
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical nanosheets built from superatomic clusters: properties, exfoliation and single-crystal-to-single-crystal intercalation†
Jonathan A.
Kephart
 a,
Catherine G.
Romero
a,
Chun-Chih
Tseng
b,
Kevin J.
Anderton
c,
Matthew
Yankowitz
bd,
Werner
Kaminsky
a and
Alexandra
Velian
*a
a,
Catherine G.
Romero
a,
Chun-Chih
Tseng
b,
Kevin J.
Anderton
c,
Matthew
Yankowitz
bd,
Werner
Kaminsky
a and
Alexandra
Velian
*a
aDepartment of Chemistry, University of Washington, Seattle, Washington 98195, USA. E-mail: avelian@uw.edu
bDepartment of Physics, University of Washington, Seattle, Washington 98195, USA
cDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
dDepartment of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA
First published on 3rd August 2020
Abstract
Tuning the properties of atomic crystals in the two-dimensional (2D) limit is synthetically challenging, but critical to unlock their potential in fundamental research and nanotechnology alike. 2D crystals assembled using superatomic blocks could provide a route to encrypt desirable functionality, yet strategies to link the inorganic blocks together in predetermined dimensionality or symmetry are scarce. Here, we describe the synthesis of anisotropic van der Waals crystalline frameworks using the designer superatomic nanocluster Co3(py)3Co6Se8L6 (py = pyridine, L = Ph2PN(Tol)), and ditopic linkers. Post-synthetically, the 3D crystals can be mechanically exfoliated into ultrathin flakes (8 to 60 nm), or intercalated with the redox-active guest tetracyanoethylene in a single-crystal-to-single-crystal transformation. Extensive characterization, including by single crystal X-ray diffraction, reveals how intrinsic features of the nanocluster, such as its structure, chirality, redox-activity and magnetic profile, predetermine key properties of the emerging 2D structures. Within the nanosheets, the strict and unusual stereoselectivity of the nanocluster's Co edges for the low symmetry (α,α,β) isomer gives rise to in-plane structural anisotropy, while the helically chiral nanoclusters self-organize into alternating Δ- and Λ-homochiral rows. The nanocluster's high-spin Co edges, and its rich redox profile make the nanosheets both magnetically and electrochemically active, as revealed by solid state magnetic and cyclic voltammetry studies. The length and flexibility of the ditopic linker was varied, and found to have a secondary effect on the structure and stacking of the nanosheets within the 3D crystals. With these results we introduce a deterministic and versatile synthetic entry to programmable functionality and symmetry in 2D superatomic crystals.
Introduction
The dimensionality of a material is a key determinant of its properties, as demonstrated especially over the past two decades with the isolation of two-dimensional (2D) atomic crystals.1 These materials have opened new frontiers in exotic physics,2 catalysis3 and nanotechnology,4 yet they face several limitations including relatively low chemical addressability, and difficulty to access high quality flakes at scale. An attractive route to circumvent these challenges is to prepare “programmable” 2D hierarchical crystals using superatomic building blocks of predetermined structural and physicochemical properties.5–12 Examples of solid state 2D hierarchical materials exist, for example the semiconductor Re6Se8Cl2, formed of layers of interlinked Chevrel-like cluster units,13,14 yet only a handful of reports of cluster-assembled 2D superatomic crystals have been synthesized thus far via a bottom-up approach.12,15–19Although wide libraries of atomically precise inorganic clusters of different sizes, compositions and properties are available,20,21 there are critical synthetic challenges to using them as superatomic building blocks. For example, access to 2D cluster-assembled materials is inhibited by the intrinsic high symmetry of the cluster cores. Typically prepared under thermodynamic control in one-pot syntheses, inorganic clusters have many chemically indistinguishable surface sites. Upon assembly, these degenerate surface sites bind indiscriminately to linkers and give rise to three dimensional structures.22–26 Synthetic strategies to deliberately lower the symmetry of the nanocluster blocks could address this challenge.25,27 For example, site-differentiation, achieved empirically under stoichiometric control, has enabled the synthesis of the low symmetry linear trans-Co6Se8(PEt3)4(CNC6H4NC)2 cluster, which in turn seeded the growth of 1D chains.16,28 More commonly though dimensional control is obtained serendipitously.18,19 For example, equipping the degenerate capping ligands with functional groups designed to interlink clusters via coordinative, π–π or H-bonding, has produced 2D cluster frameworks.9,15,17
These strategies, however, are either not deterministic or they lack versatility, and rely heavily on using the organic capping ligands as linkers, limiting further the potential to tailor the structural and electronic properties of cluster-assembled materials. For example, in-plane anisotropy, found in many inorganic solid state van der Waals materials, is a highly desirable structural feature as it provides distinct in-plane pathways for electronic and thermal transport.29 Still, deterministically encoding it in atomically precise materials remains a synthetic challenge. Most commonly, in reticular materials, in-plane anisotropy occurs serendipitously as the inorganic nodes self-assemble in situ, but it can be deliberate when using asymmetric, or mixtures of organic linkers.18,30–32 Directing in-plane anisotropy by relying on structural features of the inorganic nodes instead is desirable, but requires access to preformed blocks with distinct low symmetries. This strategy has been successfully demonstrated using anisotropic colloidal nanoparticles,33,34 but not for atomically precise inorganic clusters. This work represents, to the best of our knowledge, the first such example.
Our group developed a generalizable strategy to deliberately lower the symmetry of existing nanoclusters by decorating their inorganic cores with addressable linker-binding sites. Specifically, we recently introduced the synthesis of the helically chiral nanocluster, Fe3Co6Se8L6 (L = Ph2PN(Tol), Ph = phenyl, Tol = 4-tolyl), obtained by functionalizing the surface of the Chevrel-type cluster Co6Se8LH6 (LH = Ph2PNHTol) with three Fe edges.35 Oriented at the vertices of a triangle, the iron sites are poised to function as linker-attachment points that would impose 2D growth upon assembly with linear linkers. A quick evaluation of Fe3Co6Se8L6 as a building block for self-assembly reveals a critical limitation: the Fe edges have very low affinities for external ligands. Here, we find that by simply replacing the Fe with Co, the edge sites become not only competent attachment points for linear ditopic linkers, but also unusual inorganic stereocenters with strict stereoselectivity for a low symmetry isomer. The 2D cluster-assembled materials obtained from these nanoclusters represent exceedingly rare examples where preformed building blocks encode their key structural and physical properties, including in-plane anisotropy, quasi-chiral domains as well as rich redox activity and magnetism. Remarkably, these synthetic 2D superatomic crystals are amenable to post-synthetic manipulations, including exfoliation to ultrathin flakes, and intercalation with guest molecules in a single-crystal-to-single-crystal transformation that highlights their rich, and robust redox profile.
