the variants are displayed. Mass spectral data of each peak are shown in Fig. S2–S9.† (B) Comparison of HPLC traces for the enzymatic products (in the ferri-form) with chemical standards. (C) Steady-state kinetics of wild-type SidD. | ||
Successful reconstitution of SidD activity in vitro allowed us to further dissect its domain function. Both thiolation (T) domains are essential for SidD activity, as disabling either T domain by mutating the key serine that serves as the phosphopantetheinyl group attachment site (S801A and S1594A, designated as T01 and T02, respectively), completely abolished FSC formation. Inactivating the condensation (C) domain from module 2 by mutating the catalytic histidine residue (H999A, designated as C02) also resulted in no product formation. Interestingly, deletion of the C-terminal condensation domain (ΔCT) yielded two shunt products: linear-FSC (3) and a new compound, 4, whose molecular weight, retention time, and ability to chelate Fe3+ suggested a linear tetrameric derivative of FSC (Fig. 2A and S2–S9†). Addition of equimolar standalone CT domain restored FSC synthetase activity of SidD(ΔCT) mutant, albeit with lower efficiency compared to wild-type. These results together indicate that cyclization, but not the chain-elongation reaction, is compromised by the ΔCT truncation mutant, whereas chain-elongation is prevented by the C02 mutant. This result is consistent with the general programming rule that, CT domains found in fungal NRPS usually offload the peptidyl products through cyclization.23 This is in contrast to the role played by CT domains in CDP NRPSs, which have been shown to catalyse chain elongations in addition to cyclizations.15,16
To obtain further insights into the function of C2 and CT in SidD, we prepared cis-AMHO-derived aminoacyl-N-acetylcysteamine thioester substrates and assayed individual C domain activity in vitro.24 However, this approach was impeded by the instability of cis-AMHO and its derivatives: the cis-AMHO moiety is prone to degradation and yields N5-hydroxy-L-ornithine and Δ2-anhydro-mevalonate lactone under acidic conditions (Fig. S10†).25,26 We therefore turned to intact protein MS to provide snapshots of SidD during the assembly of FSC. This approach has been used successfully to study the enzymology of simplified polyketide synthases (PKS) and NRPS model systems.27–29 Here we applied this technique to capture the steps of an iterative biosynthetic assembly line.
In order to visualize enzyme-bound intermediates using intact protein MS, we chose the SidD(ΔCT) mutant instead of wild-type SidD. Based on our previous biochemical assays (Fig. 2), this mutant assembly line is stalled without the releasing domain (CT). This leads to accumulation of intermediates that can be captured by intact protein MS. Accordingly, in vitro reactions of SidD(ΔCT) were subject to UHPLC-ESI-Q-TOF-MS analyses (Methods see ESI†). Taking holo-SidD(ΔCT) as a starting point, after providing cis-AMHO and ATP as substrates for 15 min, we observed a new species from the deconvoluted mass spectrum (Fig. 3A). This new species corresponded to one cis-AMHO unit tethered to the assembly line (designated as intermediate I), as indicated by the mass shift of +242 Da relative to the mass of SidD(ΔCT) in the holo-form. Rather than building up more intermediates, prolonged incubation (1 h) resulted in complete disappearance of intermediate I (Fig. 3A).
