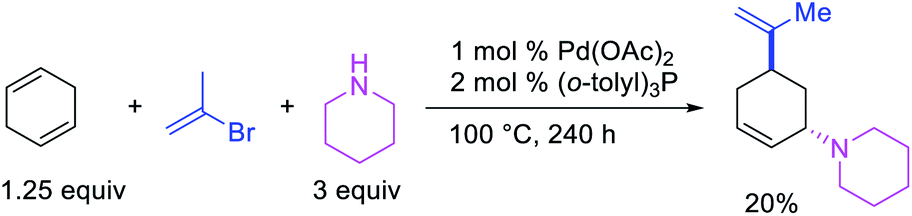DOI:
10.1039/D0SC03634J
(Minireview)
Chem. Sci., 2020,
11, 9757-9774
Walking metals: catalytic difunctionalization of alkenes at nonclassical sites
Received
1st July 2020
, Accepted 6th September 2020
First published on 7th September 2020
Abstract
Migration of metals along a carbon chain is triggered by two of the most common organometallic elementary steps – β-hydride (β-H) elimination and alkene hydrometallation. This process heralds a new future for creating bonds at carbon sites that fall outside the tenets of the conventional wisdom for reactivity and bond formation, and provides an opportunity to leverage β-H elimination to advance the very reaction of alkene difunctionalization it is intrinsically predestined to disrupt. Almost four decades since its genesis, the early adventure for alkene difunctionalization by metal migration was sporadic, and its later development went on a hiatus primarily due to original impetus on arresting β-H elimination for vicinal alkene difunctionalization. With the recent surge on alkene difunctionalization, efforts have been gradually shifting to harnessing the process of β-H elimination to difunctionalize alkenes at sites other than the classical vicinal carbons, termed henceforth nonclassical reaction sites for pedagogical simplicity. In this review article, we extricate and examine the origin and the development of such reactions over the years. This review covers a wide range of reactions for the difunctionalization of alkenes at geminal (1,1), allylic (1,3) and remote (1,n) carbon sites with a variety of coupling partners. These reactions have enabled engineering of complex molecular frameworks with the generation of new carbon–carbon (C–C)/C–C, C–C/C–heteroatom (halogens, O, N, B) and C–B/C–B bonds. The development of these unique transformations is also presented with mechanistic hypotheses and experimental evidences put forward by researchers. Judged by the number of reports emerging recently, it is now strikingly evident that the field of alkene difunctionalization by metal migration has begun to gain momentum, which holds a great future prospect to develop into a synthetic method of enormous potential.
1. Introduction
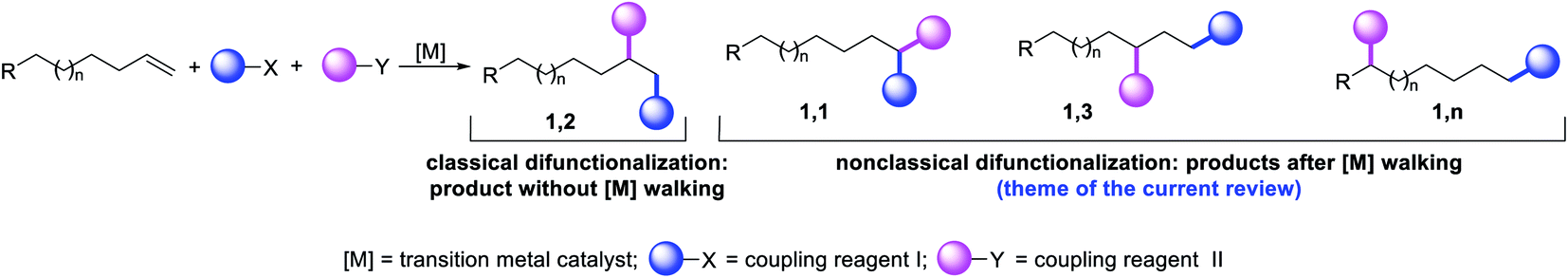
Metal-catalyzed difunctionalization of alkenes is a powerful reaction to engineer complex molecular scaffolds in one step from simple and readily available feedstock chemicals. In this process, an alkene is functionalized with two coupling reagents, which result in the formation of complex molecules with the creation of two new bonds (Scheme 1). The difunctionalization process was first developed independently by Inoue,1 Cacchi and Palmieri,2 Catellani,3 Larock,4 Yoshida5 and Heck6 in the span of a decade from the mid-seventies to early eighties as a way to intercept alkylpalladium (R-[Pd]) species generated upon alkene carbopalladation in both stoichiometric and catalytic reactions. The central premise of the method was to leverage the generation of reactive alkylmetal (R-[M]) intermediates to create two new bonds simultaneously at the sp2 hybridized carbons of alkenes prior to β-hydride (β-H) elimination from the alkylmetal species, which would otherwise derail the reaction to generate Heck products.7 Since the seminal reports, further investigations primarily focused on developing strategies to address the issue of β-H elimination in order to functionalize alkenes at classical sites i.e. the vicinal vinylic carbons (Scheme 1, 1,2-product). The last four decades have seen a steady rise in the development of this process mainly by a cyclization/coupling approach,8 and more recently by a three-component strategy (Scheme 2, Path A).9 A number of such reactions have also already been applied to the synthesis of complex molecules and pharmaceutical targets, and detailed treatments of these methodologies have appeared in a number of review articles lately.10
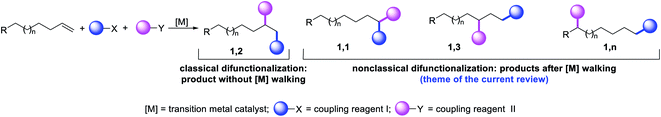 |
| | Scheme 1 Products of the classical and nonclassical alkene difunctionalization reactions. | |
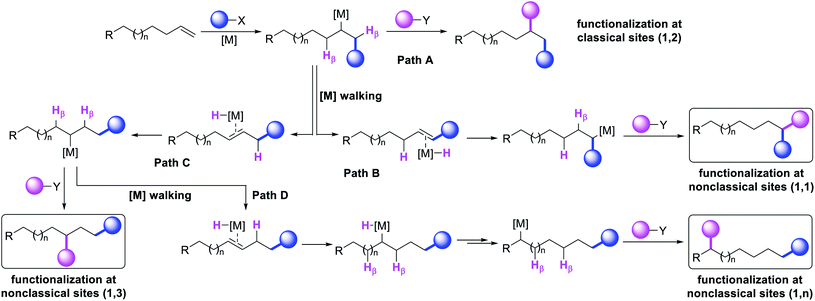 |
| | Scheme 2 Pathways for classical and nonclassical alkene difunctionalization reactions. | |
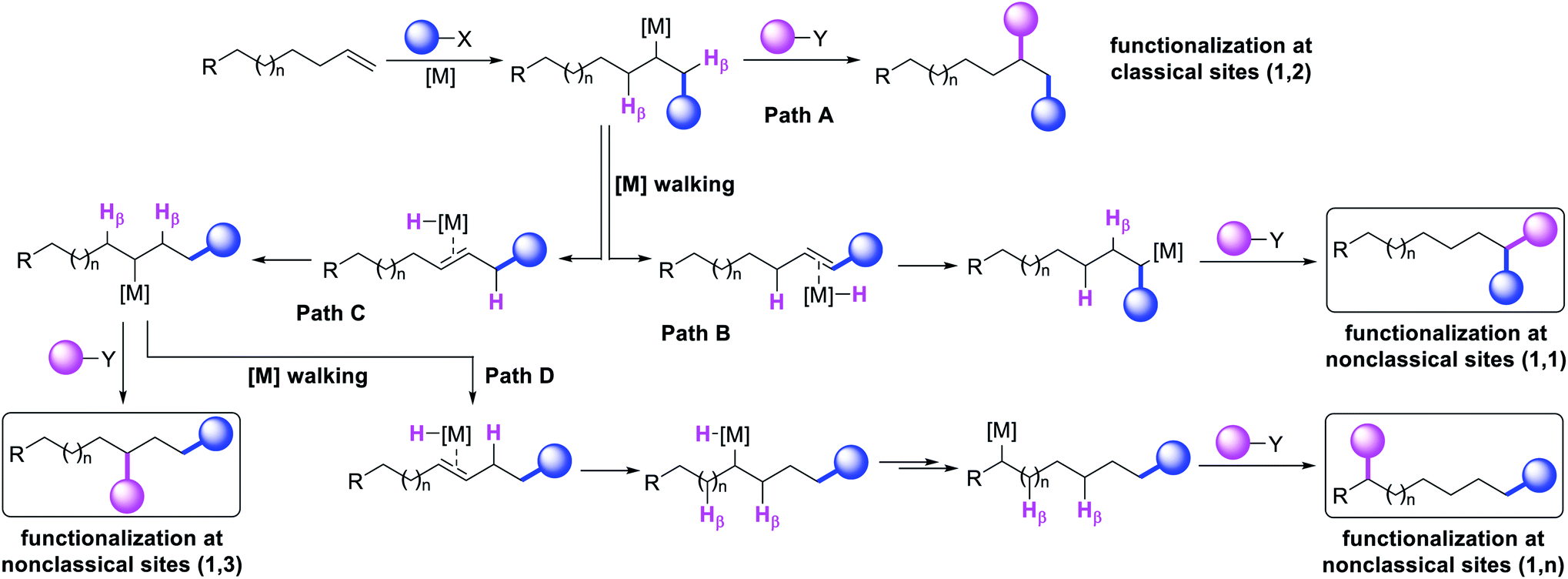
The potential emergence of the alkene difunctionalization as a powerful method in organic synthesis has also given rise to exploring opportunities to see β-H elimination through the optics of optimism, and make a stride in the development of the very reaction it intrinsically disrupts. This new course of reaction has led to the generation of unique products (Scheme 1, 1,1-, 1,3- and 1,n-) that are otherwise beyond prediction based on the traditional sense of alkene difunctionalization. The premise of the optimism is founded upon the fundamental nature of metal hydrides ([M]–H), generated after β-H elimination in the current context, to insert into alkenes to generate a new alkylmetal species, a requisite intermediate for further functionalization (Scheme 2, Paths B–D). The permutation of M–H insertion, however, could vary to generate the original (Path A) or rearranged (Paths B and C) alkylmetal species depending on the stability of the resultant alkylmetal intermediates. The original or the rearranged alkylmetal intermediates may also very well walk a long but measured distance along alkyl chains to seek further stability (Path D). The migration of the metal along the carbon chain has been popularized as a metal-walk or chain-walk process.11 In alkene difunctionalization, the fundamental nature of the metal walking process subsidizes the detrimental effects of β-H elimination by generating a new alkylmetal species, and furnishes opportunities to difunctionalize alkenes at two unique sites that are otherwise not predictable to be functionalized simultaneously in such a concerted process.12 The contemporary development of this class of alkene difunctionalization reaction is founded upon the seminal discoveries made from late seventies to mid-eighties independently by Cacchi and Palmieri,2 Larock,4 Yoshida,5 Heck6 and Bäckvall.13 These processes relied upon intercepting new alkylmetal intermediates generated by metal migration after alkene carbometallation.
In the last few years, review articles mainly focused on the development of vicinal alkene difunctionalization reactions with the mention of metal migration briefly. This review article will examine the beginning and the historical developments of these innovative reactions that difunctionalize alkenes at sites other than the classical vicinal carbons of an alkene or 1,4-carbons of a 1,3-conjugated diene. These new reaction sites are referred to as nonclassical alkene sites throughout the article for pedagogical simplicity. The non-classical alkene sites discussed in the subsequent paragraphs involve geminal (1,1), allylic (1,3), and beyond allylic (1,n) wherein two new bonds are formed either at the same sp2 hybridized alkene carbon (geminal), two different sp2 hybridized carbons of a distally separated nonconjugated diene or one bond at an sp2 hybridized alkene carbon and another bond at an sp3 hybridized carbon (such as allylic and beyond) through C–H bond cleavage.
2. 1,1-Difunctionalization
Geminal difunctionalization of alkenes is guided by the nature of the first coupling reagent that is added to alkenes to generate alkylmetal intermediates. Such reagents generally transfer aryl, vinyl and heteroatomic groups to alkenes, which function as a driving force to attract the walking metals to their vicinity since these groups propitiate the metals by forming thermodynamically stable intermediates such as π-allylmetal and π-benzylmetal complexes. Therefore, the known examples of alkene 1,1-difunctionalization reactions are based on the initial addition of aryl, vinyl and heteroatomic groups to alkenes, which have so far generated 1,1-carbon–carbon (C–C)/C–C, C–C/C–halogen, oxygen and nitrogen (C–halogen, O, N) and C–C/C–boron (C–B), C–B/C–B bonds. These reactions are discussed in details in the subsequent sections.
2.1. 1,1-C–C/C–C functionalization
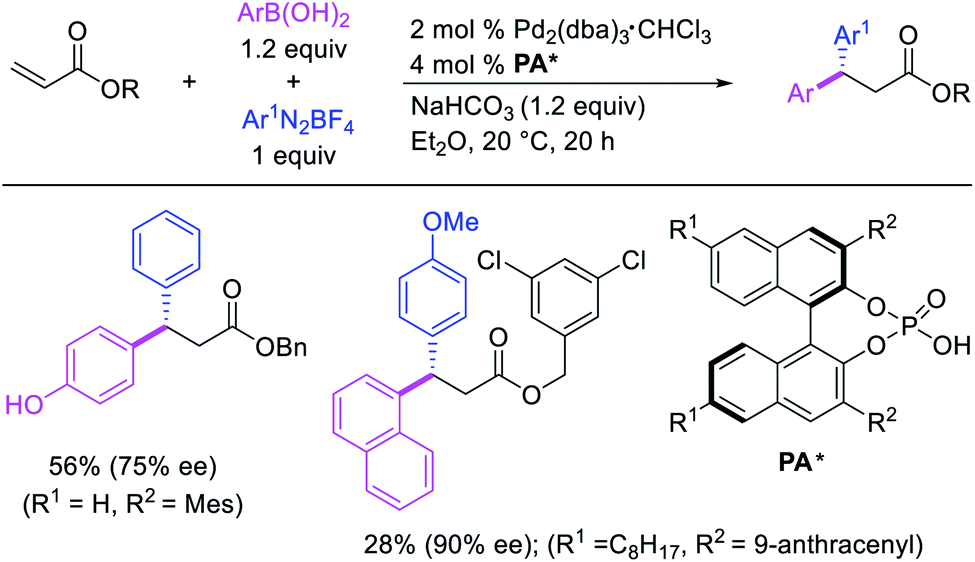
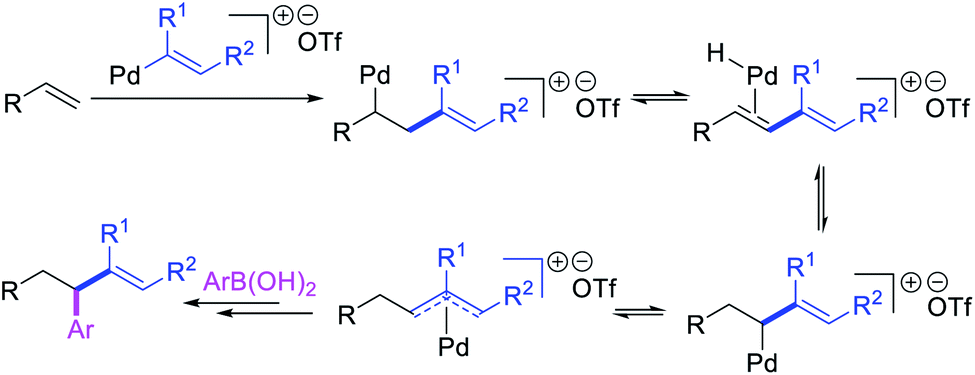
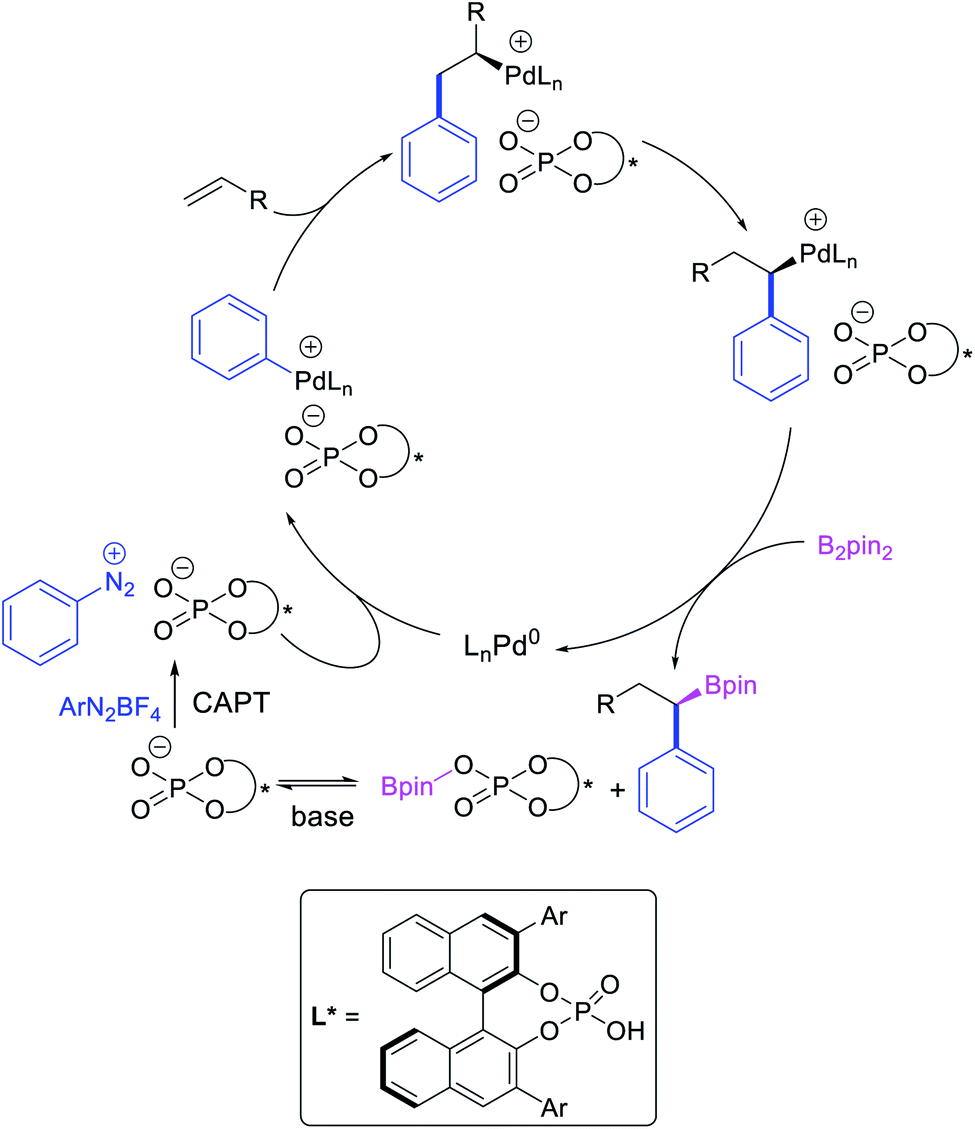
