DOI:
10.1039/D0SC03628E
(Edge Article)
Chem. Sci., 2020,
11, 10465-10482
Cancer cell-selective modulation of mitochondrial respiration and metabolism by potent organogold(III) dithiocarbamates†
Received
1st July 2020
, Accepted 4th September 2020
First published on 18th September 2020
Abstract
Metabolic reprogramming is a key cancer hallmark that has led to the therapeutic targeting of glycolysis. However, agents that target dysfunctional mitochondrial respiration for targeted therapy remains underexplored. We report the synthesis and characterization of ten (10) novel, highly potent organometallic gold(III) complexes supported by dithiocarbamate ligands as selective inhibitors of mitochondrial respiration. The structure of dithiocarbamates employed dictates the biological stability and cellular cytotoxicity. Most of the compounds exhibit 50% inhibitory concentration (IC50) in the low-micromolar (0.50–2.9 μM) range when tested in a panel of aggressive cancer types with significant selectivity for cancer cells over normal cells. Consequently, there is great interest in the mechanism of action of gold chemotherapeutics, particularly, considering that DNA is not the major target of most gold complexes. We investigate the mechanism of action of representative complexes, 1a and 2a in the recalcitrant triple negative breast cancer (TNBC) cell line, MDA-MB-231. Whole-cell transcriptomics sequencing revealed genes related to three major pathways, namely: cell cycle, organelle fission, and oxidative phosphorylation. 2a irreversibly and rapidly inhibits maximal respiration in TNBC with no effect on normal epithelial cells, implicating mitochondrial OXPHOS as a potential target. Furthermore, the modulation of cyclin dependent kinases and G1 cell cycle arrest induced by these compounds is promising for the treatment of cancer. This work contributes to the need for mitochondrial respiration modulators in biomedical research and outlines a systematic approach to study the mechanism of action of metal-based agents.
Introduction
Developing next generation metal-based drugs that exploit the cancer cell vulnerabilities will offer more effective therapies and tools to study the disease. Tumors harbor inherent complexities that present therapeutic challenges and evasion of the physiological machinery to fix the neoplastic disease state.1 The biological hallmarks of cancer, which define alterations in cell physiology to promote tumor growth include, (i) insensitivity to growth-inhibitory (antigrowth) signals, (ii) evasion of programmed cell death (apoptosis), (iii) limitless replicative potential, (iv) sustained angiogenesis, (v) tissue invasion & metastasis, (vi) avoiding immune destruction, (vii) reprogramming of energy metabolism2,3 with genetic instability and tumor-promoting inflammation as primary drivers. The complexities however present opportunities for targeted therapy. The redox properties of transition metal constructs, can influence metabolic reprogramming of cancer cells4,5 given the inherent redox activity associated with bioenergetics and mitochondrial processes such as the electron transport chain (ETC).6 Metabolically active tumors are addicted to glycolysis (known as the Warburg effect)7,8 and this has been exploited therapeutically through inhibition of glucose metabolism and the use of 2-deoxy-2-fluoro-D-glucose (FDG-PET) to detect tumors.9,10 Additionally, many tumor types depend on OXPHOS (oxidative phosphorylation), an electron transfer chain driven by substrate oxidation that is coupled to the synthesis of ATP through an electrochemical transmembrane gradient to coordinate their bioenergetic states and promote proliferation.11,12
Over 50% of cancer patients receive platinum agents worldwide.13 Platinum are particularly effective in bladder and testicular cancers.14,15 Although widely used in other cancer types such as colon and ovarian, their response rate is stymied by recurrence due to acquired and innate mechanisms.16–18 The discovery of new transition metal complexes with new biological targets and mechanism of action have become alternate strategies to improve treatment outcomes.19,20 Gold (Au) is a third-row transition metal just like platinum which can be tolerated in humans as displayed in the FDA approved drug, auranofin.21–23 Both Au(I) and (III) complexes have gained considerable interest over the years as anticancer agents with preferential targeting of mitochondria,24 thioredoxin proteasome, and inducing endoplasmic reticulum stress.24–32 Gold complexes with multifaceted target mechanisms to evade resistance pathways are attractive.
Renewed interest in Au(III) as antitumor agents in recent reports since the mid-1970s attest to their high cytotoxicity, reduced cross-resistance with cisplatin, and tolerance in tumor-bearing mice.27,33–36 Among such complexes are Au(III)-2-pyridylmethanol,37 Au(III)dichloro(N-ethylsalicylaldiminate),38 Au(III)-phosphine,39–41 cyclometalated Au(III),27,34,42–46 and Au(III)-dithiocarbamates.45,47–52 Whereas different classes of Au(III)-dithiocarbamate display promising anticancer activity in vitro and in vivo the mechanism of action is not well elucidated.45,53
Herein, we report the synthesis of Au(III) dithiocarbamates that incorporate aryl-pyridine cyclometallation [C^N] ligands to improve complex stability and different aryl/alkyl dithiocarbamate ligands for structure activity relationship (SAR). The compounds display higher potency in a panel of cancer cells than cisplatin. Using detailed HPLC, spectrometry, and cyclic voltammetry we identified the active gold(I) cytotoxic agent. We investigated the mechanism of action using a systems and functional biology approach that established induction of mitochondria membrane depolarization with subsequent modulation of oxidative phosphorylation in cancer cells. Altogether, our studies show that compound 2a exploits cancer cell vulnerability via disruption of mitochondrial respiration. These compounds exhibit significant depolarization of the mitochondrial membrane and as such, ROS production is observed as a byproduct. It is possible that compound 2a reacts with a nucleophilic side chain of its protein target within the oxidative phosphorylation machinery.
Results and discussion
Synthesis and characterization
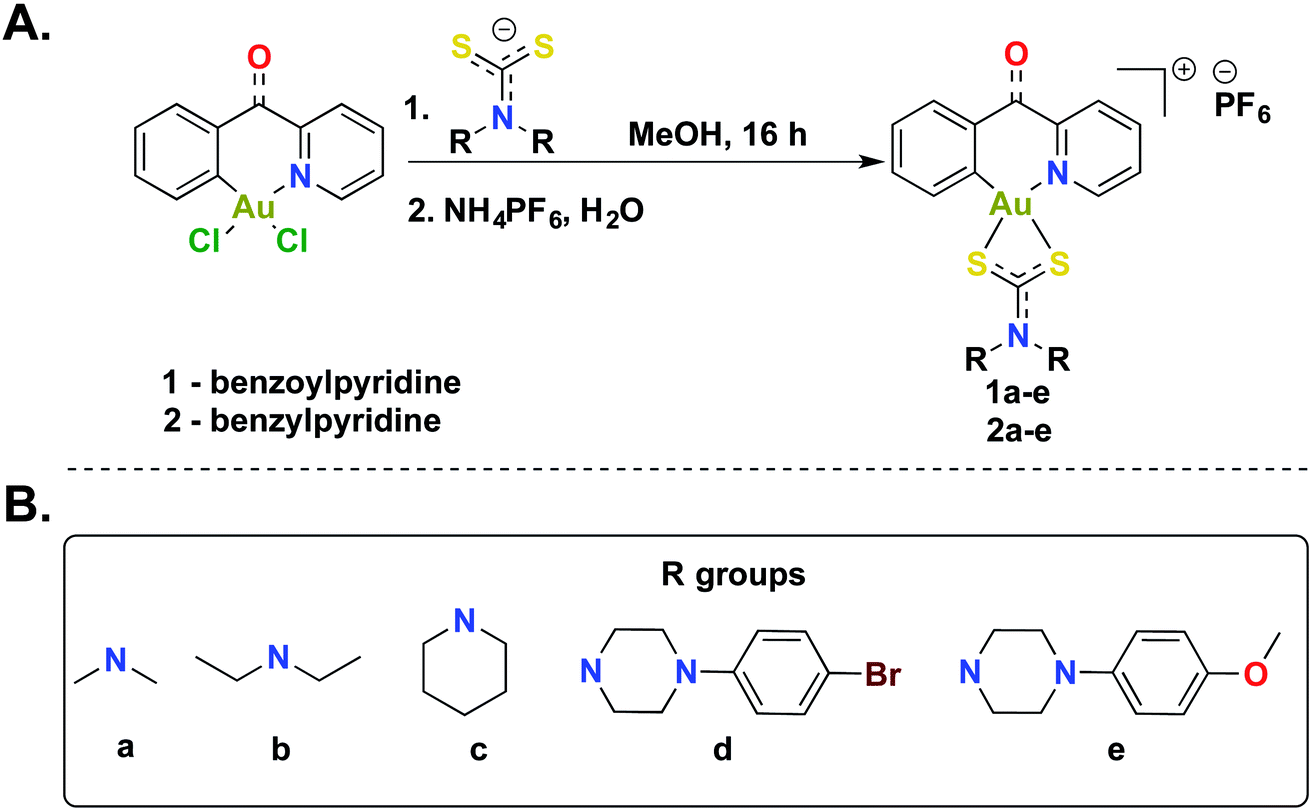
The [C^N]-cyclometalated gold(III) compounds were synthesized from previously reported methods.54 Treatment of [C^N]-Au(III)Cl2 with dithiocarbamate ligands in methanol for 16 h at room temperature followed by treatment with an aqueous saturated solution of NH4PF6 gave the desired compounds in respectable yields (Scheme 1). We characterized all 10 compounds (1a–e and 2a–e) by 1H-NMR, 13C-NMR, and 19F-NMR and high-resolution mass spectrometry (HRMS) (Fig. S1–S46†). The purity of the compounds was verified by HPLC (>95%) (Fig. S47–56†). In this study, we sought to expand the structural architecture of Au(III) dithiocarbamate complexes to include cyclic and aromatic side chains in addition to cyclometalation, which provide stabilization by strong σ-donation to the gold center.55,56 We and others have demonstrated that this stabilization strategy has positive implications on the solution chemistry, biological and electrochemical behavior of complexes.57 Two different [C^N]Au(III)Cl2 complexes with electronic variations were used in the synthesis with the aim of establishing structural diversity. Complexes 1a–e possess a carbonyl at the methine bridge of the benzylpyridine framework, whereas 2a–e does not. Studies from our laboratory show that the difference impart unique reactivity and kinetics towards nucleophiles. Alternate metal-based drugs to platinum, the first-line of chemotherapy for several cancer types are desperately needed to overcome the toxicity and resistance associated with platinum drugs.58–62 Gold compounds including auranofin have been used in preclinical and clinical trials with great promise.63–67 A more systematic design and the elucidation of the mechanism of action of gold compounds will expand the utility of well-defined gold anticancer drug candidates with reduced susceptibility to resistance and toxic side effects. In this work, we developed a small library of gold(III) compounds supported by different cyclometalated and dithiocarbamate ligands to obtain cationic complexes complemented by hexafluorophosphate ions. Structural diversity was achieved by using two different cyclometalated gold(III) starting materials and five dithiocarbamate ligands of distinct structural and electronic variety. Consequently, the library enabled us to explore the biological activity of novel gold(III) dithiocarbamates in the context of cancer as described in this report.
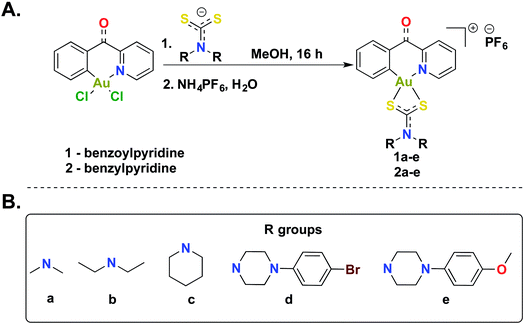 |
| | Scheme 1 Synthesis of gold(III) dithiocarbamates. (A) Synthetic scheme to generate cyclometalated (C^N) gold(III) complexes bearing dithiocarbamate ligands. (B) Diversity of dithiocarbamate ligands used in this study. | |
Single crystal X-ray diffraction
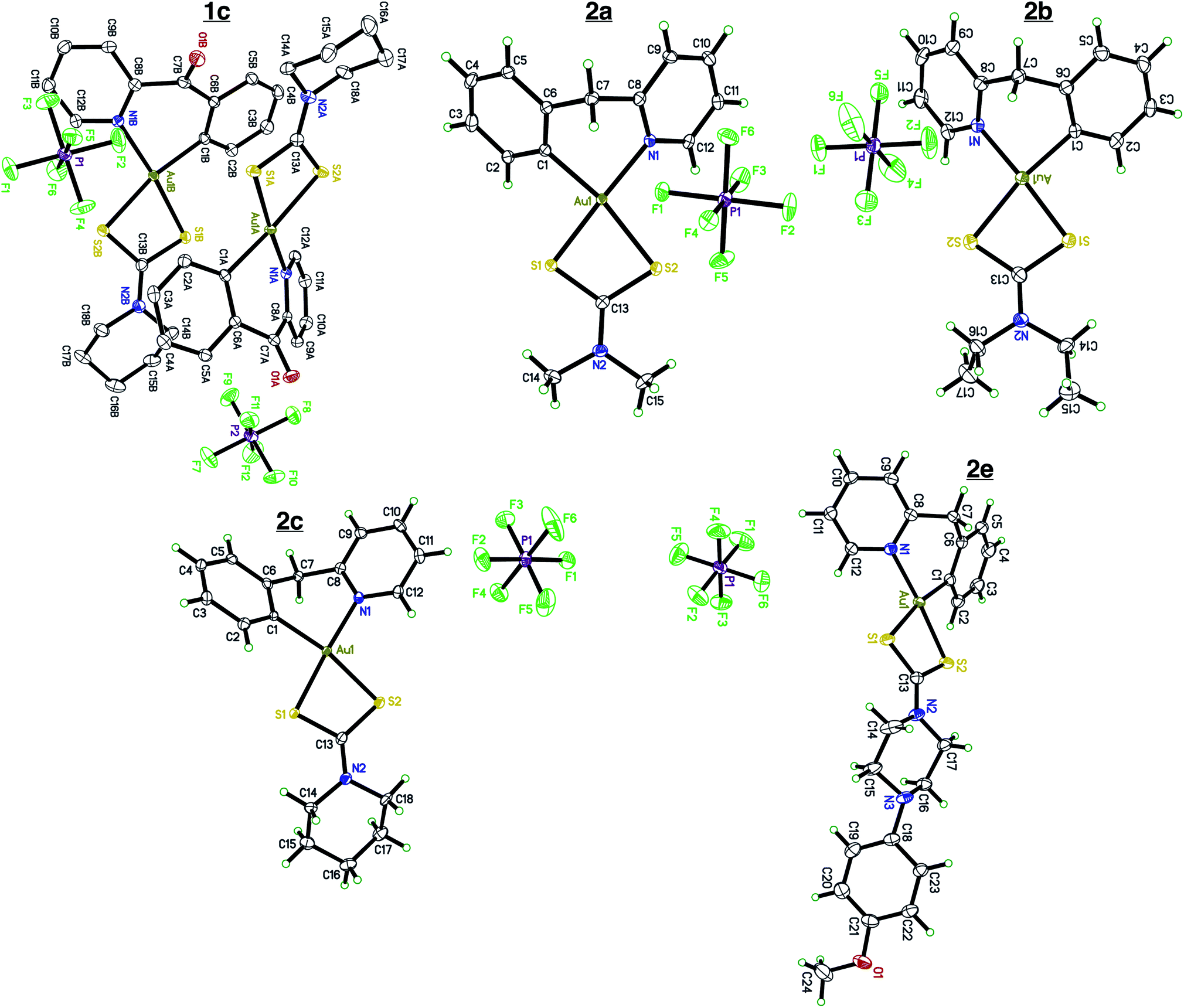
Single crystals of compounds 1c, 2a, 2b, 2c, and 2e (Fig. 1) were grown by slow diffusion of Et2O into a concentrated MeCN solution at room temperature.