Results and discussion
Synthesis and characterization of nanocluster building block 1
We illustrate the versatility of our synthetic strategy to program nanoclusters through the facile synthesis of the tricobalt variant, prepared similarly to Fe3Co6Se8L6.35 With increased affinity of the edge sites for external ligands, the tricobalt nanopropeller is isolated exclusively as the tris-pyridine adduct Co3(py)3Co6Se8L6 (1, 81% yield, Scheme 1). In nanocluster 1, each Co edge is anchored κ3 to the cluster core via two amides, and one surface selenium (Fig. 1). The slow exchange of the surface-bound pyridines in benzene-d6, and the resultant lower symmetry of the nanocluster are reflected in a very broad 1H NMR spectrum. Dissolution in pyridine-d5 dramatically speeds up ligand exchange, giving rise to seven, comparatively sharper L proton signals, suggestive of the pseudo-D3 symmetry of 1. | ||
| Scheme 1 Synthesis of tritopic nanoblock 1; LH = Ph2PNH(Tol); py = pyridine. | ||
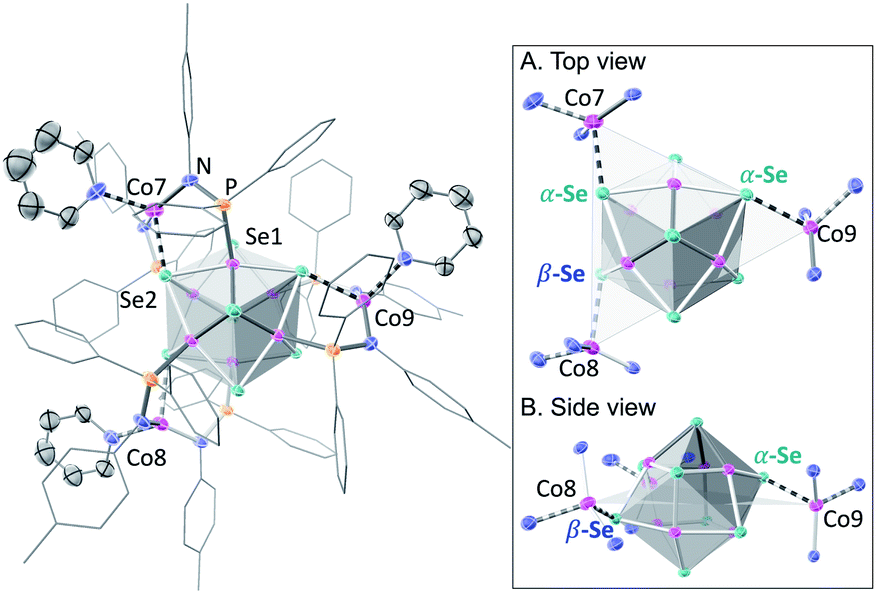 | ||
| Fig. 1 Single crystal X-ray structure of the Λ enantiomer of 1, with selected interatomic distances (Å): Co7–Se2 2.534(1), Co7⋯Se1 3.200(2), Co8–Se3 2.524(1), Co9–Se6 2.573(2). Thermal ellipsoids plotted at 50%. Disordered ligands, solvent and hydrogen atoms hidden for clarity. Inset: top (A) and side (B) view of 1 illustrating (α,α,β) orientation with respect to the plane of the Co edge sites, with two Se bound above this triangle (α) and one below (β). | ||
Nanocluster 1 contains two distinct Co environments: three exposed tetrahedral Co “edge” sites doubling as linker-attachment points, and six square pyramidal Co “core” sites. X-ray diffraction analysis reveals two remarkable structural features, consequential to using 1 as a pre-templated inorganic building block for nanomaterials. One is the intrinsic helicity of 1, isolated as a (Δ/Λ) racemic pair, stemming from the propeller geometry of its inorganic core.35 The other is the stereogenic nature of the Co edge sites. The stereoisomerism of 1 is easiest understood by analogy to isomerism in 1,2,3-trisubstituted cyclopropanes, where the three substituents are either all oriented on the same face of the ring, or one points to the opposite side from the other two. Similarly, the three edge Co atoms define a triangle that separates two Co3Se4 hemispheres of the Co6Se8 nanocube, with each edge-binding site containing an above-the-plane (α-Se) and beneath-the-plane (β-Se) selenium atom (Fig. 1). Upon coordination, pyridine formally displaces one of the Se atoms from a κ4-bound Co edge site, to yield an α- or a β-bound edge site. Each Co surface site effectively discriminates between the two structurally equivalent α, β selenium atoms, and creates three discrete edge Co stereocenters. In turn, these stereocenters orient the bound pyridine above or beneath the plane of the Co edge sites, akin to the isomers of 1,2,3-trisubstituted cyclopropane.
