
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2-Addition and cycloaddition reactions of niobium bis(imido) and oxo imido complexes†
Jade I.
Fostvedt‡
 a,
Lauren N.
Grant‡
a,
Benjamin M.
Kriegel‡
a,
Andreas H.
Obenhuber
a,
Trevor D.
Lohrey
ab,
Robert G.
Bergman
*a and
John
Arnold
*ab
a,
Lauren N.
Grant‡
a,
Benjamin M.
Kriegel‡
a,
Andreas H.
Obenhuber
a,
Trevor D.
Lohrey
ab,
Robert G.
Bergman
*a and
John
Arnold
*ab
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: arnold@berkeley.edu; rbergman@berkeley.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA 94720, USA
First published on 9th October 2020
Abstract
The bis(imido) complexes (BDI)Nb(NtBu)2 and (BDI)Nb(NtBu)(NAr) (BDI = N,N′-bis(2,6-diisopropylphenyl)-3,5-dimethyl-β-diketiminate; Ar = 2,6-diisopropylphenyl) were shown to engage in 1,2-addition and [2 + 2] cycloaddition reactions with a wide variety of substrates. Reaction of the bis(imido) complexes with dihydrogen, silanes, and boranes yielded hydrido-amido-imido complexes via 1,2-addition across Nb-imido π-bonds; some of these complexes were shown to further react via insertion of carbon dioxide to give formate-amido-imido products. Similarly, reaction of (BDI)Nb(NtBu)2 with tert-butylacetylene yielded an acetylide-amido-imido complex. In contrast to these results, many related mono(imido) Nb BDI complexes do not exhibit 1,2-addition reactivity, suggesting that π-loading plays an important role in activating the Nb–N π-bonds toward addition. The same bis(imido) complexes were also shown to engage in [2 + 2] cycloaddition reactions with oxygen- and sulfur-containing heteroallenes to give carbamate- and thiocarbamate-imido complexes: some of these complexes readily dimerized to give bis-μ-sulfido, bis-μ-iminodicarboxylate, and bis-μ-carbonate complexes. The mononuclear carbamate imido complex (BDI)Nb(NAr)(N(tBu)CO2) (12) could be induced to eject tert-butylisocyanate to generate a four-coordinate terminal oxo imido intermediate, which could be trapped as the five-coordinate pyridine or DMAP adduct. The DMAP adducted oxo imido complex (BDI)NbO(NAr)(DMAP) (16) was shown to engage in 1,2-addition of silanes across the Nb-oxo π-bond; this represents a new reaction pathway in group 5 chemistry.
Introduction
In stark contrast to their late-metal congeners, the transition metals in groups 3–5 of the periodic table possess relatively high-energy valence d-orbitals; often, complexes containing these metals are found in d0 oxidation states with no valence electrons available to engage in redox chemistry.1 As such, these transition metal complexes tend to engage in redox-neutral bond making and breaking processes, with σ-bond metathesis being the prime example. One commonly used method to expand the reactivity of early metal complexes is to employ a π-donor ligand—such as an oxo, imido, or nitrido—that forms more than one bonding interaction with the high valent metal center.2–5 The electrons contained in the metal–ligand π-bond(s) can be used to form new chemical bonds, either via cycloadditions with substrates containing additional π bonds or by 1,2-additions across the metal–ligand bond.6While this strategy has been used and described extensively in the literature, two challenges have arisen: first, in some cases, the metal–ligand interaction is too strong, and the supposedly reactive moiety instead behaves as a spectator ligand and second, dimerization can occur through the π-donor ligand, forming insoluble, unreactive multinuclear species. Due to the increasing covalency of the bonding interaction between the metal nd and the ligand 2p orbitals (relative to the more polar and reactive bonds formed in complexes of groups 3 and 4), the former issue is of particular relevance to group 5 chemistry.7 For example, previous work from our group aimed to explore the 1,2-addition and cycloaddition reactivity of d0 group 5 monoimido complexes, including (BDI)Nb(NtBu)Cl2, (BDI)Nb(NtBu)F2, and (BDI)Nb(NAr)Cl2 (BDI = N,N′-bis(2,6-diisopropylphenyl)-3,5-dimethyl-β-diketiminate; Ar = 2,6-diisopropylphenyl); none of these complexes displayed reactivity across the Nb-imido bond.7 To address both of these challenges, our group and others have introduced a second multiply-bound ligand into the early metal coordination sphere. In terms of the first challenge—unreactive spectator ligands—a second multiply-bound ligand serves to electronically saturate the symmetrically available π-bonding orbitals on the metal (a strategy termed π-loading), thus activating the π-bonded ligands by polarizing the electron density away from the metal center.7–16 Furthermore, the addition of a second π-donor ligand—specifically, one with a sterically demanding substituent attached—can impede deleterious dimerization, thus avoiding the formation of unreactive species.