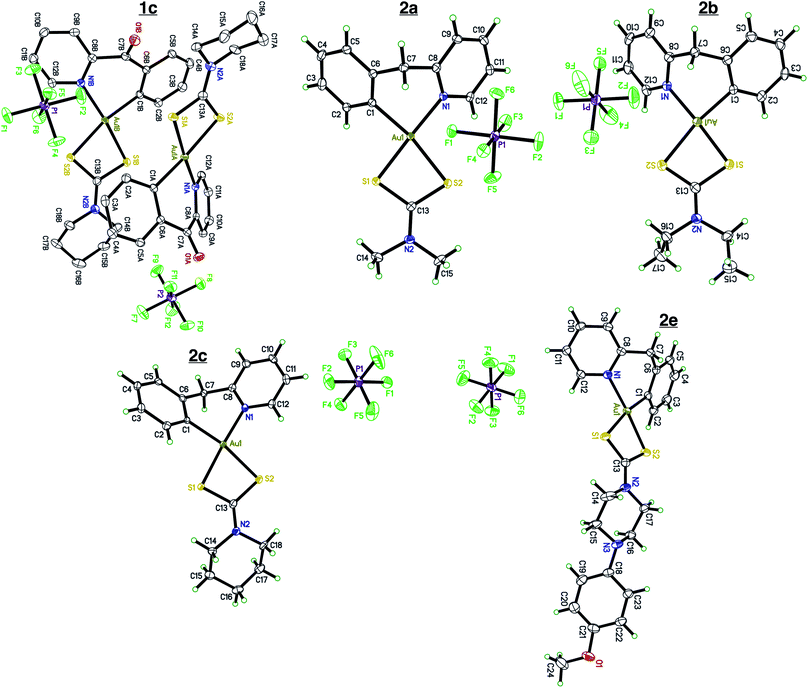 |
| | Fig. 1 X-ray crystallography. Single crystal X-ray diffraction of complexes 1c, 2a, 2b, 2c, and 2e. Complex 1c contains two cation/anion pairs in the asymmetric unit. Complex 2e crystalized out in the solid state with a molecule of Et2O. All atoms are drawn at the 50% probability level. Solvents were omitted for clarity. Full crystallographic parameters can be found in the ESI Tables S1–S5.† | |
Compounds 2a, 2b, 2c, and 2e crystallize out in the solid state with one molecule per asymmetric unit while compound 1c contains two cation/anions pair. Each cyclometalated ligand, 2-benzylpyridine and 2-benzoylpyridine have a slightly puckered twist. The bridging methine carbon allows for the formation of a 6-membered ring containing the Au atom, leading to a less strained cyclometalated system. Each molecule is distorted around the Au–C and Au–N bonds resulting in half occupancy among the two atoms. Analyzing the bonding motif of the [C^N] framework reveals a significant trans-effect on the binding dithiocarbamate ligand. The Au–S bond trans to the nitrogen of the [C^N] ligand is significantly shorter (on average 0.156 Å) than the Au–S bond trans to the carbon (Table 1). This results in a slightly distorted square planar geometry around the Au(III) center in comparison to other Au(III) bis-dithiocarbamate complexes bearing symmetrical dithiocarbamate ligands which contain four Au–S bonds equidistant to one another (∼2.33 Å).47,68
Table 1 Selected interatomic distances. Bond distances of complexes 1c, 2a, 2b, 2c, and 2e. Bond lengths were obtained from X-ray crystallography dataset
| Bond (Å) |
Compound |
|
1c
|
2a
|
2b
|
2c
|
2e
|
| Au–C |
2.042 (19) |
2.043 (3) |
2.061 (5) |
2.042 (3) |
2.045 (19) |
| Au–N |
2.062 (18) |
2.071 (2) |
2.065 (5) |
2.065 (3) |
2.069 (17) |
| Au–S(trans to N) |
2.283 (8) |
2.280 (7) |
2.278 (16) |
2.276 (9) |
2.298 (5) |
| Au–S(trans to C) |
2.376 (9) |
2.404 (8) |
2.392 (14) |
2.499 (8) |
2.379 (5) |
Photophysical and solution chemistry
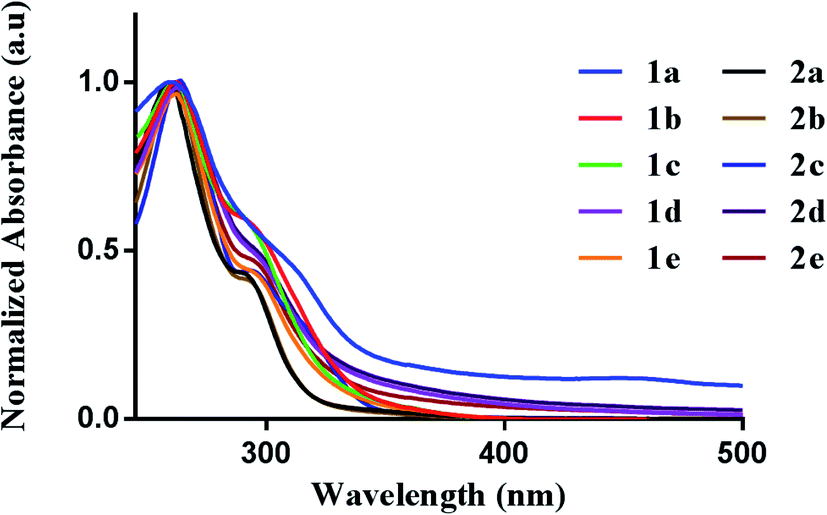
We first evaluated the photophysical properties of all ten complexes by scanning their absorption profile in DMSO (Fig. 2). A stock solution of each complex was prepared and diluted to a final concentration of (50 μM). The UV-vis profile was then obtained by scanning from 600 to 200 nm. Each complex displayed similar absorption profiles with a high-energy transition at 260 nm and a corresponding lower energy transition at 295–305 nm. The high-energy transition at 260 nm can be attributed to intraligand transitions of the 2-benzylpyridine or 2-benzoylpyridine fragments in the [C^N] ligands. The low-energy transition is attributed to ligand-to-metal-charge transfer from the donor dithiocarbamate ligand to the electrophilic Au(III) metal center.39 The different dithiocarbamate ligands utilized had minimal effect on the measured absorbance.
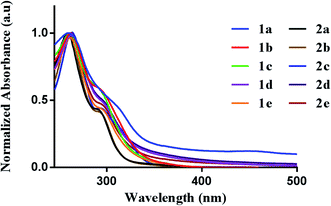 |
| | Fig. 2 Photophysical behavior. UV-vis spectra of 1a–e (50 μM) and 2a–e (50 μM) in DMSO. | |
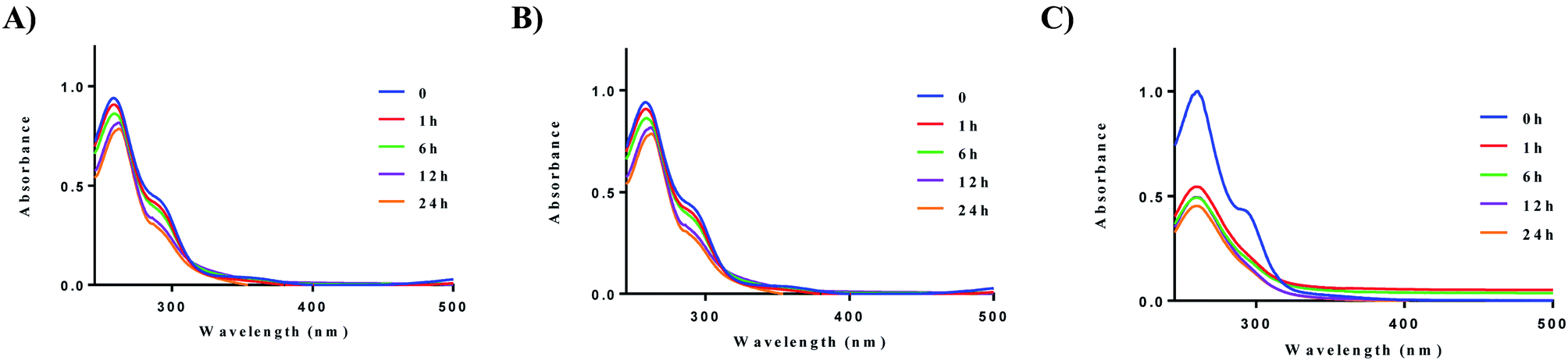
We next evaluated the stability of all ten complexes in three relevant biological media, namely, PBS (phosphate-buffered saline), DMEM (Dulbecco's modified eagle medium), and RPMI-1640 (Roswell Park Memorial Institute-1640). RPMI-1640 contain several biological nucleophiles such as amino acids and extracellular relevant levels of GSH, which are common sources of reductants. Development of new drug molecules require proper characterization of compound stability in solution. To evaluate their stability, the complexes were prepared as a stock solution in DMSO (5 mM) and diluted with PBS or DMEM at room temperature to a final concentration of 50 μM. Upon addition of the stock solution to the aqueous based medium, no precipitation was observed. Complex 2a displayed higher stability in PBS, and DMEM at 37 °C in comparison to RPMI-1640 (Fig. 3). The solution stability of the other 8 complexes, 1b–e and 2b–e can be found in the ESI (Fig. S57–S64† (PBS), Fig. S65–S72† (DMEM), and Fig. S73–S83† (RPMI-1640)).
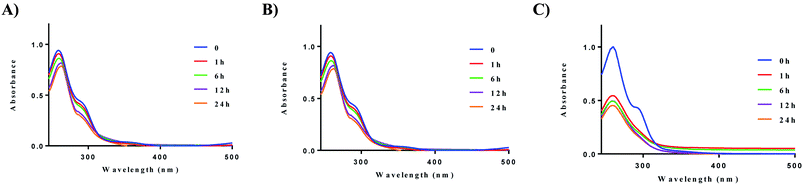 |
| | Fig. 3 Solution chemistry of 2a. (A) Stability of 2a (50 μM) in PBS over 24 h, (B) stability of 2a (50 μM) in DMEM over 24 h, (C) stability of 2a (50 μM) in RPMI-1640 over 24 h. Experiments were conducted at 37 °C. | |
After 24 h, no significant decrease in absorbance was observed for compounds 1a and 2a. The cyclic dithiocarbamate ligands used, especially complexes bearing the phenyl-substituted piperazine moiety demonstrated reduced stability in comparison to the alkyl substituted dithiocarbamates over 24 h. Although no deposition of elemental gold was observed, UV-vis analysis showed a significant decrease in absorption. We attribute the reduced stability of these complexes to the longer Au–S bond distances (2.298 Å (5)) trans to the aromatic nitrogen of the cyclometalated ligand, indicative of a weaker bond. This in addition to the other electronic factors promote facile reactivity with nucleophiles, hence the instability. Also, X-ray crystallography reveals the (4-methoxyphenyl)piperazine dithiocarbamate as an elongated moiety with a puckered square planar geometry. Elongation of the ligand likely weakens the Au–S bonds, which reduces stability to nucleophiles in the medium used. Overall, alkyl substituted gold dithiocarbamates, 1a and 2a show stability in biologically relevant media over extended periods of time and are promising candidates for further studies.
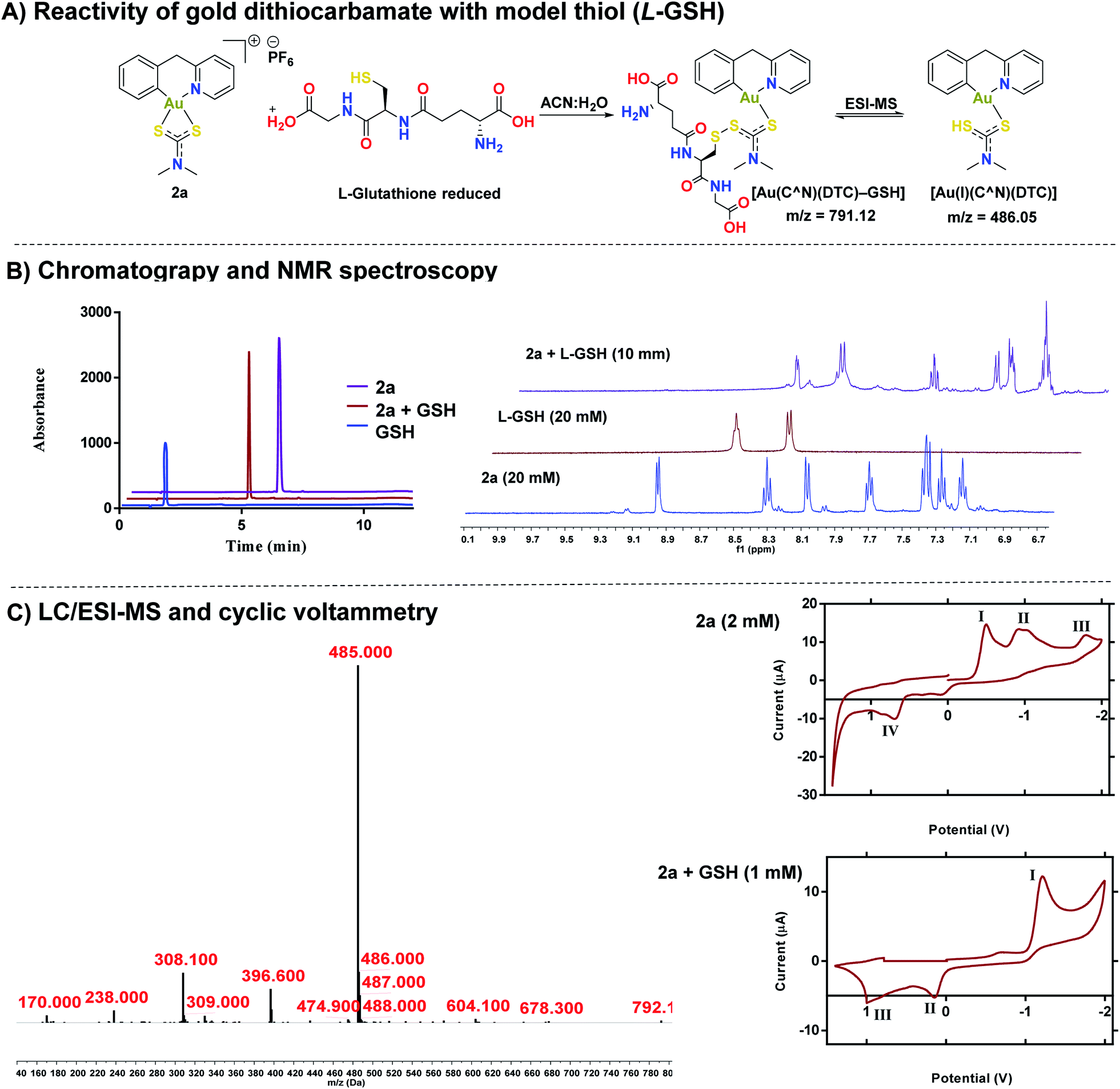
Reactivity of gold(III) dithiocarbamates with cysteine thiols
After establishing the stability of complexes, 1a or 2a in biological buffer, DMEM, and RPMI-1640 solutions, we investigated the reactivity of 2a with cysteine thiols using L-glutathione (GSH) and L-N-acetyl cysteine as a models (Fig. 4). Reactivity of 1a–1e and 2b–2e with both GSH and NAC can be found in the ESI (Fig. S84–S103†). Whereas gold dithiocarbamates are known to react with thiols,45,49,51,69 detailed studies to unravel the potential mechanism of activity do not exist. HPLC trace of the reaction solution revealed one distinct band at a different retention time (Rt – 5.00 minutes) from 2a (Rt – 6.03 minutes) or GSH (Rt – 1.72 minutes) (Fig. 4B). We subjected the reaction solution of 2a with GSH to LC-MS analysis (Fig. 4C), which supported the formation of a new species with a mass peak at m/z 791, attributable to a gold(I) disulfide species, [Au(C^N)(DTC)–GSH] (Fig. 4C), which is short-lived under ESI-MS conditions due to the labile disulfide bond formed between the thiol of L-GSH and the dithiocarbamate ligand of the gold complex (Fig. 4A). Thus, a more dominant mass peak at m/z 485, attributable to an intact gold complex with liberated L-GSH is observed. 1H NMR spectroscopy revealed the farthest downfield-shifted peak (δ = 9.03 ppm), which is the hydrogen located next to the nitrogen in the cyclometalated-ligand, was significantly shifted upfield in the adduct solution (Fig. 4B and S105†). We attribute this to the loss of coordination from the nitrogen of the cyclometalated ligand to gold, a key signature of the proposed adduct. We then confirmed the formation of the gold(I) complex by cyclic voltammetry (Fig. 4C), displaying a reduction potential of −1.20 V that is consistent with a gold(I) species.70–72 We evaluated the effect of the 2a-GSH adduct on breast cancer cells and found similar toxicity (IC50 – 0.53 μM (Fig. S110† as reported in Table 2 below. Taken together, we have further elucidated the reactivity of [C^N] gold(III) dithiocarbamates with representative thiols. Interestingly, the reduced gold(I) compound display high anticancer activity.