Several research reports have shown that the rearranged alkylmetal intermediates can be intercepted with carbon-based coupling partners, which lead to the generation of two geminal C–C bonds. The formation of 1,1-dicarbofunctionalized products by a metal-catalyzed difunctionalization of alkenes in α,β-unsaturated carbonyl compounds was first reported by Cacchi and Palmieri in 1984 (Scheme 3).2 This Pd-catalyzed reaction of activated alkenes with aryl halides was proposed to follow a double Heck reaction mechanism in which the first Heck product further underwent a second Heck reaction. In 2016, Toste, Sigman et al. also reported a Pd-catalyzed 1,1-diarylation of α,β-unsaturated esters with aryldiazonium salts and arylboronic acids, a reaction that proceeded enantioselectively in the presence of a chiral anion phase transfer (CAPT) ligand (Scheme 4).14 However, unlike Cacchi and Palmieri's 1,1-diarylation, this reaction was proposed to proceed with the formation of rearranged alkylpalladium(II) intermediates by β-H elimination from the original alkylpalladium(II) species followed by [Pd]–H reinsertion (Scheme 5).15
 |
| | Scheme 3 Pd-catalyzed 1,1-diarylation of conjugated alkenes with aryl iodides. | |
 |
| | Scheme 4 Pd-catalyzed 1,1-diarylation of conjugated alkenes with arylboronic acids and aryldiazonium salts. | |
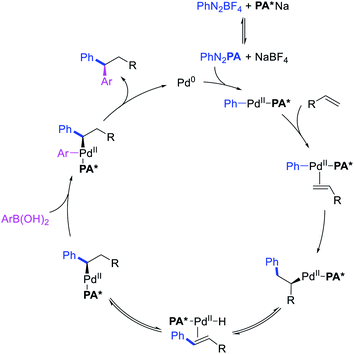 |
| | Scheme 5 Mechanism of the Pd-catalyzed 1,1-diarylation. | |
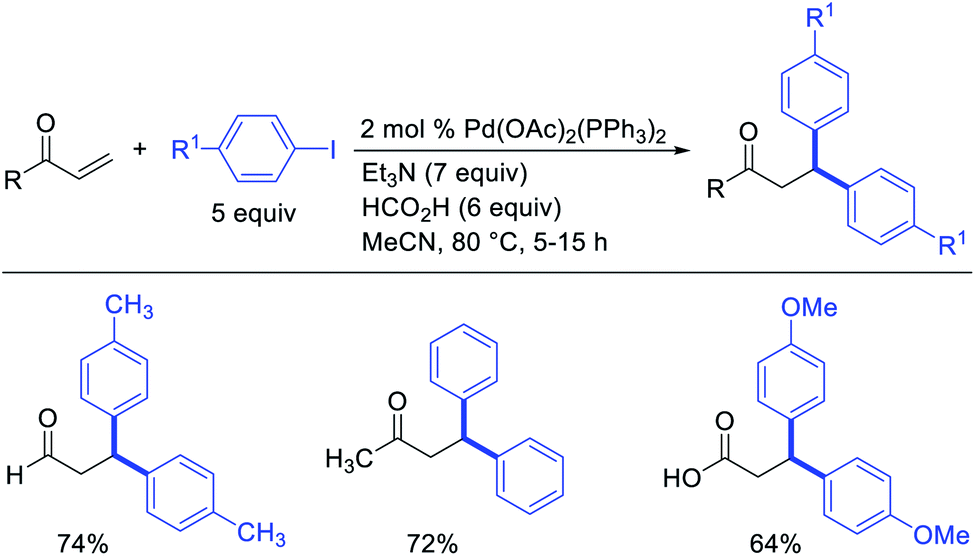
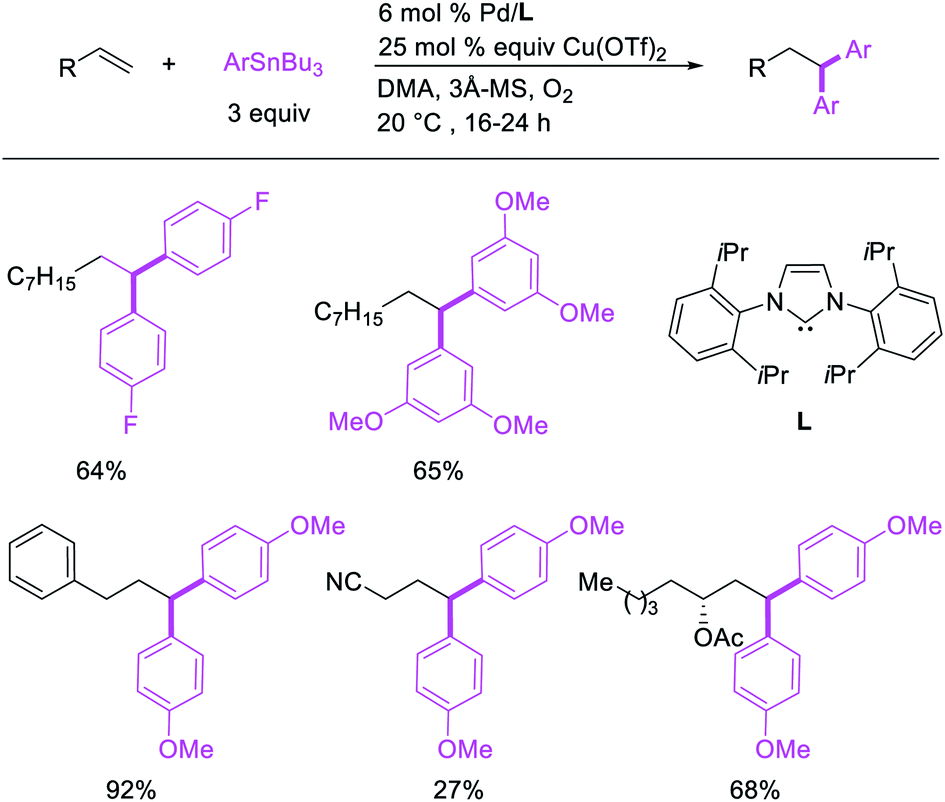
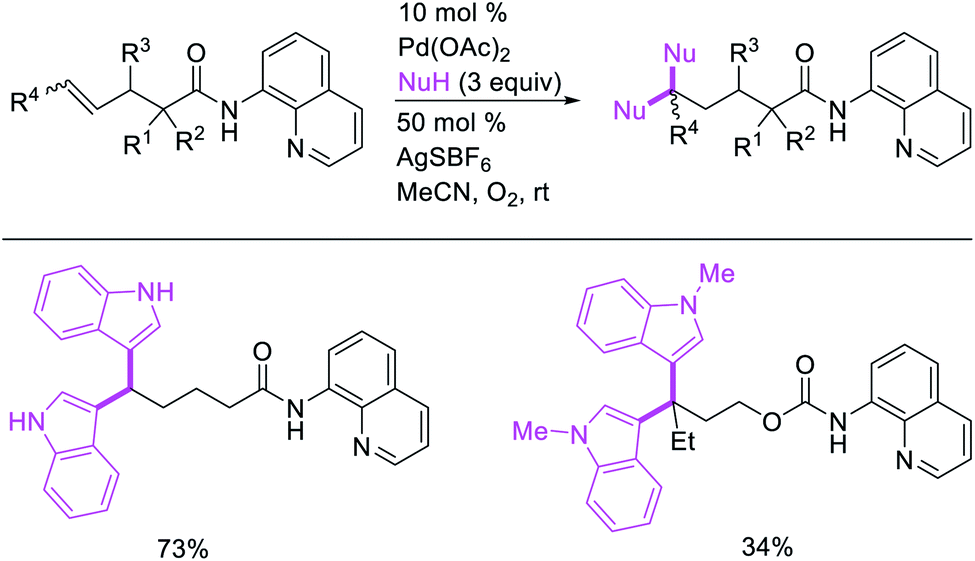
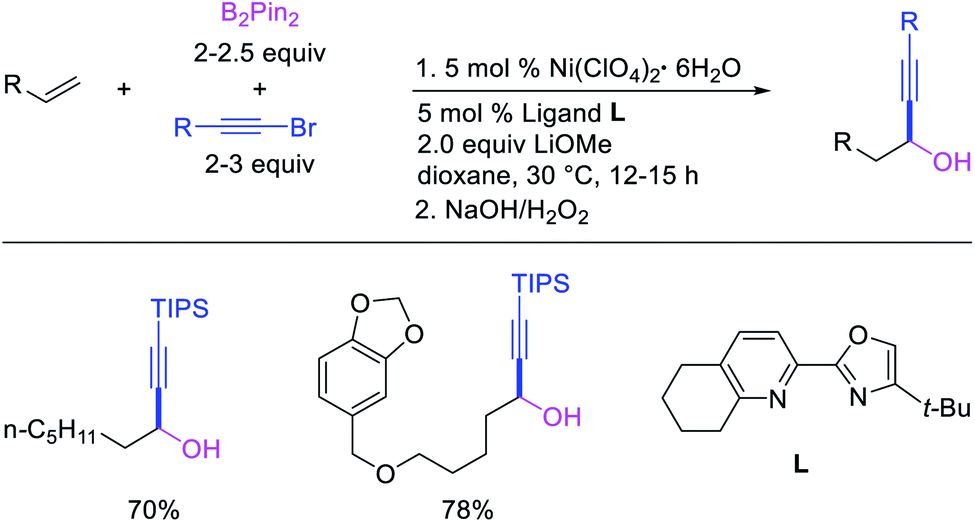
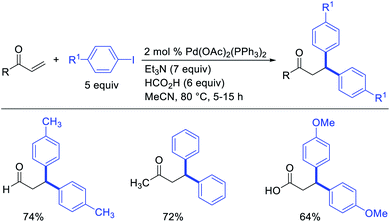
Metal-catalyzed 1,1-dicarbofunctionalization of unactivated alkenes was first reported by Bras et al. in 2008.16 The Pd-catalyzed reaction difunctionalized alkenes with 2-alkylfurans furnishing 1,1-difurylated products (Scheme 6). The 1,1-difurylated products were formed as a mixture of regioisomers at both termini of the alkene vicinal carbons. Based on selectivity and deuterium labelling experiments, studies with Heck products and ESI-MS analysis of reaction products, the authors proposed a mechanism in which two furans were activated by electrophilic C–H cleavage (Scheme 7). The C–H activation was then followed by one furyl group addition to alkenes, β-H elimination and second furyl group addition prior to reductive elimination of the C–H bond from an alkylpalladium(II)–hydride species and without generating a rearranged palladium(II) intermediates. In 2009, Sigman et al. reported a similar Pd-catalyzed alkene 1,1-diarylation of 1-nonene with ArSnBu3 in the presence of Cu(OTf)2 as a co-catalyst (Scheme 8).17 Unlike Bras' reaction, this oxidative 1,1-difunctionalization reaction was proposed to follow a β-H elimination/[Pd]–H reinsertion pathway to generate rearranged alkylpalladium(II) species prior to interception by a second molecule of ArSnBu3. The rearrangement of the alkylpalladium(II) intermediate was confirmed by the migration of a deuterium in deuterium-labeled 1-nonene. The authors later expanded the scope of the reaction to functionalize a wide range of terminal alkenes bearing functional groups (Scheme 8).18 Recently, Baik and Hong et al. implemented the bidentate coordination of 8-aminoquinoline auxiliary to 1,1-diindolylation of terminal alkenes in alkenyl 8-aminoquinolinamides (Scheme 9).19 The use of 8-aminoquinolinyl coordinating group was crucial for nucleophilic carbopalladation of the alkene and rearrangement of the alkylpalladium(II) species to form π-allylpalladacycle. A second molecule of indole functioned as an intermolecular C-nucleophile to intercept the π-allylpalladim(II) intermediate. Moreover, Yin et al. also showed in 2019 that the conditions developed for 1,1-alkynylborylation described in Section 2.3 (Scheme 34) could be modified by substituting Ni(ClO4)2·6H2O in dioxane for NiI2 with a modified pyridyloxazoline ligand in NMP in order to produce 1,1-dialkynylated products with alkynyl bromides and B2pin2 (Scheme 10).20 In this reaction, the authors proposed that the in situ generated alkynylboryl products would further undergo a Ni-catalyzed Suzuki coupling to form the dialkynylated products.
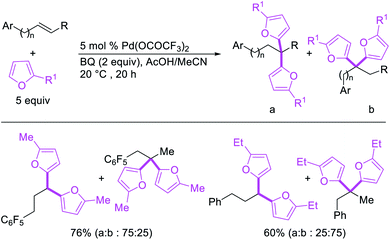 |
| | Scheme 6 Pd-catalyzed 1,1-difurylation. | |
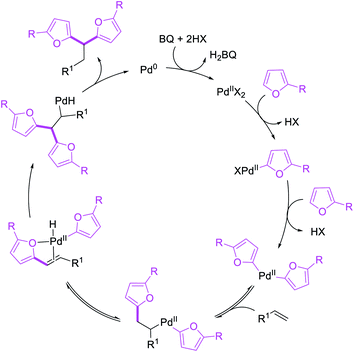 |
| | Scheme 7 Mechanism of the Pd-catalyzed 1,1-difurylation. | |
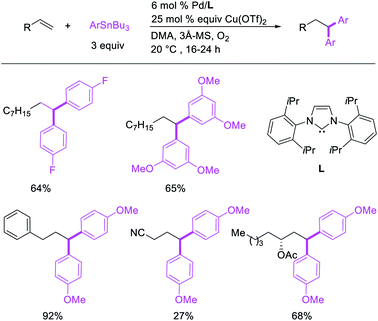 |
| | Scheme 8 Pd-catalyzed 1,1-homodiarylation with aryltin reagents. | |
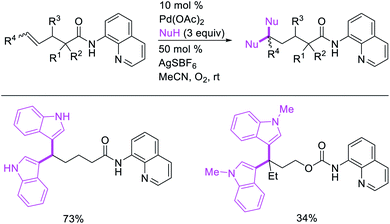 |
| | Scheme 9 Pd-catalyzed 1,1-diindolylation. | |
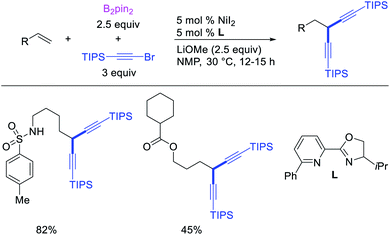 |
| | Scheme 10 Pd-catalyzed 1,1-dialkynylation. | |
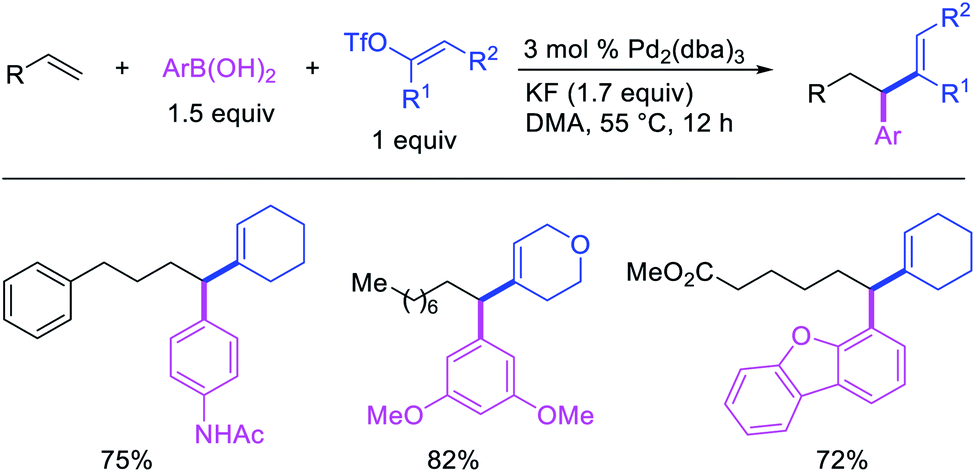
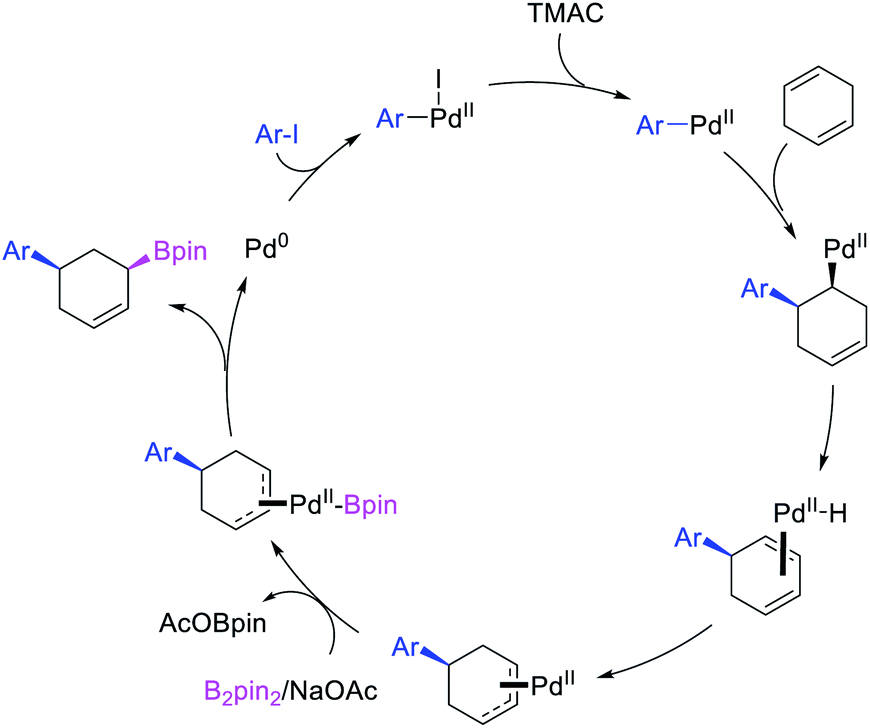
Alkene 1,1-dicarbofunctionalization was mainly reported for the addition of two identical carbon-based groups at the geminal position of alkenes as discussed above. In 2011, Sigman et al. disclosed the first example of 1,1-dicarbofunctionalization of unactivated alkenes with two different coupling partners.21 In this alkene difunctionalization, Pd2(dba)3 catalyzed a reaction of terminal alkenes with arylboronic acids and vinyl triflates (Scheme 11). The authors proposed that the reaction would proceed with the formation of π-allylpalladium(II) intermediate upon addition to alkenes of the vinylpalladium(II) species generated by oxidative addition of vinyl triflates to Pd(0) (Scheme 12). The allylpalladium(II) was then intercepted by transmetallation with arylboronic acids to form the products after reductive elimination. With the replacement of KF by NaHCO3 as a base, the authors later extended the reaction to 1,1-difunctionalize ethylene with aryl-, heteroaryl- and vinylboronic acids or their pinacol esters and vinyl triflates and nonaflates (Scheme 13).22 Additionally, the authors further demonstrated that the vinyl triflates could also be replaced with aryldiazonium salts in order to 1,1-diarylate the unactivated alkenes when DMA was exchanged for tBuOH as a solvent (Scheme 14).23 Unlike prior 1,1-diarylation reactions, this method furnished 1,1-difunctionalized products with two dissimilar aryl groups.
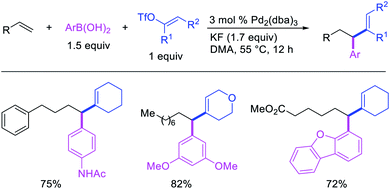 |
| | Scheme 11 Pd-catalyzed 1,1-arylvinylation. | |
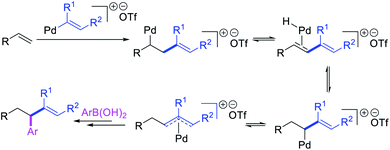 |
| | Scheme 12 Mechanism of the Pd-catalyzed 1,1-arylvinylation. | |
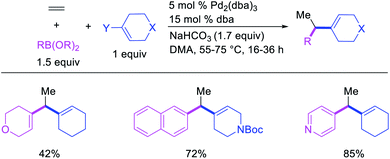 |
| | Scheme 13 Pd-catalyzed 1,1-divinylation and 1,1-diarylation of ethylene. | |
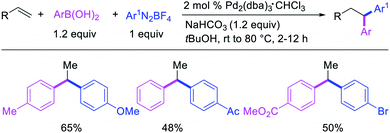 |
| | Scheme 14 Pd-catalyzed 1,1-diarylation. | |
2.2. 1,1-C–C/C–halogen, O, N functionalization
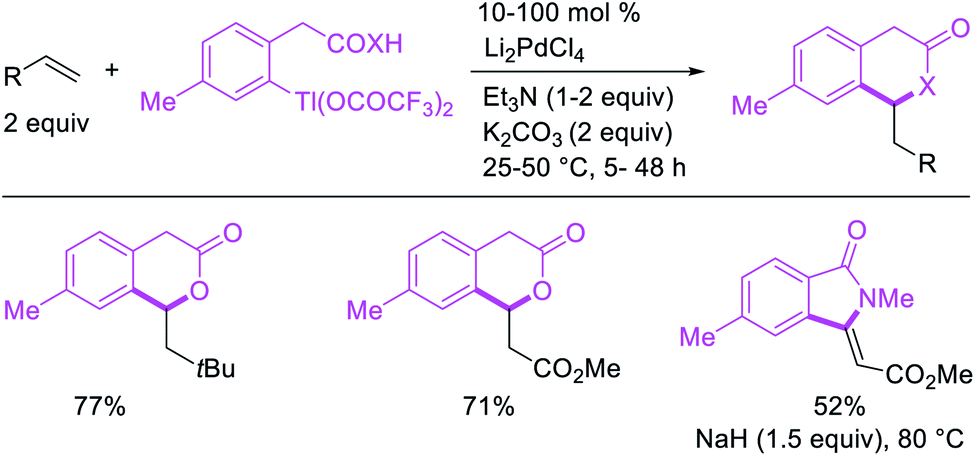
The alkene 1,1-difunctionalization by metal walking was first observed by Bäckvall et al. in 1980 during mechanistic studies on the oxidative cleavage of a carbon–palladium bond generated upon the addition of PhPd(II) complex to ethylene (Scheme 15).13 In this catalytic reaction, the 1,1-arylacetoxylation product was formed after Pd migration to benzylic position by β-H elimination from the 2-phenethylpalladium(II) species and [Pd]–H reinsertion in a manner reminiscent of the mechanism of the Wacker process. Similar 1,1-arylcarboxylation and 1,1-arylcarboamination reactions were also observed later by Larock et al.24 in 1984 during the olefination of arylthallium complexes with Li2PdCl4 in which the rearranged benzylpalladium(II) intermediate was intercepted intramolecularly with ortho-carboxylate and ortho-carboxylamide to form six-membered lactones and lactams (Scheme 16).24
 |
| | Scheme 15 Pd-catalyzed 1,1-aryloxygenation with phenylmercury reagent. | |
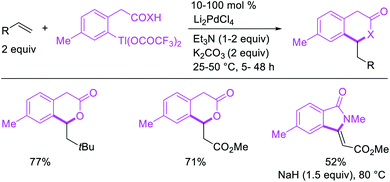 |
| | Scheme 16 Pd-catalyzed 1,1-aryloxygenation with arylthallium reagent. | |
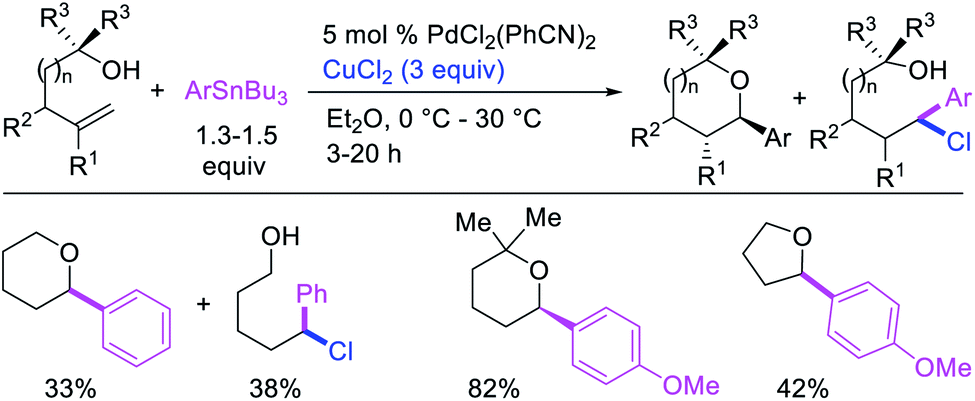
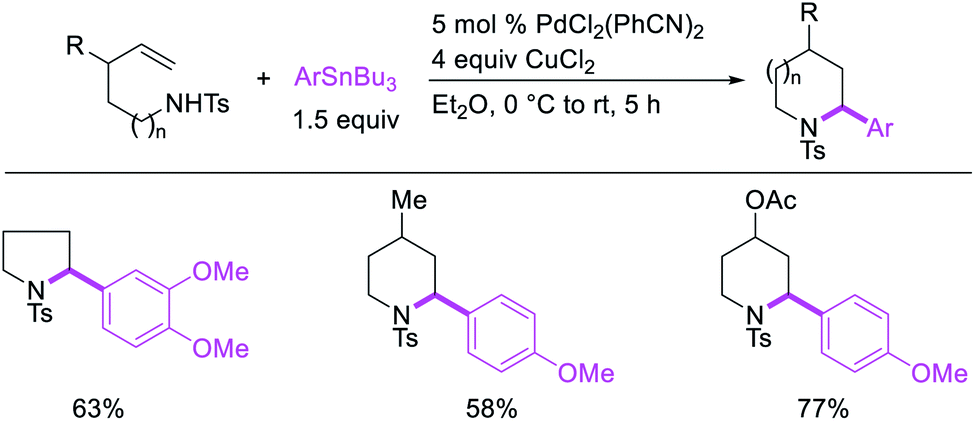
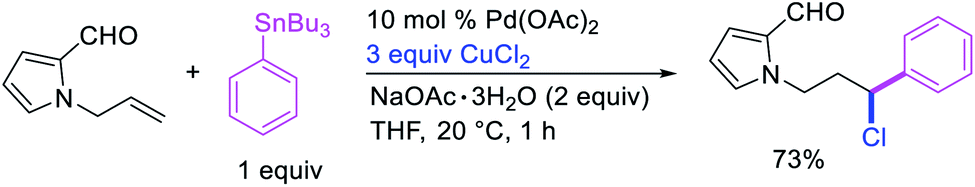
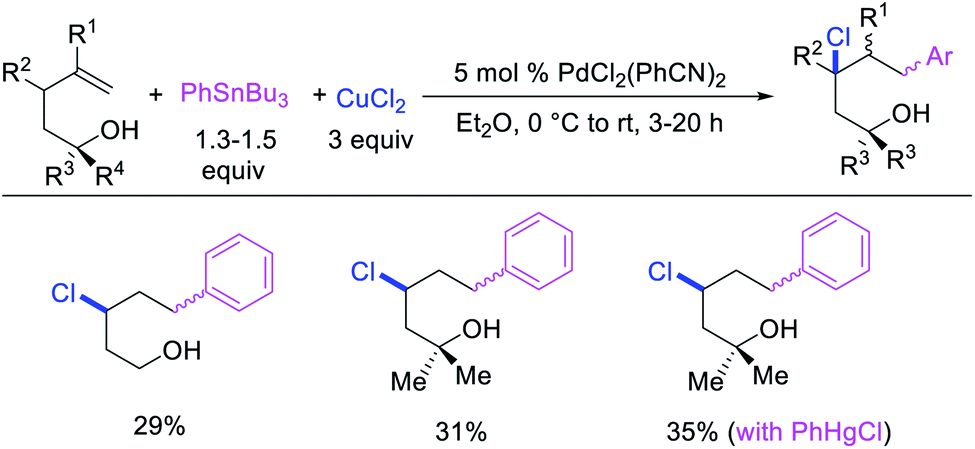
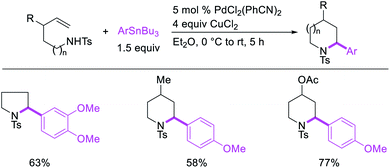
Catalytic variations of the alkene 1,1-difunctionalization reactions appeared in 1986 when Yoshida et al.5a disclosed 1,1-arylchlorination and 1,1-aryloxygenation of alkenyl alcohols with ArSnBu3 and excess of CuCl2 in the presence of 5 mol% PdCl2(PhCN)2 (Scheme 17).5a The reaction formed a mixture of both products or favoring only 1,1-arylchlorination with PhSnBu3 while 1,1-aryloxygenation was predominantly formed with 4-MeOC6H4SnBu3. The authors also observed a similar electronically biased selectivity with alkenyl tosylamides when oxygen was replaced with nitrogen as an intramolecular nucleophile (Scheme 18).5b In 2002, Jung et al. also reported a single case of similar electronically controlled Pd-catalyzed 1,1-phenylchlorination in N-allyl-2-pyrrole carbaldehyde during their studies of oxidative Heck reactions with PhSnBu3 as an unintended consequence of the addition of CuCl2 as an oxidant (Scheme 19).25 These early studies with mechanistic understanding and catalytic conditions have led to better reactions recently, which has enabled selective interception of rearranged alkylmetal intermediates for 1,1-difunctionalization reactions.