Remarkably, in the solid state, 1 is completely stereoselective for the formation of the low symmetry (α,α,β) diastereomer. Searching the literature for examples of stereoisomerism in trinuclear clusters, we find they overwhelmingly favor the formation of the high symmetry isomer (α,α,α).36–38 Low symmetry isomers could only be isolated as adducts with bulky metal electrophiles that sterically enforce the (α,α,β) orientation of the ring substituents. For example, treating the (α,α,α) isomer of the indenyl complex Ir3(CO)3(η5-C9H7)3 with the electrophile [Au(PPh3)][PF6] allows the isolation of the (α,α,β) isomer as the {Ir3[Au(PPh3)](CO)3(η5-C9H7)3}[PF6] adduct.39 While different structural phases of the same elemental composition are prevalent in both extended and molecular inorganic materials, such well-defined, localized isomerism is typically associated with organic structures; 1 represents an unusual and rare example of stereoisomerism within a well-defined inorganic cluster core.40–42
Investigating the origin of the stereoselectivity guiding the interaction of the edge Co with the Co6Se8 surface in 1, we note an analogy with how metal adatoms discriminate between different surface sites of inorganic supports, preferentially adsorbing to minimize adsorption-induced strain.43,44 In 1, the Co6Se8 support behaves as an electron reservoir, donating electron density to the Lewis-acidic edge sites through Co–Se interactions.35 In turn, these core–edge contacts induce steric distortions and electron density redistribution within the Co6Se8 core. As Se lone pairs engage in bonding interactions with the edge Co sites, their corresponding bonds to the core Co elongate from an average of 2.33 Å to 2.38 Å. The unobserved (α,α,α) isomer would place all elongated intracore Co–Se bonds in the same cluster hemisphere, compounding the structural and electronic polarization effects brought about by the distortions. In contrast, the (α,α,β) isomer distributes the edge Co–Se bonds between the two cluster hemispheres, minimizing strain and dispersing electron density more evenly within the inorganic core.
Self-assembly of anisotropic nanosheets
We hypothesized that while the presence of trigonally disposed attachment points on the nanocluster surface would give rise to a 2D material upon assembly with linear ditopic linkers, the (α,α,β) stereoselectivity would ensure in-plane structural anisotropy. To investigate the potential offered by 1 as a building block, we selected a family of ditopic, linear linkers of increasing length and structural flexibility: bpy, bpyπ and bpyσ (4,4′-bipyridine, 1,2-di(4-pyridyl)ethylene and 1,2-bi(4-pyridyl)ethane, respectively). We found that mixing the linkers with 1 in a non-coordinating solvent induced the formation of dark red crystals (Scheme 2). Each of the emerging materials, Co3(bpy)1.5Co6Se8L6 (2-bpy), Co3(bpyσ)1.5Co6Se8L6 (2-bpyσ) and Co3(bpyπ)1.5Co6Se8L6 (2-bpyπ), were isolated in good yields after 48 h (55–61%). Inspecting their single crystal X-ray structures, we discovered in each case the formation of anisotropic nanosheets that are layered to form superatomic van der Waals solids (Fig. 2a). The three laminar materials are structurally similar, with slight variations in the structure, morphology and stacking of the nanosheets in the 3D crystal, in accordance with the length and flexibility of the linkers. Each nanosheet is constructed of fused six-sided polygons in a chair-like conformation, with nanocluster vertices interconnected by linkers. Upon assembly, the inorganic nodes retain the (α,α,β) stereoisomerism of 1, successfully engendering in-plane structural anisotropy. | ||
| Scheme 2 Self-assembly of van der Waals nanosheets, 2-bpy, 2-bpyσ and 2-bpyπ, from tritopic nanoblock 1 and pyridyl-based ditopic linkers. | ||
 | ||
| Fig. 2 (a) Single crystal X-ray structure for single layers of 2-bpy, 2-bpyσ and 2-bpyπ (top and side views), illustrating the variations in tile sizes and layer corrugation. (b) In-plane anisotropy and quasi-chiral phase separation in 2-bpyσ. Linkers and nanopropeller blades are colored blue when linked to β-Se, and teal when associated with α-Se sites. | ||
The smoothness, thickness and orientation of individual layers in the 3D crystal depend on the organic linker. Within each layer, the nanopropeller nodes are tilted in unison with respect to the plane of the nanosheet, causing the formation of periodic ripples (Fig. 2a). The amplitude of the ripples is determined by the length and flexibility of the linker. With an amplitude of 1.31 Å, the ripples are most pronounced for 2-bpyπ, which is built using a long and rigid linker. In contrast, 2-bpyσ, which contains the more flexible ethane-spaced linker, exhibits a smoother nanosheet surface with a ripple amplitude of only 0.14 Å. Notably, the intersheet distance of 0.92 nm in 2-bpyσ is significantly smaller than the 1.29 nm layer thickness. Inspecting the structure of this derivative closer, we discover that neighboring sheets interdigitate to allow pairs of Co9Se8 vertices in each polygon to rest within the valley found at the center of polygons belonging to above and beneath layers. Further, the nanosheets in 2-bpyσ propagate along a 21 screw axis that runs parallel to a primary crystal facet, making this material amenable to mechanical exfoliation. In contrast, 2-bpy and 2-bpyπ, built of more rigid linkers, contain nanosheets packed along diagonal lattice planes, and their layers are significantly more separated, by 1.40 and 1.33 nm, respectively.
Within the nanosheets, the racemic mixture of helical nanopropellers undergoes quasi-chiral phase separation within the nanosheets, segregating into alternating Δ- and Λ-homochiral rows (Fig. 2b). This phenomenon, wherein a racemic mixture of enantiomers spontaneously separates into enantiomerically pure domains upon assembly, was first observed in monolayers of chiral organic molecules deposited on surfaces,45,46 and more recently also in 3D frameworks.472-bpy, 2-bpyσ and 2-bpyπ represent examples where quasi-chiral separation is a guiding force in the self-assembly of 2D frameworks. Notably, the homochiral rows are also aligned vertically in the 3D lattice of 2-bpyσ, opening intriguing possibilities for harnessing emerging optoelectronic properties associated with the individual homochiral domains within the 3D crystals.