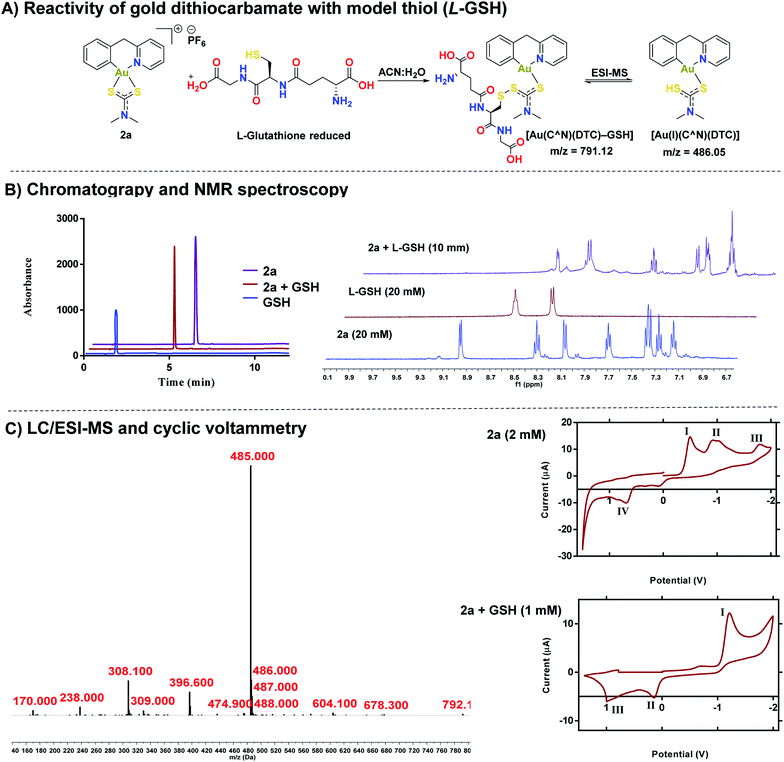 |
| | Fig. 4 Elucidation of the Au(III) – thiol adduct. (A) Proposed reaction scheme of compound 2a and GSH in an equimolar ratio, (B) HPLC chromatograms of 2a, GSH, and the reaction of 2a + GSH adduct (λ = 240 nm) (see ESI† for HPLC method and sample preparation) with 1H NMR of the same mixture in DMSO (see ESI† for full spectra), (C) LC-MS of the 2a + GSH adduct (λ = 280 nm) (see ESI† for HPLC method and sample preparation), cyclic voltammograms of 2a (I = Au(I) to Au(0) reduction,70 II = disproportionation of a one electron reduction from Au(III)/Au(II) and Au(II)/Au(I),73,74 III = Au(III) to Au(I) reduction,46,73,74 IV = oxidation of the dithiocarbamate ligand)and 2a + GSH(I = Au(I) to Au(0), II = oxidation of the dithiocarbamate ligand, III = oxidation of GSH). | |
Table 2
In vitro antiproliferative activity. IC50 values for 1a–e and 2a–e across a panel of cell lines. Cells were seeded at a density of 2000 cells per well and treated for 72 h. IC50 values are plotted as the mean ± s.e.m (n = 3). Full dose response curves can be found in the ESI
|
|
IC50 (μM), 72 h |
| MDA-MB-175 |
MDA-MB-231 |
A2780 |
RPE-NEO |
MRC5 |
|
1a
|
0.926 ± 0.10 |
0.531 ± 0.103 |
0.521 ± 0.081 |
10.29 ± 2.61 |
>50 |
|
1b
|
1.26 ± 0.307 |
0.935 ± 0.16 |
0.924 ± 0.187 |
25.4 ± 1.98 |
>50 |
|
1c
|
1.85 ± 0.194 |
0.951 ± 0.135 |
1.19 ± 0.126 |
12.11 ± 1.59 |
>50 |
|
1d
|
0.842 ± 0.671 |
1.14 ± 0.08 |
0.816 ± 0.123 |
15.13 ± 2.19 |
>50 |
|
1e
|
1.12 ± 0.431 |
1.36 ± 0.04 |
1.13 ± 0.065 |
14.8 ± 2.15 |
>50 |
|
2a
|
0.618 ± 0.080 |
0.773 ± 0.117 |
0.741 ± 0.086 |
17.1 ± 1.86 |
>50 |
|
2b
|
0.831 ± 0.099 |
2.11 ± 0.100 |
0.820 ± 0.073 |
15.1 ± 1.86 |
>50 |
|
2c
|
1.031 ± 0.091 |
1.04 ± 0.101 |
1.20 ± 0.33 |
21.1 ± 18.6 |
>50 |
|
2d
|
0.937 ± 0.399 |
0.849 ± 0.067 |
2.962 ± 0.71 |
14.5 ± 2.45 |
>50 |
|
2e
|
1.17 ± 0.487 |
1.31 ± 0.088 |
0.811 ± 0.892 |
19.2 ± 2.81 |
>50 |
Cellular uptake
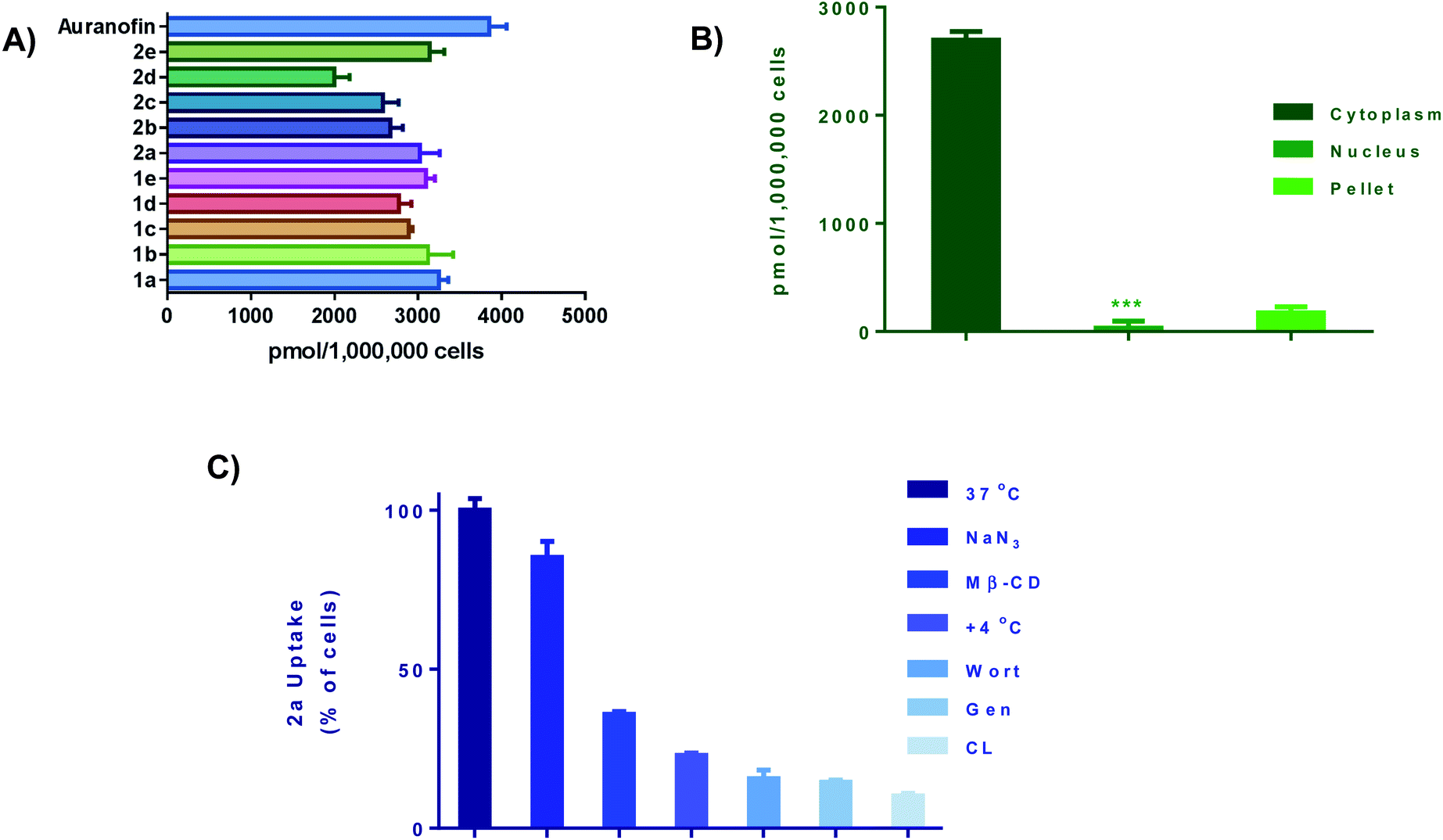
We next performed cellular uptake studies to determine what percentage of the compounds were getting into the cell after treatment. Given that most therapeutic targets are localized receptors on the plasma membrane, quantifying the amount of gold compounds in cellular fractions and investigating their mode of uptake is crucial. For whole-cell uptake studies, MDA-MB-231 were treated with all 10 compounds and Auranofin at 5 μM for 24 h (Fig. 5A). Auranofin is the only gold-based therapeutic to be clinically approved, therefor we used this drug as a benchmark for comparison.67 After treatment, the cells were washed with PBS, centrifuged, and digested with 0.5 mL of conc. HNO3 and diluted with DI water and subjected to ICP-OES analysis. All ten compounds exhibited intracellular uptake above 2000 pmol per million cells. In comparison to Auranofin, none of the complexes exhibited higher uptake, however; the complexes 1a and 2a had uptakes (3017 pmol per million cells) and (3267 pmol per million cells) respectively. Complexes 1c–e and 2c–e, which contain more hydrophobic R groups such as the piperadine and phenyl substituted piperazine handles, exhibited lower intracellular uptake than the complexes with the less hydrophobic shorter alkyl (methyl and ethyl) handles. Further evaluation revealed that >90% of the Au content was found localized in the cytoplasm (Fig. 5B) and total membrane fraction (pellet). Given that these complexes readily react with nucleophilic biological molecules and the lack of accumulation in the nuclear fraction, it is evident that the mechanism of action of this class of compounds is differentiated from the clinically available platinum agents.
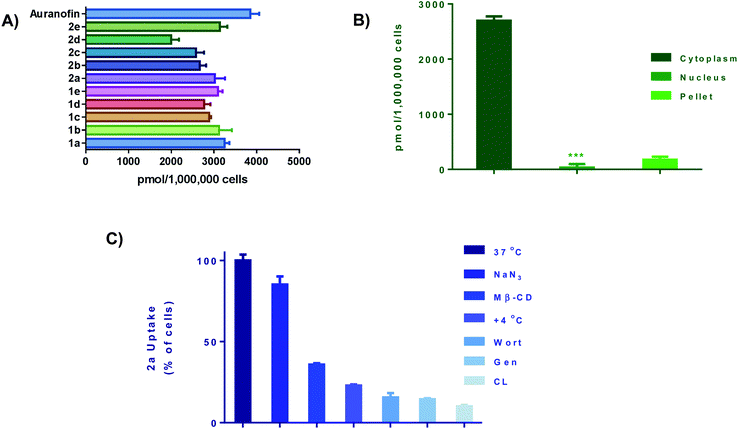 |
| | Fig. 5 Cellular uptake and uptake mechanism. (A) Intracellular uptake of 1a–e and 2a–e as well as Auranofin. Cells were seeded at a density of 1 × 106 and incubated overnight. Cells were incubated with gold drug or compounds for 24 h (5 μM), data is plotted as the mean ± s.e.m (n = 3), (B) cell fraction uptake of 2a at 5 μM for 24 hours, data are plotted as the mean ± s.e.m (n = 3), ***denotes value lower than detection limit of the instrument (5 μg L−1), (C) whole cell uptake of 2a (5 μM for 24 h) against pre-treatment with uptake inhibitors, data are plotted as the mean ± s.e.m (n = 3). Calibration curves can be found in Fig. S133–S135.† | |
To understand cell uptake mechanisms, detailed uptake studies using GF-AAS were conducted whereby MDA-MB-231 cells were incubated with gold compound, 2a after cellular pretreatment with one of a number of known inhibitors of cellular uptake. Sodium azide (NaN3), which is known to inhibit mitochondrial oxidative phosphorylation, was used as a general inhibitor of energy (ATP)-dependent (active) uptake; chlorpromazine (CL) was utilized as an inhibitor of clathrin-dependent endocytosis (CDE); β-cyclodextrin (Me-β-CD) and genestein (Gen) were used as inhibitors of clathrin-independent endocytosis (CIE); and, wortmannin (Wort) was employed as a known inhibitor of micropinocytosis. We found that the uptake of 2a was significantly decreased with all inhibitors with the exception of NaN3 (Fig. 5C). Being an ATP-dependent pathway illustrates that these compounds are not dependent on oxidative phosphorylation for uptake, despite; their ability to modulate OXPHOS. Overall, these ten Au(III) complexes demonstrate relatively high intracellular uptake which can explain the high in vitro cytotoxicity as well as the rate at which cellular bioenergetics are affected.
