
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInside polyMOFs: layered structures in polymer-based metal–organic frameworks†
Kyle C.
Bentz
 a,
Karthikeyan
Gnanasekaran
b,
Jake B.
Bailey
a,
Sergio
Ayala
Jr
a,
F. Akif
Tezcan
a,
Nathan C.
Gianneschi
*b and
Seth M.
Cohen
*a
a,
Karthikeyan
Gnanasekaran
b,
Jake B.
Bailey
a,
Sergio
Ayala
Jr
a,
F. Akif
Tezcan
a,
Nathan C.
Gianneschi
*b and
Seth M.
Cohen
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0358, USA. E-mail: scohen@ucsd.edu
bDepartments of Chemistry, Materials Science & Engineering, Biomedical Engineering, Pharmacology, International Institute for Nanotechnology, Simpson-Querrey Institute, Chemistry of Life Processes Institute, Lurie Cancer Center, Northwestern University, Evanston, Illinois 60208, USA
First published on 15th September 2020
Abstract
In this report, we explore the internal structural features of polyMOFs consisting of equal mass ratios of metal-coordinating poly(benzenedicarboxylic acid) blocks and non-coordinating poly(ethylene glycol) (PEG) blocks. The studies reveal alternating lamellae of metal-rich, crystalline regions and metal-deficient non-crystalline polymer, which span the length of hundreds of nanometers. Polymers consisting of random PEG blocks, PEG end-blocks, or non-coordinating poly(cyclooctadiene) (COD) show similar alternation of metal-rich and metal-deficient regions, indicating a universal self-assembly mechanism. A variety of techniques were employed to interrogate the internal structure of the polyMOFs, including transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), and small-angle synchrotron X-ray scattering (SAXS). Independent of the copolymer architecture or composition, the internal structure of the polyMOF crystals showed similar lamellar self-assembly at single-nanometer length scales.
Introduction
Metal–organic frameworks (MOFs) are a class of two- or three-dimensional materials composed of multi-topic organic ligands connected to metal clusters known as secondary building units (SBUs).1–3 These materials are characterized by their large surface areas, highly crystalline networks, and diverse chemical functionalities. MOFs have emerged as materials with enormous potential for solving a variety of important problems including greenhouse and toxic gas sequestration and degradation,4–9 water management,5,6,10 and energy storage.11–13 However, a major impediment to widespread implementation of MOFs for real-world applications is the inherent form factor of MOFs as crystalline, granular powders with highly limited processability.2 In contrast, traditional amorphous or semicrystalline polymers are highly processable and durable materials. Thus, hybridization of MOFs and polymers has the potential to create a class of materials with unprecedented properties.14–17A bottom-up approach to hybridize MOFs and traditional amorphous or semicrystalline polymers involves the transformation of polymer ligands directly into crystalline frameworks through the incorporation of metal ions. This approach yields materials termed polyMOFs.7,18 The first reports on polyMOFs focused on simple homopolymers obtained through step growth polymerization of dihydroxybenzene diesters and dibromo alkanes. Upon hydrolysis polyethers with 1,4-benzene dicarboxylic acid (bdc) monomers were obtained and the resulting linear, amorphous polymers were converted into highly crystalline, porous frameworks with Zn2+ or Cu2+-based SBUs under solvothermal conditions. In subsequent reports, polyMOFs with robust Zr4+-based UiO-66-type frameworks (polyUiO-66) were developed with varying spacer lengths in the polymer backbone between bdc units,19 isoreticular expansion was demonstrated as a means to markedly increase surface areas in polyMOFs,20 and block polymer-based polyMOFs have been synthesized.21,22
UiO-66 synthesized from small-molecule ligands often gives octahedral shaped crystals. By contrast, polyUiO-66, depending on the polymer composition, yields a large variety of exotic morphologies.22 While imaging techniques such as scanning electron microscopy (SEM) can provide an understanding of the bulk morphology of polyMOF nano- and microcrystals, obtaining information about the internal architecture of the polyMOF crystals is considerably more challenging. Herein, we investigated the interior structure of polyMOFs through a variety of techniques including transmission electron microscopy (TEM) of ultramicrotomed sections of polyMOF coupled with energy-dispersive X-ray spectroscopy (EDS), small-angle synchrotron X-ray scattering (SAXS), scanning electron microscopy (SEM), and X-ray diffraction (PXRD). Our results show that all block polymer derived polyUiO-66 particles are composed of phase segregated alternating metal-rich and metal poor regions forming a hierarchical architecture that span the length of hundreds of nanometers.