The π-loading strategy has proven successful in expanding redox-neutral reaction pathways available to high-valent early metal complexes. Indeed, the ability of group 4 cyclopentadienide imido complexes to engage in C–H activations of methane and other hydrocarbons, as well as cycloadditions of unsaturated substrates, was first realized in the late 1980s.17,18 Since then, further work in investigating reactions across terminal imido groups in high-valent early transition metal complexes has led to the development of a number of catalytic processes involving nitrene transfers,19–21, including imine and azide metatheses,22–25 hydroaminations of alkenes and alkynes,26–31 and carboaminations to give α,β-unsaturated imines.32–34 In addition, Ti imido,35–37 Zr imido,38 and V bis(imido) complexes14 have recently been shown to carry out multicomponent coupling to give carbodiimides, pyrroles, pyrazoles, and other nitrogen-containing heterocycles.
While there are numerous examples of nitrene transfer chemistry involving π-loaded metal imido complexes, catalytic reactions without nitrene transfer are rare. High-valent transition metal complexes of groups 3–5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39–44 and actinide45,46 imido complexes have been shown to activate dihydrogen, silanes and/or boranes across imido bonds, but in all of these cases, only stoichiometric reactivity could be observed. A cationic V bis(imido) complex was shown to catalytically hydrogenate alkynes to alkenes via a 1,2-addition and 1,2-elimination pathway.8 In the realm of related group 4 terminal chalcogenide complexes,47–53 a handful of Ti and Zr oxo and sulfido complexes were shown to react stoichiometrically with dihydrogen and silanes via a 1,2-addition pathway.54–56 In contrast, while many examples of group 5 mono(imido) and mono(oxo) complexes have been reported,57–64 only a few have been shown to exhibit 1,2-addition reactivity,8,43,65,66 suggesting that these systems could benefit from activation of metal–ligand multiple bonds via a π-loading strategy.
39–44 and actinide45,46 imido complexes have been shown to activate dihydrogen, silanes and/or boranes across imido bonds, but in all of these cases, only stoichiometric reactivity could be observed. A cationic V bis(imido) complex was shown to catalytically hydrogenate alkynes to alkenes via a 1,2-addition and 1,2-elimination pathway.8 In the realm of related group 4 terminal chalcogenide complexes,47–53 a handful of Ti and Zr oxo and sulfido complexes were shown to react stoichiometrically with dihydrogen and silanes via a 1,2-addition pathway.54–56 In contrast, while many examples of group 5 mono(imido) and mono(oxo) complexes have been reported,57–64 only a few have been shown to exhibit 1,2-addition reactivity,8,43,65,66 suggesting that these systems could benefit from activation of metal–ligand multiple bonds via a π-loading strategy.
We are interested in developing π-loaded Nb bis(imido) and oxo imido systems that can carry out complex hydrofunctionalizations of unsaturated substrates, such as hydroborations and hydrosilations, using 1,2-addition and elimination reactions across imido or oxo groups. We have previously described the synthesis of Nb bis(imido) complexes:21,67 π-loading effects in these compounds were shown to engender reactivity across their Nb–N π-bonds7,21 relative to the chemistry observed with related mono(imido) Nb systems.7,57–64 Here we describe their 1,2-addition and [2 + 2] cycloaddition68 reactivity with a variety of small molecule substrates including dihydrogen, silanes, boranes, carbon disulfide, and carbon dioxide. Moreover, we describe the utility of these bis(imido) complexes in accessing isolobal Nb terminal oxo imido complexes and subsequently demonstrate unprecedented reactivity across a Nb-oxo group.