Anticancer activity in vitro
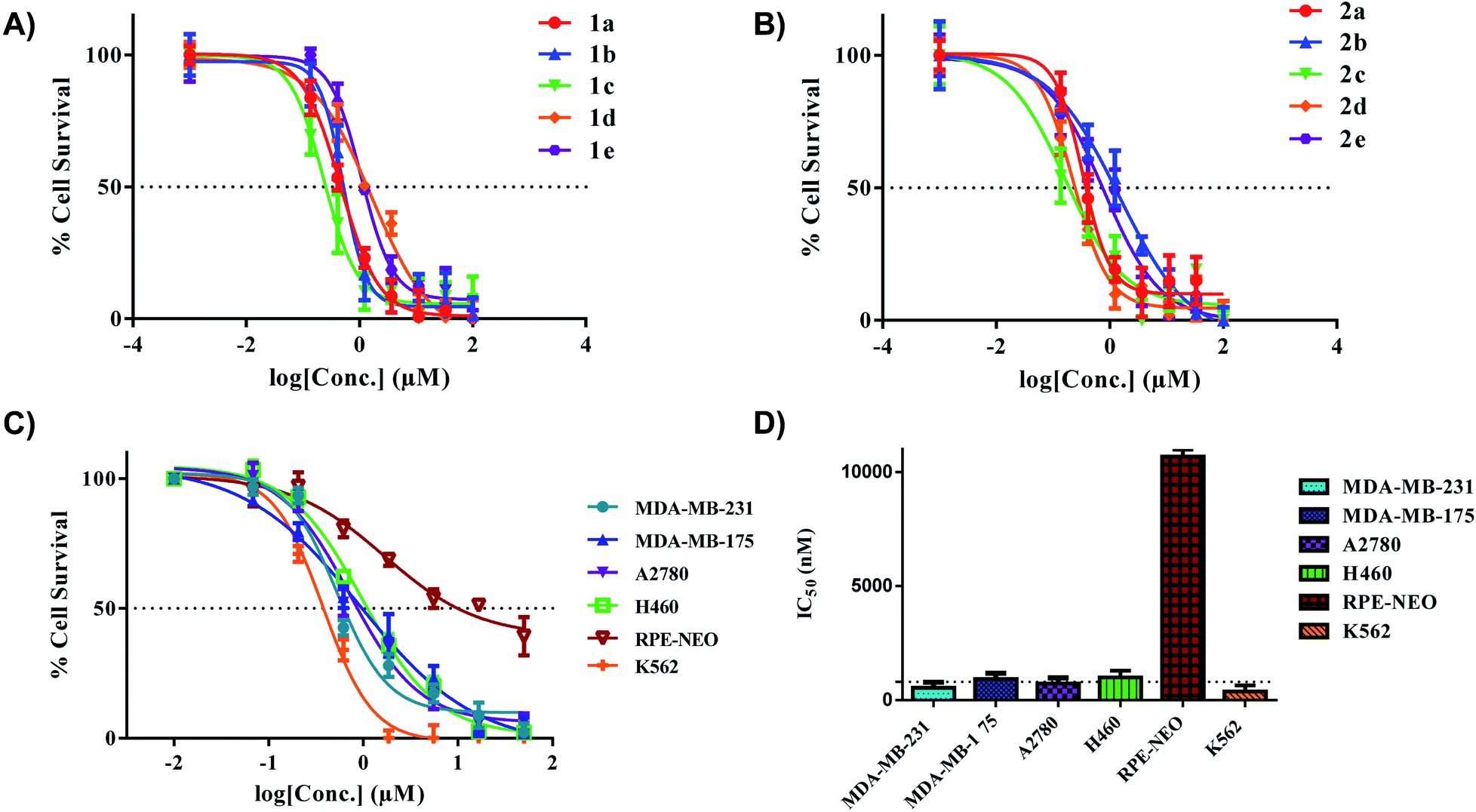
All the gold(III) dithiocarbamate compounds display remarkable cell killing potential in a panel of breast, ovarian, lung and leukemia cancers. Preliminary studies focused on different cancer cell lines (MDA-MB-231, MDA-MB-175, and A2780), an immortalized normal retinal epithelial cell line (RPE-NEO) and human fetal lung fibroblasts (MRC5) (Fig. S111–S129†). These adherent cells were exposed to a serial-diluted concentration of all ten compounds for 72 h. The cells were subsequently treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solutions, dissolved with DMSO and the absorbance at 570 nm was measured with a plate-reader. Our initial screen showed promising half-maximal inhibitory concentration (IC50) values for all complexes across the three cancer cell lines (Table 2). Comparison of IC50 values with uptake revealed no significant correlation.
Specifically, the complexes exhibited high toxicity (0.5–1.5 μM) towards the TNBC cell line MDA-MB-231 (Fig. 6A and B). These complexes collectively are more cytotoxic towards the cell line MDA-MB-231 in comparison to the [C^N]Au(III)DTC complexes prepared by Bochmann et al.45 The different [C^N] backbone presented here proves to be a critical component in drug design and overall improvement of cytotoxic efficacy. The cytotoxicity of complexes 1a and 2a towards TNBC's is quite promising as these cell lines are typically cisplatin resistant.75,76 Drug resistance is an ever-increasing problem in medicine so developing therapeutics for refractory tumours is of great importance.77 To test the selectivity of compounds 1a and 2a for cancer cells over normal cells, we evaluated these complexes in the normal retinal epithelial cells using the MTT assay. We found up to ∼30-fold selectivity for cancer cells over normal cells, indicative of compounds with potential for reduced side effects.
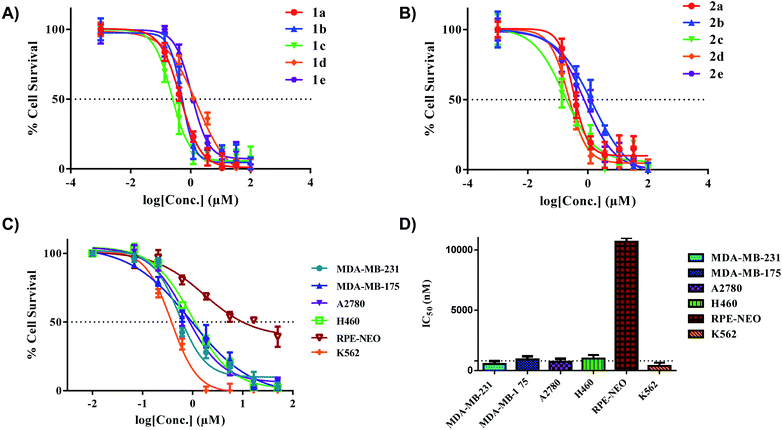 |
| | Fig. 6
In vitro activity of complexes 1a–e and 2a–e in multiple cancer cell lines. (A) IC50 plot for complexes 1a–e in MDA-MB-231 (72 hours), (B) IC50 plot for complexes 2a–e in MDA-MB-231 (72 hours), (C) extended panel of cell lines for complex 2a, (D) representative bar graph illustrating the IC50 values of 2a across a panel of cancer cell lines. The dotted line indicates a threshold of 1 μM. % Cell survival was determined with MTT (adherent cells) and CellTiter-Glo (suspended cells). Data are plotted as the mean ± s.e.m (n = 3). | |
With the completion of preliminary cytotoxicity screening, we set out to explore the efficacy of compound 2a in different cancer tissue type beyond breast cancer. Both H460 (human large cell lung carcinoma) and K562 (human chronic myelogenous leukemia) were utilized (Fig. 6C and D). We found comparable cytotoxicity in these cell lines to those in our preliminary screen. With 2a displaying IC50 values of 1.5 μM in H460 and 1.0 μM in K562. With a promising candidate in hand, we pursued further biological testing to gain mechanistic insight of this class of Au(III) dithiocarbamates.
Differential gene expression and biological pathway analysis
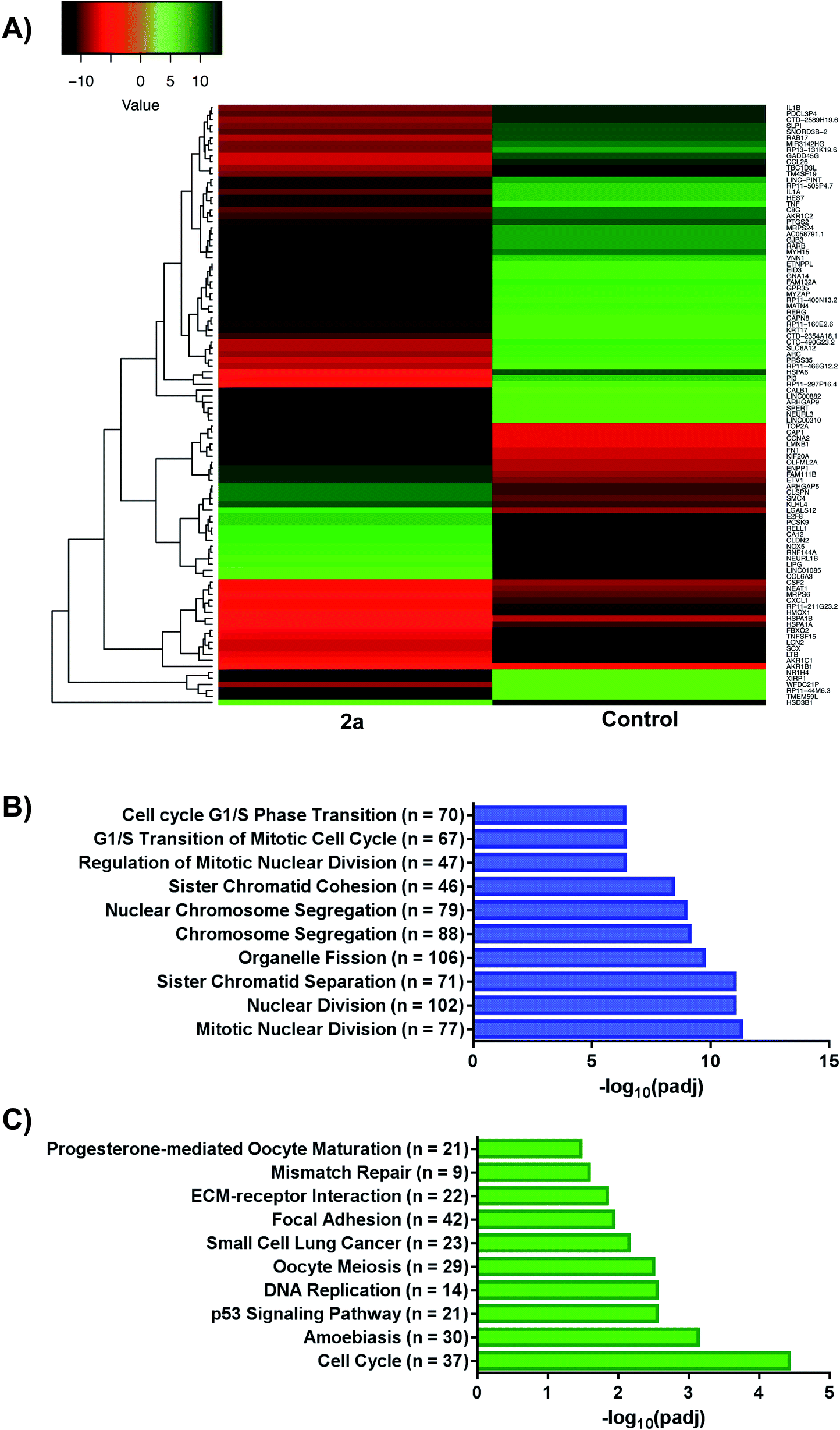
We investigated the whole-cell effect of compound 2a by analysing differentially expressed genes (DEG) from RNA-seq. We treated MDA-MB-231 cells with 1 μM of 2a for 12 h followed by the isolation of high-purity RNA for Illumina Hi-seq. We found 3019 DEG with 1596 upregulated and 1423 downregulated genes in response to 2a (Fig. 7A). Subsequent use of gene ontology (GO)78,79 (Fig. 7B) and Kyoto encyclopaedia of genes and genomes (KEGG)80–82 (Fig. 7C) pathway analysis software led to potential processes perturbed by 2a. The pathway analysis software employed are an extensive library database capable of integrating chemical and biological pathway perturbation processes and is well suited for drug development studies. For the identified processes, we examined the corresponding downstream canonical pathways and corroborated activated or inhibited pathways with functional biology experiments (vide infra). Notable pathways identified include, mitotic nuclear division, organelle fission, cell-cycle, progesterone oocyte maturation, and focal adhesion processes. Interestingly, a significant number of these processes are regulated by the mitochondria. It is well established that a number of aggressive tumor types including TNBCs exhibit impaired mitochondria function, which present a vulnerability.83,84 We therefore, hypothesized that this class of gold(III) dithiocarbamates disrupt mitochondria function in MDA-MB-231 cells. This was tested by detailed mitochondrial investigations.
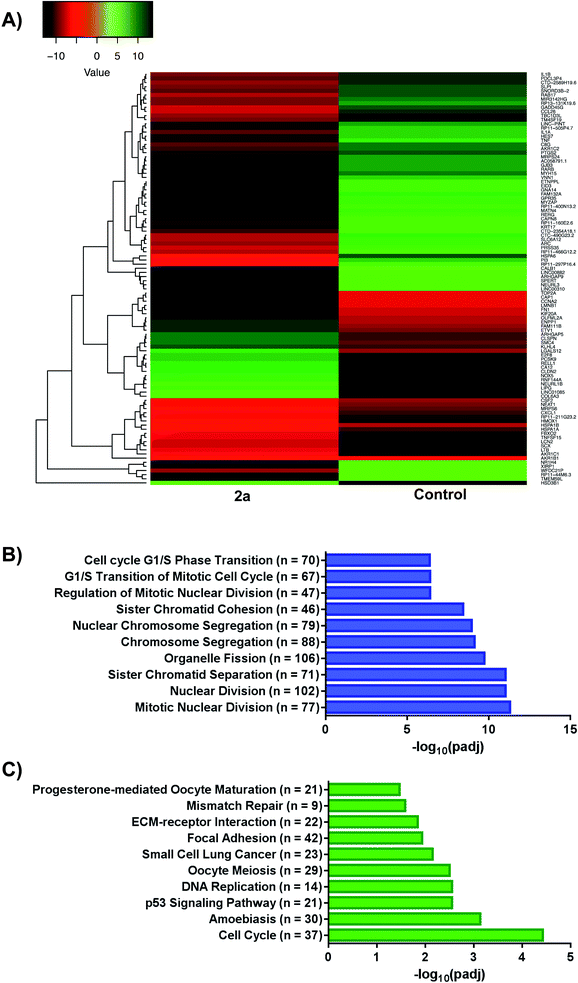 |
| | Fig. 7 RNA-sequencing and pathway analysis. (A) Representative heat map of differentially expressed genes (DEGs) in response to 2a, (B and C) gene ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) plots outlining varying pathways perturbed upon treatment with 2a. For GO; GeneRatio = n/2005 and = BgRatio n/17107. For KEGG; GeneRatio = n/727 and BgRatio = n/5824. | |
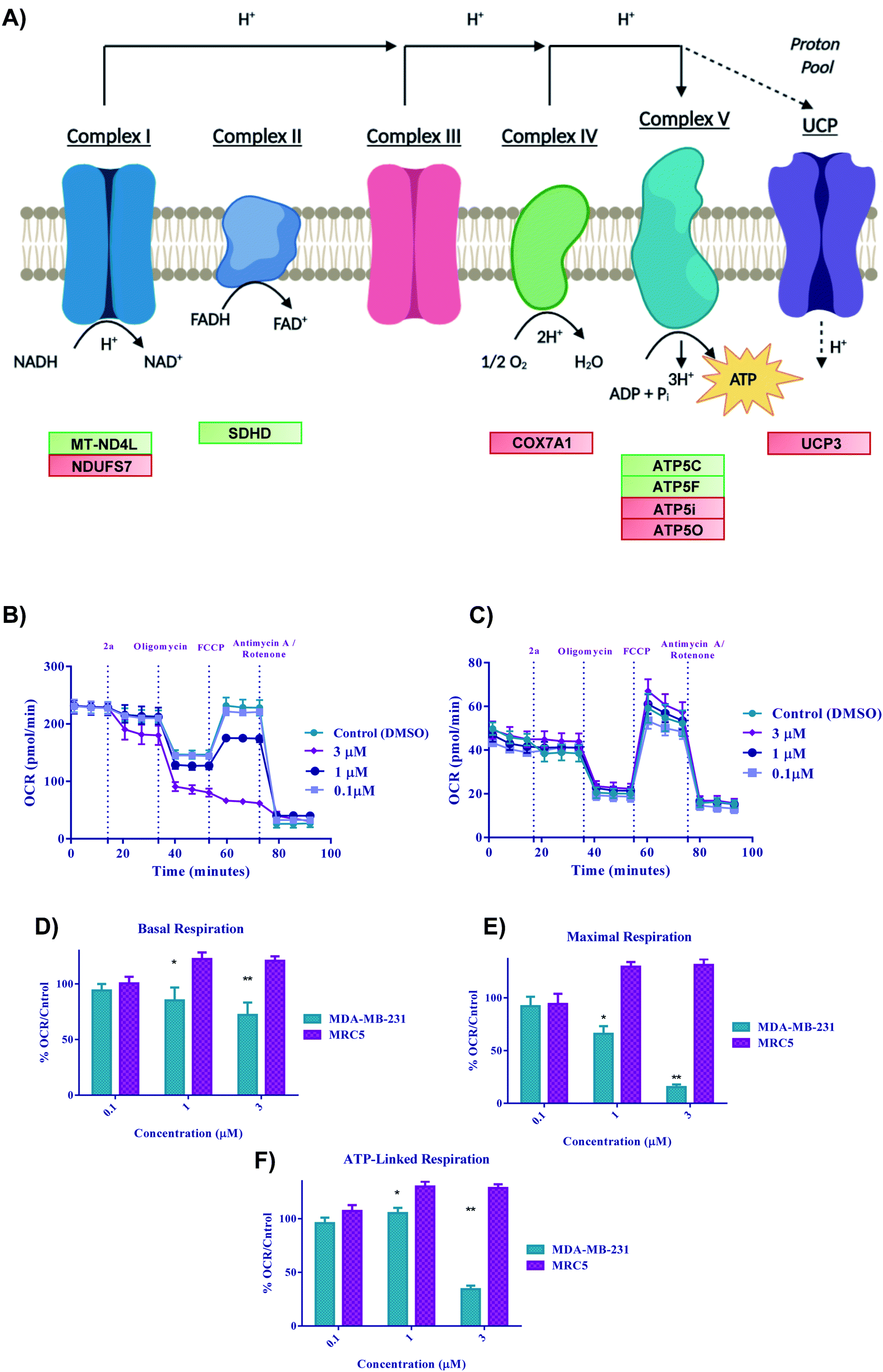
Gold(III) dithiocarbamate disrupts TNBC cell metabolism
The RNA-seq showed transcriptional suppression of key genes involved in the catalytic conversion of long-chain fatty acids such as ACSL4. Further analysis of DEG revealed the modulation of several genes within the mitochondria respiratory chain or oxidative phosphorylation. Activated DEG included NDUFS7, [complex I], COX7A1 gene [complex IV], and ATP5O, ATP5I genes [complex V] whereas the inhibited DEG were MT-ND4L [complex I], SDHD gene [complex II], and ATP5C, ATP5F [complex V]. Moreover, an uncoupling protein related gene, UCP3 was found to be upregulated in response to 2a. These findings prompted functional biology experiments to further corroborate the effect of 2a or its mechanism of action in TNBC. We examined the effect of 2a on mitochondrial membrane potential, assayed by JC-1 dye.85 Following an extended 2a treatment and JC-1 staining (Fig. 8), we found that 2a exhibits strong depolarization of the MMP (J-monomers) in comparison to the DMSO treatment (J-aggregates) in MDA-MB-231 cells. We used carbonyl cyanide m-chlorophenyl hydrazine (CCCP), an uncoupler, as a positive control to validate the experiment. The MMP is a key driving force in ATP synthesis and overall a key factor in maintaining redox homeostasis throughout the cell.86 Uncoupling of the mitochondrial membrane results in acute cell death as the mitochondrial become dysfunctional in this aggregative state.