Results and discussion
A range of polymer architectures were employed to construct polyMOFs for this study including homopolymers, block polymers, and statistical copolymers (Fig. 1).22 Acyclic diene metathesis polymerization (ADMET) was used to synthesize all polymers, which proceeds through a step-growth mechanism.23,24 In general, the simple alkanes used in this study allow for random copolymerization, although the kinetics of polymerization for the larger PEG macromonomers may be slower at lower conversions. The ligand-containing monomer was a diene-functionalized benzenedicarboxylic ester derivative and alkene end-capped PEG or cyclooctadiene (COD) were used as co(macro)monomers for copolymer synthesis. In the first system investigated, a random block polymer was synthesized from the copolymerization of the diene ligand and alkene end-capped PEG at a 10% molar ratio of PEG (pbdc-8a-PEG4k-10%; 8a refers to the 8 carbon atoms between bdc groups with ‘a’ referring to the deprotected carboxylic acid moieties on the bdc ligand) (Fig. 1a). The second system consists of an internal block of pbdc-8a with terminal PEG groups synthesized by introducing the PEG macromonomers at the end of the polymerization (pbdc-8a-PEG2k-OMe) (Fig. 1b). Lastly, a statistical copolymer of equal molar ratios of bdc and COD monomers was synthesized (pbdc-8a-COD1:1) (Fig. 1c). Across all three systems the molecular weight was held constant at ∼17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (as determined by GPC, Fig. S1–S4†). Finally, all polymers were converted to polyUiO-66 by solvothermal coordination with ZrCl4 in diethylformamide in the presence of formic acid modulator (Fig. 1d) (additional synthetic details can be found in the ESI†).
000 g mol−1 (as determined by GPC, Fig. S1–S4†). Finally, all polymers were converted to polyUiO-66 by solvothermal coordination with ZrCl4 in diethylformamide in the presence of formic acid modulator (Fig. 1d) (additional synthetic details can be found in the ESI†).
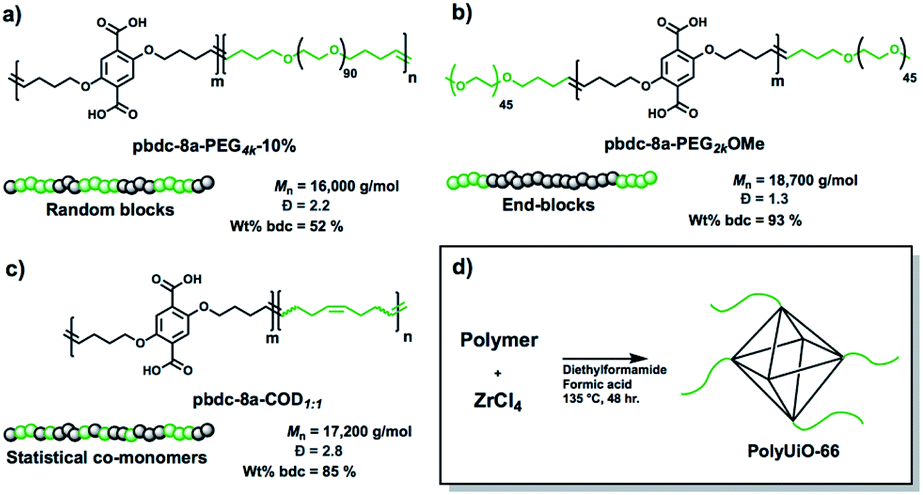 | ||
| Fig. 1 Schematic representation of polymers used in this study with names, molecular weights, dispersities, and weight percent of ligand block pbdc-8a. (a) Random block polymer, (b) end-blocks, and (c) statistical copolymers. (d) Synthesis conditions for conversion of copolymer ligands to polyUiO-66. | ||
The metalation of block copolymers to form polyUiO-66 resulted in unusual crystal morphologies and self-assembly (vide infra). SEM and TEM of the bulk crystals reveal platelet-like structures of polyUiO-66 formed from the random block polymer pbdc-8a-PEG4k-10% (Fig. 2a–c and S5†). However, TEM of ultramicrotomed sections (Fig. 2d) of the polyUiO-66 reveal an additional ordering in the form of layers with an average spacing of 3.5 nm to 4.5 nm in width and hundreds of nanometers in length (Fig. 2e). The overall width of the layered assembly varied between 70 nm and 120 nm, which matches the thickness of the flat, platelet-like features in the bulk crystal (Fig. 2a). The fast Fourier transform (FFT) of these layered channels quantified the spacings to have ordering at lengths of 4.2, 2.6, and 1.5 nm (Fig. 2f). The scattering form factor measured from the small-angle X-ray scattering (SAXS, Fig. 2g) shows a d-spacing of ∼4.5 nm in the high-q region, which matches with the layered channels observed in TEM and FFT analysis. Powder X-ray diffraction confirmed the presence of the UiO-66 framework with a primary reflection at a d-spacing of 1.2 nm (Fig. 2h).