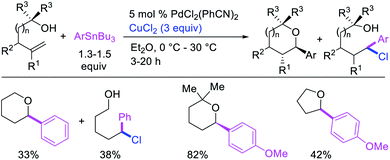 |
| | Scheme 17 Pd-catalyzed 1,1-aryloxygenation and 1,1-arylchlorination. | |
 |
| | Scheme 18 Pd-catalyzed 1,1-arylamination. | |
 |
| | Scheme 19 Pd-catalyzed 1,1-phenylchlorination. | |
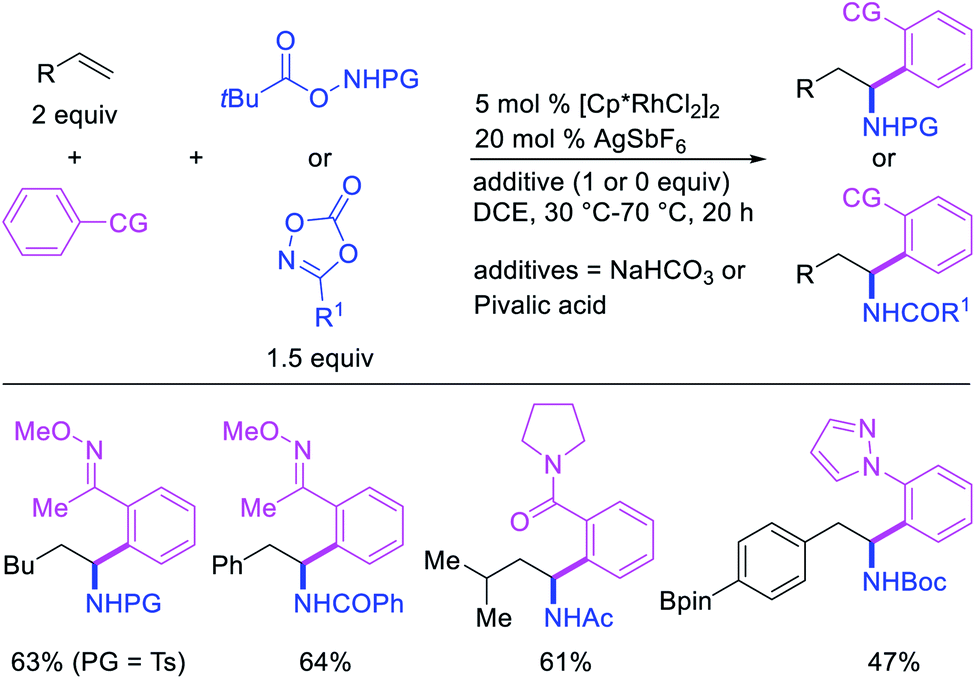
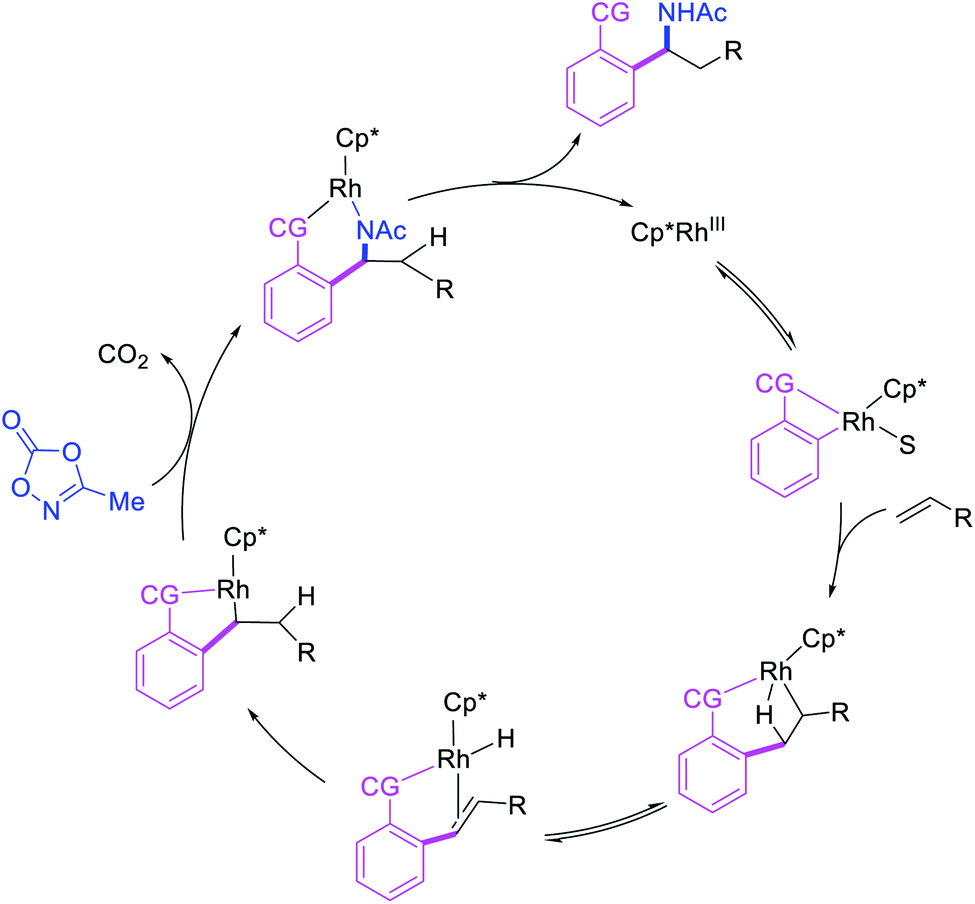
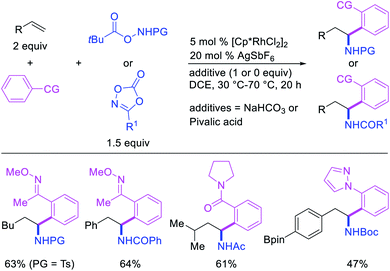
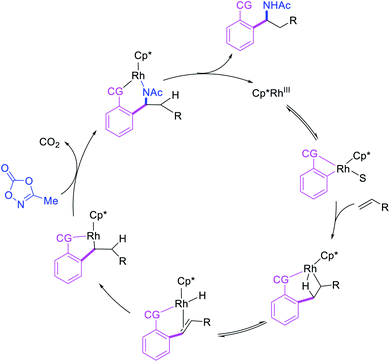
In 2017, Ellman et al. disclosed a Rh-catalyzed 1,1-arylamination of alkenes (Scheme 20).26 In this reaction, ortho-C–H bond in a wide range of arenes bearing a coordinating group was added to alkenes in the presence of aminating reagents to furnish the 1,1-difunctionalized products. Mechanistically, the author proposed that the Ar–Rh intermediates generated upon C–H activation would undergo migratory insertion of alkenes to form a benzylrhodium species, which subsequently underwent Rh migration by β-H elimination/Rh–H reinsertion (Scheme 21). The migrated Rh-species was then intercepted by the aminating reagents to form the 1,1-arylaminated products.
 |
| | Scheme 20 Rh-catalyzed 1,1-arylamination of alkenes via C–H activation. | |
 |
| | Scheme 21 Mechanism of the Rh-catalyzed alkene 1,1-arylamination via C–H activation. | |
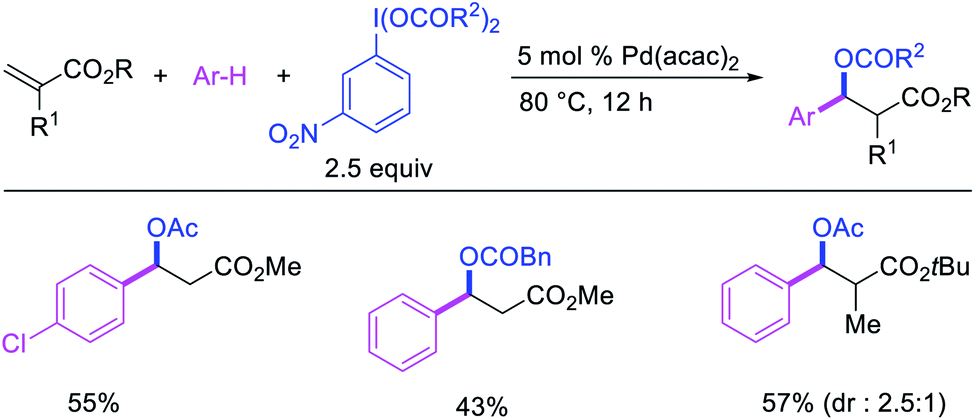
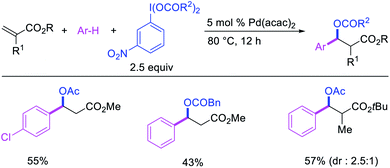
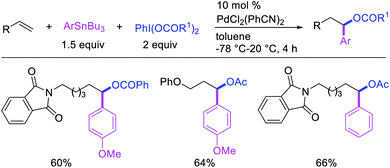
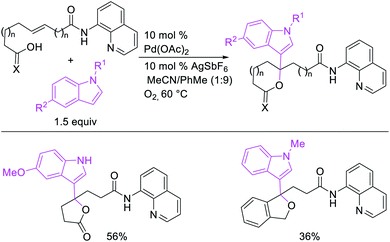
In 2009, Moran et al. utilized hypervalent iodine reagents and arenes as an oxygen and aryl sources for a Pd-catalyzed 1,1-aryloxygenation reaction of the α,β-unsaturated alkenes in acrylate esters (Scheme 22).27 The authors proposed that the reaction would proceed by β-H elimination/[Pd]–H reinsertion and reoxidation of the rearranged Pd(II) intermediate to Pd(IV) with the hypervalent iodine reagent followed by reductive elimination of the final products. Building up their group's chemistry on Pd-catalyzed reactions with hypervalent reagents, Sanford et al. also demonstrated that PhI(OCOR)2 in combination with ArSnBu3 reagents would selectively difunctionalize unactivated alkenes to generate 1,1-aryloxygenation products (Scheme 23).28 More recently, Baik and Hong et al. reported a Pd-catalyzed 1,1-aryloxygenation of unactivated alkenes in β,γ-alkenyl-8-aminoquinolinamides bearing a terminal hydroxyl group, which formed five-and six-membered oxygen heterocycles (Scheme 24).19 This reaction proceeded under the conditions developed for 1,1-diindolylation described in Section 2.1 but the rearranged alkylpalladium(II) intermediate was intercepted with an intramolecular O-nucleophile rather than a second molecule of indole.
 |
| | Scheme 22 Pd-catalyzed 1,1-aryloxygenation via C–H activation. | |
 |
| | Scheme 23 Pd-catalyzed 1,1-aryloxygenation involving hypervalent iodine reagents. | |
 |
| | Scheme 24 Pd-catalyzed 1,1-aryloxygenation using indoles. | |
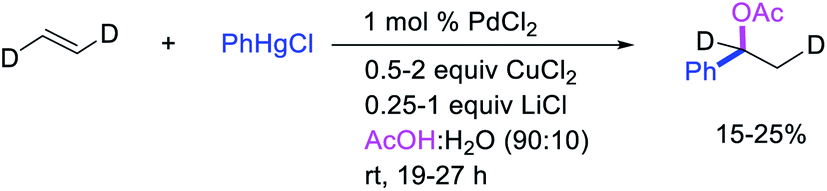
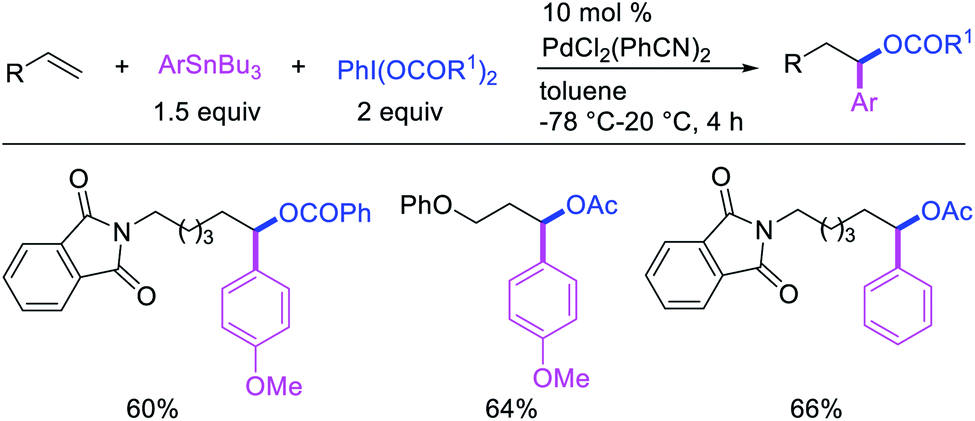
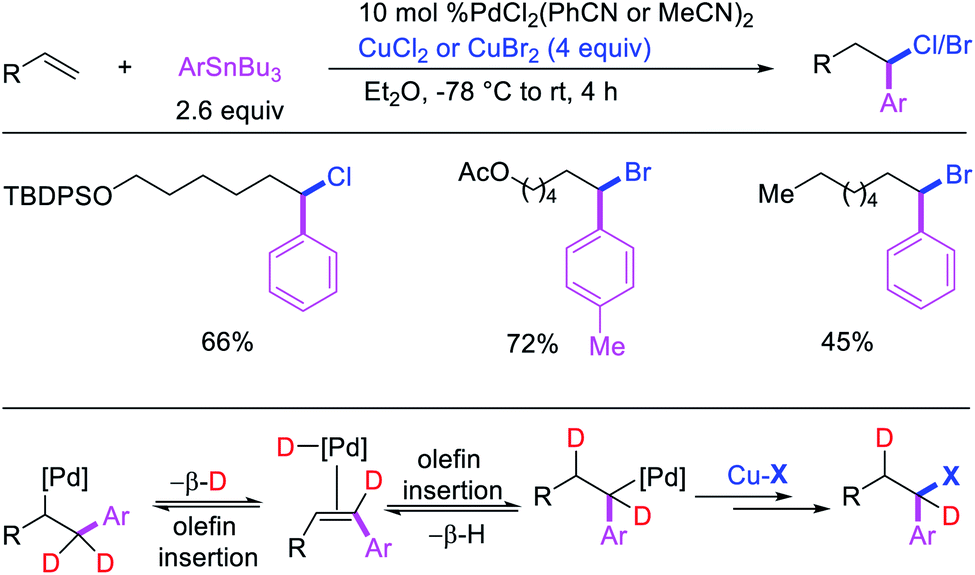
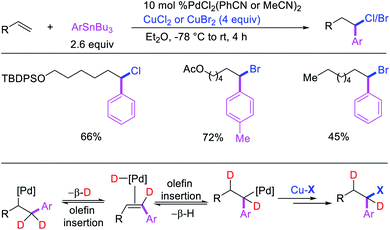
Highly selective Pd-catalyzed 1,1-arylhalogenation reactions of unactivated alkenes were disclosed by Sanford et al. with the selection of appropriate electrophilic reagents. In 2008, the authors reported 1,1-arylchlorination of unactivated terminal alkenes at the terminal vinylic carbon in a three-component process with ArSnBu3 and excess of CuCl2 at −78 °C (Scheme 25).29 As further synthetic appeal, the authors also demonstrated that CuCl2 could be replaced with CuBr2 to generate 1,1-arylbrominated products.30 The high selectivity for 1,1-difunctionalization was attributed to the mild electrophilicity of CuX2 (X = Cl, Br) which reacted with a slower rate for oxidative cleavage of the alkylpalladium(II) bond than the rate of β-H elimination/[Pd]–H reinsertion to generate a more stable benzylpalladium(II) intermediate and prime the rearranged intermediate for benzylic halogenation (Scheme 25). The Pd walking was further supported by a deuterium-labeling experiment in which a deuterium cleanly migrated from the terminal position of 1-octene-(1,1-d2) to the homobenzylic position of the 1,1-arylhalogenated product.