Solid state magnetic analysis
One trait that distinguishes 1 from other reported superatomic building blocks is the high-spin electronic configuration of the Co edge sites. Here, we find that within the nanosheets, the individual magnetic properties of the nanoblock 1 are unaltered. This is illustrated in Fig. 3a, which depicts the virtually identical magnetic responses of nanoblock 1 and nanosheet 2-bpyπ. Similar results are observed for the other two derivatives, 2-bpy and 2-bpyσ, with minor deviations at high temperatures for the latter (Fig. S9†). Given the similarity between the magnetic response of the nanosheets with the monomeric precursor, we focused our interpretation of these properties on those of 1. Featuring a triangle of high-spin Co(II) sites that inscribes a hexametallic Co6Se8 core, formally (CoIII)4(CoII)2, nanopropeller 1 offers a rare opportunity to study the magnetic interaction between paramagnetic ions mediated through a well-defined inorganic support.48 Within the nanoblock 1, the magnetic properties are dominated by the three S = 3/2 Co(II) edge sites. Above 150 K, the magnetic susceptibility of 1 is greater than expected for three non-interacting Co(II) ions (7.2 vs. 5.6 cm3 mol−1 K, g = 2.0). This discrepancy is expected for the distorted-tetrahedral Co(II) edges, and stems from unquenched orbital angular momentum contributions to the spin-only susceptibility (g = 2.28).49,50 Reduced magnetization data, which resists saturation up to a maximum field of 7 T at 1.8 K, corroborates this interpretation and confirms the presence of a high-spin ground state (Fig. 3b). Below 150 K, the magnetic susceptibility decreases steadily, reaching a minimum of 2.9 cm3 mol−1 K at 2 K. We interpret this behavior to be a combined result of weak antiferromagnetic exchange between the three Co edge sites, also reflected in the negative Weiss constant (θ = −10.5 K),51 and localized anisotropy of the Co(II) edges. As intra- and intermolecular edge site Co⋯Co distances are prohibitively long for through-space magnetic exchange (>9 Å),52 we propose the antiferromagnetic coupling is mediated through the diamagnetic Co6Se8 core.53 Considering that the previously reported Re5OsSe8 cluster core, diamagnetic and isostructural with Co6Se8, has been shown to prevent the magnetic interaction between surface bound S = 1/2 Cu(II) ions, antiferromagnetic coupling between the edge S = 3/2 Co in 1 was initially surprising. The different behavior observed in 1 may stem in part from the increased spin response of the Co(II) ions. We propose that the high-spin Co(II) sites interact through low-lying Van Vleck paramagnetic spin states within the Co6Se8 core, which resemble magnetic interactions reported in Co6Te8 and Ni9Te6 clusters.54 Alternatively, the edge Co may interact with the core via intervalence charge transfer processes that may be enabled at low temperature. | ||
| Fig. 3 Post-assembly, 1 preserves its individual magnetic properties within the nanosheets. (a) Plotted are the variable-temperature dc magnetic susceptibility (χMT, filled circles), the reciprocal molar susceptibility (1/χM, hollow circles) data for nanoblock 1 (blue) and nanosheet 2-bpyπ (teal), collected between 2 to 300 K, at 0.1 T, and the linear fit (25 to 150 K) used to obtain the Weiss constant. (b) Variable-temperature, variable-field reduced magnetization data for 1, collected upon warming from 1.8 to 10 K at each applied field. | ||
Electrochemical analysis
Nanocluster 1 exhibits a total of five quasi-reversible one-electron redox events spanning 2.5 V and six oxidation states, between −2 and +3 (Fig. 4). Randles–Sevcik analysis confirms that each redox process is chemically reversible within the diffusion-limit.35,55,56 Interestingly, the robust binding of pyridine to the Co edge sites is reflected in an electrochemical profile completely insensitive to the presence of a coordinating solvent. The first two oxidation events in 1 occur at similar potentials to those observed for Fe3Co6Se8L6 and the parent cluster Co6Se8LH6, suggesting these remain localized on the Co6Se8 core, and do not involve the edge Co sites. Additionally, in Fe3Co6Se8L6 we determined that the LUMO, estimated electrochemically as the first reduction event, was localized on the Co6Se8 core and stabilized relative to Co6Se8LH6. We attributed this stabilization to the Lewis acidity of the edge sites that withdraw electron density from the core through the binding Se. In 1, donation from the electron rich pyridine ligands effectively decreases the Lewis acidity of the Co edge sites, which in turn leads to a larger HOMO–LUMO gap than that of the Fe derivative (1.01 vs. 0.79 eV), but still markedly smaller than that of parent cluster (1.44 eV).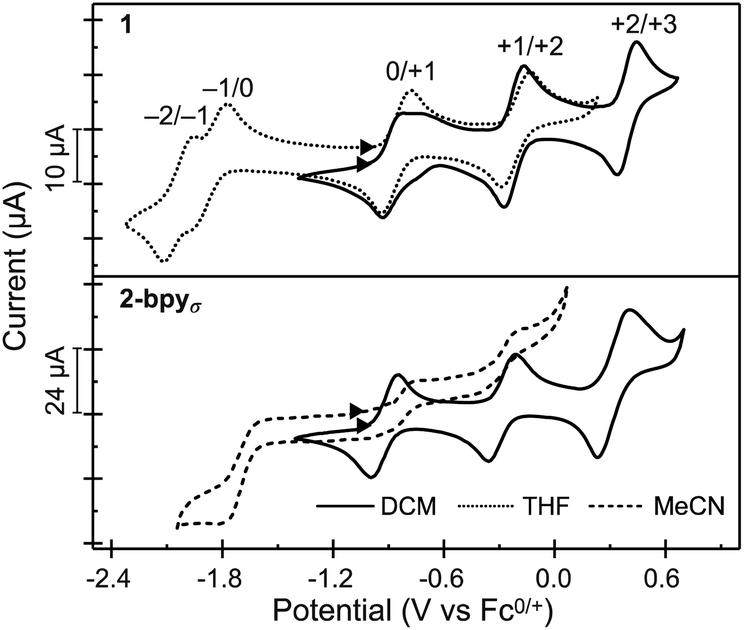 | ||