 | ||
| Fig. 3 Snapshots of late-stage biosynthetic intermediates. Deconvoluted intact protein mass spectra of (A) SidD(ΔCT) mutant acting in cis (B) split SidD(ΔCT) mutant (C2dAT2 + A1T1) acting in trans. Mass shifts corresponding to biosynthetic steps are highlighted with arrows, and proposed intermediates are displayed. Peaks labeled with asterisks indicate N-terminal gluconoylation, a known post-translational modification of recombinant heterologous proteins in E. coli.11 Exact measured and observed masses are detailed in Table S5.† | ||
When we repeated this analysis using Fe3+-containing cis-AMHO substrates, we observed a new species with a +1021 Da mass shift relative to holo-SidD(ΔCT), corresponding to loading of four cis-AMHO units onto the assembly line plus a chelated ferric ion (designated as intermediate IV). We interpreted this intermediate as most likely T1 charged with one cis-AMHO while T2 tethered with a linear trimeric depsipeptide coordinating to a Fe3+ ion (Fig. 3A). However, because SidD has two phosphopantetheinyl (Ppant) arms, it is uncertain as to which T domain each proposed peptidyl intermediate is tethered and different combinations of intermediates can be envisaged (e.g. dimeric depsipeptides appended to both T1 and T2, with only one of these dimeric depsipeptide chelating the Fe3+ ion). To resolve this ambiguity, we performed a similar assay, using a split version of the SidD construct (A1T1 + C2dAT2) in which module 1 and module 2 must interact in trans, such that the mass changes on individual Ppant arm can be tracked unambiguously. As shown in Fig. 3B, a +779 Da mass shift relative to the mass of C2dAT2 in the holo-form was clearly observed for a new species corresponding to three cis-AMHO units attached to C2dAT2 plus a coordinated Fe3+ ion. The low population of this new species may be attributed to less efficient crosstalk between two modules acting in trans as compared to the intact bimodule construct. Nevertheless, this result confirmed our interpretation of intermediate IV, and these snapshots together strongly support our proposal that CT domain is solely responsible for cyclization and product release, and is not involved in chain elongation steps as seen in fungal CDP NRPSs.15,16
Successful trapping of the trimeric intermediate IV achieved only in the presence of Fe3+ ions indicates that shielding of the hydroxamate moieties from the oligomeric peptidyl intermediate through coordination to Fe3+ ion can suppress the nonenzymatic thioester cleavage leading to 3 (and 4). Indeed, when Fe3+-containing cis-AMHO was supplied as substrates, no product was observed in our LC-MS analysis (Fig. 2A), which is consistent with the assembly line stalled at the stage of intermediate IV. It is likely that in the absence of CT domain, a nearby free hydroxamate group may act as a general base to assist direct hydrolytic cleavage of the scissile thioester bond leading to spontaneous release of the peptidyl intermediates 3 and 4; or help cleave the thioester bond through an intramolecular displacement mechanism driven by its nucleophilicity (Fig. S11†).
Encouraged by the successful visualization of intermediate IV, we next applied this methodology to intercept intermediates at early stages by further inactivating T1 and C2 domains from the SidD(ΔCT) mutant, respectively. When the reaction mixture of SidD(ΔCT, C02) mutant was subject to MS analysis, two species were observed (Fig. 4A). One species represents intermediate I while the other one has an additional mass shift of +242 Da relative to intermediate I, which indicates addition of totally two cis-AMHO units to SidD (designated as intermediate II).
 | ||
| Fig. 4 Snapshots of early-stage biosynthetic intermediates. Deconvoluted intact protein mass spectra of (A) SidD(ΔCT, C02) mutant and (B) SidD(ΔCT, T01) mutant. Exact measured and observed masses are detailed in Table S5.† | ||
Since no dipeptidyl-shunt products were observed in our in vitro assays, intermediate II was best interpreted as one cis-AMHO unit tethered on each T domain. This interpretation is also consistent with the proposed role of C2; namely that it C2 carries out chain-elongation reactions. When the assay mixture of SidD(ΔCT, T01) mutant was analysed, only one major species corresponding to one cis-AMHO unit tethered was observed (Fig. 4B). Since now only T2 has the obligatory Ppant arm for substrate tethering, this covalently-bound monomeric cis-AMHO unit must be attached to T2 (intermediate I′). This result also unambiguously proves that direct intermodular substrate loading between A1 and T2 has occurred and bypassed T1. Thus, the queuing model hypothesis, in which loading of T2 is T1-dependent (activated cis-AMHO substrate is transferred from T1 to T2 through transthioesterification), can be ruled out (Fig. S12†).