 |
| | Scheme 25 Pd-catalyzed 1,1-arylhalogenation with CuX2 as halide source. | |
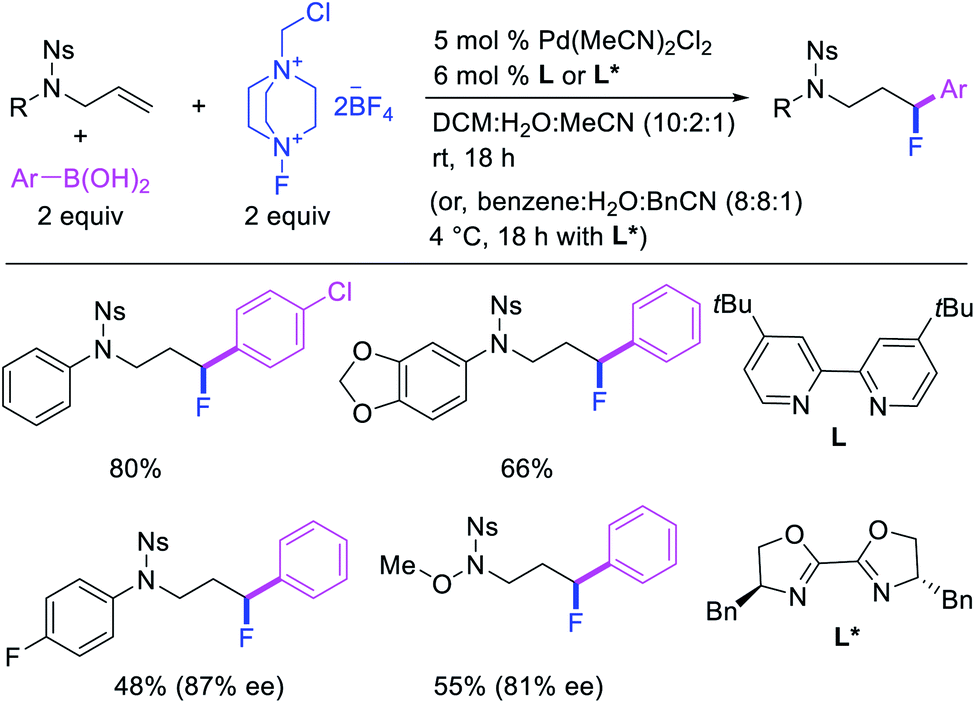
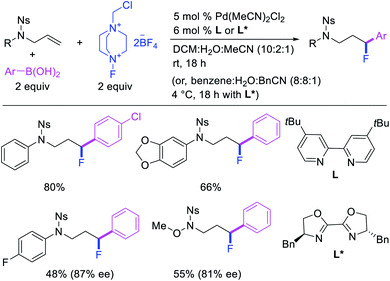
In 2015, Toste et al. utilized Selectfluor as a strong electrophilic fluorinating reagent in the presence of a Pd/tBuBiPy catalyst for 1,1-arylfluorination of N-allylnosylamides (Scheme 26).31 The reaction proceeded by a β-H elimination/[Pd]–H reinsertion sequence to rearrange the original alkylpalladium(II) species to a benzylpalladium(II) intermediate, which was likely subsequently oxidized to Pd(IV) by Selectfluor prior to reductive elimination.32 The authors further demonstrated that tBuBiPy ligand could be replaced with a chiral, nonracemic bisoxazoline ligand for enantioselective 1,1-arylfluorination, which remarkably furnished highly enantioenriched products.
 |
| | Scheme 26 Pd-catalyzed 1,1-arylfluorination. | |
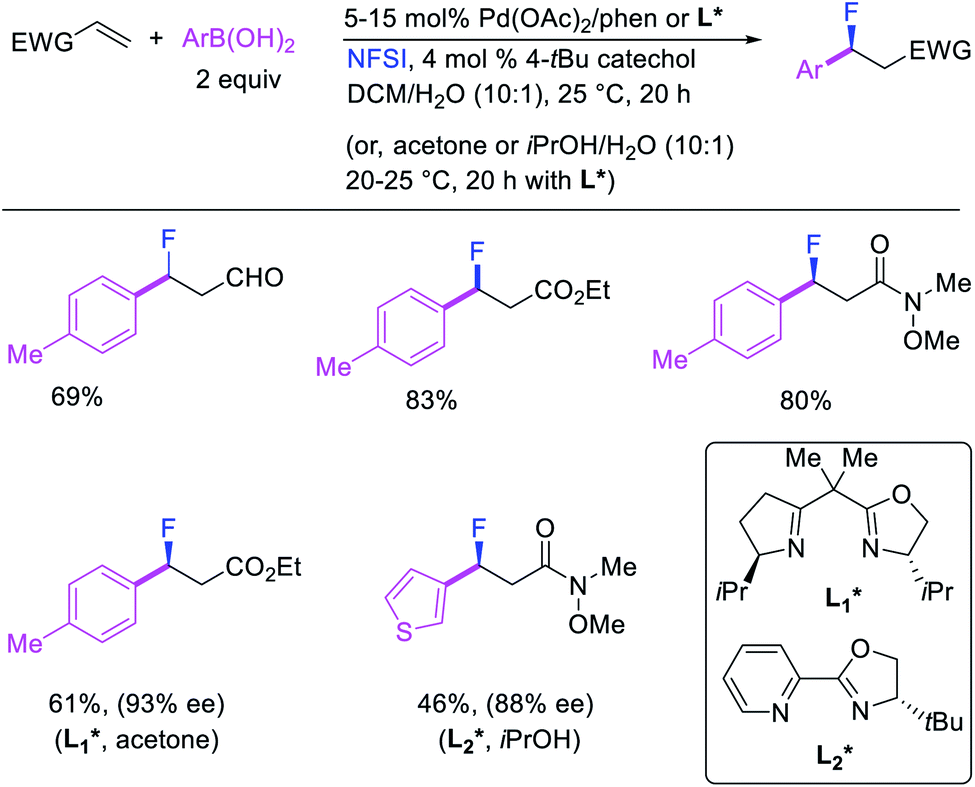
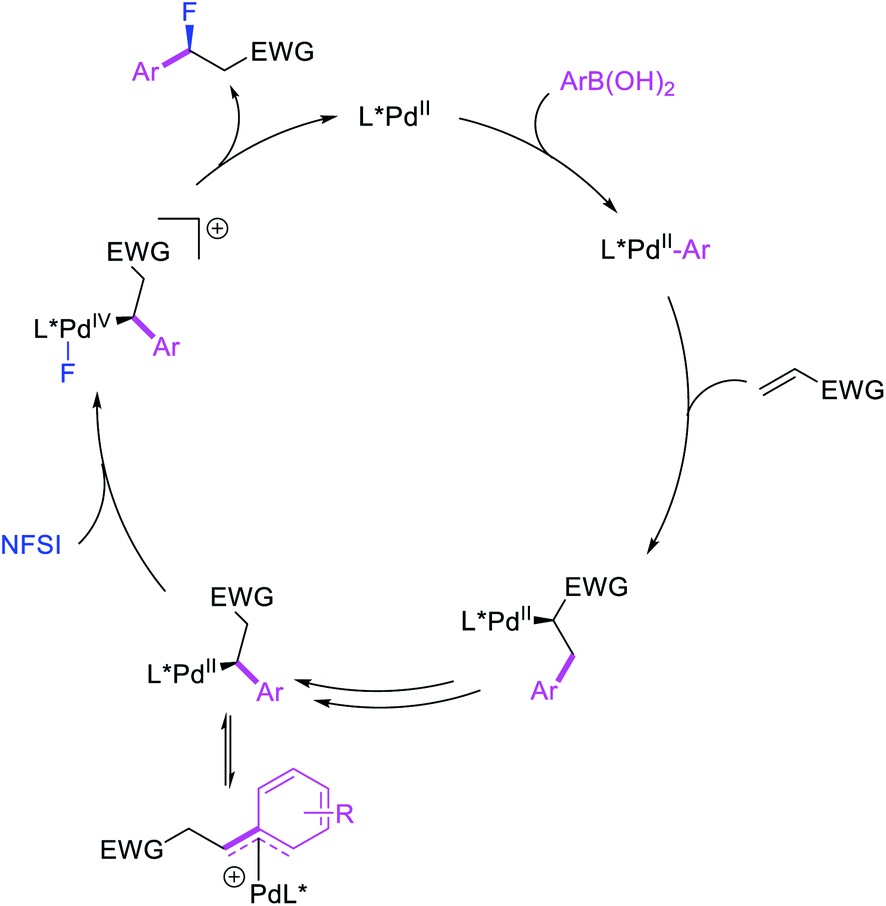
More recently, Pozo, Toste and Fustero et al. developed a Pd/phen-catalyzed 1,1-arylfluorination of α,β-unsaturated carbonyl compounds with arylboronic acids and N-fluorobenzenesulfonamide (NFSI) (Scheme 27).33 The reaction could utilize a variety of α,β-unsaturated esters, acids, amides, nitriles, ketones and aldehydes to functionalize the activated alkenes. The authors further demonstrated that the use of chiral, nonracemic bisoxazoline and pyridyloxazoline ligands could also render the reaction enantioselective and furnish the products with good enantioselectivity. The authors proposed that the reaction would likely proceed via an oxidative Heck-type mechanism through the formation of Pd(IV) fluoride intermediates (Scheme 28).
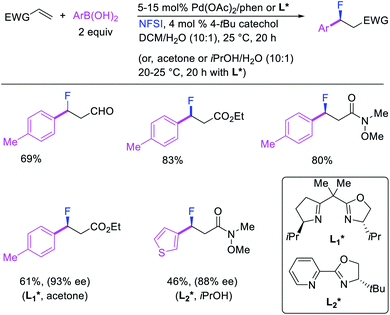 |
| | Scheme 27 Pd-catalyzed 1,1-arylfluorination of activated alkenes. | |
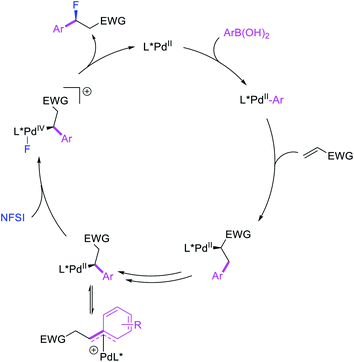 |
| | Scheme 28 Mechanism of the 1,1-arylfluorination. | |
2.3. 1,1-C–C/C–B and C–B/C–B functionalization
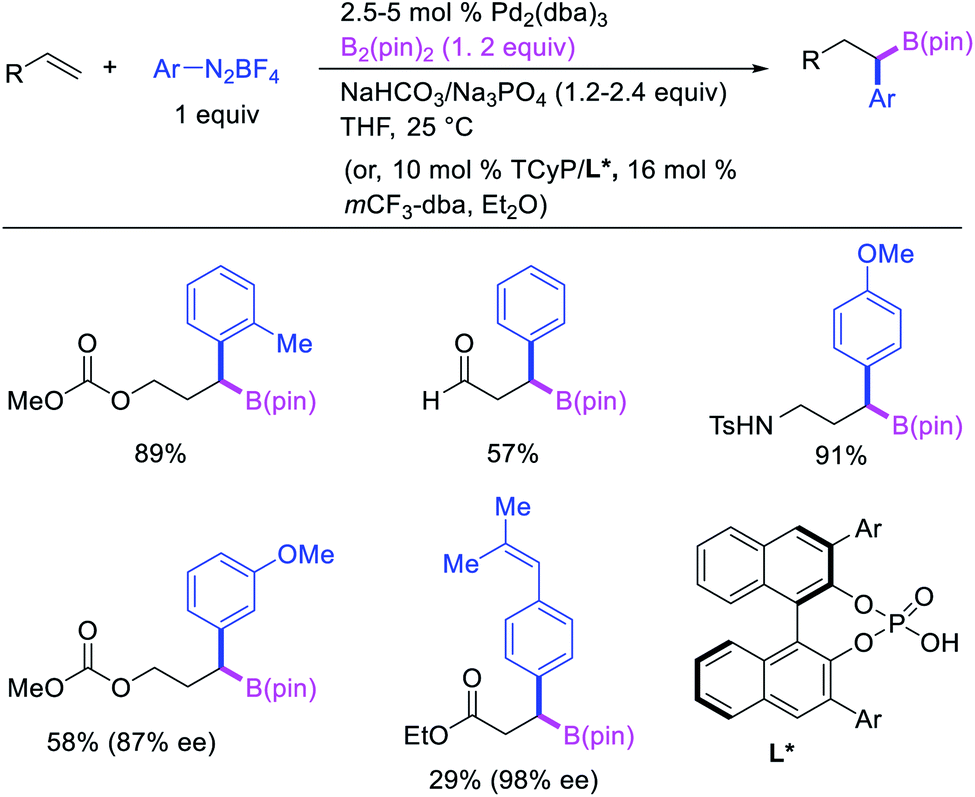
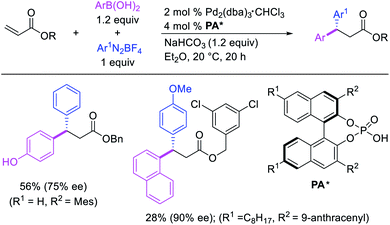
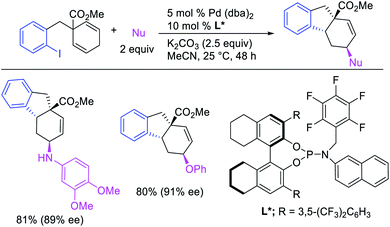
Geminal difunctionalization of an alkene with the formation of C–C and C–B bonds was first reported by Toste et al. in early 2015.34 The reaction of both unactivated alkenes and activated alkenes in α,β-unsaturated carbonyl compounds with aryldiazonium salts and bis(pinacolato)diboron was catalyzed by Pd2(dba)3, which furnished 1,1-arylborylated products under “ligand-free” conditions (Scheme 29). The authors also developed an enantioselective variant of the reaction in which they implemented cooperative catalysis between a TCyP chiral anion phase transfer (CAPT) catalyst and the Pd catalyst, an unprecedented approach which produced 1,1-arylborylated products in good to excellent enantioselectivity. The reaction was proposed to proceed with the formation of a cationic arylpalladium(II) intermediate with the chiral phosphate as a counter anion, which subsequently followed a migration insertion, β-H elimination and [Pd]–H reinsertion sequence to set the stereocenter (Scheme 30).
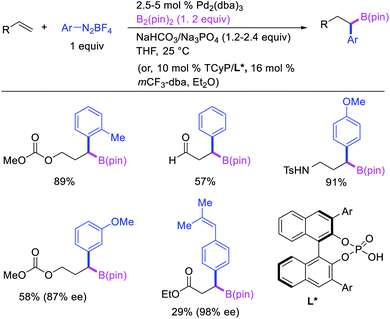 |
| | Scheme 29 Pd/CAPT-catalyzed 1,1-arylborylation. | |
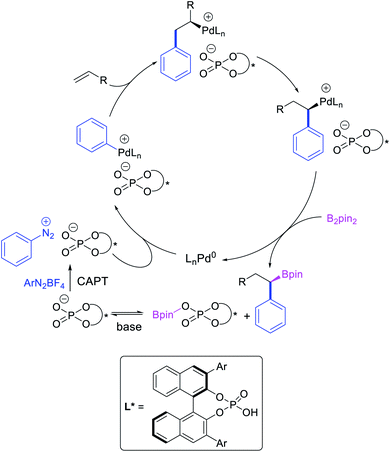 |
| | Scheme 30 Mechanism of the Pd/CAPT-catalyzed 1,1-arylborylation. | |
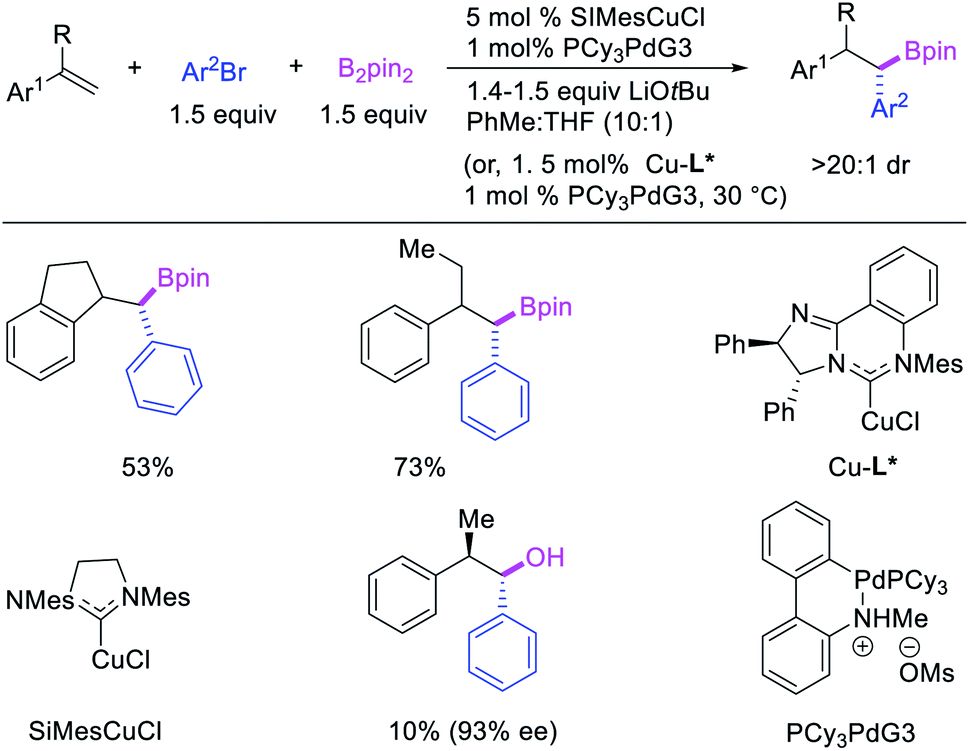
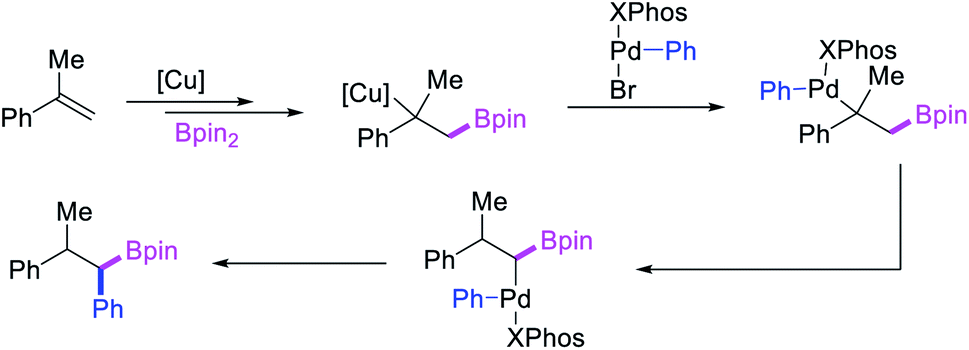
In 2019, Brown et al. developed a Cu/Pd cooperative catalysis for 1,1-arylboration of vinyl arenes with B2pin2 and ArBr (Scheme 31).35 The reaction was catalyzed by a combination of SIMesCuCl and XPhosPdG3 in which Cu and Pd activated B2pin2 and ArBr, respectively. A single case of a reaction was also demonstrated to proceed with enantiocontrol. The impressive cooperative catalysis was further confirmed by generating reactive species in situ in stoichiometric reactions wherein alkyl-CuSIMes species, formed upon borocupration of α-methylstyrene, transmetallated with PhPdBr (Scheme 32). The resultant alkyl-Pd(II) species further underwent Pd walking proximal to the boron substituent by β-H elimination and [Pd]–H reinsertion steps prior to reductive elimination to form geminally arylborated products.