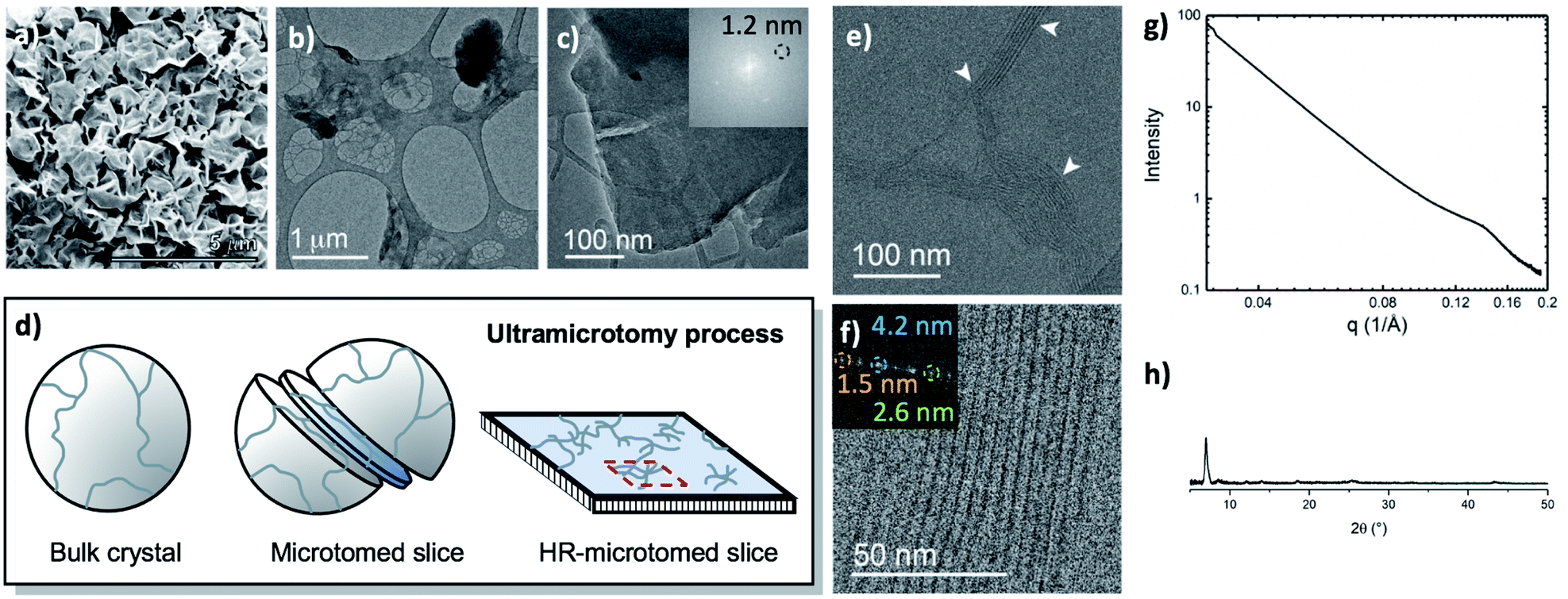 | ||
| Fig. 2 Morphological analysis of polyUiO-66 derived from pbdc-8a-PEG4k-10%: (a) SEM of bulk crystals; (b and c) TEM of bulk crystals dispersed in methanol. Inset shows the spacing corresponds to UiO-66 crystals; (d) schematic representation of ultramicrotomy process of polyMOF crystals; (e and f) TEM of ultramicrotomed section revealing the layered morphology of polyUiO-66 (white arrows in (e) denote layered assemblies); (g) SAXS scattering profile of the bulk crystals showing a spacing at 4.5 nm; (h) PXRD powder pattern of the bulk material shows the spacing of UiO-66 at 1.2 nm. | ||
We further explored the layered assembly of polyMOFs using energy dispersive X-ray spectroscopy (EDS) and high-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 3). We identified that these structures correspond to alternating layers of Zr-rich and Zr-deficient regions (Fig. 3a–c). Further analysis by HAADF-STEM (Fig. 3d–g) and subsequent intensity-weighted line analysis (Fig. 3h) revealed the bright region corresponds to electron scattering spanning the width of approximately two Zr SBU clusters separated by a spacing of ∼1.5 nm, attributed to the polymer ligand. The local minima in the line analysis represents a lower density scattering of the pores between the Zr clusters and the global minima arise from the scattering completely free of Zr atoms with a spacing of ∼2.6 nm (Fig. 3h). These observations suggest that polyMOFs self-assembles into two-dimensional layered morphologies with the non-coordinating PEG block forming alternating lamellae with crystalline polyUiO-66 with an average total width of approximately 4.2 nm.
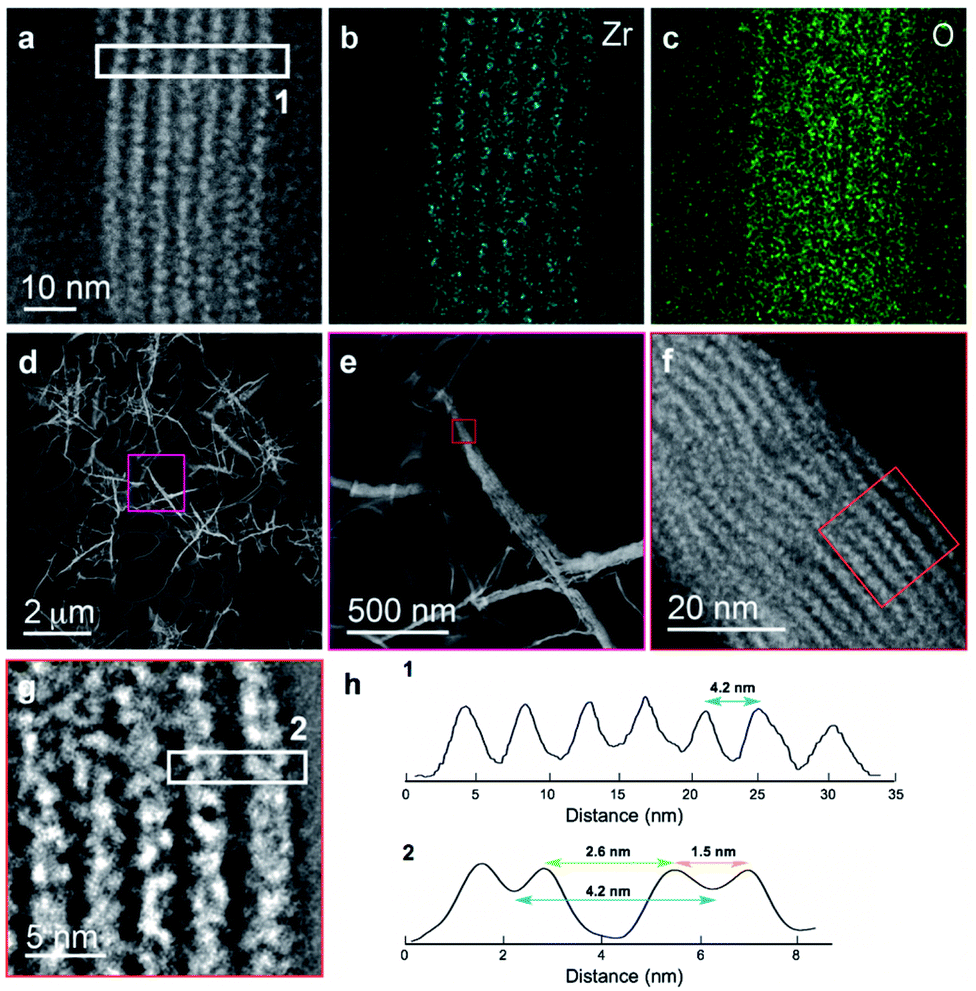 | ||
| Fig. 3 Elemental mapping and HAADF-STEM of layered assembly of polyUiO-66 derived from pbdc-8a-PEG4k-10%. (a) HAADF-STEM of polyUiO-66 derived from pbdc-8a-PEG4k-10%; (b and c) zirconium and oxygen mapping of layered assembly illustrates the assembly of individual UiO-66 moieties forming layered architecture; (d–h) HAADF-STEM of layered assemblies in polyUiO-66 from pbdc-8a-PEG4k-10% and the corresponding line profiles from boxes denoted (1) and (2) in (a) and (g). | ||
Based on the above observations, we reconstructed the layered assembly of polyMOF to develop a model for the molecular-level understanding of the structure (Fig. 4). In the HAADF-STEM images of the pbdc-8a-PEG4k-10% system, the metal-rich layered spacings are on average 1.5 nm wide, and the metal-deficient spacings are on average 2.6 nm wide. If an area 4.2 nm wide (the combined width of the metal-rich and metal-deficient sections) and approximately 6 nm in length is considered, we