| Fig. 4 Solution and solid state cyclic voltammograms of 1 and 2-bpyσ, respectively, recorded in DCM (solid lines), THF (dotted lines) or MeCN (dashed lines) solutions of TBAPF6 (0.1 M), at a scan rate of 200 mV s−1. The data for 2-bpyσ was recorded by dropcasting crystalline material on a glass carbon disk electrode. | ||
To probe the electronic states of the assembled nanosheets, we dropcast crystalline material on a glassy carbon working electrode. Fig. 4 depicts the comparison between the electrochemical profile of the nanocluster 1, acquired in solution, and that of the 2-bpyσ nanosheets. We discover that the rich redox behavior of the nanopropeller nodes is preserved upon assembly, and the nanosheets exhibit three well-resolved oxidation processes and one irreversible reduction event, all at potentials effectively coinciding with those of nanocluster 1. The irreversibility of the −1/0 event is not surprising, and we attribute it to the decrease in the Co edge binding affinity for the pyridyl linker, which in turn would be associated with the dissolution of the nanosheet. A similar redox-state controlled affinity for ligands was observed for the triiron nanocluster Fe3Co6Se8L6. Finally, we found the linker has a minimal impact on the electrochemical behavior of the cluster nodes. 2-bpy and 2-bpyπ exhibit similar redox profiles to that of 2-bpyσ, though slight variations in the half-wave potentials, peak-to-peak separation and reversibility of peaks are observed, and documented in Table S3† and Fig. S15a.†
Mechanical exfoliation of 2-bpyσ into ultrathin nanosheets
The individual nanosheets assembled from nanocluster 1 are structurally robust, and can be isolated as ultrathin atomic crystals, behaving akin to solid state inorganic van der Waals 2D materials.12 Indeed, 2-bpyσ can be successfully exfoliated using the Scotch tape method (Fig. 5c and d).57 Optical microscopy imaging of the exfoliated crystals reveals their layered structure, along with a well-defined, thickness-dependent color contrast. Atomic force microscopy analysis shows the exfoliated flakes depicted in Fig. 5d have large lateral dimensions (6–15 μm), flat surfaces, and a well-defined lamellar structure with steps ranging between 10 and 30 nm. The thickness of these flakes ranges from 60 nm to as low as 8 nm (ca. 8 layers). Examples of bottom-up assembled 2D materials that could be exfoliated or prepared as few or mono-layers primarily include organic58,59 or hybrid60–62 reticular frameworks. However, 2D cluster-assembled materials—attractive for their promise of function programmability—that have been successfully exfoliated are exceedingly scarce. In addition to 2-bpyσ, there are only two other examples, a 2D framework interconnected through the capping ligands by zinc-carboxylate nodes, and a hybrid nanosheet in which individual clusters are sandwiched between sheets of photopolymerized C60.15,17 | ||
| Fig. 5 The laminar structure of 2-bpyσ revealed by imaging pre- and post-mechanical exfoliation of a single crystal. (a) Optical microscopy image of a bulk single crystal. (b) Scanning electron microscopy image focused on laminar edge of the bulk crystal. (c) Optical microscopy of a mechanically exfoliated flake. (d) Atomic force microscopy characterization of the exfoliated flake, including height profiles and a zoom-in inset of a thin flake. | ||
Redox intercalation
Redox intercalation into layered materials is a powerful strategy to post-synthetically alter structural, electronic, or magnetic properties.63 For example, ion intercalation has been used to switch between insulating and semiconductor behaviors in a 2D metal–organic framework,64 or to tune the conductive properties of the cluster-assembled donor–acceptor solid.65 Testing our ability to predictably introduce dopants via nondestructive post-synthetic techniques, we investigated the intercalation of redox-active tetracyanoethylene (TCNE) guest molecules into the lattice of 2-bpyσ (Fig. 6a).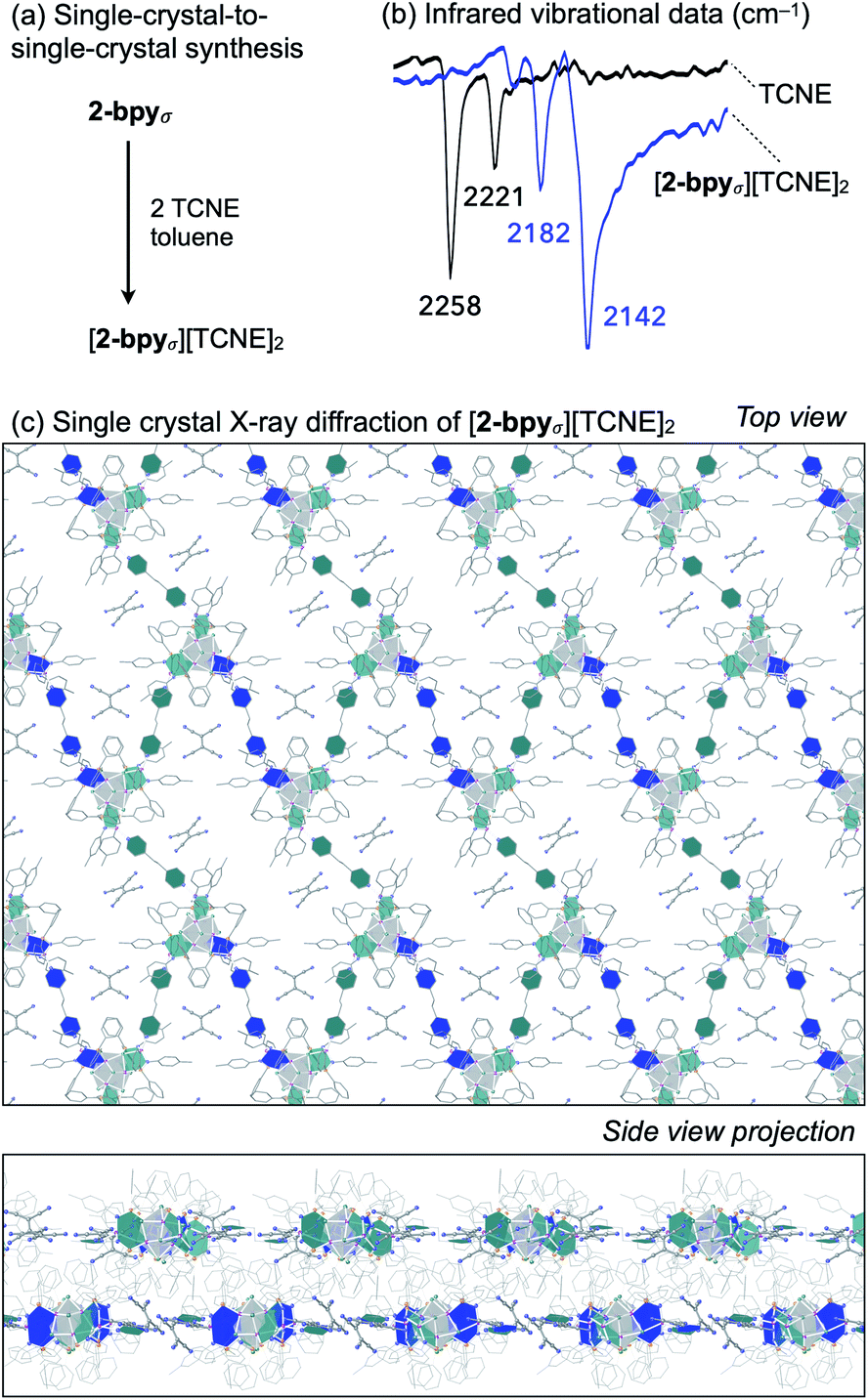 | ||
| Fig. 6 (a) Single-crystal-to-single-crystal synthesis of [2-bpyσ][TCNE]2via redox intercalation. (b) ATR-FTIR analysis of [2-bpyσ][TCNE]2 and TCNE, highlighting the CN stretching region. (c) Single crystal X-ray structure of [2-bpyσ][TCNE]2 (top and side views). | ||
A crop of crystals of 2-bpyσ (1 equiv.) were immersed in a solution of TCNE (5 equiv.) in n-pentane/toluene for 24 h. Before collecting the product (41 mg, 95%), excess TCNE was removed by repeatedly soaking the crystals in toluene (3 × 3 mL) and n-pentane (3 × 3 mL), and volatiles were removed under reduced pressure. Infrared vibrational analysis revealed the crystals exhibit only CN stretching frequencies diagnostic of the mono-reduced [TCNE]˙− anion (νCN = 2182, 2142 cm−1),66 indicating the redox reaction had occurred (Fig. 6b). Thermodynamic considerations derived from the electrochemical analysis of 2-bpyσ (Fig. 4) indicate excess TCNE will lead to the dioxidation of each cluster node, as TCNE−/0 = −0.27 V vs. Fc0/+; this is indeed verified by solid state analysis. The change in oxidation state from neutral is also reflected in the 1H NMR spectrum of the nanoclusters, obtained by dissolving the intercalated nanosheets in d5-pyridine.