Results and discussion
1,2-Addition across Nb-imido π-bonds
A solution of the bis(imido) complex (BDI)Nb(NtBu)2 (BDI = N,N′-bis(2,6-diisopropylphenyl)-3,5-dimethyl-β-diketiminate) in C6D6 lightened in color from orange to yellow upon addition of an atmosphere of H2; 1H NMR spectroscopy indicated partial conversion to the Nb amido-hydrido complex 1, the result of 1,2-addition of H2 across one of the imido groups (Scheme 1). Compound 1 displayed characteristic Nb–H (broad) and N–H (sharp) singlets at 9.1 and 7.8 ppm, respectively, with Cs symmetry in solution being evident. The ratio of 1 to starting material in solution varied based on the H2 pressure, indicating that an equilibrium was established between the complexes (Keq ≈ 8.0 at 20 °C under 1 atmosphere of H2). The reaction could be driven almost completely to the H2 addition product by addition of ca. 3.8 atmosphere of H2 to an NMR tube containing (BDI)Nb(NtBu)2.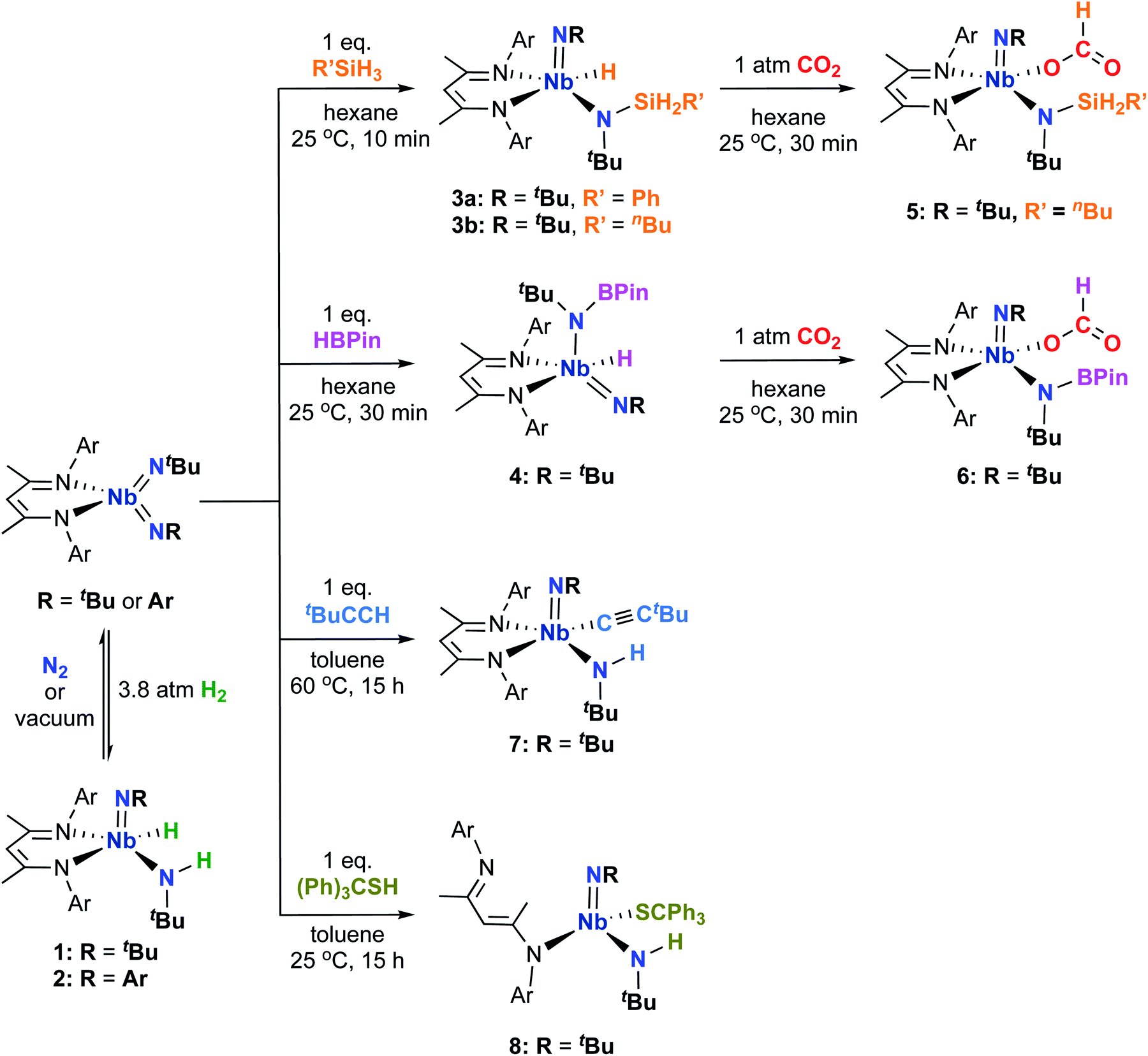 | ||
| Scheme 1 1,2-addition of element-hydrogen bonds across Nb-imido π-bonds to yield complexes 1, 2, 3ab, 4, 7, and 8, and subsequent insertion of CO2 to give complexes 5 and 6. | ||
Although the removal of H2, either by exposing solutions of 1 to nitrogen atmosphere or by placing it under vacuum, resulted in conversion back to (BDI)Nb(NtBu)2, X-ray quality crystals of 1 could be grown by adding a dihydrogen atmosphere to a highly concentrated solution of (BDI)Nb(NtBu)2 in hexamethyldisiloxane (HMDSO) and slowly cooling the resulting