show how the polymer chains may reasonably pack into this space and how the pbdc-8a and PEG segregate to give the layered spacings. The UiO-66 unit cell, outlined by a green dashed line in the figure, spans a width of 2.1 nm. As the metal-rich domain in the pbdc-8a-PEG4k-10% system is on average 1.5 nm wide, we conclude that on average, the metal-rich domain is approximately two Zr6 SBUs wide. Additionally, the width of the metal-deficient domain is about 2.6 nm, into which PEG of 4000 g mol−1 can pack to fill the remaining space of the considered volume. PXRD patterns were measured for free semi-crystalline PEG, which showed distinctive features at 2θ values of 20° and 25° (Fig. S6†). These semi-crystalline, PEG-associated reflections are also present in the PXRD pattern of the non-metallated block polymer pbdc-8a-PEG4k-10%. However, in the polyUiO-66 material derived from pbdc-8a-PEG4k-10% these PEG-associated reflections are absent, suggesting the PEG in the polyUiO-66 sample is largely amorphous. To validate this observation, polyUiO-66 derived from the pbdc-8a homopolymer was blended with free, semi-crystalline PEG4k in the same weight percent as is found in the pbdc-8a-PEG4k-10% block polymer. In this physically mixed sample, the crystalline PEG features are clearly observed (Fig. S6†). These PXRD experiments confirm that in the polyUiO-66 derived from pbdc-8a-PEG4k-10% that the PEG segments are non-crystalline as depicted in Fig. 4. Given that a single polymer chain of pbdc-8a-PEG4k-10% is 16000 g mol−1 and is 52 wt% bdc, an average polymer contains 25 bdc2− groups and 2 PEG blocks per chain. In the area considered, we show how 32 Zr6 SBUs organize, which corresponds to 190 bdc2− groups and thus ∼7 polymer chains. We also highlight in red the potential path that one polymer chain may take in this system. It is highly likely, both statistically and entropically,25 that multiple polymer chains contribute ligands to each SBU, which is reflected in the reconstruction. It was recently determined, through solid-state 1H NMR studies, that a significant number of bdc groups along the polymer backbone in these materials remain uncoordinated (i.e., not bound to SBUs). This finding suggests that individual polymer chains extend throughout the material and that multiple polymer chains contribute to the binding at each SBU.26 Indeed, layered spacings similar to these have been observed in assemblies of Zr–phosphonate and polyphenylmaleimide,27 demonstrating the tendency for polymers to self-assemble into layers in the presence of layered metal-oxides. However, this is the first demonstration of such layered assemblies in polymer-MOF hybrid materials.