Single crystal X-ray analysis of the intercalated nanosheets revealed the crystallinity of the parent material was preserved, the reaction proceeding via a single-crystal-to-single-crystal transformation. Identified as [2-bpyσ][TCNE]2, we draw attention to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of [Co9Se8L6]2+ nodes and [TCNE]˙− guest molecules (Fig. 6c). The oxidized product is largely isostructural with the neutral starting material, maintaining the P21/n space group, but it does have an increased interlayer distance, from 9.2 to 9.6 Å. Two pairs of crystallographically distinct [TCNE]˙− anions occupy each pore of the nanosheet. We note that upon dioxidation, the Co(edge)–Se interaction weakens, elongating by 0.05 Å (avg.), while the linker-Co(edge) bonds strengthen, contracting by 0.05 Å (avg.). Other structural changes that occur within the inner core of the nodes upon oxidation include a contraction of the trans-Co⋯Co interatomic distances within the Co6Se8 unit from 4.12 to 3.99 Å (avg.), and an elongation of the Co–P average bond lengths from 2.14 to 2.19 Å, both of which are tell-tale structural changes incurred upon oxidation of Co6Se8 clusters.65,67
:2 ratio of [Co9Se8L6]2+ nodes and [TCNE]˙− guest molecules (Fig. 6c). The oxidized product is largely isostructural with the neutral starting material, maintaining the P21/n space group, but it does have an increased interlayer distance, from 9.2 to 9.6 Å. Two pairs of crystallographically distinct [TCNE]˙− anions occupy each pore of the nanosheet. We note that upon dioxidation, the Co(edge)–Se interaction weakens, elongating by 0.05 Å (avg.), while the linker-Co(edge) bonds strengthen, contracting by 0.05 Å (avg.). Other structural changes that occur within the inner core of the nodes upon oxidation include a contraction of the trans-Co⋯Co interatomic distances within the Co6Se8 unit from 4.12 to 3.99 Å (avg.), and an elongation of the Co–P average bond lengths from 2.14 to 2.19 Å, both of which are tell-tale structural changes incurred upon oxidation of Co6Se8 clusters.65,67
Summary and conclusions
With this report we introduce a deterministic and modular synthetic strategy to access anisotropic 2D superatomic crystals, and demonstrate these are post-synthetically addressable for further engineering their physical and chemical properties. Our unique strategy to circumvent the pervasive synthetic limitations to interlink clusters relies on equipping the inorganic surface of the nanocluster with three chemically accessible linker-attachment sites. The intrinsic features of the resulting designer nanocluster block predetermine the key structural and physical properties of the emergent materials, including dimensionality, in-plane anisotropy, quasi-chiral domains, magnetism and redox-activity. Notably, the rich redox profile and layered structure uniquely poises this new class of cluster-assembled materials for use as atomically defined anodes in battery materials.68 The ease of mechanical exfoliation to ultrathin superatomic crystals, and single-crystal-to-single-crystal intercalation with TCNE demonstrates the emerging nanosheets are structurally and chemically robust, providing a potent strategy to encrypt desired functionality in superatomic 2D materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr T. A. Betley (Harvard University), Dr J.-H. Chu, and P. Malinowski (UW Physics) for assistance with magnetic measurements, and D. Rogers (UW Chemistry) for assistance with X-ray diffraction measurements. This research was supported by a National Science Foundation (NSF) through a Faculty Early Career Development Program award (NSF CAREER 1944843), and through a Seed Grant from the Materials Research Science and Engineering Center at UW (DMR 1719797 AM005). J. A. K. was supported in part through a UW Clean Energy Institute fellowship. C. G. R. is grateful for support from the Clean Energy Bridge to Research REU (NSF 1559787).References
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- M. Yankowitz, S. Chen, H. Polshyn, Y. Zhang, K. Watanabe, T. Taniguchi, D. Graf, A. F. Young and C. R. Dean, Science, 2019, 363, 1059–1064 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- G. Fiori, F. Bonaccorso, G. Iannaccone, T. Palacios, D. Neumaier, A. Seabaugh, S. K. Banerjee and L. Colombo, Nat. Nanotechnol., 2014, 9, 768–779 CrossRef CAS PubMed.