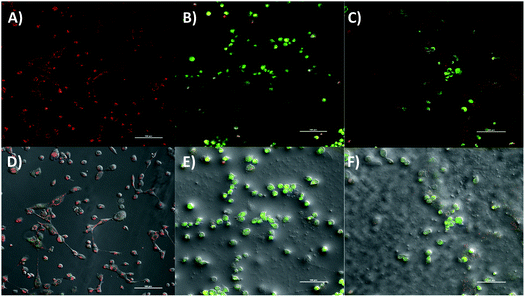 |
| | Fig. 8 Mitochondrial membrane potential of 2a in MDA-MB-231 cells. (A) DMSO vehicle, (B) 2a at 10 μM for 6 h, (C) CCCP as a positive control at 100 μM for 30 minutes, (D–F) corresponding bright field image: DMSO vehicle, 2a, and CCCP. Compound 2a significantly depolarizes the mitochondria membrane upon treatment within 6 h. MDA-MB-231 cells were seeded at a density of 5 × 105 cells per glass bottom dish with a #1.5 coverslip. J-Monomers shown as green fluorescence (exc. 488 nm) and J-aggregates shown as red fluorescence (exc. 520 nm). Images are representative of three independent treatments. | |
Bioenergetics
With the previous biological data pointing towards metabolic changes, we sought to explore further the effect 2a had on redox metabolism. The ETC is a complicated biological system that is constantly changing in response to external stress in order to achieve redox homeostasis within the cell. With the DEG data alluding to involvement of ETC genes (Fig. 9A), we then decided to look in depth at the effect of complexes on these biological parameters. We next performed oxygen consumption rate (OCR) experiments by Seahorse 96XF to quantify the effect of 2a on mitochondrial bioenergetics or stress (MitoStress) (Fig. 9B and C).87 A series of known inhibitors of the various parts of the ETC that allow for the measurement of specific parameters were used. We conducted a pneumatic injection of 2a into wells containing adhered MDA-MB-231 cells followed by subsequent injections of oligomycin, a complex V inhibitor, to view the basal OCR; FCCP, an uncoupler used to observe the maximum OCR, and rotenone/antimycin A, a complex I/III inhibitor to completely shut down the ETC. The first step was to determine the optimal MDA-MB-231 cell density and FCCP concentration, which was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and (0.6 μM) FCCP (ESI†) respectively. At a concentration of 3 μM, the basal OCR is diminished by more than 20% after only 17 minutes from time of injection of 2a (Fig. 9D). We found that at a low concentration of 2a (1 μM), maximal OCR (Fig. 9E) is diminished by more than 25% within 50 minutes of treatment compared to the control. The overall decrease in maximal OCR at 1 and 3 μM implies acute depletion of mitochondrial respiration. ATP linked respiration was also calculated and found to be decreased by more than 80% at 3 μM treatment (Fig. 9F). Overall, the rapid decline in OCR suggests that these complexes are severely impacting the ETC and subsequently OXPHOS, thus causing cell death. Despite the acute dose-dependent OCR depletion induced by 2a in MDA-MB-231 cancer cells (Fig. 9B), 2a did not impact the OCR of human fetal lung fibroblast cells, MRC5 (Fig. 9C). These data indicate that 2a causes rapid irreversible inhibition of OXPHOS in the TNBC, MDA-MB-231 but not in the normal lung epithelial fibroblast, MRC5.
000 cells per well and (0.6 μM) FCCP (ESI†) respectively. At a concentration of 3 μM, the basal OCR is diminished by more than 20% after only 17 minutes from time of injection of 2a (Fig. 9D). We found that at a low concentration of 2a (1 μM), maximal OCR (Fig. 9E) is diminished by more than 25% within 50 minutes of treatment compared to the control. The overall decrease in maximal OCR at 1 and 3 μM implies acute depletion of mitochondrial respiration. ATP linked respiration was also calculated and found to be decreased by more than 80% at 3 μM treatment (Fig. 9F). Overall, the rapid decline in OCR suggests that these complexes are severely impacting the ETC and subsequently OXPHOS, thus causing cell death. Despite the acute dose-dependent OCR depletion induced by 2a in MDA-MB-231 cancer cells (Fig. 9B), 2a did not impact the OCR of human fetal lung fibroblast cells, MRC5 (Fig. 9C). These data indicate that 2a causes rapid irreversible inhibition of OXPHOS in the TNBC, MDA-MB-231 but not in the normal lung epithelial fibroblast, MRC5.
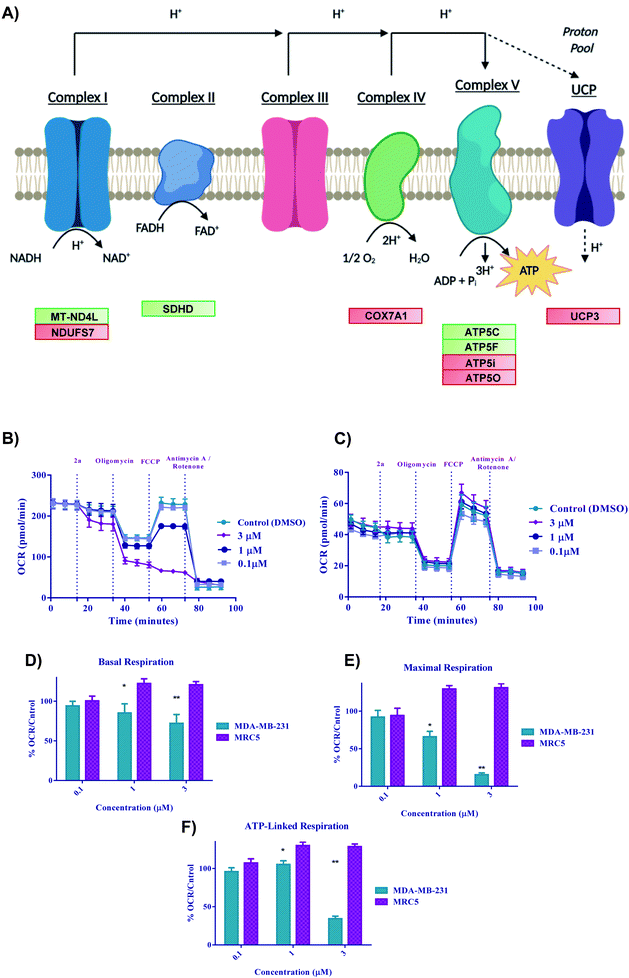 |
| | Fig. 9 Bioenergetics of 2a in cells. (A) Schematic illustration of the electron transport chain and corresponding DEGs for 2a, (B–F) Bioenergetic monitoring of MDA-MB-231 (green bars) and MRC5 (purple bars) cells following acute in vitro treatment with complex 2a. MDA-MD-231 were seeded at a density of 3 × 104 and MRC5 at a density of 5 × 104 and analyzed with a Seahorse XF96 analyzer to assess the effect on key bioenergetic parameters. (B and C) Mitochondria stress test was performed with pneumatic injections of compound 2a at concentrations ranging from (0.1–3 μM) and response to injections of oligomycin, FCCP, and antimycin A/rotenone. Data is plotted as the mean ± s.e.m (8 technical replicates). (D–F) Key bioenergetic parameters have significant statistical response to treatment with compound 2a at concentrations as low as 3 μM within 30 minutes in MDA-MB-231 with selectivity over MRC5 cells. Data are plotted as the mean ± s.e.m, *p < 0.05, **p < 0.01. | |
Apoptosis evaluation
To further assess the mechanistic pathway upon treatment with 2a, we analysed the apoptotic effect in MDA-MB-231.
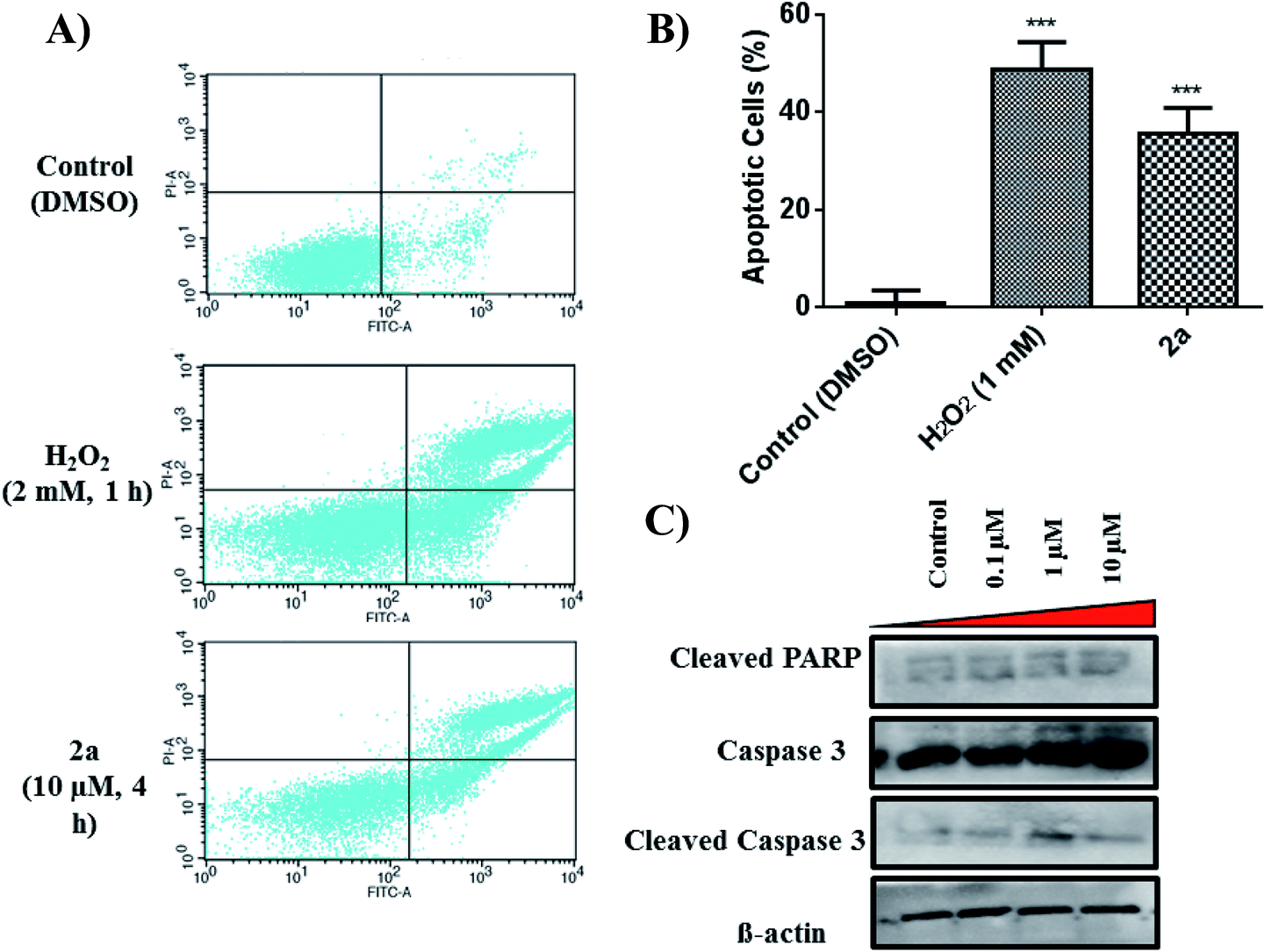
Apoptosis is a common cell death pathway for chemotherapeutics which can be characterized by distinct morphological features and biochemical mechanisms.88,89 Apoptosis occurs normally in healthy cells to maintain a healthy population of cells during aging or development of tissues. Some transition metal-based drugs can trigger apoptosis due to inhibition of p53 dependant pathway (a tumour suppressor gene).90,91 Populations of apoptotic cells can be determined by containing cells with Annexin V and PI.92,93 Cells undergoing apoptosis contain ample amount of phosphatidylserine (PS) which can be bound by Annexin V. The Annexin is then labelled with FITC, a green fluorogenic dye which can be visualized by fluorescence activated cell sorting (FACS). PI is used to stain damaged DNA to distinguish apoptotic from necrotic cells. Such staining gives four separate quadrants upon analysis; (i) lower left, healthy cells which are negative for both markers, (ii) lower right, pre-apoptotic cells which are positive for FITC but not for PI, (iii) upper right, apoptotic cells which are positive for both markers, (iv) upper left, necrotic cells which are positive for only PI. Fig. 10A illustrates that 2a at 10 μM induced significant apoptosis of MDA-MB-231 cells. Experimental data shows a 36% increase (extrapolated from Fig. 10B) in apoptotic cells in comparison to the control in just 4 h, indicative of apoptosis as a possible mode of cell death. It is possible that 2a triggers either caspase dependant- or caspase independent-apoptosis.94,95 Immunoblotting was used to assess the effect of 2a on proteins involved in caspase related apoptosis. In a concentration dependant manner, both caspase 3 and cleaved caspase 3 were found to be upregulated in comparison to the control (Fig. 10C). Upregulation of these proteins is indicative of a caspase dependant apoptotic pathway. Caspase 3 is considered to be an executioner caspase in apoptosis as it coordinates the destruction of cellular structures including DNA fragmentation and degradation of the cytoskeleton.96,97 Immunoblotting of cleaved PARP in response to 2a showed unaltered protein expression, which is likely due to an uninvolved role of PARP DNA damage response.98–102