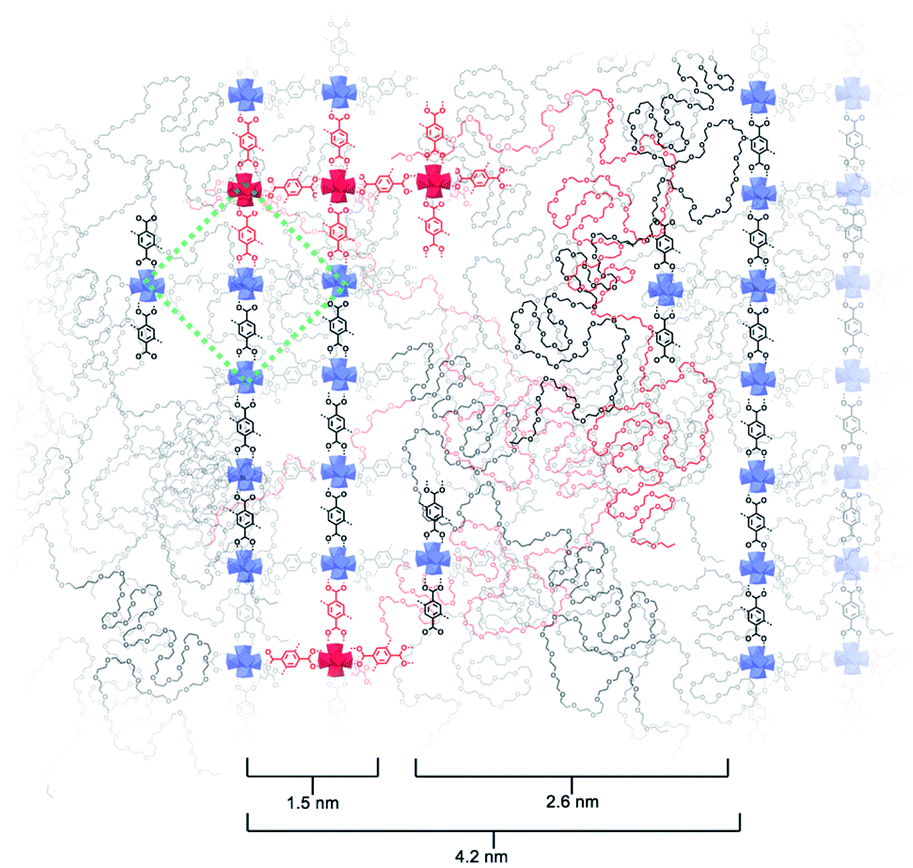 | ||
| Fig. 4 Reconstruction of layered assemblies in pbdc-8a-PEG4k-10%. The schematic represents an area occupied by roughly 7 polymer chains, which in turn contain approximately 190 bdc2− ligands and 14 PEG blocks. The MOF-dominant region is, on average, five Zr-based SBUs wide. UiO-66 unit cell drawn in green dashed lines for reference. A possible path that one polymer chain may navigate through this material is highlighted in red. | ||
In contrast to the random block polymer pbdc-8a-PEG4k-10%, polyMOFs derived from pbdc-8a-PEG2k-OMe display a different bulk morphology with particle size of ∼3 microns and less faceted (Fig. 5a–c). Remarkably, ultramicrotomed TEM analysis (Fig. 5d, e and S7†) of this system shows a similar characteristic lamellar pattern with an average spacing of 4.2 nm, 2.6 nm, and 1.5 nm. SAXS scattering profiles show deviations from linearity near a q-range of 0.1 Å−1, with a peak center near 0.14 Å−1 corresponding to a d-spacing of 4.3 nm (Fig. 5f). PXRD of the bulk crystal also shows high levels of crystallinity in this system (Fig. 5g). Although similar self-assembly into alternating lamellae was observed in this system, the lamellar regions are in general more ordered and display less curvature than in the case of the random block polymer (i.e. pbdc-8a-PEG4k-10%). Curved lamellae may result as a means to minimize free energy of assembly to accommodate polymers of shorter or longer length due to the polymer dispersity and because the random block polymer (i.e. pbdc-8a-PEG4k-10%) contained approximately 50 wt% non-coordinating PEG blocks.28 Far less material needs to be accommodated within and around the crystalline framework when the end-block polymer is used to generate the polyUiO-66 thereby allowing for a more ordered crystal network because that polymer only contains 7 wt% non-coordinating blocks.