- P. Alivisatos, P. F. Barbara, A. W. Castleman, J. Chang, D. A. Dixon, M. L. Klein, G. L. McLendon, J. S. Miller, M. A. Ratner, P. J. Rossky, S. I. Stupp and M. E. Thompson, Adv. Mater., 1998, 10, 1297–1336 CrossRef.
- E. A. Doud, A. Voevodin, T. J. Hochuli, A. M. Champsaur, C. Nuckolls and X. Roy, Nat. Rev. Mater., 2020, 5, 371–387 CrossRef.
- Z. Ji, C. Trickett, X. Pei and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 13618–13622 CrossRef CAS PubMed.
- N. E. Horwitz, J. Xie, A. S. Filatov, R. J. Papoular, W. E. Shepard, D. Z. Zee, M. P. Grahn, C. Gilder and J. S. Anderson, J. Am. Chem. Soc., 2019, 141, 3940–3951 CrossRef CAS PubMed.
- S. A. Baudron, P. Batail, C. Coulon, R. Clérac, E. Canadell, V. Laukhin, R. Melzi, P. Wzietek, D. Jérome, P. Auban-Senzier and S. Ravy, J. Am. Chem. Soc., 2005, 127, 11785–11797 CrossRef CAS PubMed.
- B. D. Yuhas, A. L. Smeigh, A. P. S. Samuel, Y. Shim, S. Bag, A. P. Douvalis, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2011, 133, 7252–7255 CrossRef CAS PubMed.
- M. J. Turo, L. Chen, C. E. Moore and A. M. Schimpf, J. Am. Chem. Soc., 2019, 141, 4553–4557 CrossRef CAS PubMed.
- L. G. Beauvais, M. P. Shores and J. R. Long, Chem. Mater., 1998, 10, 3783–3786 CrossRef CAS.
- B. Choi, K. Lee, A. Voevodin, J. Wang, M. L. Steigerwald, P. Batail, X. Zhu and X. Roy, J. Am. Chem. Soc., 2018, 140, 9369–9373 CrossRef CAS PubMed.
- E. J. Telford, J. C. Russell, J. R. Swann, B. Fowler, X. Wang, K. Lee, A. Zangiabadi, K. Watanabe, T. Taniguchi, C. Nuckolls, P. Batail, X. Zhu, J. A. Malen, C. R. Dean and X. Roy, Nano Lett., 2020, 20, 1718–1724 CrossRef CAS PubMed.
- A. M. Champsaur, J. Yu, X. Roy, D. W. Paley, M. L. Steigerwald, C. Nuckolls and C. M. Bejger, ACS Cent. Sci., 2017, 3, 1050–1055 CrossRef CAS PubMed.
- A. M. Champsaur, C. Mézière, M. Allain, D. W. Paley, M. L. Steigerwald, C. Nuckolls and P. Batail, J. Am. Chem. Soc., 2017, 139, 11718–11721 CrossRef CAS PubMed.
- K. Lee, B. Choi, I. J.-L. Plante, M. V. Paley, X. Zhong, A. C. Crowther, J. S. Owen, X. Zhu and X. Roy, Angew. Chem., Int. Ed., 2018, 57, 6125–6129 CrossRef CAS PubMed.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed.
- R.-W. Huang, X.-Y. Dong, B.-J. Yan, X.-S. Du, D.-H. Wei, S.-Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2018, 57, 8560–8566 CrossRef CAS PubMed.
- S. A. Claridge, A. W. Castleman, S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 CrossRef CAS PubMed.
- A. W. Cook and T. W. Hayton, Acc. Chem. Res., 2018, 51, 2456–2464 CrossRef CAS PubMed.
- T. Wu, R. Khazhakyan, L. Wang, X. Bu, S.-T. Zheng, V. Chau and P. Feng, Angew. Chem., Int. Ed., 2011, 50, 2536–2539 CrossRef CAS PubMed.
- M. P. Shores, L. G. Beauvais and J. R. Long, J. Am. Chem. Soc., 1999, 121, 775–779 CrossRef CAS.
- S. Jin and F. J. DiSalvo, Chem. Mater., 2002, 14, 3448–3457 CrossRef CAS.
- Z.-Y. Wang, M.-Q. Wang, Y.-L. Li, P. Luo, T.-T. Jia, R.-W. Huang, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2018, 140, 1069–1076 CrossRef CAS PubMed.
- R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- N. Zheng, X. Bu, H. Lu, L. Chen and P. Feng, J. Am. Chem. Soc., 2005, 127, 14990–14991 CrossRef CAS PubMed.
- A. M. Champsaur, A. Velian, D. W. Paley, B. Choi, X. Roy, M. L. Steigerwald and C. Nuckolls, Nano Lett., 2016, 16, 5273–5277 CrossRef CAS PubMed.
- C. Wang, G. Zhang, S. Huang, Y. Xie and H. Yan, Adv. Opt. Mater., 2020, 8, 1900996 CrossRef CAS.
- N. Huang, L. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. Jiang, Nat. Commun., 2016, 7, 12325 CrossRef PubMed.
- G. S. Papaefstathiou, T. Friščić and L. R. MacGillivray, J. Am. Chem. Soc., 2005, 127, 14160–14161 CrossRef CAS PubMed.