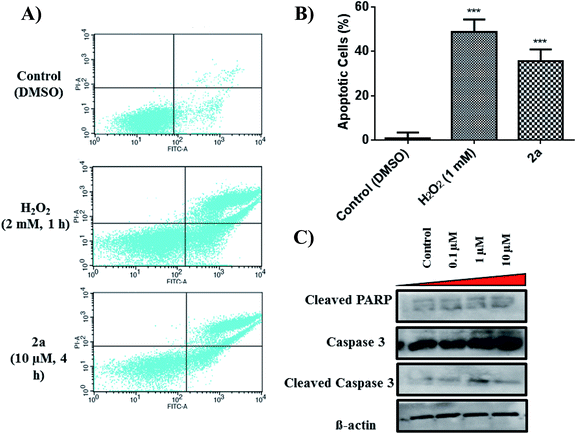 |
| | Fig. 10 Effect of 2a on apoptosis. (A) Quadrants displaying apoptotic population of MDA-MB-231 within 4 h of treatment with 2a. Cells were seeded at a density of 5 × 105 per well. Data is representative of three individual experiments. H2O2 was used a positive control, (B) bar graph illustrating the early-stage apoptotic cell population. Data are plotted as the mean ± s.e.m (n = 3), ***p < 0.001. (C) Immunoblots of MDA-MB-231 treated in a concentration dependant manner. | |
ROS analysis
We examined the status of intracellular ROS induced by 2a. For this experiment, we employed the used 2′,7′-dichlorofluoresceindiacetate (DCF-DA),103 a fluorogenic dye with an excitation/emission wavelength of 495/525 nm. DCF-DA enters into the cell and is subsequently deacetylated by cellular esterases where it is then oxidized by ROS to produce a fluorescent compound that can detected with flow cytometry (FACS) using the FITC channel.104
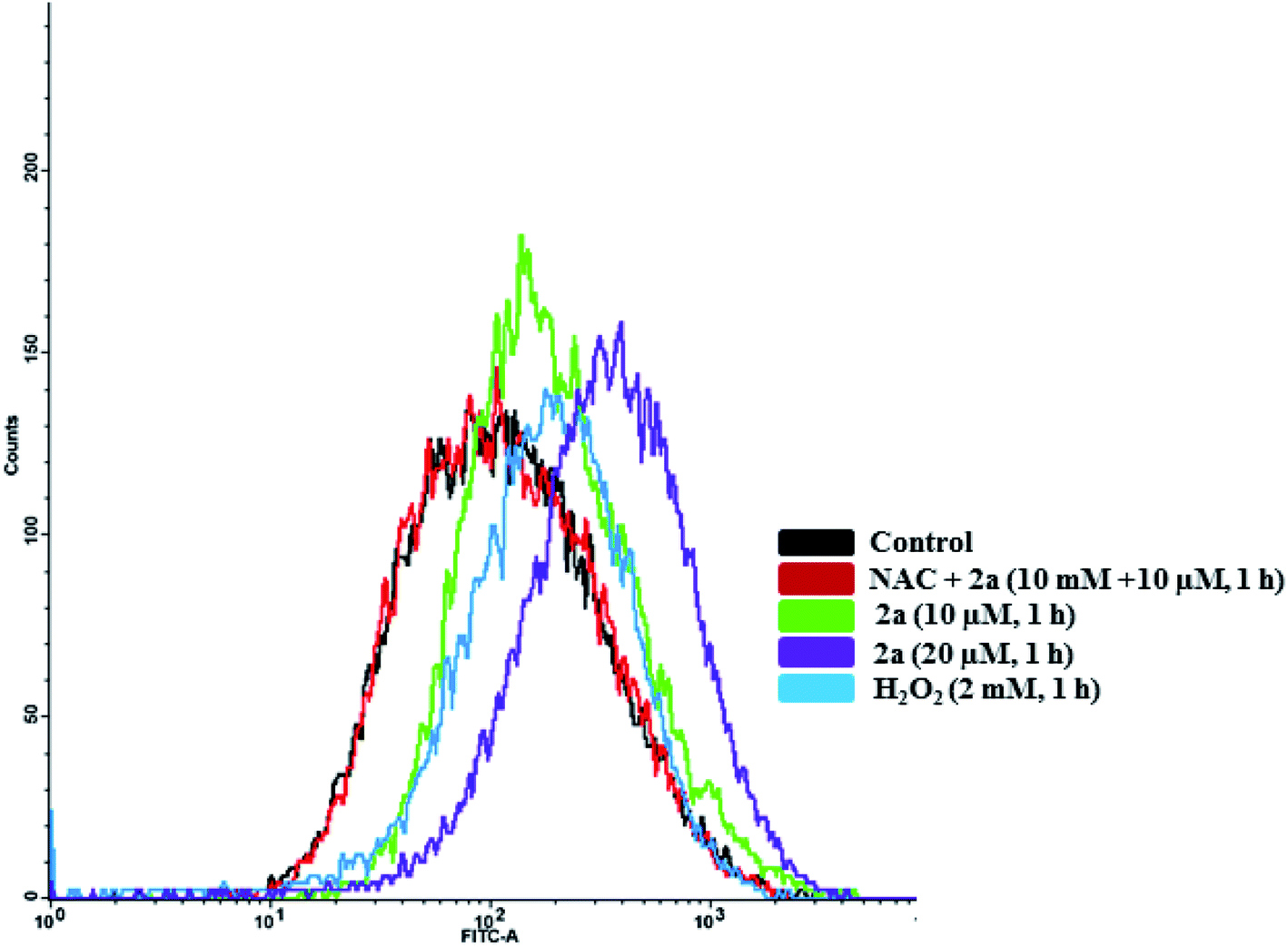
To quantify the amount of ROS produced, we subjected MDA-MB-231 cells to 2a at 10 and 20 μM to assess concentration dependence. Within 1 hour of treatment, we observed a 1.3× increase in ROS in cells treated with 2aversus DMSO (Fig. 11). At twice the concentration, 20 μM, we observed a 1.5× increase in ROS. This shows a slight concentration dependence as well as very fast imbalance of the cell homeostasis. When compared to the H2O2 control, 2a produces similar ROS levels in MDA-MB-231 cells (Fig. 11). This suggests that ROS production has a key role in the cell death pathway of these complexes. To further solidify that the DCF-DA fluorescence is from ROS and not other reactive species such as RNS, we pre-treated cells with 10 mM N-acetyl cysteine for 2 h. NAC is a natural ROS scavenger and a key component in the formation of glutathione (GSH), which is a powerful antioxidant.105,106 After pre-treatment, the cells were subjected to the same concentration of 2a (10 μM) for 1 h. We observed using FACS that there was no increase in ROS levels in comparison to the control. This reveals that there is a significant amount of ROS being produced in the cells upon treatment and could be a product of OXPHOS inhibition.
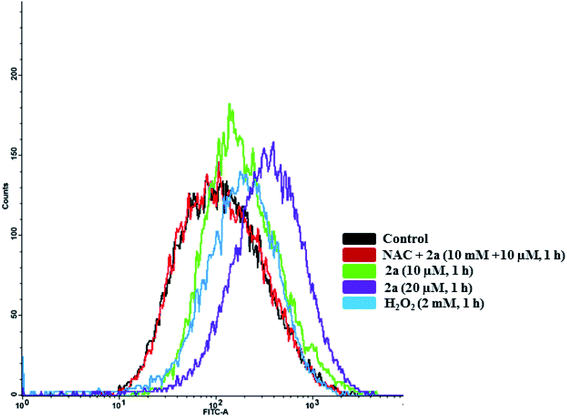 |
| | Fig. 11 Intracellular ROS induced by 2a. Compound 2a induces significant increase in ROS at 10 μM in 1 h. ROS accumulation in MDA-MB-231 cells were monitored with DCF-DA over 1 h. Cells were seeded at a density of 5 × 105 and added compounds from a 5 mM stock solution in DMSO. H2O2 was used a positive control (30 minutes, 1 mM). Cells were pre-treated with 10 mM N-acetyl cysteine (NAC) for 2 h prior to addition of the compounds. | |
Cell cycle analysis
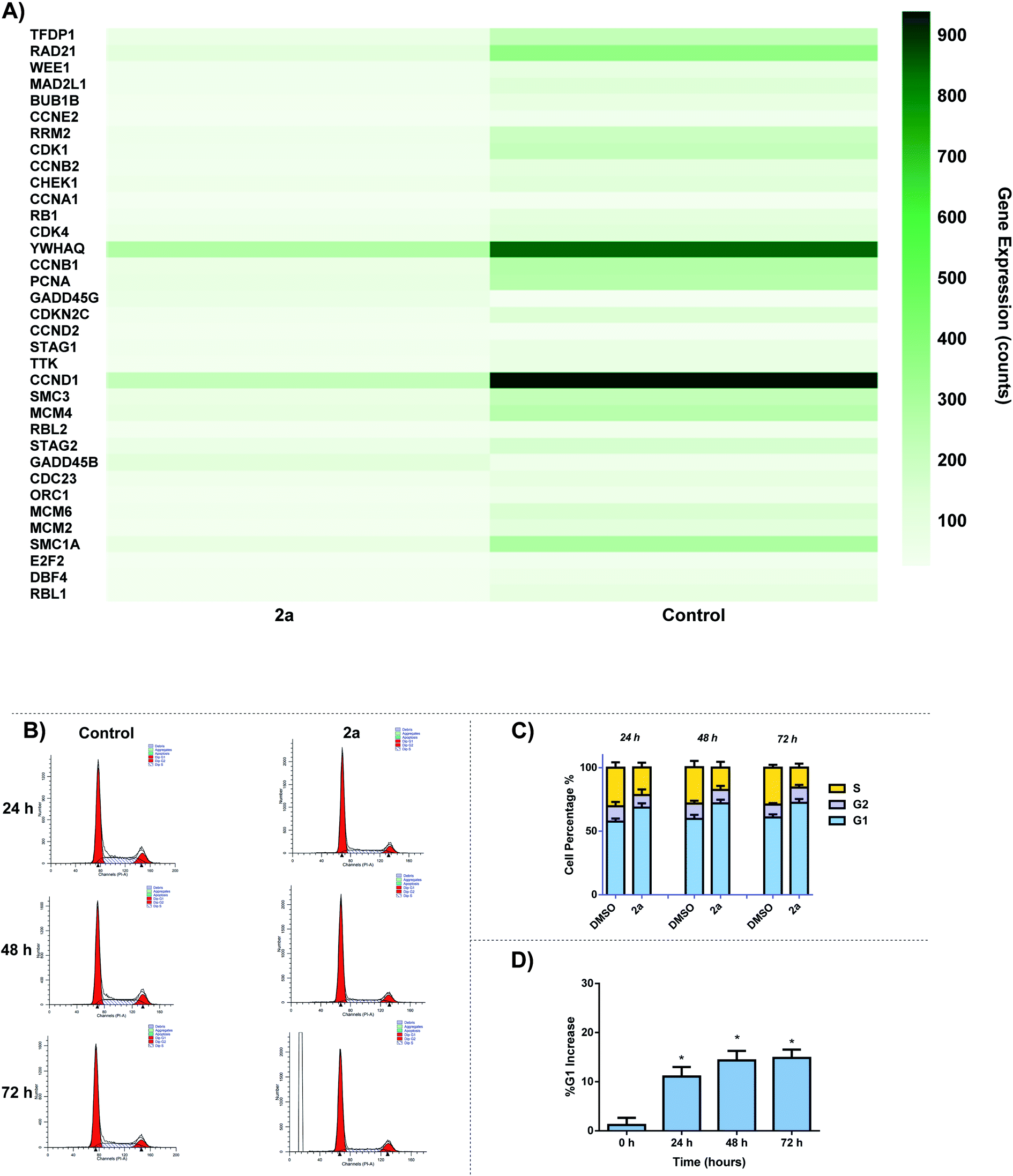
The effect of 2a on cell cycle was studied by flow cytometry. We observed a time-dependent increase in G0/G1 cell cycle population over 24–72 h period. Analysis of RNA-seq data revealed the down regulation of several cell cycle related genes including cyclin D1 and cyclin dependent kinases (CDK1, CDK4, CCND1) (Fig. 12A). There are several small molecule drug candidates in clinical trials as inhibitors of CDK and induce G1 cell cycle arrest.107–110 There are several small molecule drug candidates in clinical trials as inhibitors of CDK4/6 and induce G1 cell cycle arrest.107–110 Thus, our finding is relevant for the design of gold based therapies for refractory tumors such as TNBC.107–110 After 24 h, a 10% increase can be seen for G1 cell population as well as a 6% decrease in S phase in comparison to the control (Fig. 12B and C). Over the course of 72 h, there is a significant increase in G1 phase, suggesting that 2a is arresting the cell cycle at the G1 phase. This data enables us to glean insights into the mechanism of action of gold dithiocarbamates, which is clearly differentiated from cisplatin.111 The mitochondria control cellular ATP production, it is therefore possible that inhibition of mitochondria respiration and consequently low ATP levels lead to cell cycle arrest.112–114
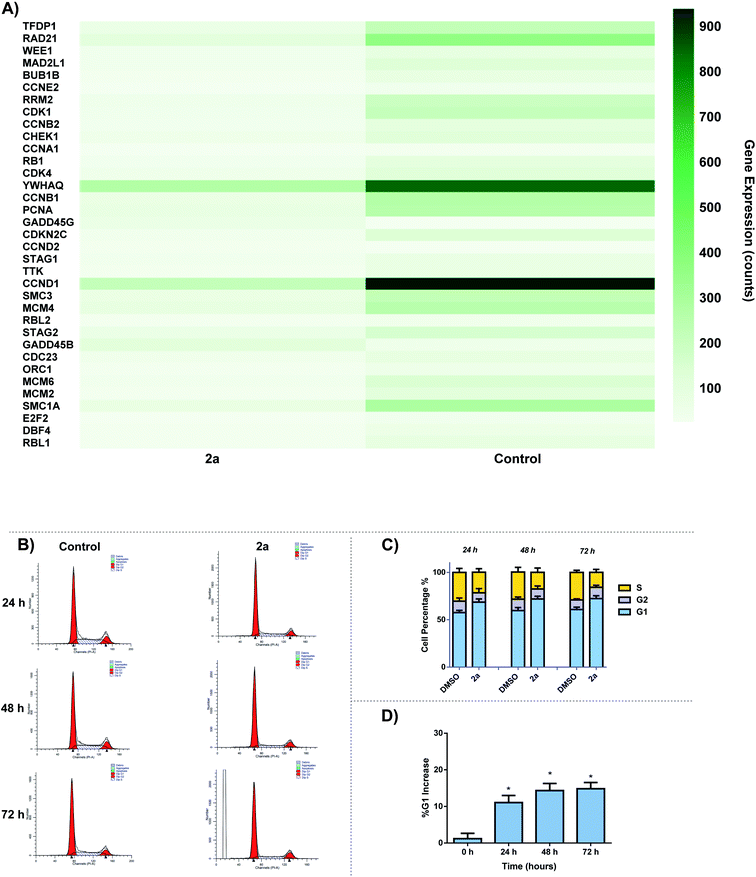 |
| | Fig. 12 Cell-cycle effect by 2a. (A) DEGs associated with cell cycle in response to 2a, (B) representative histogram of the cycle; control (DMSO vehicle) vs.2a, MDA-MB-231 cells were seeded at a density of 2 × 105 cells per well and treated with 2a at 0.1 μM for 24, 48, and 72 h. (C) Bar graph detailing the change in G1, G2/M, and S phase over 24, 48, and 72 h, data are plotted as the mean ± s.e.m (n = 3), (D) bar graph illustrating the % increase in G1 over 72 h after treatment with 2a, data are plotted as the mean ± s.e.m (n = 3) *p < 0.05. | |
Conclusion
We have taken advantage of structural diversity of organogold(III) dithiocarbamates to investigate their anticancer potential and mechanism of action. Solution chemistry reveal that the active stable specie of these compounds in the presence of biological thiols is a neutral organogold(I), which was characterized by NMR spectroscopy, Mass spectrometry and electrochemistry. This points to a mode of action that may involve reactivity to cysteine thiols. These complexes exhibit high in vitro cytotoxicity across a panel of cancer cell lines, with inhibitory concentration in the range of 500–2900 nM. The compounds display a 30-fold selectivity for cancer cells over the normal RPE-Neo cells. In addition, the compounds display high cellular uptake in cancer cells of >1000 pmol per million cells. Using whole-cell transcriptomics, we explored the global effects of lead compound 2a on MDA-MB-231 cells and found that mitochondrial processes related to oxidative phosphorylation, cell cycle, and organelle fission processes were impacted. We utilized Seahorse XF96, to measure oxygen consumption rate of cancer or normal cells in response to 2a and found selective inhibition of mitochondrial respiration in cancer cells over normal cells (MRC5) at low concentrations (0.1–3 μM). Thus, these agents will be useful as targeted therapy for cancers that shift metabolism and rely on OXPHOS for proliferation. We further validate the cell death pathway by investigating key biological markers including, cell cycle, ROS production, apoptosis, and MMP depolarization. All of these combine to give a unique outlook on the mechanism of action of Au(III) complexes that has not yet been reported. Overall, this in-depth study provides key information for a foundation to build upon in designing future Au(III) drug candidates.