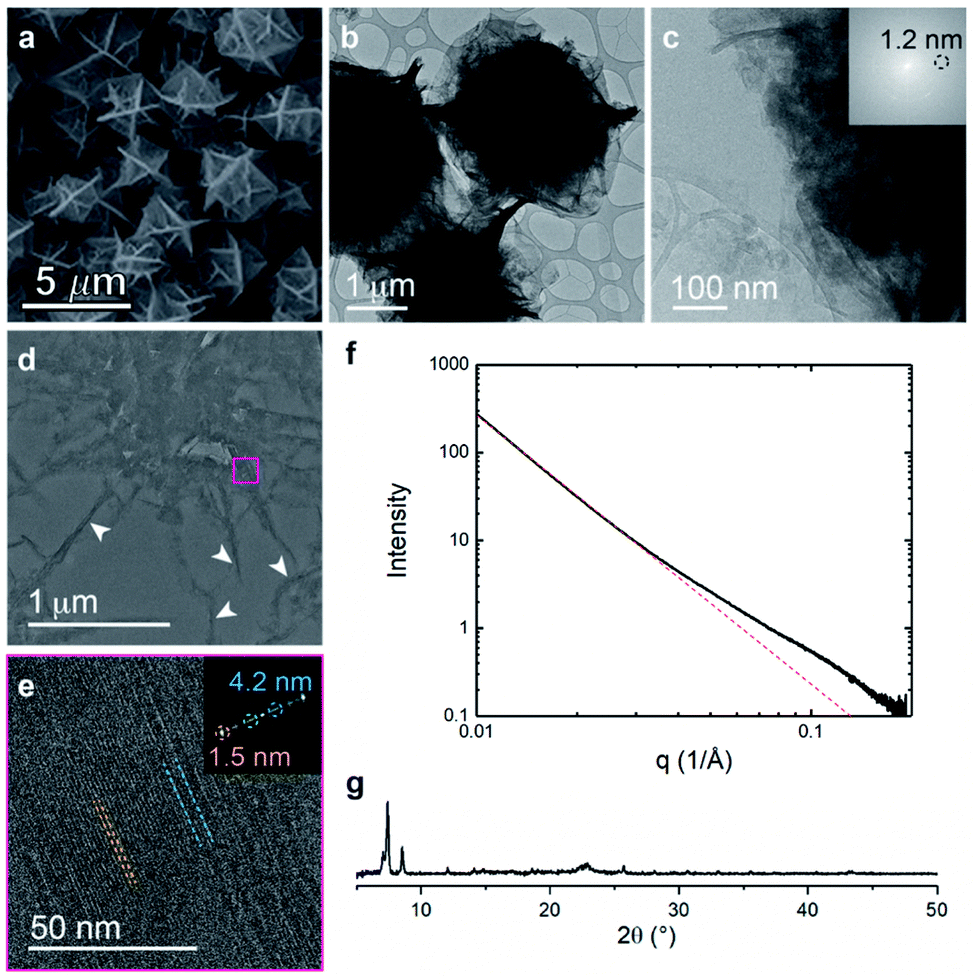 | ||
| Fig. 5 Morphological analysis of polyUiO-66 derived from pbdc-8a-PEG2k-OMe: (a) SEM of bulk crystals; (b and c) TEM of bulk crystals dispersed in methanol. Inset shows the spacing corresponds to UiO-66 crystals; (d and e) TEM of ultramicrotomed section revealing the layered morphology of polyUiO-66 (white arrows in (d) denote layered assemblies); (f) SAXS scattering profile of the bulk crystals showing a d-spacing of 4.3 nm; (g) PXRD powder pattern of the bulk material shows the spacing of UiO-66 at 1.2 nm. | ||
We also observed pore-like structures in the polyUiO-66 derived from pbdc-8a-PEG2k-OMe when viewed top-down suggesting the presence of nanochannels (Fig. S8†). In a previous report, it was hypothesized that assembly by block copolymers prior to MOF formation was responsible for templating the resulting morphologies observed after conversion to crystalline material.22 While the polyMOF synthesis solvent diethylformamide (DEF) is a good solvent for both blocks, there is still a thermodynamic driving force towards energy minimization that induces self-assembly in this system, as well as the other polymers used in this study.29 In particular, when the pbdc-8a-PEG2k-OMe polymer was examined by dynamic light scattering (DLS), large assemblies of ∼100 nm were observed in solution prior to addition of the metal salt. However, these assemblies require the addition of metal salt and elevated temperatures to induce the formation of larger, higher-order assemblies. These solvated assemblies may coalesce and pack during the crystallization process to yield the self-assembled nanochannels (Fig. S8†). While the other polymer systems in this study were also found to assemble in solution, the observation of nanochannels is unique to this system. The triblock architecture of the pbdc-8a-PEG2k-OMe may help facilitate packing between these assemblies, with PEG chains being preferentially located on the assembly exterior.30,31
The final system composed of a statistical copolymer of pbdc and cyclooctadiene (pbdc-8a-COD1:1,Fig. 1c), is a true statistical copolymer, which is in contrast to the PEG systems (Fig. 6). Here, the bulk crystal morphology is less faceted and consists of roughly spherical particles with wrinkled surfaces (Fig. 6a and b). As in the PEG systems, self-assembled lamellae of alternating metal-rich and metal-poor domains are observed, although the lamellae are more curved in this system (Fig. 6c–e and S9†). The SAXS scattering profile shows a very broad range of diffraction with d-spacings ranging from 25 nm to 6 nm (Fig. 6f). As with the other systems, the COD-based polyUiO-66 shows a high degree of crystallinity as confirmed by PXRD (Fig. 6g). We hypothesize that because the non-metal coordinating components are distributed much more evenly throughout the pbdc-8a-COD1:1 polymer than in the PEG-containing systems, this causes the high levels of curvature in the self-assembled lamellae. Whereas blocks of PEG can phase separate outside of the crystalline layers, statistical copolymers of COD are likely located more frequently between and near individual unit cells of the polyUiO-66 network.