- X.-H. Bu, W. Weng, J.-R. Li, W. Chen and R.-H. Zhang, Inorg. Chem., 2002, 41, 413–415 CrossRef CAS PubMed.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- Y. Liu, K. Deng, J. Yang, X. Wu, X. Fan, M. Tang and Z. Quan, Chem. Sci., 2020, 11, 4065–4073 RSC.
- J. A. Kephart, B. S. Mitchell, A. Chirila, K. J. Anderton, D. Rogers, W. Kaminsky and A. Velian, J. Am. Chem. Soc., 2019, 141, 19605–19610 CAS.
- F. A. Cotton, R. Llusar and C. T. Eagle, J. Am. Chem. Soc., 1989, 111, 4332–4338 CrossRef CAS.
- M. P. Robben, P. H. Rieger and W. E. Geiger, J. Am. Chem. Soc., 1999, 121, 367–375 CrossRef CAS.
- B. K. Park, C. Y. Lee, J. Jung, J. H. Lim, Y.-K. Han, C. S. Hong and J. T. Park, Angew. Chem., Int. Ed., 2007, 46, 1436–1439 CrossRef CAS PubMed.
- M. C. Comstock, T. Prussak-Wieckowska, S. R. Wilson and J. R. Shapley, Organometallics, 1997, 16, 4033–4040 CrossRef CAS.
- X. Fan, J. Wang, K. Wu, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 1320–1323 CrossRef CAS PubMed.
- C. B. Williamson, D. R. Nevers, A. Nelson, I. Hadar, U. Banin, T. Hanrath and R. D. Robinson, Science, 2019, 363, 731–735 CrossRef CAS PubMed.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-E. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS PubMed.
- N. Nilius, E. D. L. Rienks, H.-P. Rust and H.-J. Freund, Phys. Rev. Lett., 2005, 95, 066101 CrossRef PubMed.
- W. Xu and J. B. Adams, Surf. Sci., 1995, 339, 241–246 CrossRef CAS.
- Y. Cai and S. L. Bernasek, J. Am. Chem. Soc., 2003, 125, 1655–1659 CrossRef CAS PubMed.
- T. Huang, Z. Hu, A. Zhao, H. Wang, B. Wang, J. Yang and J. G. Hou, J. Am. Chem. Soc., 2007, 129, 3857–3862 CrossRef CAS PubMed.
- F. Zhang, T. Yajima, Y.-Z. Li, G.-Z. Xu, H.-L. Chen, Q.-T. Liu and O. Yamauchi, Angew. Chem., Int. Ed., 2005, 44, 3402–3407 CrossRef CAS PubMed.
- E. G. Tulsky, N. R. M. Crawford, S. A. Baudron, P. Batail and J. R. Long, J. Am. Chem. Soc., 2003, 125, 15543–15553 CrossRef CAS PubMed.
- J. R. Pilbrow, J. Magn. Reson., 1978, 31, 479–490 CAS.
- S. Sottini, G. Poneti, S. Ciattini, N. Levesanos, E. Ferentinos, J. Krzystek, L. Sorace and P. Kyritsis, Inorg. Chem., 2016, 55, 9537–9548 CrossRef CAS PubMed.
- R. L. Carlin, Magnetochemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 1986 Search PubMed.
- T. Mochizuki, T. Nogami and T. Ishida, Inorg. Chem., 2009, 48, 2254–2259 CrossRef CAS PubMed.
- D. Fenske, J. Ohmer and J. Hachgenei, Angew. Chem., Int. Ed., 1985, 24, 993–995 CrossRef.
- T. T. M. Palstra, M. L. Steigerwald, A. P. Ramirez, Y.-U. Kwon, S. M. Stuczynski, L. F. Schneemeyer, J. V. Waszczak and J. Zaanen, Phys. Rev. Lett., 1993, 71, 1768–1771 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- R. H. Sánchez, A. M. Willis, S.-L. Zheng and T. A. Betley, Angew. Chem., Int. Ed., 2015, 54, 12009–12013 CrossRef PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- R. Zhang, J. Liu, Y. Gao, M. Hua, B. Xia, P. Knecht, A. C. Papageorgiou, J. Reichert, J. V. Barth, H. Xu, L. Huang and N. Lin, Angew. Chem., Int. Ed., 2020, 59, 2669–2673 CrossRef CAS PubMed.
- Y. Quan, G. Lan, Y. Fan, W. Shi, E. You and W. Lin, J. Am. Chem. Soc., 2020, 142, 1746–1751 CrossRef CAS PubMed.
- J. Wan, S. D. Lacey, J. Dai, W. Bao, M. S. Fuhrer and L. Hu, Chem. Soc. Rev., 2016, 45, 6742–6765 RSC.
- N. Ogihara, N. Ohba and Y. Kishida, Sci. Adv., 2017, 3, e1603103 CrossRef PubMed.
- E. S. O'Brien, M. T. Trinh, R. L. Kann, J. Chen, G. A. Elbaz, A. Masurkar, T. L. Atallah, M. V. Paley, N. Patel, D. W. Paley, I. Kymissis, A. C. Crowther, A. J. Millis, D. R. Reichman, X.-Y. Zhu and X. Roy, Nat. Chem., 2017, 9, 1170–1174 CrossRef PubMed.
- J. S. Miller, Angew. Chem., Int. Ed., 2006, 45, 2508–2525 CrossRef CAS PubMed.
- B. Choi, J. Yu, D. W. Paley, M. T. Trinh, M. V. Paley, J. M. Karch, A. C. Crowther, C.-H. Lee, R. A. Lalancette, X. Zhu, P. Kim, M. L. Steigerwald, C. Nuckolls and X. Roy, Nano Lett., 2016, 16, 1445–1449 CrossRef CAS PubMed.
- A. P. Aydt, B. Qie, A. Pinkard, L. Yang, Q. Cheng, S. J. L. Billinge, Y. Yang and X. Roy, ACS Appl. Mater. Interfaces, 2019, 11, 11292–11297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data including CIF files for 1, 2-bpy, 2-bpyσ, 2-bpyπ, and [2-bpyσ][TCNE]2. CCDC 2012111, 2012112, 2012110, 2012109 and 2012108. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03506h |
| This journal is © The Royal Society of Chemistry 2020 |