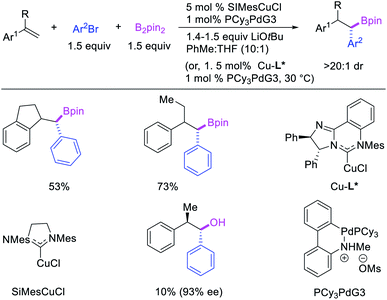 |
| | Scheme 31 Pd/Cu-catalyzed 1,1-arylboration. | |
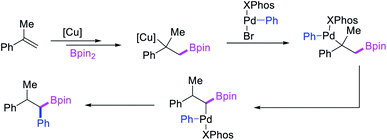 |
| | Scheme 32 Mechanism of the Pd/Cu-catalyzed 1,1-arylboration. | |
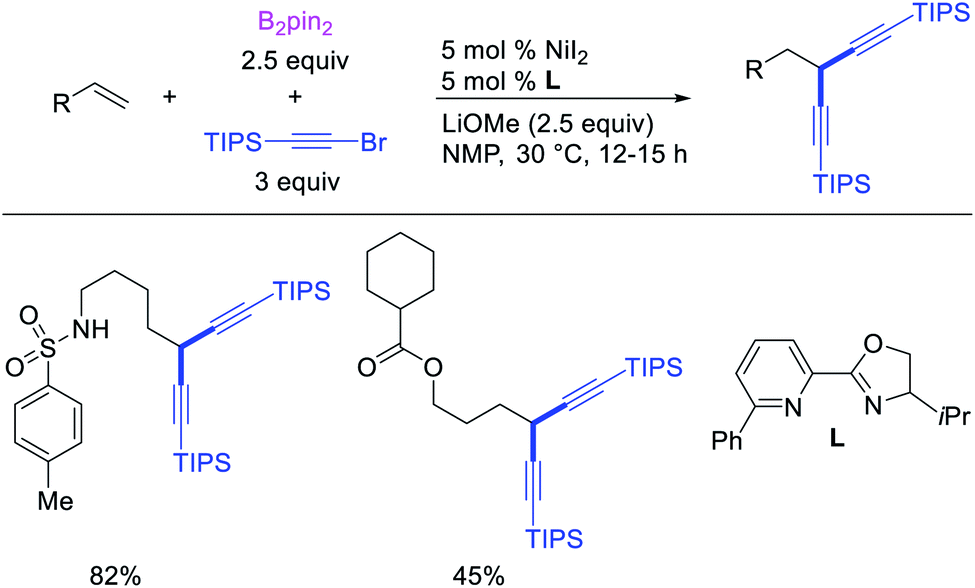
Recent studies have shown that Ni-catalysts are also effective in following a β-H elimination/[Ni]–H reinsertion sequence for 1,1-carboboration reactions. In 2019, Yin et al. demonstrated that a tridentate ligand-bound Ni(II) complex could perform 1,1-benzylboration of unactivated alkenes with benzyl bromides and B2pin2 (Scheme 33).36 The authors further showed with the replacement of the tridentate ligand by an appropriate pyridyloxazoline ligand, Ni(ClO4)2·6H2O could catalyze the reaction of alkynyl bromides and B2pin2 with unactivated alkenes to generate 1,1-alkynylborated products, which were further oxidized to alcohols prior to isolation (Scheme 34).20
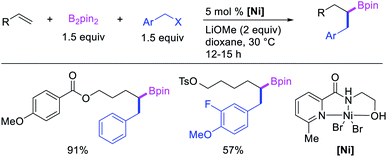 |
| | Scheme 33 Ni-catalyzed 1,1-benzylboration. | |
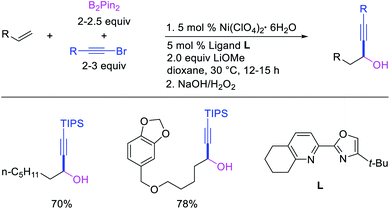 |
| | Scheme 34 Ni-catalyzed 1,1-alkynylboration (hydroxylation). | |
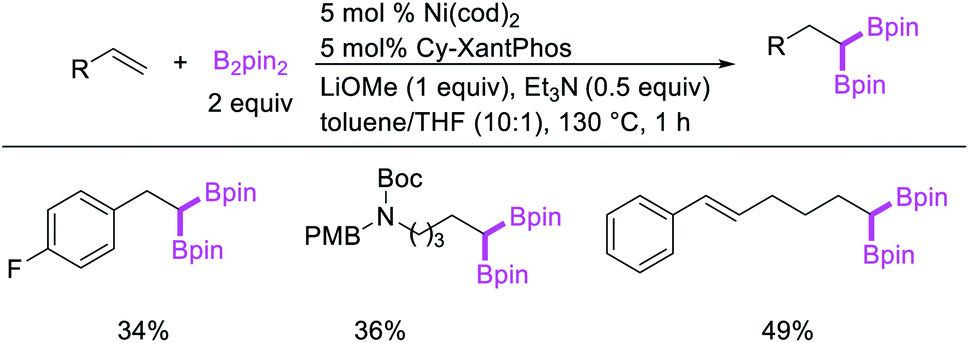
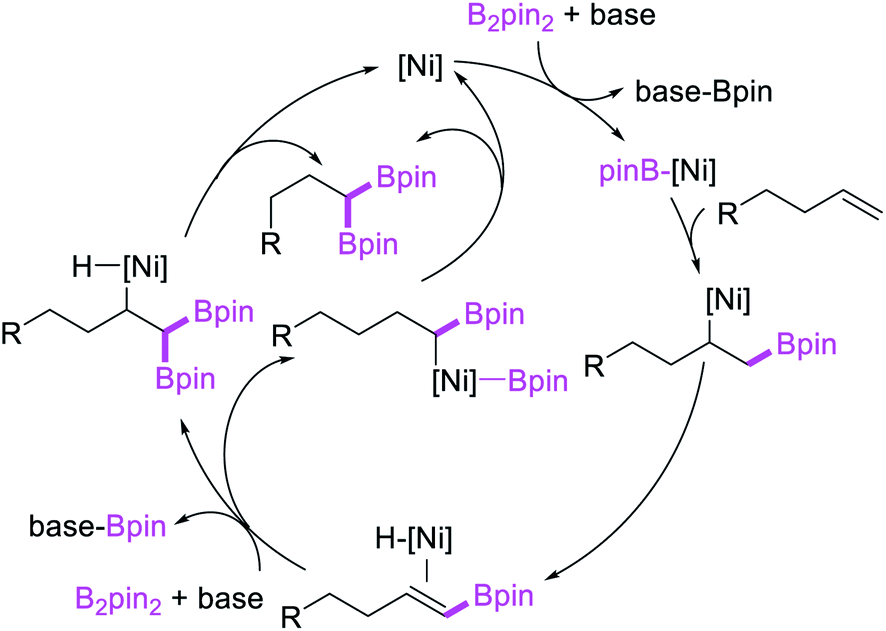
In 2017, Fu et al. developed a Ni/Cy-XantPhos-catalyzed 1,1-diboration of terminal alkenes with B2pin2 to furnish 1,1-diborylalkanes (Scheme 35).37 The reaction proceeded selectively with both unactivated alkenes and vinylarenes. The authors proposed that the reaction would proceed via boronickelation and β-H elimination steps followed by two possible insertion processes for the incorporation of the second boron group (Scheme 36).
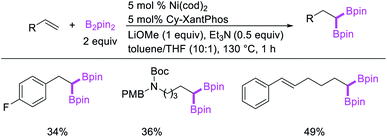 |
| | Scheme 35 Ni-catalyzed 1,1-diboration. | |
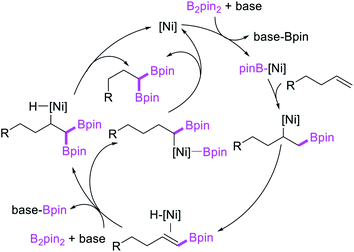 |
| | Scheme 36 Mechanism of the Ni-catalyzed 1,1-diboration. | |
3. 1,3-Difunctionalization
The alkene 1,3-difunctionalized products are observed after the formation of a rearranged alkylmetal intermediate from the original alkylmetal species, generated upon alkene carbometallation, by a β-H elimination/[M]–H reinsertion pathway. The formation of the 1,3-difunctionalized products are also thermodynamically driven by the generation of stable alkylmetal species like π-allylmetal intermediates analogous to the generation of alkylmetal intermediates for 1,1-difunctionalization. Therefore, alkene 1,3-difunctionalization is observed in substrates where molecular features such as alkenes or heteroatoms are already present that can stabilize the initially generated alkylmetal species such as by forming π-allylmetals and stable metallacycles. Thus far these reactions have generated 1,1-C–C/C–C, C–C/C–Hal, O, N, C–C/C–B and C–B/C–B bonds. These reactions are discussed in details in the subsequent sections.
3.1. 1,3-C–C/C–C functionalization
In 1982, Larock et al.4 and Heck et al.6 independently reported the migration of palladium after alkene carbopalladation, which could be subsequently intercepted by nucleophiles to generate 1,3-difunctionalized products in a three-component fashion. The π-allylpalladium(I) intermediates under Heck's reaction conditions were intercepted by amines, and the reaction is discussed in details in Section 3.2. Under Larock's conditions, norbornadiene was carbopalladated with a thiophenylpalladium(II) complex, derived from the reaction of a thiophenylmercury complex and PdCl2, which generated a nortricyclylpalladium(II) complex upon the migration of palladium prompted by the presence of the additional alkene (Scheme 37). The nortricyclylpalladium(II) intermediate could then be reacted with alkynyllithium to form 1,3-alkynylarylated product. In 1991, Larock et al. also disclosed a single case of catalytic three-component 1,3-dicarbofunctionalization of 1,4-cyclohexadiene with phenyl iodide and diethyl malonate (Scheme 38).38 Although the practical implication of the three-component 1,3-difunctionalization method was less significant at the moment due to the formation of the product in low yields (Heck) and the limited substrate scope or the requirement of stoichiometric amounts of palladium (Larock), Larock and Heck's independent observations invited a new trend in catalytic three-component difunctionalization of alkenes. Later, Larock extensively extended the method to three-component 1,n-difunctionalization (discussed in Section 4.1) and two-component 1,3-cyclization/coupling reactions.
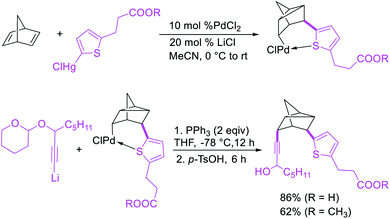 |
| | Scheme 37 Pd-catalyzed 1,3-alkynylarylation. | |
 |
| | Scheme 38 Pd-catalyzed 1,3-difunctionalization of 1,4-cyclohexadiene. | |
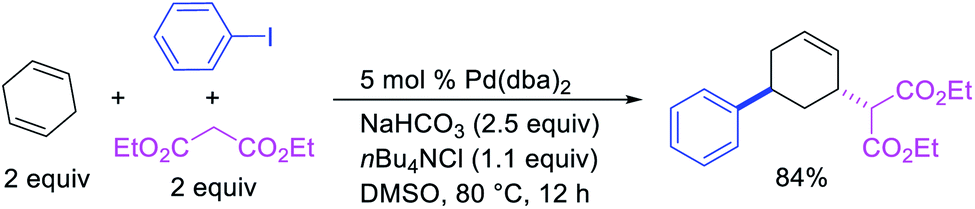
Larock et al. expanded the Pd-catalyzed 1,3-dicarbofunctionalization of 1,4-cyclohexadiene to cyclization/coupling (Scheme 39).39 In this two-component variant, 1,4-cyclohexadiene derivatives bearing tethered aryl and vinyl iodides underwent a Pd-catalyzed cyclization followed by the migration of Pd to generate π-allylpalladium(II) intermediates (Scheme 40). These Pd-complexes subsequently proceeded with anti-nucleophilic attack by diethyl malonate to generate 1,3-difunctionalized products. While the two-component reaction could functionalize a number of substituted 1,4-cyclohexadiene derivatives, the scope of the carbon nucleophile was limited to diethyl malonate. In 2019, Wu et al. developed an enantioselective version of the 1,3-cyclization/coupling process (Scheme 41).40 In this reaction, the authors used catalytic Pd(dba)2 in combination with a chiral nonracemic phosphoramidite ligand, which furnished products in good to excellent enantioselectivity with different malonate nucleophiles.
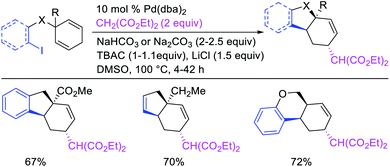 |
| | Scheme 39 Pd-catalyzed 1,3-difunctionalization involving cyclization. | |
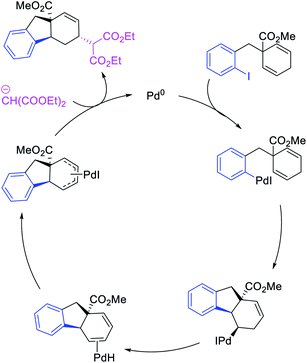 |
| | Scheme 40 Mechanism of the Pd-catalyzed 1,3-difunctionalization. | |
 |
| | Scheme 41 Pd-catalyzed enantioselective 1,3-difunctionalization. | |
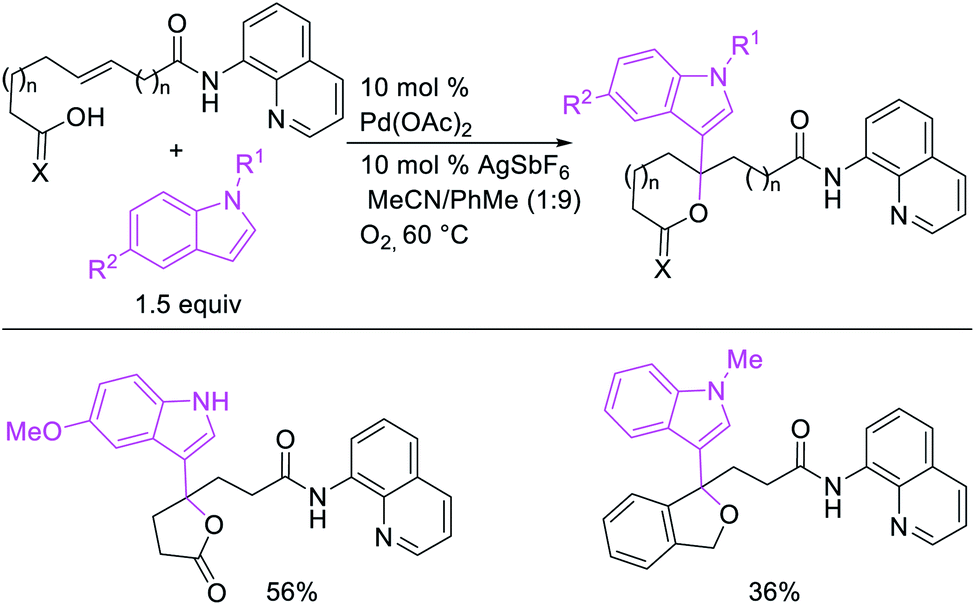
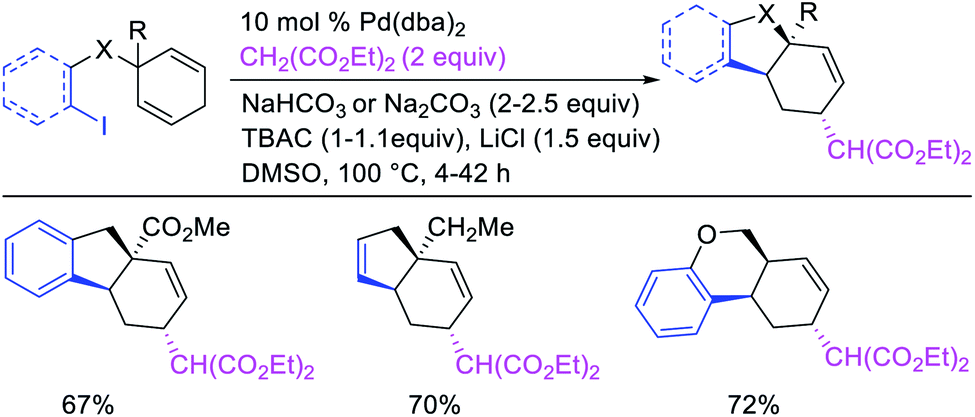
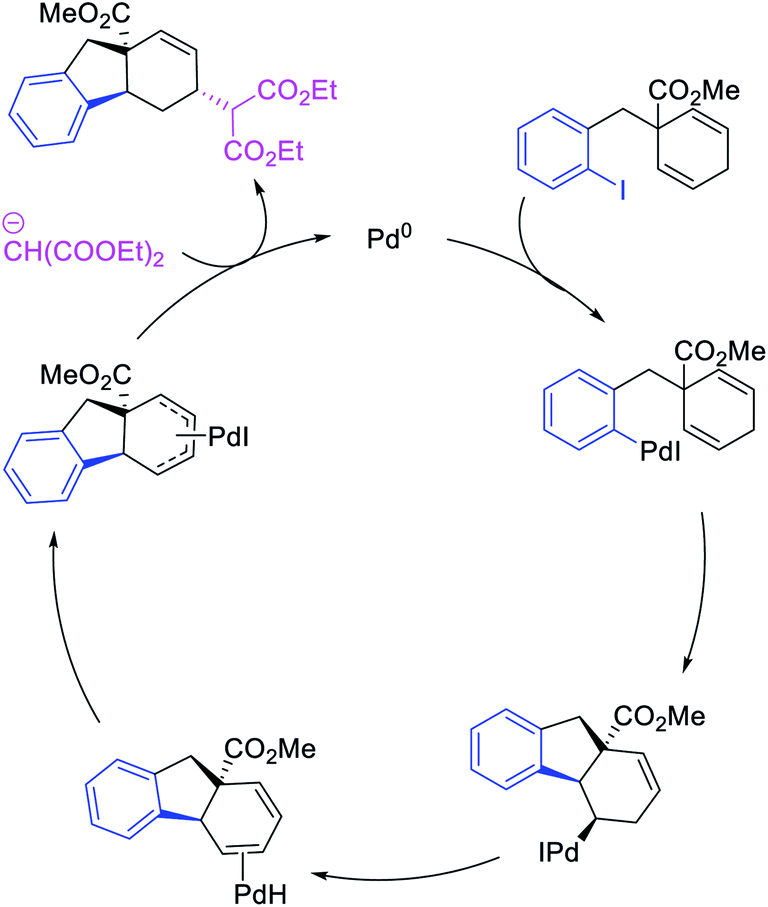
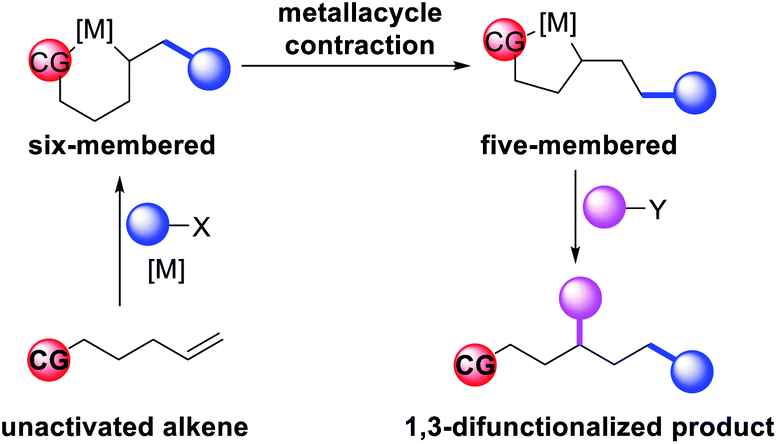
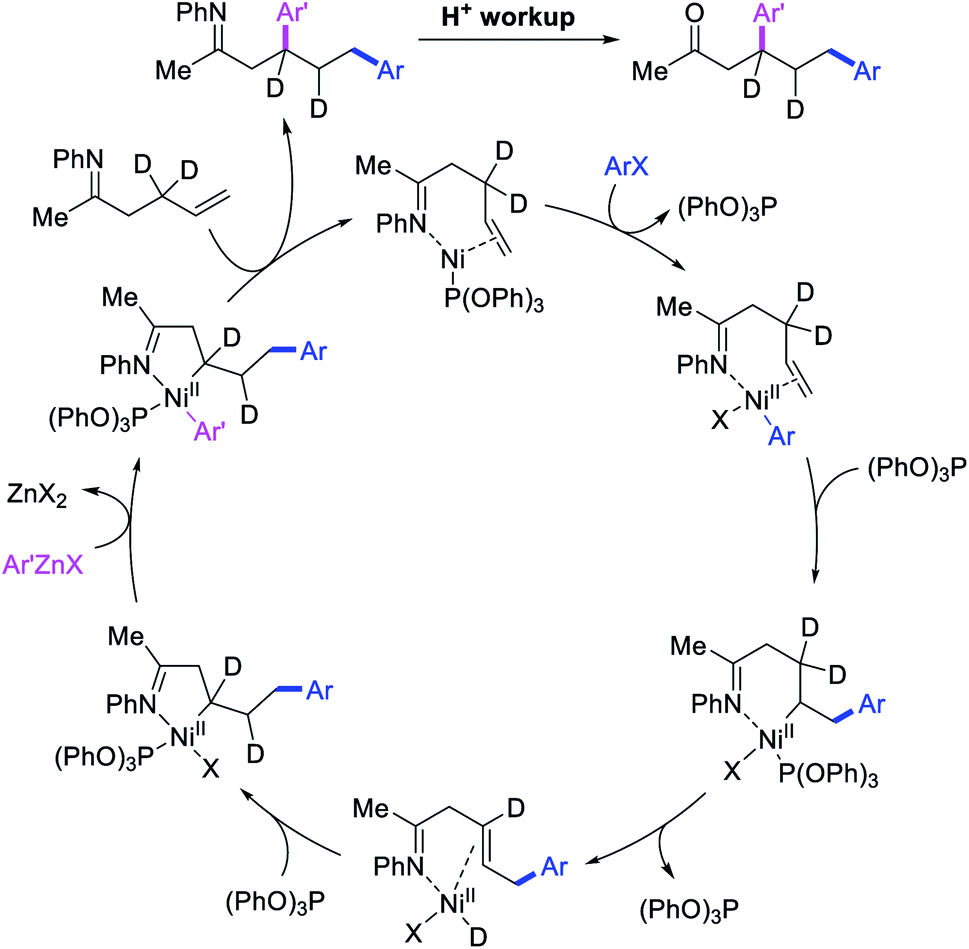
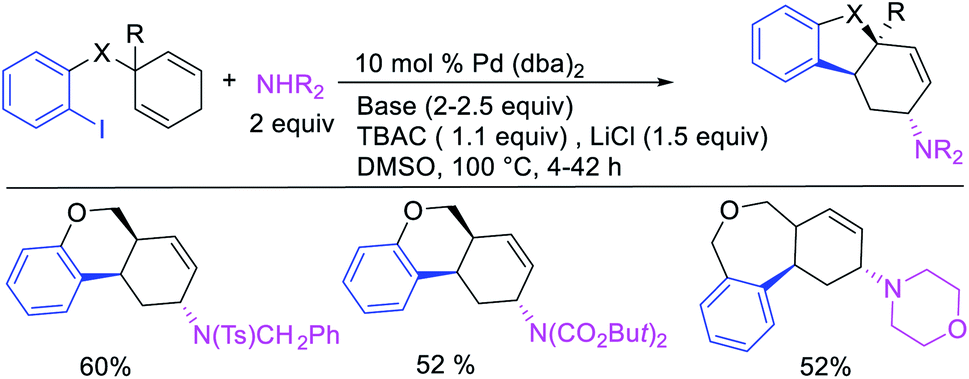
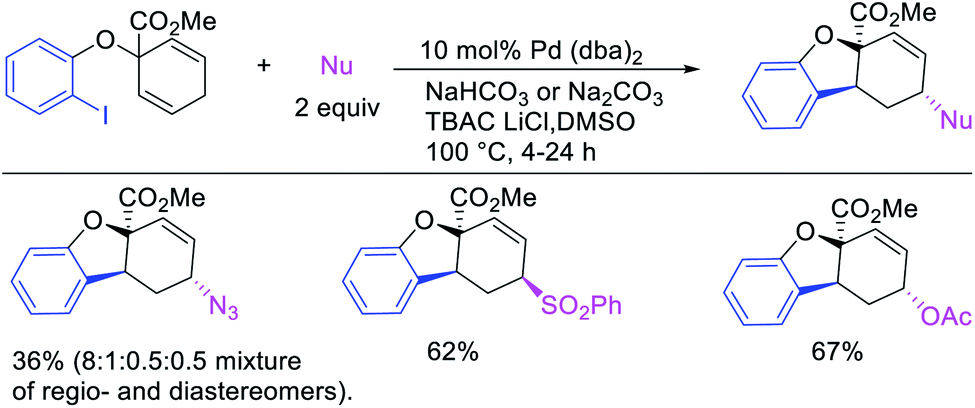
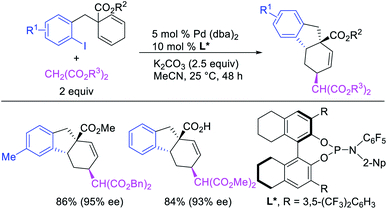
In 2018, we implemented a new approach for regioselective 1,3-dicarbofunctionalization of unactivated alkenes using a heteroatom coordination group.41 This unprecedented strategy relies upon the formation of an alkylmetal species as a six-membered transient metallacycle upon alkene carbometallation, which then undergoes metallacycle contraction prompted by the heteroatom coordination due to the propensity of the fluxional and less stable larger-sized metallacycle to form a more stable five-membered metallacycle (Scheme 42). The metallacycle contraction occurs by metal walking via β-H elimination and [M]–H reinsertion steps. The stable five-membered metallacycle is then intercepted by transmetallation with organic nucleophiles followed by reductive elimination to generate 1,3-difunctionalized products. Based upon this concept, we reported a Ni-catalyzed regioselective β,δ-diarylation of unactivated γ,δ-alkenes in carbonyl compounds with aryl iodides and arylzinc reagents (Scheme 43).41a In this reaction, a ketimine was used as a coordinating group and triphenylphosphite as an electron-deficient ligand to stabilize the alkylnickel(II) species generated after alkene carbonickellation as a six-membered nickellacycle, which then underwent nickellacycle contraction to generate a five-membered nickellacycle (Scheme 44). A deuterium migration experiment confirmed that the contraction of the nickellacycle proceeded by β-H elimination and [Ni]–H reinsertion steps. In addition, a crossover experiment, which tested the fidelity of [Ni]–H to remain bound to the in situ generated alkene, further provided strong evidence for metallacycle contraction by metal-walking rather than a double-Heck as a pathway for product formation.