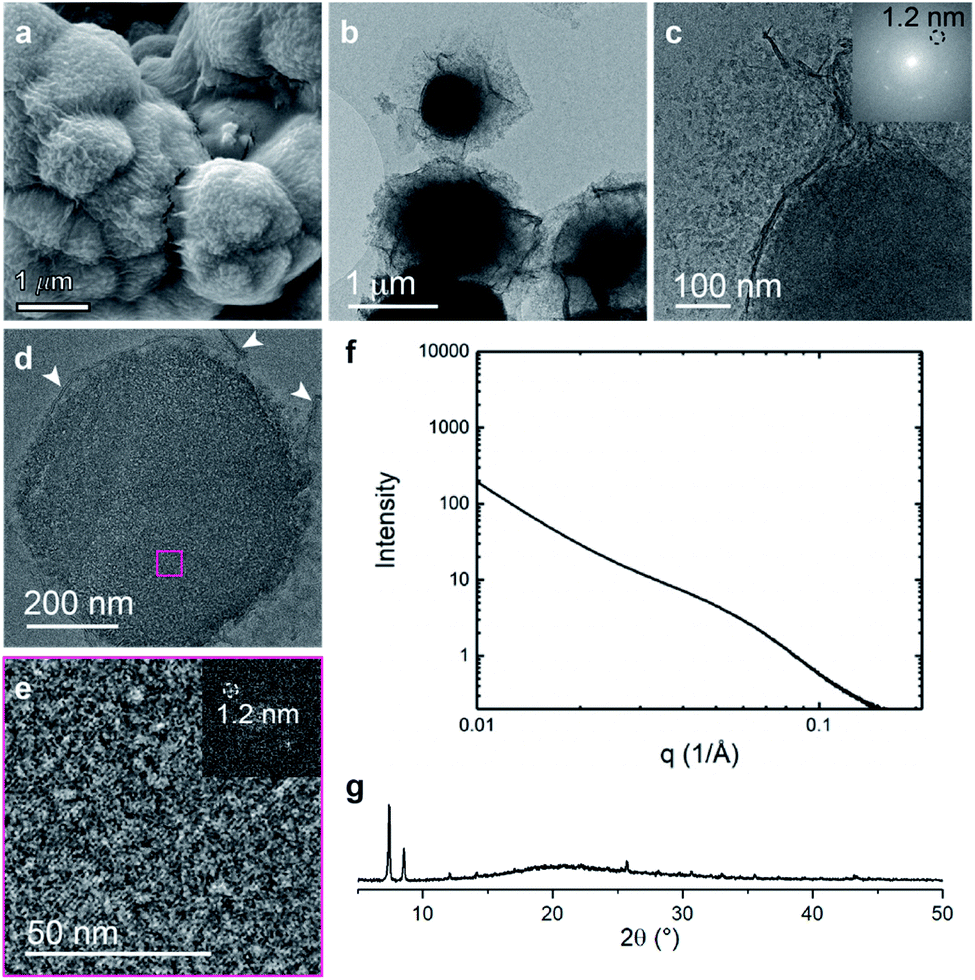 | ||
| Fig. 6 Morphological analysis of polyUiO-66 derived from pbdc-8a-COD1:1: (a) SEM of bulk crystals. (b and c) TEM of bulk crystals dispersed in methanol. Inset shows the spacing corresponds to UiO-66 crystals; (d and e) TEM of ultramicrotomed section. Arrows indicate the alternating layered assembly of metal rich and metal deficient structures. FFT shows the spacing at 1.2 nm that corresponds to UiO-66 (white arrows in (d) denote layered assemblies). (f) SAXS scattering profile of the bulk crystals showing a spacing at 4.5 nm; (g) PXRD powder pattern of the bulk material shows the spacing of UiO-66 at 1.2 nm. | ||
Overall, in polyMOFs constructed from polymers containing poly(benzenedicarboxylic acid) with both non-coordinating blocks located in the interior, termini, and containing statistical copolymers, a universal feature is observed wherein alternating lamellae of crystalline, metal-rich and non-crystalline metal-deficient domains are formed. The PEG and polyCOD chains are excluded from the crystalline domains because the polyMOF pore interior is unable to accommodate non-metal coordinated polymer. We hypothesize that lamellae are the preferred morphology due to the polyMOF structure, which prefers to form highly faceted morphologies. In general, the bulk crystal morphologies in the PEG-containing systems are highly faceted (Fig. 2a and 5a), and correspondingly, the interior morphology of the self-assembled lamellae display the lowest degree of curvature. In contrast, the polyCOD system displays bulk morphologies that are the most spherical (Fig. 6a) and thus the corresponding lamellae have the highest degree of curvature among the systems examined in this study. Furthermore, there appears to be a general preference in these specific systems for lamellae thickness on the single nanometer length scale, likely due to the roughly similar overall molecular weight of the polymers used to construct the polyMOFs. It is likely that higher molecular weight non-coordinating blocks would lead to correspondingly larger distances between layers. However, the polymer architecture plays an important role in the organization of the lamellae. For example, in the PEG-end block polymer, pbdc-8a-PEG2k-OMe, the lamellae are highly linear, but in the random co-monomer polymer, pbdc-8a-COD1:1, highly curved lamellae are observed as a consequence of less ordering and higher dispersity in the polymer molecular weight. As a control, TEM of thin films of polyUiO-66 from homo-pbdc-8a were examined as well (Fig. S10†). Unlike all of the copolymer systems, no self-assembled lamellae were observed.