Taken together, we propose a working model for SidD (Fig. 5). The only functional A domain from module 1 (A1) must aminoacylate T domains from both modules. However, whether loading of each module is in an ordered or random sequence remains unresolved. Once both T domains are charged with cis-AMHO, chain elongation will be catalysed by C2 to give a dimeric cis-AMHO intermediate (tethered to T2). Meanwhile, the vacant T1 will be charged with another cis-AMHO unit by A1. The CT domain is likely to dictate the chain-length by favouring a 36-membered ring closure cyclization reaction on a trimeric intermediate bound on T2. Similar chain-length determining role of CT has been proposed in CDP NRPSs.15,16 Hence, if the dimer intermediate reaches CT, it will be rejected and return to C2 to condense another cis-AMHO unit from T1, yielding the desired on-pathway trimer intermediate that can be offloaded by CT through cyclization to yield FSC (1). It is noteworthy that in this biosynthetic model, the C2 domain must accommodate substrates of different chain lengths appended to T2 for each of the condensation events (i.e. monomeric cis-AMHO vs. dimeric cis-AMHO). Although this scenario would require the C2 domain to catalyse chain elongation using different acyl-acceptors, i.e. the terminal hydroxyl moiety from substrates of vastly different chain lengths (12-membered vs. 24-membered), similar iterative use of C domain has been proposed for asperlicin biosynthesis.30
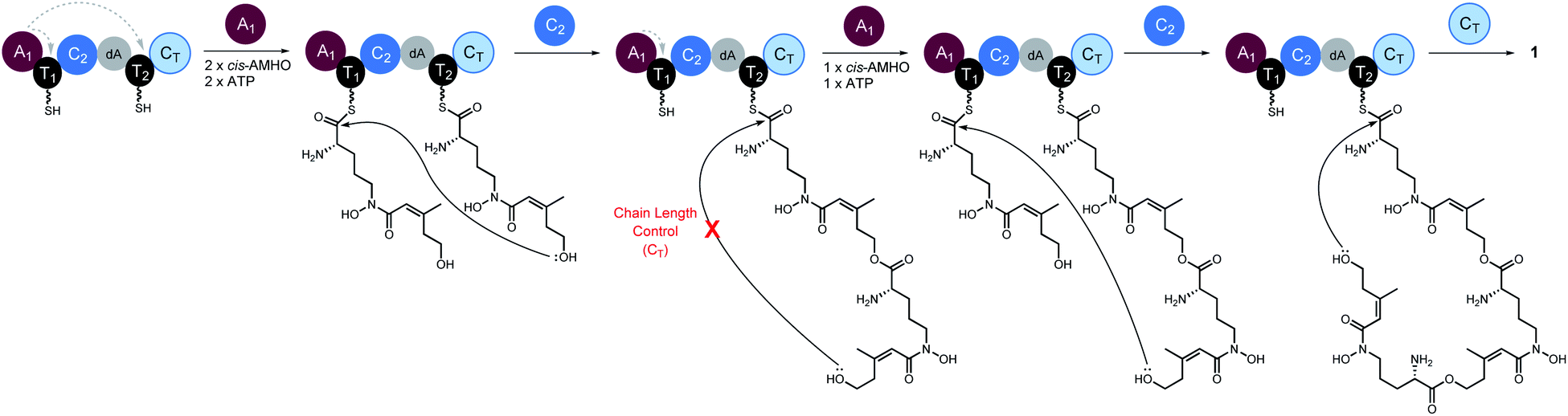 | ||
| Fig. 5 Proposed biosynthetic model for SidD-catalysed formation of FSC (1). Grey dashed arrows indicate productive interactions between the A1 domains and the T1/T2 domains. Our data suggest that the CT domain, not the central C2 domain, determines the chain-length of the nascent peptidyl chain permitted for cyclisation. A subtle variation of this model involving a chain back-transfer step cannot be ruled out and is outlined in Fig. S13.† | ||
Our proposed working model could also explain the formation of shunt products 3 and 4. In the absence of CT, the SidD assembly line will progress and stall when the trimer intermediate is reached. Hydroxamate-assisted, slow non-enzymatic thioester cleavage will occur, eventually releasing the stalled intermediate as shunt product 3. Furthermore, the long-lived trimer intermediate could now have a chance to complete an aberrant chain-elongation reaction presumably at C2 with a rate comparable to that of the hydrolytic releasing of 3, which could explain the formation of derailed product 4 (Fig. S13†). Formation of the longer-than-desired product 4 in the absence of CT further reinforced the idea that CT, rather than C2 is the chain-length determining domain. Offloading either 3 or 4 will free up T2 and enable multiple turnovers of the substrate.
A subtle variation on this pathway could also be envisioned, in which the dimeric cis-AMHO intermediate bound to T2 following the first condensation event is “back transferred” onto T1via a trans-thioesterification step, followed by loading of T2 with another cis-AMHO unit. This would mean that C2 always utilises monomeric cis-AMHO from the T2 domain (Fig. S14†). Although such “chain back-transfer” step cannot be ruled out by our current data, it is unprecedented for NRPS enzymology. It is worth noting, that although “pass-back chain extension” has been observed in fungal CDP NRPSs and bacterial PKS–NRPS hybrids,15,31 it is different from the “chain back-transfer” proposed here. The aforementioned, “chain back-transfer” does not involve chain elongation, whilst “pass-back chain extension” is essentially a chain elongation step except the direction is not forward. This difference also renders SidD distinct from the closely related CDP NRPSs. The CT domains from CDP NRPSs not only catalyze chain cyclization to offload the final cyclicdepsipeptides, but also catalyses “pass-back chain extension” to enable iterative use of the whole assembly line; whereas the CT domain from SidD is solely involved in cyclization since elongated products are still observed in the absence of CT.