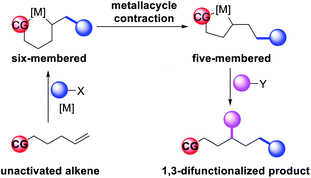 |
| | Scheme 42 Concept of metallacycle contraction for alkene 1,3-difunctionalization. | |
 |
| | Scheme 43 Ni-catalyzed 1,3-difunctionalization by metallacycle contraction. | |
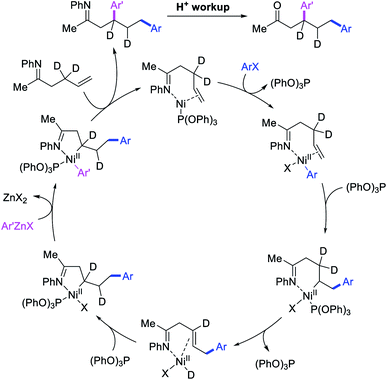 |
| | Scheme 44 Mechanism of the Ni-catalyzed 1,3-difunctionalization by metallacycle contraction. | |
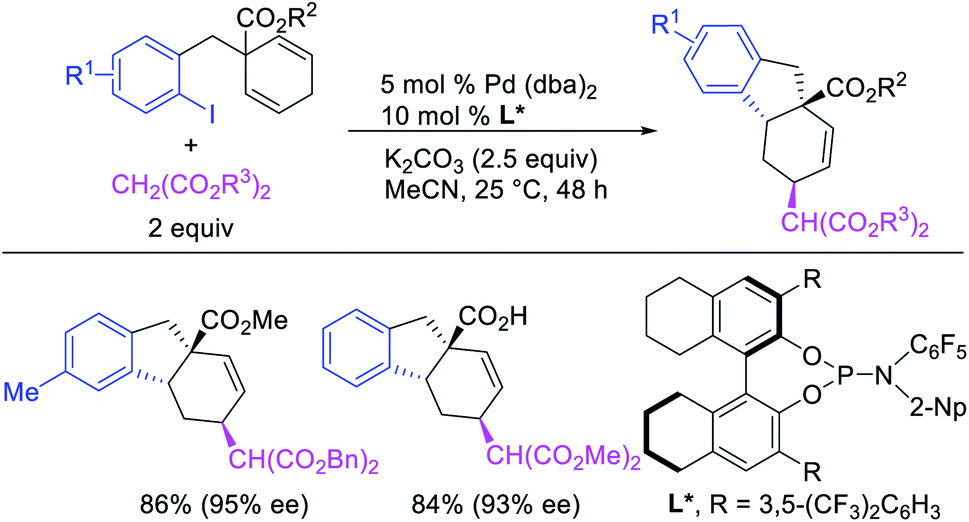
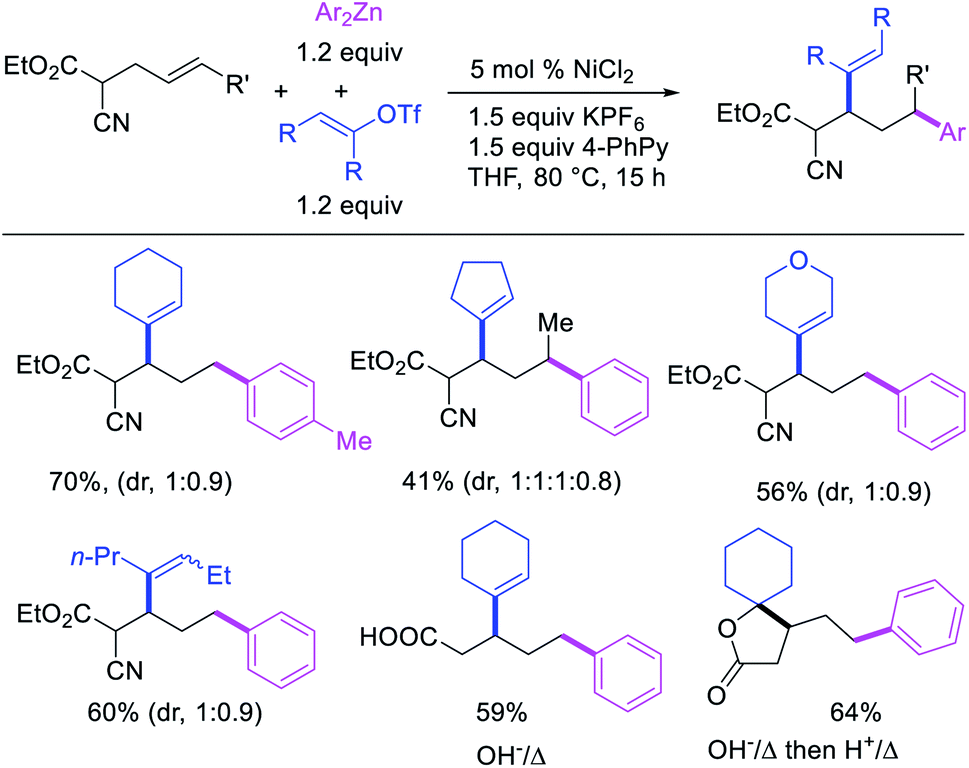
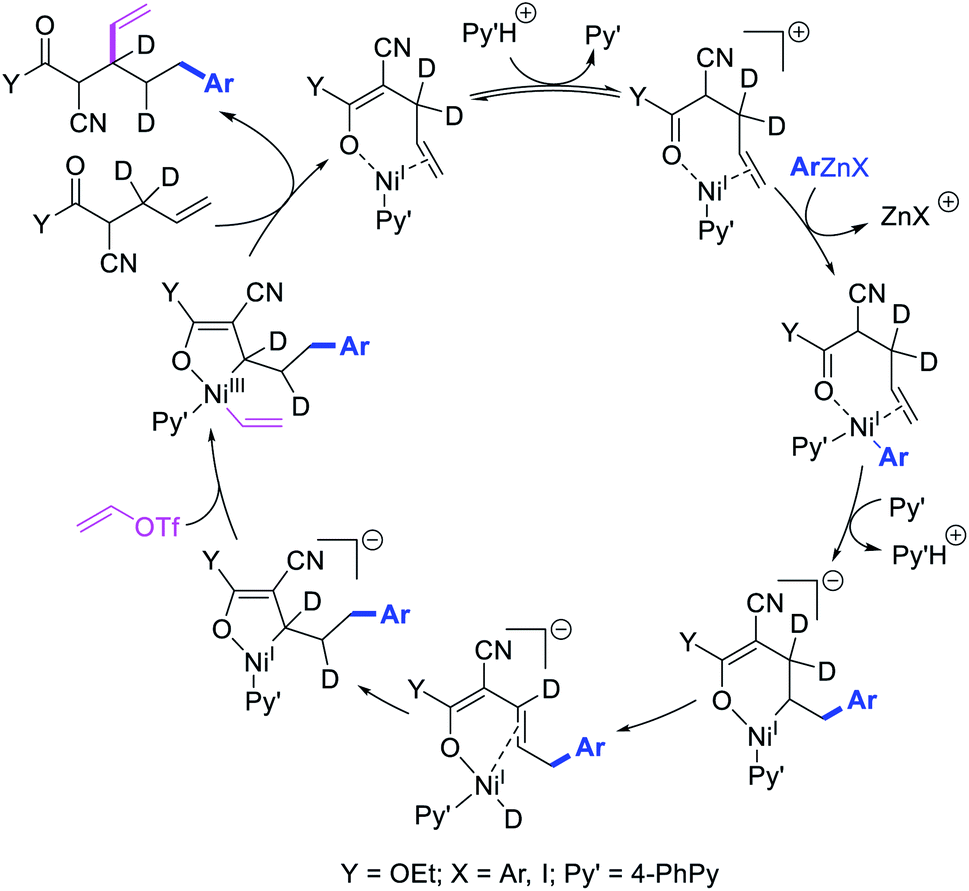
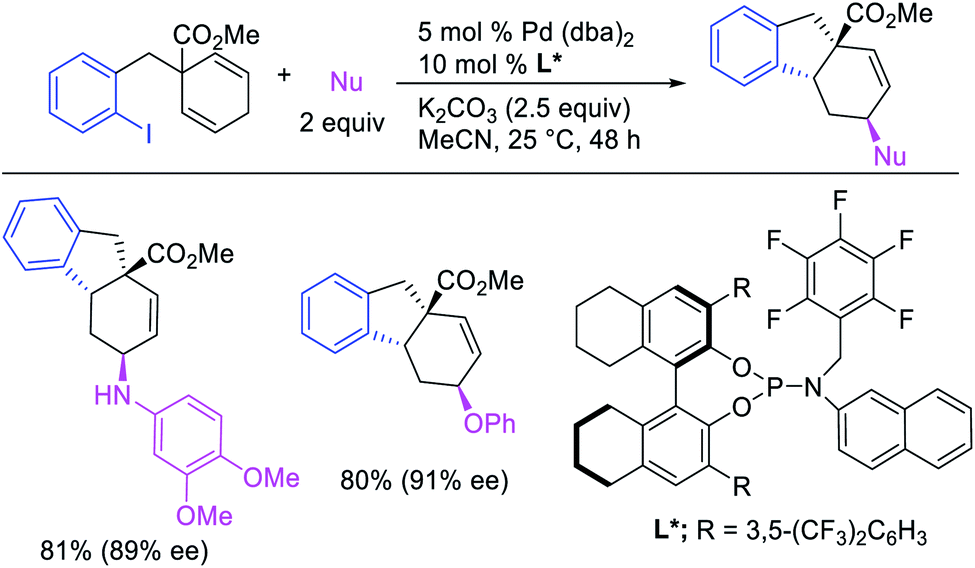
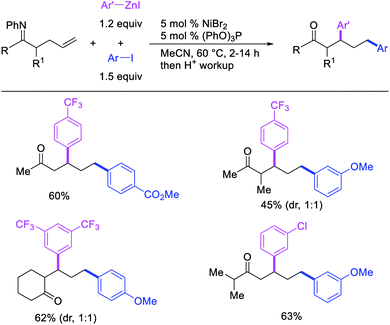
Recently, we also expanded the concept of metallacycle contraction for 1,3-difunctionalization to substrates where simple functional groups functioned as a coordinating group to promote metallacycle contraction.41b In this Ni(I)-catalyzed regioselective β,δ-arylvinylation reaction, the ester group of γ,δ-alkenyl nitrile-ester substrates acted as a coordinating group (Scheme 45). The reaction furnished products with a wide range of γ,δ-alkenyl nitrile-esters including disubstituted internal alkenes, vinyl triflates and arylzinc reagents. The products were also formed in a regio-reversed fashion by a transmetallation-initiated Ni(I)/Ni(III) catalytic cycle unlike most other known alkene difunctionalization reactions, which would generally proceed by oxidative addition-initiated pathways. The resultant β,δ-arylvinylated products could further be readily elaborated to a variety of synthetically useful complex products such as aliphatic α-cyanoesters, α-cyanocarboxylic acids, dicarboxylic acids, dicarboxylic acid monoamides, monocarboxylic acids, nitriles, and spirolactones. The ester group of the nitrile-ester substrates stabilized the six-membered transient metallacycles and prompted their contraction to five-membered rings (Scheme 46). In addition to the ester coordinating group, success of the reaction was also critically dependent on cation generators like KPF6 or a CuI as cocatalyst, α-proton (α-H) for substrate enolization, and substituted pyridine as a base and a potential ligand, all of which enabled in situ formation and stabilization of cationic Ni-species and anionic Ni-enolates during the catalytic progression of the reaction. The metallacycle contraction by a β-H elimination/[Ni]–H pathway, the requirement for α-H and the presence of Ni-enolates were also supported by control, deuterium migration and crossover experiments.
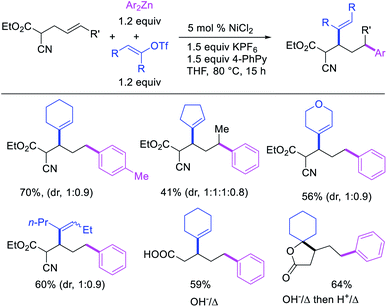 |
| | Scheme 45 Ni-catalyzed 1,3-difunctionalization assisted by simple functional group. | |
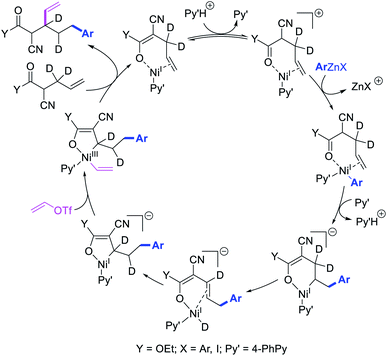 |
| | Scheme 46 Mechanism of the Ni-catalyzed 1,3-difunctionalization. | |
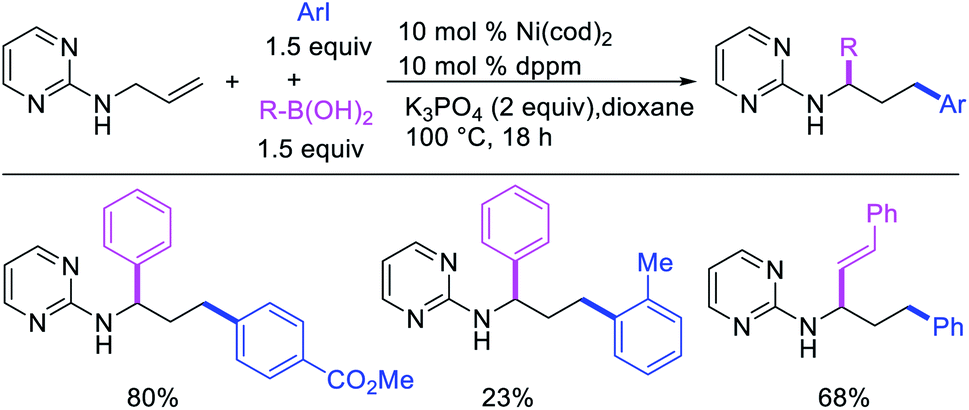
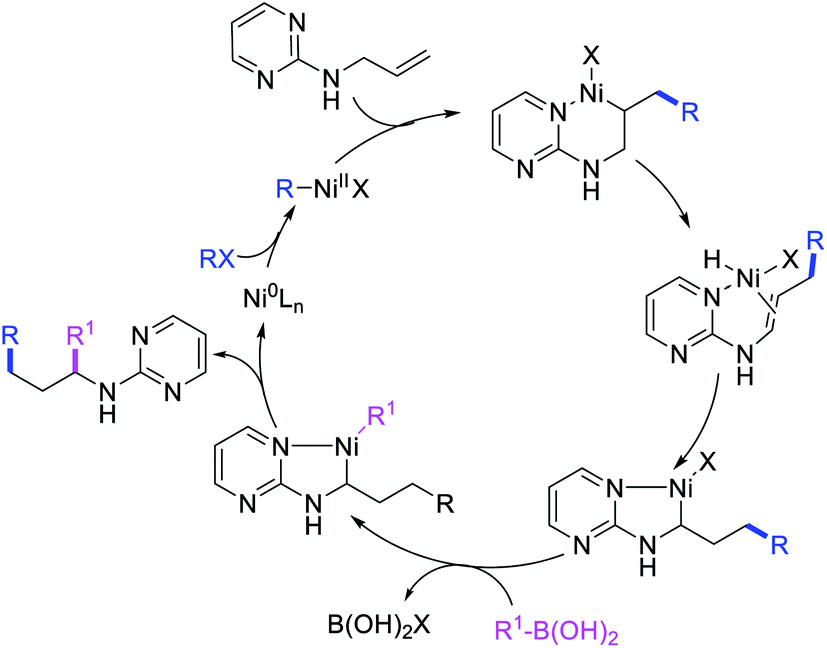
Concurrent to our work in 2018, Zhao et al. also disclosed a Ni-catalyzed regioselective diarylation and arylvinylation of the allyl group in N-allyl-2-pyrimidinamine with aryl iodides and aryl- and vinylboronic acids (Scheme 47).42 In this reaction, the pyrimidine moiety functioned as a coordinating group to from a nickelallacycle, which then rearranged to a five-membered metallacycle by metal-walking via β-H elimination and [Ni]–H reinsertion steps prior to interception with arylboronic acids followed by reductive elimination (Scheme 48). Radical trap and deuterium labelling experiments provided evidence for metal migration and non-radical nature of the reaction.
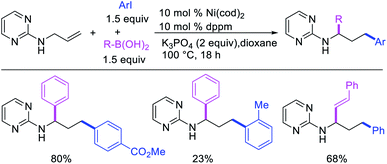 |
| | Scheme 47 Ni-catalyzed 1,3-difunctionalization of N-allyl group. | |
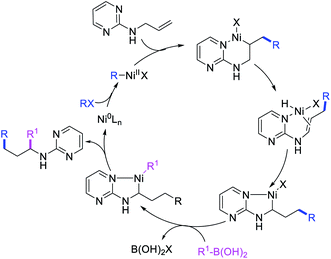 |
| | Scheme 48 Mechanism of the Ni-catalyzed 1,3-difunctionalization of N-allyl group. | |
3.2. 1,3-C–C/C–Hal, O, N, B functionalization
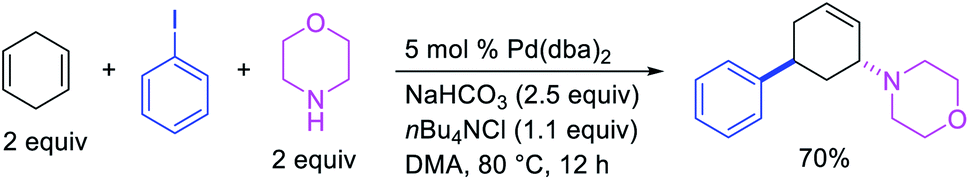
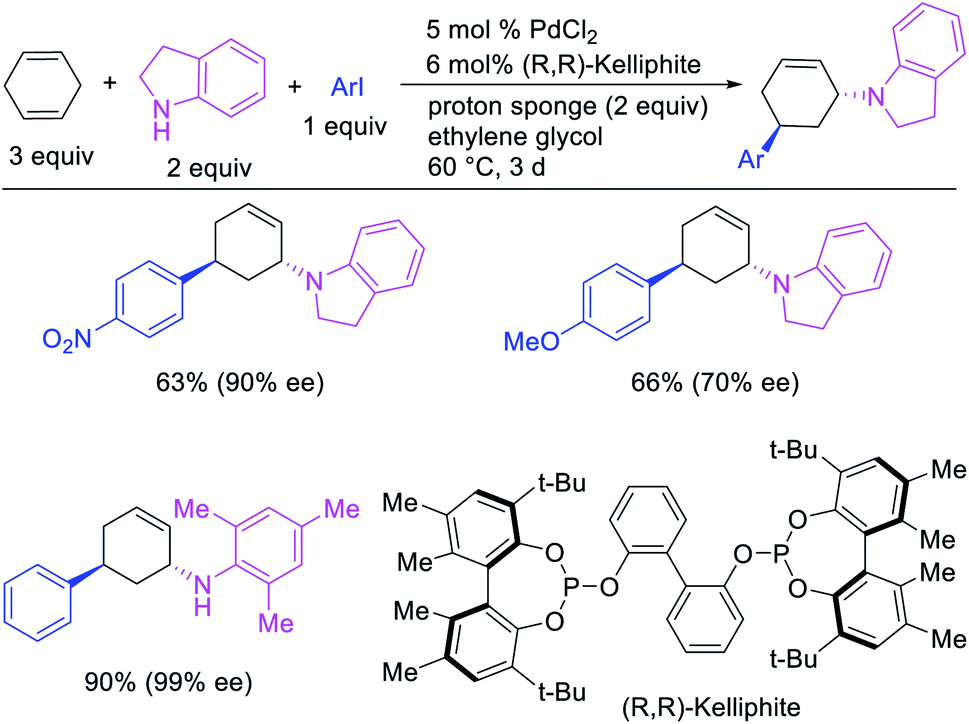
In 1982, Heck et al. reported the first example of a three-component 1,3-carboamination reaction that proceeded with the formation of rearranged alkylpalladium(II) intermediate after metal walking (Scheme 49).6 In this reaction, Pd-catalyzed the addition of vinyl bromide and piperidine to 1,4-cyclohexadiene, which produced the 1,3-vinylaminated product in 20% yield. The product was formed with trans stereochemistry after the anti-attack of the π-allylpalladium generated after metal migration by the amine. In 1994, Larock et al. disclosed an alkene 1,3-carboamination reaction of a non-conjugated diene (Scheme 50).43 In this Pd-catalyzed reaction, 1,4-cyclohexadiene was arylaminated at 1,3-position with phenyl iodide and morpholine. A broader scope of this reaction along with a high level of enantioselectivity has recently been reported by Zhou et al. (Scheme 51).44 The reaction was catalyzed by PdCl2 and (R,R)-kelliphite ligand, and proceeded with a variety of aryl iodides, non-conugated 1,4-cyclohexadiene and 1,8-cyclooctadiene derivatives, and primary and secondary alkyl and arylamines.