Conclusions
Using a combination of TEM, EDS mapping, SAXS, and SEM, the interior structure of block copolyMOFs has been examined. In systems composed of block polymers of pbdc-8a and PEG, and pbdc-8a and polycyclooctadiene, uniquely layered structures of amorphous polymer and crystalline polyUiO-66 were observed. These phase-separated lamellae likely form due to exclusion of non-coordinated polymer from the polyMOF pores and as a means to minimize free energy of the polymer assemblies. The lamellar morphology appears to be a general phenomenon, regardless of the polymer architectures reported here, but features such as layer spacing and curvature are affected by changes in the non-coordinating block molecular weight and architecture. Future work is aimed at evaluation of the kinetics of self-assembly, as well as computational modeling of these complex systems to validate the hypotheses of the mechanism for the self-assembled morphologies. We expect these systems will open new avenues for the fabrication of nano-templated metal-containing materials, in addition to being of tremendous interest for the fundamental study of complex self-assembled systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Army Research Office, Department of Army Material command, under award no. W911NF-15-1-0189. Synthesis of polyMOFs was supported by a grant from the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under Award No. DE-FG02-08ER46519. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. Research reported in this publication was supported in part by instrumentation provided by the Office of the Director, National Institutes of Health, under Award Number S10OD026871. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. K. G. acknowledges a postdoctoral fellowship from the Human Frontier Science Program (LT000869/2018-C). J. B. B. and F. A. T. were supported by the Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences, Biomolecular Materials Program (DE-SC0003844). SAXS experiments were performed at APS, a Department of Energy Office of Science User Facility operated by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. K. C. B. and acknowledges support from the Research Corporation for Scientific Advancement (RCSA) through the Cottrell Fellowship Initiative, which is partially funded by a National Science Foundation award to the RCSA (CHE-2039044).References
- S. M. T. Abtab, D. Alezi, P. M. Bhatt, A. Shkurenko, Y. Belmabkhout, H. Aggarwal, Ł. J. Weseliński, N. Alsadun, U. Samin, M. N. Hedhili and M. Eddaoudi, Chem, 2018, 4, 94–105 Search PubMed.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 Search PubMed.
- M. Kalaj, M. S. Denny Jr, K. C. Bentz, J. M. Palomba and S. M. Cohen, Angew. Chem., Int. Ed., 2019, 58, 2336–2340 Search PubMed.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 Search PubMed.
- A. J. Rieth, S. Yang, E. N. Wang and M. Dincă, ACS Cent. Sci., 2017, 3, 668–672 Search PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920–925 Search PubMed.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 Search PubMed.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244–245, 444–456 Search PubMed.
- N. Hanikel, M. S. Prévot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 Search PubMed.
- M. Gutierrez, B. Cohen, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2016, 18, 27761–27774 Search PubMed.
- A. M. Ullman, J. W. Brown, M. E. Foster, F. Léonard, K. Leong, V. Stavila and M. D. Allendorf, Inorg. Chem., 2016, 55, 7233–7249 Search PubMed.
- H. Yang and X. Wang, Adv. Mater., 2019, 31, 1800743 Search PubMed.
- K. C. Bentz and S. M. Cohen, Angew. Chem., Int. Ed., 2018, 57, 14992–15001 Search PubMed.
- M. Kalaj, K. C. Bentz, S. Ayala Jr, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020, 120, 8267–8302 Search PubMed.
- Y. Zhang, X. Feng, S. Yuan, J. Zhou and B. Wang, Inorg. Chem. Front., 2016, 3, 896–909 Search PubMed.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 Search PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 6152–6157 Search PubMed.
- S. Ayala Jr, Z. Zhang and S. M. Cohen, Chem. Commun., 2017, 53, 3058–3061 Search PubMed.
- G. E. M. Schukraft, S. Ayala Jr, B. L. Dick and S. M. Cohen, Chem. Commun., 2017, 53, 10684–10687 Search PubMed.
- M. J. MacLeod and J. A. Johnson, Polym. Chem., 2017, 8, 4488–4493 Search PubMed.
- S. Ayala Jr, K. C. Bentz and S. M. Cohen, Chem. Sci., 2019, 10, 1746–1753 Search PubMed.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Macromolecules, 1991, 24, 2649–2657 Search PubMed.
- M. D. Schulz and K. B. Wagener, Macromol. Chem. Phys., 2014, 215, 1936–1945 Search PubMed.
- R. Krämer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 Search PubMed.
- P. G. M. Mileo, S. Yuan, S. Ayala Jr, P. Duan, R. Semino, S. M. Cohen, K. Schmidt-Rohr and G. Maurin, J. Am. Chem. Soc., 2020, 142, 10863–10868 Search PubMed.
- P. Kohli and G. J. Blanchard, Langmuir, 1999, 15, 1418–1422 Search PubMed.
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 Search PubMed.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 Search PubMed.
- J. Grumelard, A. Taubert and W. Meier, Chem. Commun., 2004, 1462–1463 Search PubMed.
- I. W. Wyman and G. Liu, Polymer, 2013, 54, 1950–1978 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/d0sc03651j |
| This journal is © The Royal Society of Chemistry 2020 |