Our mechanistic study of SidD also provides valuable insights into the biosynthesis of other fungal siderophores, in particular, the coprogen-type siderophores. Nps6 shares several striking similarities with SidD: (1) they adopt almost identical domain architectures (except in Nps6 tandem repeated T domains are present in module 2; and (2) they utilize very similar building blocks (SidD activates cis-AMHO while NPS6 activates trans-AMHO), yet they make distinct products: coprogen is a partially linear depsipeptide while FSC is a bona fide macrocycle. Based on our study of SidD, we expect that Nps6 should iteratively use A1 to aminoacylate T domains in both of its modules (i.e. three T domains in total). The tandem T domains in Nps6 module 2 are likely employed to increase overall flux.32 Assuming the CT domain in Nps6 also catalyses cyclization and chain release to forge the diketopiperazine scaffold, then the C2 domain must be iteratively used to account for the extra chain elongation in this bimodular NRPS. Intriguingly, if this proposal is correct, the C2 domain from Nps6 would be a C domain with dual-function; catalysing both ester bond and amide bond formation. We hypothesise that when the Nps6 C2 domain catalyses a second chain elongation event, it attaches T1-tethered trans-AMHO to the amino group of T2-tethered trans-AMHO dimers instead of the hydroxy group at the end of the growing chain as proposed for the SidD C2 domain. Then a CT-catalysed intra-molecular cyclisation would simultaneously yield the diketopiperazine moiety and release the coporgen product from the NRPS (Fig. S15†). Although this biosynthetic proposal is consistent with our working model for SidD, the exact molecular mechanism of Nps6 undoubtedly warrants more in-depth biochemical investigation in the future.
Conclusions
We have provided evidence supporting an iterative NRPS mechanism for SidD, including an iterative A domain, iterative chain-elongation C domain, and a chain-releasing CT domain catalysing cyclization and determining product chain-length, respectively. The iterative use of A domains to incorporate repeat units of hydroxamate amino acids is a hallmark for fungal siderophore-producing NRPS and a remarkable strategy to improve atom economy for biosynthetic assembly-lines. Understanding the molecular mechanism underpinning such programmed and highly efficient iterative domain usage may guide future design and engineering of novel and smaller NRPS assembly-lines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NIH 1R35GM118056 to YT. YH is a Life Sciences Research Foundation fellow sponsored by the Mark Foundation for Cancer Research. M. J. is the recipient of a BBSRC Future Leader Fellowship (BB/R01212/1). The Bruker MaXis II instrument used in this study was funded by the BBSRC (BB/M017982/1).Notes and references
- C. D. Kaplan and J. Kaplan, Chem. Rev., 2009, 109, 4536–4552 CrossRef CAS.
- S. C. Andrews, A. K. Robinson and F. Rodríguez-Quiñones, FEMS Microbiol. Rev., 2003, 27, 215–237 CrossRef CAS.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413–451 CrossRef CAS.
- A. H. T. Hissen and M. M. Moore, J. Biol. Inorg Chem., 2005, 10, 211–220 CrossRef CAS.
- H. Haas, Nat. Prod. Rep., 2014, 31, 1266–1276 RSC.
- A. C. W. Franken, B. E. Lechner, E. R. Werner, H. Haas, B. C. Lokman, A. F. J. Ram, C. A. M. J. J. van den Hondel, S. de Weert and P. J. Punt, Brief. Funct. Genomics, 2014, 13, 482–492 CrossRef CAS.
- H. Haas, M. Eisendle and B. G. Turgeon, Annu. Rev. Phytopathol., 2008, 46, 149–187 CrossRef CAS.
- T. Schwecke, K. Göttling, P. Durek, I. Dueñas, N. F. Käufer, S. Zock-Emmenthal, E. Staub, T. Neuhof, R. Dieckmann and H. von Döhren, ChemBioChem, 2006, 7, 612–622 CrossRef CAS.
- K. E. Bushley, D. R. Ripoll and B. G. Turgeon, BMC Evol. Biol., 2008, 8, 328 CrossRef.