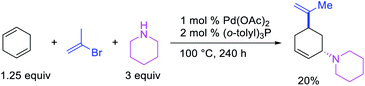 |
| | Scheme 49 Pd-catalyzed 1,3-vinylamination. | |
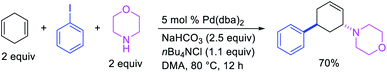 |
| | Scheme 50 Pd-catalyzed 1,3-arylamination. | |
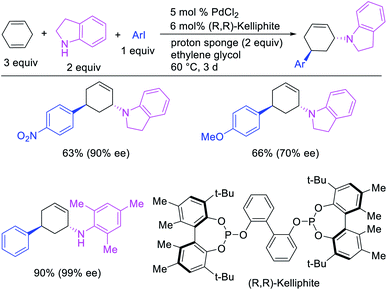 |
| | Scheme 51 Pd-catalyzed enantioselective 1,3-arylamination. | |
In late nineties, Larock et al. expanded the scope of the Pd-catalyzed three-component 1,3-difunctionalization reactions to two-component cyclization/coupling processes.39 In these methods, a variety of substituted 1,4-cyclohexadienes tethered to vinyl iodides and the ortho-position of aryl iodides were 1,3-carboaminated and 1,3-carbooxygenated with LiOAc and different primary and secondary amines and amides to furnish complex molecules with 4-, 5- and 6-membered carbocycles (Scheme 52). In addition, the authors also demonstrated that the reaction could utilize azides and sulfinates as nucleophiles although the products with azides were generally formed as a regioisomeric mixture due to the attack by azide to both electrophilic carbons of the π-allylpalladium(II) intermediates (Scheme 53).39b These reactions proceeded by the same mechanistic pathway as described for carbon nucleophiles in Scheme 40 (Section 3.1).
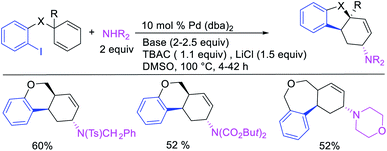 |
| | Scheme 52 Pd-catalyzed 1,3-arylamination by cyclization. | |
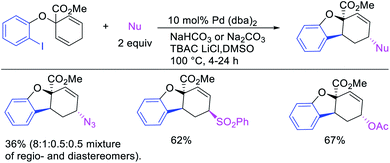 |
| | Scheme 53 Pd-catalyzed 1,3-arylheterofunctionalization by cyclization. | |
In 2011, Sanford et al. demonstrated that the Pd-catalyzed reaction condition developed for 1,1-aryloxygenation could be applied to difunctionalize 2,3-dihydrofuran to generate 1,3-aryloxygenated product (Scheme 54).28 Similarly, Wu et al. also expanded the scope of their enantioselective 1,3-cyclization/coupling reaction with carbon nucleophiles discussed in Section 3.1 (Scheme 41) to nitrogen and oxygen nucleophiles (Scheme 55).40 The Pd(dba)2/chiral phosphoramidite-catalyzed reaction generated 1,3-cyclization/amination and 1,3-cyclization/oxygenation products in good to excellent enantioselectivities.
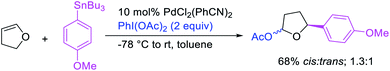 |
| | Scheme 54 Pd-catalyzed 1,3-aryloxygenation. | |
 |
| | Scheme 55 Pd-catalyzed enantioselective 1,3-cyclization/amination and oxgenation. | |
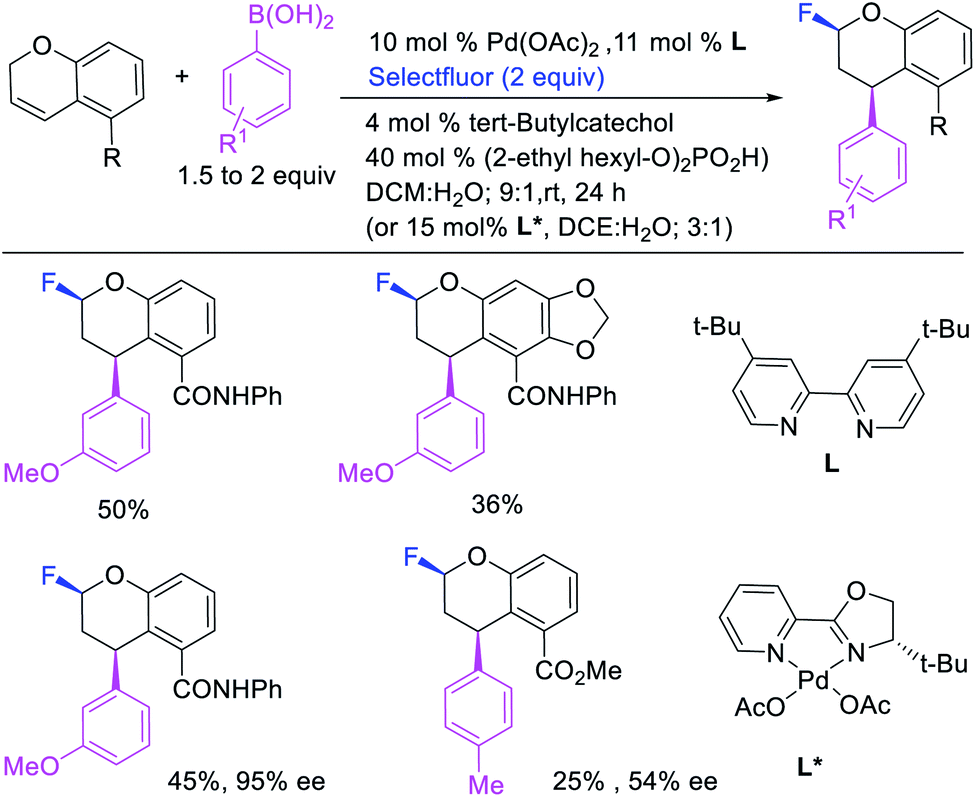
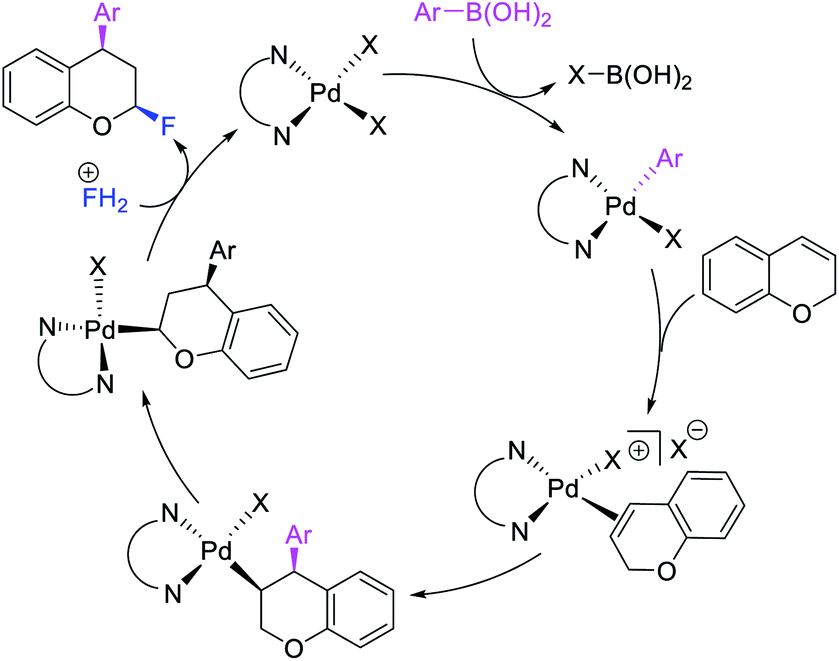
Over the years, only two examples of 1,3-carbohalogenation reactions have been reported. Alkene 1,3-carbohalogenation was first reported by Yoshida et al. in 1986 during arylchlorination of alkenyl alcohols with ArSnBu3 and excess of CuCl2 in the presence of 5 mol% PdCl2(PhCN)2 (Scheme 56).5a The 1,3-arylchlorination products were observed with PhSnBu3 in 29–35% yields along with 1,1-difunctionalized products. Recently, Toste et al. reported a synthetically useful three-component 1,3-arylfluorination of [2H]-chromene derivatives (Scheme 57).45 The Pd-catalyzed 1,3-difunctionalization reaction required 4,4′-di-tert-butyl-2,2′-dipyridyl (dbbpy) as a ligand and proceeded with arylboronic acids as aryl sources and Selectfluor as a fluoride donor. The authors further rendered the process enantioselective by substituting dbbpy for a chiral, nonracemic pyridyloxazoline ligand. The authors proposed that the reaction was catalyzed by a Pd(II) catalyst, which initiated the reaction through transmetalation with arylboronic acids followed by migratory insertion of the alkene (Scheme 58). The resultant alkylpalladium(II) intermediate then would undergo metal walking prior to oxidation by Selectfluor to generate the 1,3-arylfluorination products.
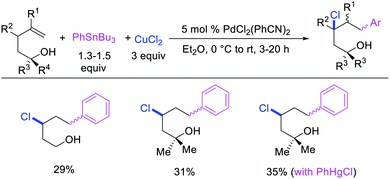 |
| | Scheme 56 Pd-catalyzed 1,3-arylchlorination. | |
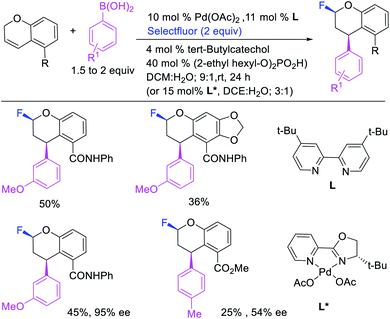 |
| | Scheme 57 Pd-catalyzed 1,3-arylfluorination. | |
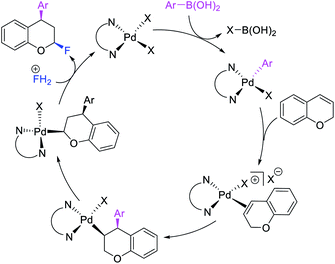 |
| | Scheme 58 Mechanism of the Pd-catalyzed 1,3-arylfluorination. | |
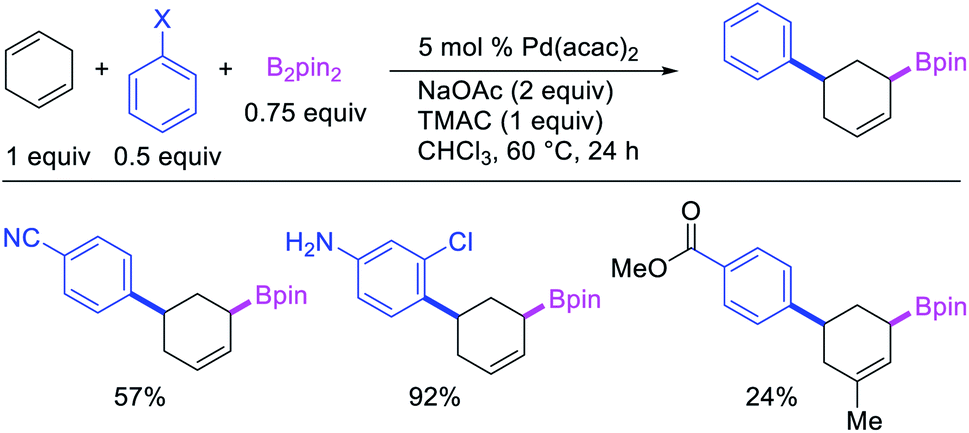
In 2019, Yin et al. disclosed a Pd-catalyzed 1,3-arylboration of non-conjugated dienes. In this reaction, 1,4-cyclohexadiene and 1-methyl-1,4-cyclohexadiene were arylborated with aryl halides and B2pin2 (Scheme 59).46 The products were formed with cis-stereochemistry since the reaction proceeded by metal migration by β-H elimination and [Pd]–H reinsertion with the retention of Pd on the same face of the diene and π-allylpalladium(II) species during metal walking (Scheme 60).
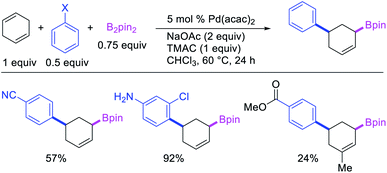 |
| | Scheme 59 Pd-catalyzed 1,3-arylboration. | |
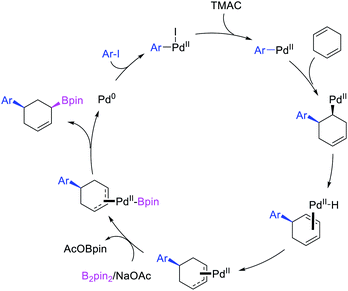 |
| | Scheme 60 Mechanism of the Pd-catalyzed 1,3-arylboration. | |
4. 1,n-Difunctionalization
An additional benefit of the alkene difunctionalization by metal-walking is the ability to place a second new bond at a measured distance along the carbon chain. Unlike the single stage metal walking for 1,1- and 1,3-difunctionalization reactions, the long distance (1,n) migration of metals ensues with several iterations of β-H elimination and hydrometallation steps along the carbon backbone of a hydrocarbon until the emigrating metal is thermodynamically satisfied to settle. Irrespective of the initial adductive reagents, the metal walk is incentivized by a distally located stabilizing group such as an arene or a terminal alkene that propitiates the migrating metal by forming thermodynamically stable intermediates such as π-benzylmetal and π-allylmetal complexes. Therefore, the reported examples of alkene 1,n-difunctionalization reactions are observed in nonconjugated dienes and alkenylalkanes bearing a terminal arene. Thus far long distance metal walking has generated C–C/C–C and C–C/C–X (N, O and B) bonds. These reactions are discussed in details in the subsequent sections.
4.1. 1,n-C–C/C–C and C–C/C–O, N, B functionalization
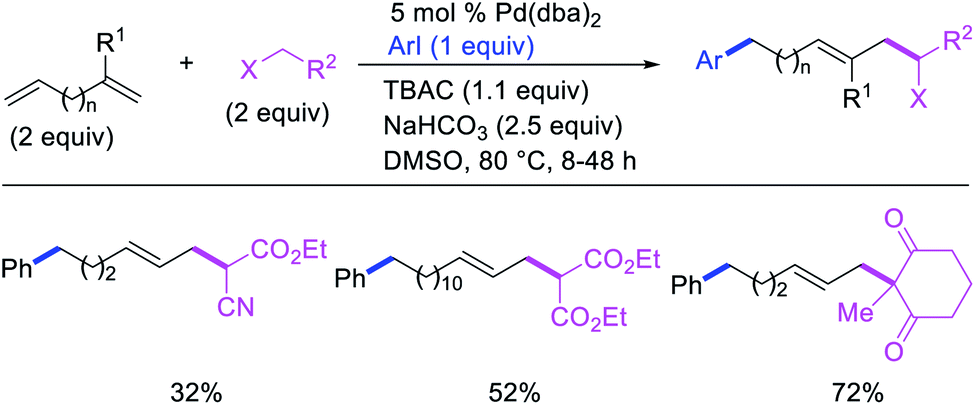
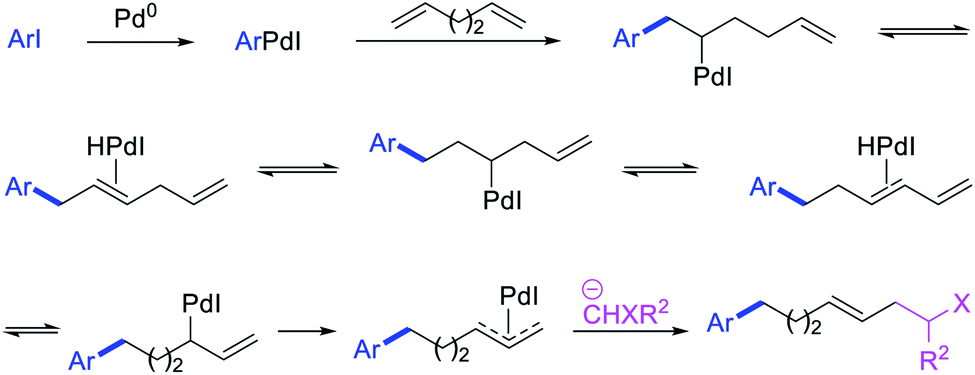
In 1991, Larock et al. disclosed the first example of Pd-catalyzed 1,n-difunctionalization of nonconjugated 1,n-dienes with aryl iodides and 1,3-dicarbonyl compounds (Scheme 61).38 The reaction proceeded with a variety of electron-deficient and rich aryl iodides and 1,3-diketones, 1,3-ketoesters, 1,3-diesters and 1,3-ester nitriles. The authors proposed that the reaction would proceed via Pd migration through a series of β-H elimination/[Pd]–H reinsertion steps until the alkylpalladium(II) species would be stabilized as π-allylpalladium(II) intermediate at the other terminal of the diene chain (Scheme 62). It was shown that the Pd could migrate along up to 14 carbon-chain length with 10 carbons between the nonconjugated alkenes. The π-allylpalladium(II) intermediate would then be intercepted by nucleophiles.