Funding sources
We are grateful to the University of Kentucky for funding. The authors acknowledge support of the Center for Pharmaceutical Research and Innovation (NIH P20 GM130456).
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to acknowledge all of those who helped contribute to the project: Samuel Ofori who gratefully cultured the MRC5 cell line used in this manuscript. We would like to thank all of the facilities at the University of Kentucky who provided support in completion of the experiments detailed in this manuscript. The UK NMR Center supported by NSF (CHE-997738) and the UK X-ray facility supported by the MRI program from NSF (CHE-1625732). For the flow cytometry experiments we would like to thank Greg Bauman PhD (UK Flow Cytometry and Immune Function core supported by the Office of the Vice President of Research, the Markey Cancer Center, and NCI Center Core Support Grant (P30 CA177558). For microscopy, we would like to thank Thomas Wilkop PhD and Mr James Schwartz (UK Light Microscopy Core) for their assistance. We would also like to thank Tomoko Sengoku PhD and Mr Michael Alstott for the support with our redox metabolism experiments, supported by the shared resource(s) of the University of Kentucky Markey Cancer Center (P30CA177558). We are grateful for the use of Dr Steven Van Lanen's laboratory (UK College of Pharmacy) for the use of their LC-MS.
References
- M. De Palma and D. Hanahan, The biology of personalized cancer medicine: facing individual complexities underlying hallmark capabilities, Mol. Oncol., 2012, 6(2), 111–127 Search PubMed.
- D. Hanahan and R. A. Weinberg, The Hallmarks of Cancer, Cell, 2000, 100(1), 57–70 Search PubMed.
-
D. D. Hanahan and R. A. Weinberg, Biological Hallmarks of Cancer, in Holland-Frei Cancer Medicine, 2017, pp. 1–10 Search PubMed.
- J. M. Hearn, I. Romero-Canelón, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(29), E3800–E3805 Search PubMed.
- J. J. Soldevila-Barreda and P. J. Sadler, Approaches to the design of catalytic metallodrugs, Curr. Opin. Chem. Biol., 2015, 25, 172–183 Search PubMed.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, The potent oxidant anticancer activity of organoiridium catalysts, Angew. Chem., Int. Ed. Engl., 2014, 53(15), 3941–3946 Search PubMed.
- M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science, 2009, 324(5930), 1029–1033 Search PubMed.
- M. V. Liberti and J. W. Locasale, The Warburg Effect: How Does it Benefit Cancer Cells?, Trends Biochem. Sci., 2016, 41(3), 211–218 Search PubMed.
- E. Hamilton, M. Fennell and D. M. Stafford, Modification of Tumour Glucose Metabolism for Therapeutic Benefit, Acta Oncol., 1995, 34(3), 429–433 Search PubMed.
- S.-C. Choi, A. A. Titov, G. Abboud, H. R. Seay, T. M. Brusko, D. C. Roopenian, S. Salek-Ardakani and L. Morel, Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells, Nat. Commun., 2018, 9(1), 4369 Search PubMed.
- T. M. Ashton, W. G. McKenna, L. A. Kunz-Schughart and G. S. Higgins, Oxidative Phosphorylation as an Emerging Target in Cancer Therapy, Clin. Cancer Res., 2018, 24(11), 2482 Search PubMed.
- P. E. Porporato, N. Filigheddu, J. M. B.-S. Pedro, G. Kroemer and L. Galluzzi, Mitochondrial metabolism and cancer, Cell Res., 2018, 28(3), 265–280 Search PubMed.
- J. Chen, Z. Wang, T. Zou, J. Cui, J. Yin, W. Zheng, W. Jiang, H. Zhou and Z. Liu, Pharmacogenomics of platinum-based chemotherapy response in NSCLC: a genotyping study and a pooled analysis, Oncotarget, 2016, 7(34), 55741–55756 Search PubMed.
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7(8), 573–584 Search PubMed.
- L. H. Einhorn, Treatment of testicular cancer: a new and improved model, J. Clin. Oncol., 1990, 8(11), 1777–1781 Search PubMed.
- K. Matsuo, Y. G. Lin, L. D. Roman and A. K. Sood, Overcoming platinum resistance in ovarian carcinoma, Expert Opin. Invest. Drugs, 2010, 19(11), 1339–1354 Search PubMed.
- R. Pokhriyal, R. Hariprasad, L. Kumar and G. Hariprasad, Chemotherapy Resistance in Advanced Ovarian Cancer Patients, Biomarkers Cancer, 2019, 11, 1179299X19860815 Search PubMed.
- R. K. Mehmood, Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments, Oncol. Rev., 2014, 8(2), 256 Search PubMed.
- I. Romero-Canelón and P. J. Sadler, Next-Generation Metal Anticancer Complexes: Multitargeting via Redox Modulation, Inorg. Chem., 2013, 52(21), 12276–12291 Search PubMed.
- T. W. Hambley, Metal-Based Therapeutics, Science, 2007, 318(5855), 1392 Search PubMed.
- D. T. Felson, J. J. Anderson and R. F. Meenan, The comparative efficacy and toxicity of second-line drugs in rheumatoid arthritis results of two metaanalyses, Arthritis Rheum., 1990, 33(10), 1449–1461 Search PubMed.
- M. Chaffman, R. N. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Auranofin. A preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis, Drugs, 1984, 27(5), 378–424 Search PubMed.
- W. Fiskus, N. Saba, M. Shen, M. Ghias, J. Liu, S. D. Gupta, L. Chauhan, R. Rao, S. Gunewardena, K. Schorno, C. P. Austin, K. Maddocks, J. Byrd, A. Melnick, P. Huang, A. Wiestner and K. N. Bhalla, Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia, Cancer Res., 2014, 74(9), 2520 Search PubMed.
- P. J. Barnard and S. J. Berners-Price, Targeting the mitochondrial cell death pathway with gold compounds, Coord. Chem. Rev., 2007, 251(13), 1889–1902 Search PubMed.
- K.-C. Tong, C.-N. Lok, P.-K. Wan, D. Hu, Y. M. E. Fung, X.-Y. Chang, S. Huang, H. Jiang and C.-M. Che, An anticancer gold(III)-activated porphyrin scaffold that covalently modifies protein cysteine thiols, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(3), 1321 Search PubMed.
- M. Pia Rigobello, L. Messori, G. Marcon, M. Agostina Cinellu, M. Bragadin, A. Folda, G. Scutari and A. Bindoli, Gold complexes inhibit mitochondrial thioredoxin reductase: consequences on mitochondrial functions, J. Inorg. Biochem., 2004, 98(10), 1634–1641 Search PubMed.
- T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chemical biology of anticancer gold(iii) and gold(i) complexes, Chem. Soc. Rev., 2015, 44(24), 8786–8801 Search PubMed.
- D. Saggioro, M. P. Rigobello, L. Paloschi, A. Folda, S. A. Moggach, S. Parsons, L. Ronconi, D. Fregona and A. Bindoli, Gold(III)-Dithiocarbamato Complexes Induce Cancer Cell Death Triggered by Thioredoxin Redox System Inhibition and Activation of ERK Pathway, Chem. Biol., 2007, 14(10), 1128–1139 Search PubMed.
- R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Stable Anticancer Gold(III)–Porphyrin Complexes: Effects of Porphyrin Structure, Chem.–Eur. J., 2010, 16(10), 3097–3113 Search PubMed.
- T. Gamberi, F. Magherini, T. Fiaschi, I. Landini, L. Massai, E. Valocchia, L. Bianchi, L. Bini, C. Gabbiani, S. Nobili, E. Mini, L. Messori and A. Modesti, Proteomic analysis of the cytotoxic effects induced by the organogold(iii) complex Aubipyc in cisplatin-resistant A2780 ovarian cancer cells: further evidence for the glycolytic pathway implication, Mol, 2015, 11(6), 1653–1667 Search PubMed.
- N. Shaik, A. Martínez, I. Augustin, H. Giovinazzo, A. Varela-Ramírez, M. Sanaú, R. J. Aguilera and M. Contel, Synthesis of Apoptosis-Inducing Iminophosphorane Organogold(III) Complexes and Study of Their Interactions with Biomolecular Targets, Inorg. Chem., 2009, 48(4), 1577–1587 Search PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Cationic, linear Au(i) N-heterocyclic carbene complexes: synthesis, structure and anti-mitochondrial activity, Dalton Trans., 2006, 30, 3708–3715 Search PubMed.
- K. D. Mjos and C. Orvig, Metallodrugs in Medicinal Inorganic Chemistry, Chem. Rev., 2014, 114(8), 4540–4563 Search PubMed.
- C. I. Yeo, K. K. Ooi and E. R. T. Tiekink, Gold-Based Medicine: A Paradigm Shift in Anti-Cancer Therapy?, Molecules, 2018, 23(6), 1410 Search PubMed.
- R. A. Alderden, M. D. Hall and T. W. Hambley, The Discovery and Development of Cisplatin, J. Chem. Educ., 2006, 83(5), 728 Search PubMed.
- V. Milacic, D. Chen, L. Ronconi, K. R. Landis-Piwowar, D. Fregona and Q. P. Dou, A Novel Anticancer Gold(III) Dithiocarbamate Compound Inhibits the Activity of a Purified 20S Proteasome and 26S Proteasome in Human Breast Cancer Cell Cultures and Xenografts, Cancer Res., 2006, 66(21), 10478–10486 Search PubMed.
- B. Bruni, M. Ferraroni, P. Orioli and G. Speroni, A Biologically Active Gold Complex: Trichloro[(2-pyridyl)methanol-N]gold(III), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52(6), 1423–1424 Search PubMed.
- P. Calamai, A. Guerri, L. Messori, P. Orioli and G. Paolo Speroni, Structure and DNA binding properties of the gold(III) complex [AuCl2(esal)], Inorg. Chim. Acta, 1999, 285(2), 309–312 Search PubMed.
- J. H. Kim, E. Reeder, S. Parkin and S. G. Awuah, Gold(I/III)-Phosphine Complexes as Potent Antiproliferative Agents, Sci. Rep., 2019, 9(1), 12335 Search PubMed.
- T. Srinivasa Reddy, S. H. Privér, V. V. Rao, N. Mirzadeh and S. K. Bhargava, Gold(i) and gold(iii) phosphine complexes: synthesis, anticancer activities towards 2D and 3D cancer models, and apoptosis inducing properties, Dalton Trans., 2018, 47(43), 15312–15323 Search PubMed.
- J. C. Lima and L. Rodríguez, Phosphine-Gold(I) Compounds as Anticancer Agents: General Description and Mechanisms of Action, Anti-Cancer Agents Med. Chem., 2011, 11, 921–928 Search PubMed.
- S. Jürgens, V. Scalcon, N. Estrada-Ortiz, A. Folda, F. Tonolo, C. Jandl, D. L. Browne, M. P. Rigobello, F. E. Kühn and A. Casini, Exploring the C^N^C theme: Synthesis and biological properties of tridentate cyclometalated gold(III) complexes, Bioorg. Med. Chem., 2017, 25(20), 5452–5460 Search PubMed.
- M. Frik, J. Fernández-Gallardo, O. Gonzalo, V. Mangas-Sanjuan, M. González-Alvarez, A. Serrano del Valle, C. Hu, I. González-Alvarez, M. Bermejo, I. Marzo and M. Contel, Cyclometalated Iminophosphorane Gold(III) and Platinum(II) Complexes. A Highly Permeable Cationic Platinum(II) Compound with Promising Anticancer Properties, J. Med. Chem., 2015, 58(15), 5825–5841 Search PubMed.
- S. Carboni, A. Zucca, S. Stoccoro, L. Maiore, M. Arca, F. Ortu, C. Artner, B. K. Keppler, S. M. Meier-Menches, A. Casini and M. A. Cinellu, New Variations on the Theme of Gold(III) C∧N∧N Cyclometalated Complexes as Anticancer Agents: Synthesis and Biological Characterization, Inorg. Chem., 2018, 57(23), 14852–14865 Search PubMed.
- M. R. M. Williams, B. Bertrand, D. L. Hughes, Z. A. E. Waller, C. Schmidt, I. Ott, M. O'Connell, M. Searcey and M. Bochmann, Cyclometallated Au(iii) dithiocarbamate complexes: synthesis, anticancer evaluation and mechanistic studies, Metallomics, 2018, 10(11), 1655–1666 Search PubMed.
- S. Gukathasan, S. Parkin and S. G. Awuah, Cyclometalated Gold(III) Complexes Bearing DACH Ligands, Inorg. Chem., 2019, 58(14), 9326–9340 Search PubMed.
- L. Ronconi, L. Giovagnini, C. Marzano, F. Bettìo, R. Graziani, G. Pilloni and D. Fregona, Gold Dithiocarbamate Derivatives as Potential Antineoplastic Agents: Design, Spectroscopic Properties, and in Vitro Antitumor Activity, Inorg. Chem., 2005, 44(6), 1867–1881 Search PubMed.
- V. Milacic, D. Chen, L. Ronconi, K. R. Landis-Piwowar, D. Fregona and Q. P. Dou, A Novel Anticancer Gold(III) Dithiocarbamate Compound Inhibits the Activity of a Purified 20S Proteasome and 26S Proteasome in Human Breast Cancer Cell Cultures and Xenografts, Cancer Res., 2006, 66(21), 10478 Search PubMed.
- M. Altaf, M. Monim-Ul-Mehboob, A.-N. Kawde, G. Corona, R. Larcher, M. Ogasawara, N. Casagrande, M. Celegato, C. Borghese, Z. H. Siddik, D. Aldinucci and A. A. Isab, New bipyridine gold(III) dithiocarbamate-containing complexes exerted a potent anticancer activity against cisplatin-resistant cancer cells independent of p53 status, Oncotarget, 2017, 8(1), 490–505 Search PubMed.