- S. Oide, W. Moeder, S. Krasnoff, D. Gibson, H. Haas, K. Yoshioka and B. G. Turgeon, Plant Cell, 2006, 18, 2836–2853 CrossRef CAS.
- K. Reiber, E. P. Reeves, C. M. Neville, R. Winkler, P. Gebhardt, K. Kavanagh and S. Doyle, FEMS Microbiol. Lett., 2005, 248, 83–91 CrossRef CAS.
- M. Schrettl, E. Bignell, C. Kragl, Y. Sabiha, O. Loss, M. Eisendle, A. Wallner, H. N. Arst, K. Haynes and H. Haas, PLoS Pathog., 2007, 3, 1195–1207 CAS.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, ChemBioChem, 2002, 3, 490–504 CrossRef CAS.
- R. D. Süssmuth and A. Mainz, Angew. Chemie. Int. Ed., 2017, 56, 3770–3821 CrossRef.
- D. Yu, F. Xu, S. Zhang and J. Zhan, Nat. Commun., 2017, 8, 1–11 CrossRef.
- C. Steiniger, S. Hoffmann, A. Mainz, M. Kaiser, K. Voigt, V. Meyer and R. D. Süssmuth, Chem. Sci., 2017, 8, 7834–7843 RSC.
- C. J. B. Harvey, M. Tang, U. Schlecht, J. Horecka, C. R. Fischer, H. C. Lin, J. Li, B. Naughton, J. Cherry, M. Miranda, Y. F. Li, A. M. Chu, J. R. Hennessy, G. A. Vandova, D. Inglis, R. S. Aiyar, L. M. Steinmetz, R. W. Davis, M. H. Medema, E. Sattely, C. Khosla, R. P. S. Onge, Y. Tang and M. E. Hillenmeyer, Sci. Adv., 2018, 4, eaar5459 CrossRef.
- H. Oberegger, M. Eisendle, M. Schrettl, S. Graessle and H. Haas, Curr. Genet., 2003, 44, 211–215 CrossRef CAS.
- Y. Hai, A. M. Huang and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10348–10353 CrossRef CAS.
- Y. Hai, M. Jenner and Y. Tang, J. Am. Chem. Soc., 2019, 141, 16222–16226 CrossRef CAS.
- L. E. N. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585–1595 CrossRef CAS.
- E. Brandenburger, M. Gressler, R. Leonhardt, G. Lackner, A. Habel, C. Hertweck, M. Brock and D. Hoffmeister, Appl. Environ. Microbiol., 2017, 83, e01478-17 CrossRef.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS.
- D. E. Ehmann, J. W. Trauger, T. Stachelhaus and C. T. Walsh, Chem. Biol., 2000, 7, 765–772 CrossRef CAS.
- H. Diekmann and H. Zahner, Eur. J. Biochem., 1967, 3, 213–218 CrossRef CAS.
- A. Munawar, J. W. Marshall, R. J. Cox, A. M. Bailey and C. M. Lazarus, ChemBioChem, 2013, 14, 388–394 CrossRef CAS.
- A. von Tesmar, M. Hoffmann, J. Pippel, A. A. Fayad, S. Dausend-Werner, A. Bauer, W. Blankenfeldt and R. Müller, Cell Chem. Biol., 2017, 24, 1216–1227.e8 CrossRef CAS.
- M. Jenner, X. Jian, Y. Dashti, J. Masschelein, C. Hobson, D. M. Roberts, C. Jones, S. Harris, J. Parkhill, H. A. Raja, N. H. Oberlies, C. J. Pearce, E. Mahenthiralingam and G. L. Challis, Chem. Sci., 2019, 10, 5489–5494 RSC.
- J. Masschelein, P. K. Sydor, C. Hobson, R. Howe, C. Jones, D. M. Roberts, Z. Ling Yap, J. Parkhill, E. Mahenthiralingam and G. L. Challis, Nat. Chem., 2019, 11, 906–912 CrossRef CAS.
- X. Gao, W. Jiang, G. Jiménez-Osés, M. S. Choi, K. N. HouK, Y. Tang and C. T. Walsh, Chem. Biol., 2013, 20, 870–878 CrossRef CAS.
- J. J. Zhang, X. Tang, T. Huan, A. C. Ross and B. S. Moore, Nat. Chem. Biol., 2020, 16, 42–49 CrossRef CAS.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03627g |
| ‡ Present address: Department of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, California, 93106, USA. |
| This journal is © The Royal Society of Chemistry 2020 |