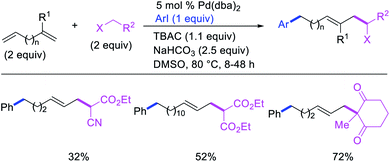 |
| | Scheme 61 Pd-catalyzed 1,n-difunctionalization. | |
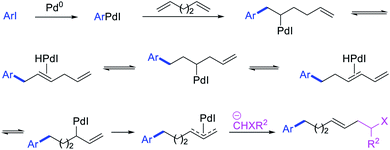 |
| | Scheme 62 Mechanism of the Pd-catalyzed 1,n-difunctionalization. | |
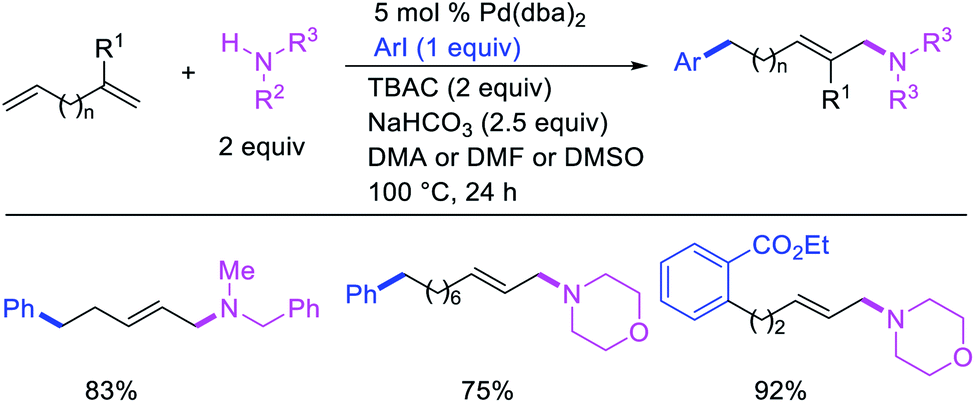
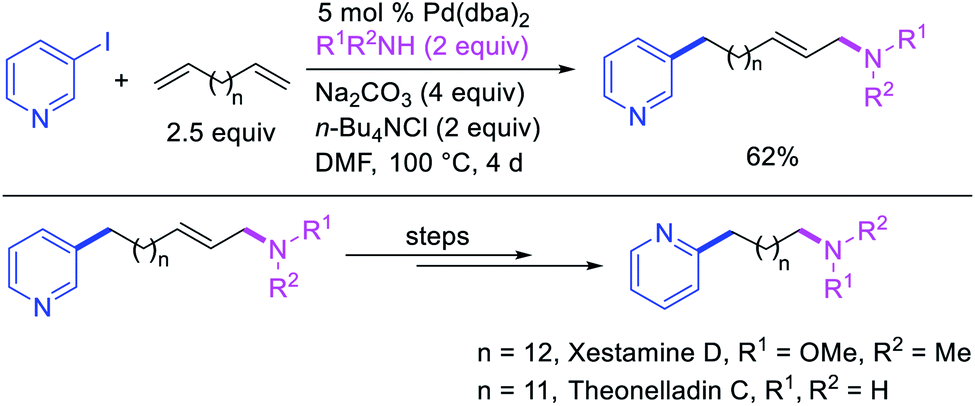
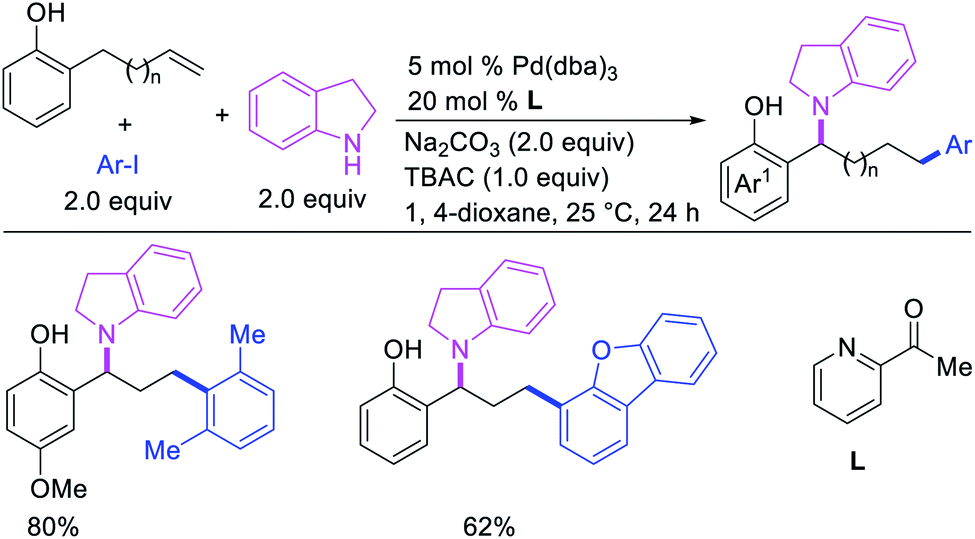
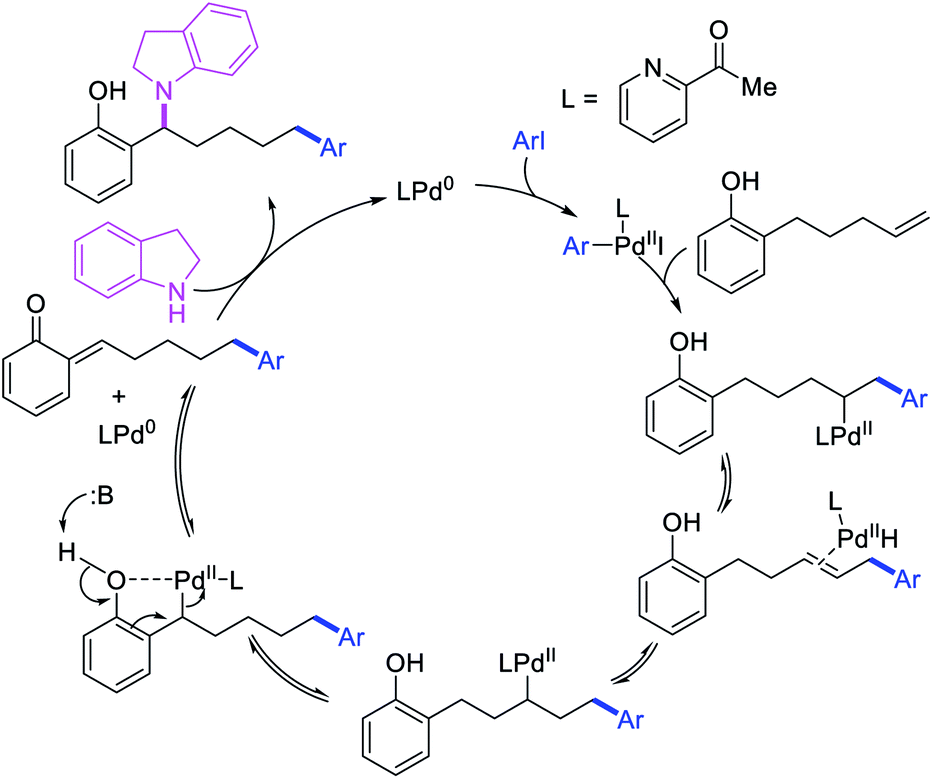
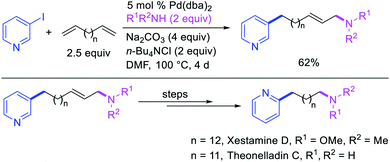
Larock et al. later extended the scope of 1,n-difunctionalization to nitrogen nucleophiles (Scheme 63)43 as well as 3-iodopyridine (Scheme 64).47 The 1,n-arylamination reaction enabled to install an aryl and an amino group at two terminals of a carbon chain. The ability to use 3-iodopyridine in 1,n-arylamination enabled the authors to synthesize pyridine alkaloids. In 2019, Yao et al. also reported a Pd-catalyzed method for the 1,n-arylamination of terminal alkenylalkanes tethered to the ortho and para positions of phenols (Scheme 65).48 The reaction required 2-acetylpyridine as a ligand. The authors proposed that the alkylpalladium(II) species would migrate to the benzylic position where the ortho hydroxy group would assist in stabilizing the η1-benzylpalladium(II) species (Scheme 66). The benzylpalladium(II) species underwent a base-assisted quinone methide formation, which then functioned as a Michael acceptor for nitrogen nucleophiles.
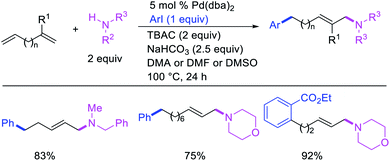 |
| | Scheme 63 Pd-catalyzed 1,n-arylamination. | |
 |
| | Scheme 64 Pd-catalyzed 1,n-heteroarylamination. | |
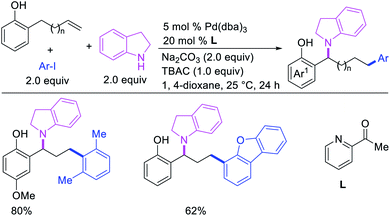 |
| | Scheme 65 Pd-catalyzed 1,n-aryliodolylation. | |
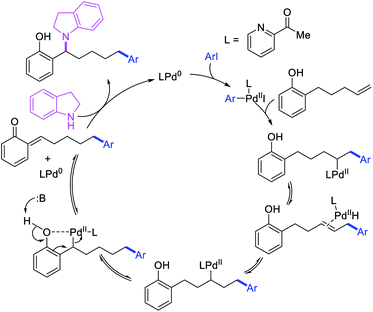 |
| | Scheme 66 Mechanism of the Pd-catalyzed 1,n-aryliodolylation. | |
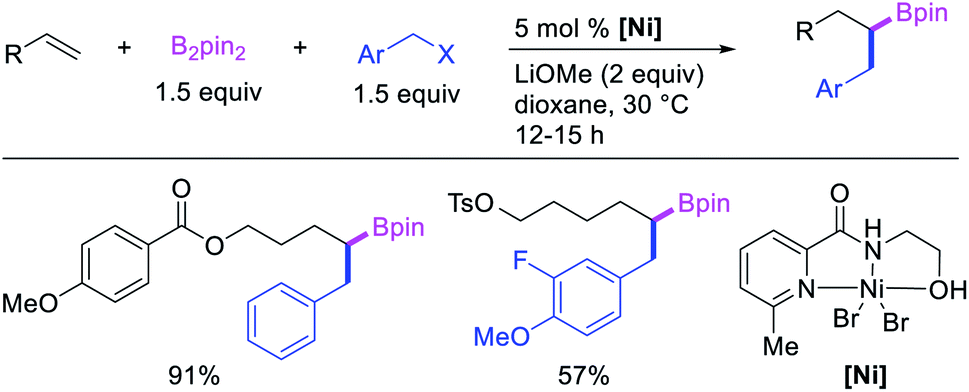
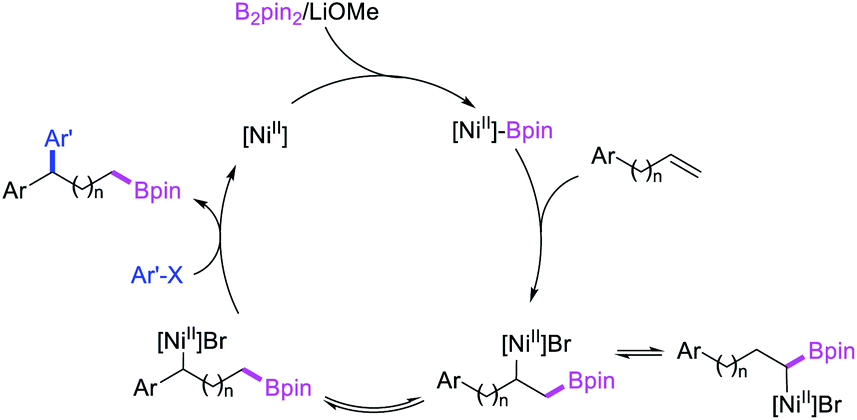
In 2019, Yin et al. reported a Ni-catalyzed 1,n-arylborylation of arylalkenes with aryl bromides and B2pin2 in the presence of a pyridyloxazoline ligand (Scheme 67).49 The reaction proceeded well for the arylboration of terminal alkenes with a range of aryl bromides. Based on control experiments and deuterium labeling, the authors proposed that the reaction would be initiated by boronickelation of alkenes followed by the migration of Ni to the benzylic position where the alkylnickel is likely to be stabilized as π-benzylnickel(II) species (Scheme 68). The π-benzylnickel(II) intermediate would then react with aryl bromides to furnish the 1,n-arylborylated products.
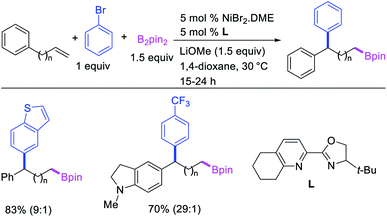 |
| | Scheme 67 Ni-catalyzed 1,n-arylborylation. | |
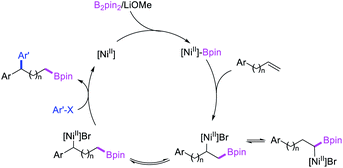 |
| | Scheme 68 Mechanism of the Ni-catalyzed 1,n-arylborylation. | |
5. Conclusion and future perspective
Metal-catalyzed difunctionalization of alkenes with two coupling partners is emerging as a formidable process to synthesize complex molecules expeditiously from readily available chemicals. Four decades in the making, the majority of research focused on solving the issue of β-H elimination from alkylmetal species generated upon alkene carbometallation to produce vicinally difunctionalized products. With the recent surge in research on alkene difunctionalization, a new optimism is on the horizon to exploit β-H elimination to advance the very process of alkene difunctionalization it is fundamentally primed to terminate. The new conception is founded upon the intrinsic nature of metal–hydrides ([M]–H), generated by β-H elimination, to undergo addition across an alkene to create a new alkylmetal species, the very reaction intermediate required for alkene difunctionalization. During the β-H elimination/[M]–H reinsertion process, the metals can emigrate short to long distances along a carbon chain until they reach the carbon of their choice largely guided by their need to attain thermodynamic stability. Therefore, unlike the conventional alkene difunctionalization that creates two bonds at the classical vicinal carbons, the new process can install bonds at carbon sites that are not within the domain of traditional wisdom for reactivity and bond formation. Over the years, the migrating metals have been intercepted with various reagents at 1, 3 and n-carbon sites generating 1,1-, 1,3- and 1,n-difunctionalized products. These products can be made with the formation of a combination of C–C and C–C, C–C and C–heteroatom including halogen, oxygen, nitrogen and boron, and C–B and C–B bonds. Despite having four decades of history, its development has, however, only progressed very slowly and the scope of the method is still limited. One of the most powerful aspects of the alkene difunctionalization by metal migration is its ability to deposit a second coupling group at a desired site along a carbon chain. However, the current methods are limited by the requirement of stabilizing factors such as heteroatomic, benzylic and alkenyl groups. Such a limitation could be addressed with the development of ligand sets amenable to hydrometallation and stabilization of the resultant alkylmetal intermediates. In addition, the current methods are also largely limited to the creation of a few C–C/C–C, C–C/C–X and C–X/C–X bonds with carbon sources mainly derived from C(sp)- and C(sp2)-hybridized reagents. Difunctionalization of alkenes at nonclassical sites with C(sp3)-hybridized sources will perhaps be the most fundamentally challenging task to address since the new alkyl entrants will compete with the incumbent alkyl group for β-H elimination and metal migration. With the recent renewed interest in developing such methods, one can only be optimistic to address the issue by mechanistic understanding, and the momentum in investigations for alkene difunctionalization by metal walking can be anticipated to rise. New discoveries are likely to be unraveled in the years to come and be applied toward expeditious synthesis of complex natural products and pharmaceuticals.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the NIH NIGMS (R35GM133438), the NSF (CHE-1554299) and The Pennsylvania State University for support of this work.
References
-
(a) H. Hiroshi, A. Mannosuke and I. Naoto, Bull. Chem. Soc. Jpn., 1974, 47, 1683 CrossRef;
(b) H. Hiroshi and I. Naoto, Bull. Chem. Soc. Jpn., 1975, 48, 2898 CrossRef;
(c) H. Horino and N. Inoue, J. Chem. Soc., Chem. Commun., 1976, 500 RSC;
(d) H. Horino and N. Inoue, Heterocycles, 1978, 11, 281 CrossRef CAS.
- S. Cacchi and G. Palmieri, Synthesis, 1984, 1984, 575 CrossRef.
-
(a) M. Catellani, G. P. Chiusoli, W. Giroldini and G. Salerno, J. Organomet. Chem., 1980, 199, C21 CrossRef CAS;
(b) M. Catellani and G. Paolo Chiusoli, Tetrahedron Lett., 1982, 23, 4517 CrossRef CAS;
(c) M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1982, 233, C21 CrossRef CAS.
-
(a) R. C. Larock, J. P. Burkhart and K. Oertle, Tetrahedron Lett., 1982, 23, 1071 CrossRef CAS;
(b) R. C. Larock, D. R. Leach and S. M. Bjorge, Tetrahedron Lett., 1982, 23, 715 CrossRef CAS.
-
(a) Y. Tamaru, M. Hojo, H. Higashimura and Z.-i. Yoshida, Angew. Chem., Int. Ed., 1986, 25, 735 CrossRef;
(b) Y. Tamaru, M. Hojo, S. Kawamura and Z. Yoshida, J. Org. Chem., 1986, 51, 4089 CrossRef CAS.
- D. D. Bender, F. G. Stakem and R. F. Heck, J. Org. Chem., 1982, 47, 1278 CrossRef CAS.
-
(a) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906 CrossRef CAS;
(b) Q. Lin and T. Diao, J. Am. Chem. Soc., 2019, 141, 17937 CrossRef CAS;
(c) G.-F. Yu, P. Wang, X. Bao and Y. Wang, Asian J. Org. Chem., 2020, 9, 793 CrossRef CAS;
(d) N. Li, R. Chang, W. Yang, Z. Zhang and Z. Guo, Organometallics, 2020, 39, 2057 CrossRef CAS.
-
(a) S. R. Chemler, S. D. Karyakarte and Z. M. Khoder, J. Org. Chem., 2017, 82, 11311 CrossRef CAS;
(b) A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC;
(c) J. P. Wolfe, Synlett, 2008, 2008, 2913 CrossRef.
-
(a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS;
(b) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70 CrossRef CAS;
(c) T. Yang, X. Chen, W. Rao and M. J. Koh, Chem, 2020, 6, 738 CrossRef CAS;
(d) S. Kc, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801 CrossRef CAS;
(e) S. KC, R. K. Dhungana, N. Khanal and R. Giri, Angew. Chem., Int. Ed., 2020, 59, 8047 CrossRef CAS;
(f) P. Basnet, S. Kc, R. K. Dhungana, B. Shrestha, T. J. Boyle and R. Giri, J. Am. Chem. Soc., 2018, 140, 15586 CrossRef CAS;
(g) B. Shrestha, P. Basnet, R. K. Dhungana, S. Kc, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653 CrossRef CAS;
(h) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657 CrossRef CAS;
(i) J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878 CrossRef CAS;
(j) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef;
(k) W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812 CrossRef CAS;
(l) P. Gao, L.-A. Chen and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 10653 CrossRef CAS;
(m) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS.
-
(a) R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314 CrossRef CAS;
(b) R. Giri and S. Kc, J. Org. Chem., 2018, 83, 3013 CrossRef CAS;
(c) J. T. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Aldrichimica Acta, 2018, 51, 21 CAS;
(d) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327 CrossRef CAS;
(e) H.-C. Guo and J.-A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354 CrossRef CAS;
(f) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC;
(g) Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990 CrossRef CAS;
(h) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542 CrossRef CAS.
- H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153 CrossRef CAS.
- U. Dutta, S. Porey, S. Pimparkar, A. Mandal, J. Grover, A. Koodan and D. Maiti, Angew. Chem., Int. Ed. DOI:10.1002/anie.202005664.
- J. E. Bäckvall and R. E. Nordberg, J. Am. Chem. Soc., 1980, 102, 393 CrossRef.
- E. Yamamoto, M. J. Hilton, M. Orlandi, V. Saini, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 15877 CrossRef CAS.
- M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688 CrossRef CAS.
- E. Thiery, D. Harakat, J. Le Bras and J. Muzart, Organometallics, 2008, 27, 3996 CrossRef CAS.
- K. B. Urkalan and M. S. Sigman, Angew. Chem., Int. Ed., 2009, 48, 3146 CrossRef CAS.
- E. W. Werner, K. B. Urkalan and M. S. Sigman, Org. Lett., 2010, 12, 2848 CrossRef CAS.
- J. Jeon, H. Ryu, C. Lee, D. Cho, M.-H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 10048 CrossRef CAS.
- Y. Li, H. Wei, D. Wu, Z. Li, W. Wang and G. Yin, ACS Catal., 2020, 10, 4888 CrossRef CAS.
- L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784 CrossRef CAS.
- V. Saini and M. S. Sigman, J. Am. Chem. Soc., 2012, 134, 11372 CrossRef CAS.
- V. Saini, L. Liao, Q. Wang, R. Jana and M. S. Sigman, Org. Lett., 2013, 15, 5008 CrossRef CAS.
- R. C. Larock, C. L. Liu, H. H. Lau and S. Varaprath, Tetrahedron Lett., 1984, 25, 4459 CrossRef CAS.
- J. P. Parrish, Y. C. Jung, S. I. Shin and K. W. Jung, J. Org. Chem., 2002, 67, 7127 CrossRef CAS.
- S. Maity, T. J. Potter and J. A. Ellman, Nat. Catal., 2019, 2, 756 CrossRef CAS.
- A. Rodriguez and W. J. Moran, Eur. J. Org. Chem., 2009, 2009, 1313 CrossRef.
- A. D. Satterfield, A. Kubota and M. S. Sanford, Org. Lett., 2011, 13, 1076 CrossRef CAS.
- D. Kalyani and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 2150 CrossRef CAS.
- D. Kalyani, A. D. Satterfield and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 8419 CrossRef CAS.
- Y. He, Z. Yang, R. T. Thornbury and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 12207 CrossRef CAS.
- E. P. A. Talbot, T. d. A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101 CrossRef CAS.
- J. Miró, C. del Pozo, F. D. Toste and S. Fustero, Angew. Chem., Int. Ed., 2016, 55, 9045 CrossRef.
- H. M. Nelson, B. D. Williams, J. Miró and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3213 CrossRef CAS.
- A. M. Bergmann, S. K. Dorn, K. B. Smith, K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2019, 58, 1719 CrossRef CAS.
- Y. Li, H. Pang, D. Wu, Z. Li, W. Wang, H. Wei, Y. Fu and G. Yin, Angew. Chem., Int. Ed., 2019, 58, 8872 CrossRef CAS.
- L. Li, T. Gong, X. Lu, B. Xiao and Y. Fu, Nat. Commun., 2017, 8, 345 CrossRef.
- R. C. Larock, Y. D. Lu, A. C. Bain and C. E. Russell, J. Org. Chem., 1991, 56, 4589 CrossRef CAS.
-
(a) X. Han and R. C. Larock, Synlett, 1998, 1998, 748 CrossRef;
(b) R. C. Larock and X. Han, J. Org. Chem., 1999, 64, 1875 CrossRef CAS.
- Y. Zhang, H.-C. Shen, Y.-Y. Li, Y.-S. Huang, Z.-Y. Han and X. Wu, Chem. Commun., 2019, 55, 3769 RSC.
-
(a) P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. Kc, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782 CrossRef CAS;
(b) R. K. Dhungana, S. Kc, P. Basnet, V. Aryal, L. J. Chesley and R. Giri, ACS Catal., 2019, 9, 10887 CrossRef CAS.
- W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC.
- R. C. Larock, Y. Wang, Y. Lu and C. A. Russell, J. Org. Chem., 1994, 59, 8107 CrossRef CAS.
- D. Zhu, Z. Jiao, Y. R. Chi, T. P. Gonçalves, K.-W. Huang and J. S. Zhou, Angew. Chem., Int. Ed., 2020, 59, 5341 CrossRef CAS.
- R. T. Thornbury, V. Saini, T. d. A. Fernandes, C. B. Santiago, E. P. A. Talbot, M. S. Sigman, J. M. McKenna and F. D. Toste, Chem. Sci., 2017, 8, 2890 RSC.
- H. Pang, D. Wu, H. Cong and G. Yin, ACS Catal., 2019, 9, 8555 CrossRef CAS.
- Y. Wang, X. Dong and R. C. Larock, J. Org. Chem., 2003, 68, 3090 CrossRef CAS.
- C. Han, Z. Fu, S. Guo, X. Fang, A. Lin and H. Yao, ACS Catal., 2019, 9, 4196 CrossRef CAS.
- W. Wang, C. Ding, Y. Li, Z. Li, Y. Li, L. Peng and G. Yin, Angew. Chem., Int. Ed., 2019, 58, 4612 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*