- L. Cattaruzza, D. Fregona, M. Mongiat, L. Ronconi, A. Fassina, A. Colombatti and D. Aldinucci, Antitumor activity of gold(III)-dithiocarbamato derivatives on prostate cancer cells and xenografts, Int. J. Cancer, 2011, 128(1), 206–215 Search PubMed.
- L. Ronconi, C. Marzano, P. Zanello, M. Corsini, G. Miolo, C. Maccà, A. Trevisan and D. Fregona, Gold(III) Dithiocarbamate Derivatives for the Treatment of Cancer: Solution Chemistry, DNA Binding, and Hemolytic Properties, J. Med. Chem., 2006, 49(5), 1648–1657 Search PubMed.
- A. A. A. Sulaiman, M. Altaf, A. A. Isab, A. Alawad, S. Altuwaijri and S. Ahmad, Synthesis, Characterization, and in vitro Cytotoxicity of Gold(I) Complexes of 2-(Diphenylphosphanyl)ethylamine and Dithiocarbamates, Z. Anorg. Allg. Chem., 2016, 642(24), 1454–1459 Search PubMed.
- F. K. Keter, I. A. Guzei, M. Nell, W. E. v. Zyl and J. Darkwa, Phosphinogold(I) Dithiocarbamate Complexes: Effect of the Nature of Phosphine Ligand on Anticancer Properties, Inorg. Chem., 2014, 53(4), 2058–2067 Search PubMed.
- J. H. Kim, R. T. Mertens, A. Agarwal, S. Parkin, G. Berger and S. G. Awuah, Direct intramolecular carbon(sp2)–nitrogen(sp2) reductive elimination from gold(III), Dalton Trans., 2019, 48(18), 6273–6282 Search PubMed.
- A. C. Ekennia, D. C. Onwudiwe, C. Ume and E. E. Ebenso, Mixed Ligand Complexes of N-Methyl-N-phenyl Dithiocarbamate: Synthesis, Characterisation, Antifungal Activity, and Solvent Extraction Studies of the Ligand, Bioinorg. Chem. Appl., 2015, 2015, 913424 Search PubMed.
- R.-G. Xiong, J.-L. Zuo and X.-Z. You, A Novel Dimeric Zinc Complex: Bis{μ- [(dimercaptomethylene)propanedinitrilato-S,S‘]}tetrakis(4-methylpyridine)dizinc(II)−Chloroform, Inorg. Chem., 1997, 36(11), 2472–2474 Search PubMed.
- G. Hogarth, Metal-dithiocarbamate complexes: chemistry and biological activity, Mini-Rev. Med. Chem., 2012, 12(12), 1202–1215 Search PubMed.
- T. Makovec, Cisplatin and beyond: molecular mechanisms of action and drug resistance development in cancer chemotherapy, Radiol. Oncol., 2019, 53(2), 148–158 Search PubMed.
- C. A. Rabik and M. E. Dolan, Molecular mechanisms of resistance and toxicity associated with platinating agents, Cancer Treat. Rev., 2007, 33(1), 9–23 Search PubMed.
-
V. Karavasilis, A. Reid, R. Sinha and J. S. De Bono, CHAPTER 17 - Cancer drug resistance, in Cancer Drug Design and Discovery, ed. S. Neidle, Academic Press, New York, 2008, pp. 405–423 Search PubMed.
- M. Kartalou and J. M. Essigmann, Mechanisms of resistance to cisplatin, Mutat. Res., 2001, 478(1), 23–43 Search PubMed.
- N. P. E. Barry and P. J. Sadler, Exploration of the medical periodic table: towards new targets, Chem. Commun., 2013, 49(45), 5106–5131 Search PubMed.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Gold compounds as anticancer agents: chemistry, cellular pharmacology, and preclinical studies, Med. Res. Rev., 2010, 30(3), 550–580 Search PubMed.
- C. Nardon, G. Boscutti and D. Fregona, Beyond Platinums: Gold Complexes as Anticancer Agents, Anticancer Res., 2014, 34(1), 487–492 Search PubMed.
- T. Lazarević, A. Rilak and Ž. D. Bugarčić, Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives, Eur. J. Med. Chem., 2017, 142, 8–31 Search PubMed.
- A. S. Mansfield, A. P. Fields, A. Jatoi, Y. Qi, A. A. Adjei, C. Erlichman and J. R. Molina, Phase I dose escalation study of the PKCI inhibitor aurothiomalate for advanced non-small-cell lung cancer, ovarian cancer, and pancreatic cancer, Anticancer Drugs, 2013, 24(10), 1079–1083 Search PubMed.
- C. Roder and M. J. Thomson, Auranofin: repurposing an old drug for a golden new age, Drugs in R&D, 2015, 15(1), 13–20 Search PubMed.
- L. Giovagnini, L. Ronconi, D. Aldinucci, D. Lorenzon, S. Sitran and D. Fregona, Synthesis, Characterization, and Comparative in Vitro Cytotoxicity Studies of Platinum(II), Palladium(II), and Gold(III) Methylsarcosinedithiocarbamate Complexes, J. Med. Chem., 2005, 48(5), 1588–1595 Search PubMed.
- F. Magherini, T. Fiaschi, E. Valocchia, M. Becatti, A. Pratesi, T. Marzo, L. Massai, C. Gabbiani, I. Landini, S. Nobili, E. Mini, L. Messori, A. Modesti and T. Gamberi, Antiproliferative effects of two gold(I)-N-heterocyclic carbene complexes in A2780 human ovarian cancer cells: a comparative proteomic study, Oncotarget, 2018, 28042–28068 Search PubMed.
- A. D. Goolsby and D. T. Sawyer, Electrochemistry of gold(I) and its complexes in acetonitrile, Anal. Chem., 1968, 40(13), 1978–1983 Search PubMed.
- L. D. Burke and P. F. Nugent, The electrochemistry of gold: I the redox behaviour of the metal in aqueous media, Gold Bull., 1997, 30(2), 43–53 Search PubMed.
- H.-S. Lo, N. Zhu, V. K.-M. Au and V. W.-W. Yam, Synthesis, characterization, photophysics and electrochemistry of polynuclear copper(I) and gold(I) alkynyl phosphine complexes, Polyhedron, 2014, 83, 178–184 Search PubMed.
- U. Koelle and A. Laguna, Electrochemistry of Au-complexes, Inorg. Chim. Acta, 1999, 290(1), 44–50 Search PubMed.
- L. Messori, F. Abbate, G. Marcon, P. Orioli, M. Fontani, E. Mini, T. Mazzei, S. Carotti, T. O'Connell and P. Zanello, Gold(III) Complexes as Potential Antitumor Agents: Solution Chemistry and Cytotoxic Properties of Some Selected Gold(III) Compounds, J. Med. Chem., 2000, 43(19), 3541–3548 Search PubMed.
- D. P. Hill, A. Harper, J. Malcolm, M. S. McAndrews, S. M. Mockus, S. E. Patterson, T. Reynolds, E. J. Baker, C. J. Bult, E. J. Chesler and J. A. Blake, Cisplatin-resistant triple-negative breast cancer subtypes: multiple mechanisms of resistance, BMC Cancer, 2019, 19(1), 1039 Search PubMed.
- D. P. Hill, A. Harper, J. Malcolm, M. S. McAndrews, S. M. Mockus, S. E. Patterson, T. Reynolds, E. J. Baker, C. J. Bult, E. J. Chesler and J. A. Blake, Cisplatin-resistant triple-negative breast cancer subtypes: multiple mechanisms of resistance, BMC Cancer, 2019, 19(1), 1039 Search PubMed.
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Drug resistance in cancer: an overview, Cancers, 2014, 6(3), 1769–1792 Search PubMed.
- T. G. O. Consortium, The Gene Ontology project in 2008, Nucleic
Acids Res., 2007, 36, D440–D444 Search PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nat. Genet., 2000, 25(1), 25–29 Search PubMed.
- M. Kanehisa and S. Goto, KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Res., 2000, 28(1), 27–30 Search PubMed.
- M. Kanehisa, Toward understanding the origin and evolution of cellular organisms, Protein Sci., 2019, 28(11), 1947–1951 Search PubMed.
- M. Kanehisa, Y. Sato, M. Furumichi, K. Morishima and M. Tanabe, New approach for understanding genome variations in KEGG, Nucleic Acids Res., 2018, 47(D1), D590–D595 Search PubMed.
- M. Guha, S. Srinivasan, P. Raman, Y. Jiang, B. A. Kaufman, D. Taylor, D. Dong, R. Chakrabarti, M. Picard, R. P. Carstens, Y. Kijima, M. Feldman and N. G. Avadhani, Aggressive triple negative breast cancers have unique molecular signature on the basis of mitochondrial genetic and functional defects, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864(4 Pt A), 1060–1071 Search PubMed.
- H. Pelicano, W. Zhang, J. Liu, N. Hammoudi, J. Dai, R.-H. Xu, L. Pusztai and P. Huang, Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential, Breast Cancer Res., 2014, 16(5), 434 Search PubMed.
- S. T. Smiley, M. Reers, C. Mottola-Hartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele Jr and L. B. Chen, Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1, Proc. Natl. Acad. Sci. U. S. A., 1991, 88(9), 3671–3675 Search PubMed.
- D. R. Green and J. C. Reed, Mitochondria and Apoptosis, Science, 1998, 281(5381), 1309–1312 Search PubMed.
-
A. S. Divakaruni, A. Paradyse, D. A. Ferrick, A. N. Murphy and M. Jastroch, Chapter Sixteen - Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data, in Methods in Enzymology, ed. A. N. Murphy and D. C. Chan, Academic Press, 2014, vol. 547, pp. 309–354 Search PubMed.
- A. Letai, Apoptosis and Cancer, Annual Review of Cancer Biology, 2017, 1(1), 275–294 Search PubMed.
- G. Ichim and S. W. G. Tait, A fate worse than death: apoptosis as an oncogenic process, Nat. Rev. Cancer, 2016, 16(8), 539–548 Search PubMed.
- B. J. Aubrey, G. L. Kelly, A. Janic, M. J. Herold and A. Strasser, How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression?, Cell Death Differ., 2018, 25(1), 104–113 Search PubMed.
- J. S. Fridman and S. W. Lowe, Control of apoptosis by p53, Oncogene, 2003, 22(56), 9030–9040 Search PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutellingsperger, A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V, J. Immunol. Methods, 1995, 184(1), 39–51 Search PubMed.
- I. Vermes, C. Haanen and C. Reutelingsperger, Flow cytometry of apoptotic cell death, J. Immunol. Methods, 2000, 243(1), 167–190 Search PubMed.
- C. Wang and R. J. Youle, The role of mitochondria
in apoptosis, Annu. Rev. Genet., 2009, 43, 95–118 Search PubMed.
- N. Yadav, S. Kumar, T. Marlowe, A. K. Chaudhary, R. Kumar, J. Wang, J. O'Malley, P. M. Boland, S. Jayanthi, T. K. S. Kumar, N. Yadava and D. Chandra, Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents, Cell Death Differ., 2015, 6(11), e1969 Search PubMed.
- E. A. Slee, C. Adrain and S. J. Martin, Executioner Caspase-3, -6, and -7 Perform Distinct, Non-redundant Roles during the Demolition Phase of Apoptosis, J. Biol. Chem., 2001, 276(10), 7320–7326 Search PubMed.
- O. Julien and J. A. Wells, Caspases and their substrates, Cell Death Differ., 2017, 24(8), 1380–1389 Search PubMed.
- G. Graziani and C. Szabó, Clinical perspectives of PARP inhibitors, Pharmacol. Res., 2005, 52(1), 109–118 Search PubMed.
- C. J. Lord and A. Ashworth, PARP inhibitors: Synthetic lethality in the clinic, Science, 2017, 355(6330), 1152–1158 Search PubMed.
- X. Jiang, W. Li, X. Li, H. Bai and Z. Zhang, Current status and future prospects of PARP inhibitor clinical trials in ovarian cancer, Cancer Manage. Res., 2019, 11, 4371–4390 Search PubMed.
- M. Robson, S.-A. Im, E. Senkus, B. Xu, S. M. Domchek, N. Masuda, S. Delaloge, W. Li, N. Tung, A. Armstrong, W. Wu, C. Goessl, S. Runswick and P. Conte, Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation, N. Engl. J. Med., 2017, 377(6), 523–533 Search PubMed.
- H. Farmer, N. McCabe, C. J. Lord, A. N. J. Tutt, D. A. Johnson, T. B. Richardson, M. Santarosa, K. J. Dillon, I. Hickson, C. Knights, N. M. B. Martin, S. P. Jackson, G. C. M. Smith and A. Ashworth, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy, Nature, 2005, 434(7035), 917–921 Search PubMed.
- G. Diaz, S. Liu, R. Isola, A. Diana and A. M. Falchi, Mitochondrial localization of reactive oxygen species by dihydrofluorescein probes, Histochem. Cell Biol., 2003, 120(4), 319–325 Search PubMed.
- C. P. LeBel, H. Ischiropoulos and S. C. Bondy, Evaluation of the probe 2',7'-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress, Chem. Res. Toxicol., 1992, 5(2), 227–231 Search PubMed.
- S.-Y. Sun, N-acetylcysteine, reactive oxygen species and beyond, Cancer Biol. Ther., 2010, 9(2), 109–110 Search PubMed.
- M. Halasi, M. Wang, T. S. Chavan, V. Gaponenko, N. Hay and A. L. Gartel, ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors, Biochem. J., 2013, 454(2), 201–208 Search PubMed.
- V. Malínková, J. Vylíčil and V. Kryštof, Cyclin-dependent kinase inhibitors for cancer therapy: a patent review (2009 – 2014), Expert Opin. Ther. Pat., 2015, 25(9), 953–970 Search PubMed.
- G. Mariaule and P. Belmont, Cyclin-dependent kinase inhibitors as marketed anticancer drugs: where are we now? A short survey, Molecules, 2014, 19(9), 14366–14382 Search PubMed.
- P. M. Fischer and A. Gianella-Borradori, CDK inhibitors in clinical development for the treatment of cancer, Expert Opin. Invest. Drugs, 2003, 12(6), 955–970 Search PubMed.
- M. Guha, Cyclin-dependent kinase inhibitors move into Phase III, Nat. Rev. Drug Discovery, 2012, 11(12), 892–894 Search PubMed.
- C. M. Sorenson and A. Eastman, Mechanism ofcis-Diamminedichloroplatinum(II)-induced Cytotoxicity: Role of G2 Arrest and
DNA Double-Strand Breaks, Cancer Res., 1988, 48(16), 4484–4488 Search PubMed.
- T. Finkel and P. M. Hwang, The Krebs cycle meets the cell cycle: Mitochondria and the G1S transition, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(29), 11825–11826 Search PubMed.
- S. Mandal, P. Guptan, E. Owusu-Ansah and U. Banerjee, Mitochondrial Regulation of Cell Cycle Progression during Development as Revealed by the tenured Mutation in Drosophila, Dev. Cell, 2005, 9(6), 843–854 Search PubMed.
- X. Du, X. Fu, K. Yao, Z. Lan, H. Xu, Q. Cui and E. Yang, Bcl-2 delays cell cycle through mitochondrial ATP and ROS, Cell Cycle, 2017, 16(7), 707–713 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All synthetic procedures, chemical experiments, biological experiments and characterization. The crystallographic data for compounds 1c, 2a, 2b, 2c, and 2e was deposited in the Cambridge Crystallographic Data Centre under accession codes 1889819–1889821 and 1986842–1986843. CCDC 1889819–1889821, 1986842 and 1986843. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03628e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a and
Samuel G.
Awuah
a and
Samuel G.
Awuah