reaction mixture. Crystalline 1 was found to be stable under a nitrogen atmosphere at −40 °C for multiple weeks and under vacuum for ca. 30 min. Dissolution in C6D6 under nitrogen resulted in rapid effervescence of H2 gas; 1H NMR spectroscopy showed that the compound was converted back to starting material within minutes. The solid-state structure of 1 (Fig. 1, left, and Table S5, ESI†) showed a distorted square-based pyramidal geometry, with the imido moiety occupying the apical position. The Nb-bound hydride was located in the difference map and refined isotropically. Disorder in the crystal structure between the imido and amido groups made the determination of amido Nb–N–C angles somewhat tenuous, but these metrics (Nb–N(3)–C(30) = 176.8(5)° for the imido group and Nb–N(4)–C(34) = 127.5(4)° for the amido group) are consistent with a structure containing a linear imido and a bent amido. The Nb–H stretch, observed in the IR spectrum at 1652 cm−1, is consistent with similar data on known Nb terminal hydrides.69–71
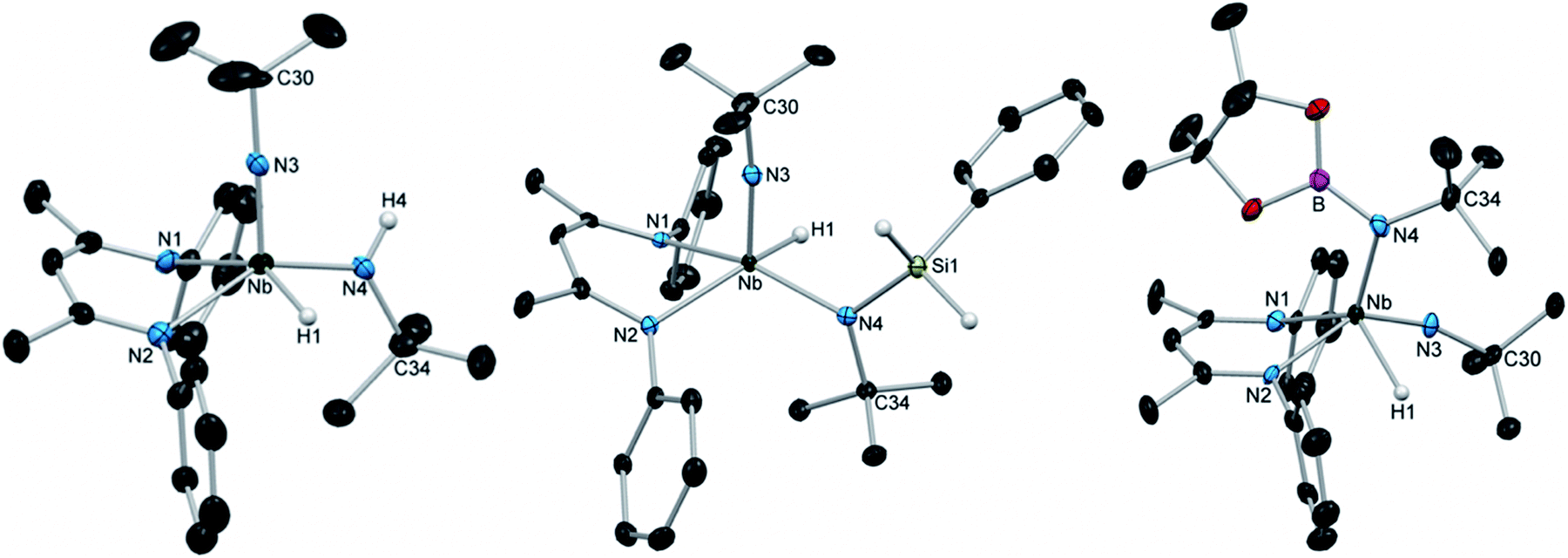 | ||
| Fig. 1 Crystal structures of 1 (left), 3a (center), and 4 (right) with 50% probability thermal ellipsoids. Selected H atoms, aryl iPr groups, and lattice solvent are excluded. | ||
Similarly, the bis(imido) complex (BDI)Nb(NtBu)(NAr) (Ar = 2,6-diisopropylphenyl) also reacted reversibly with dihydrogen across the tert-butylimido moiety, establishing an equilibrium between the starting material and 2 (Keq ≈ 2.5 at 20 °C under 1 atmosphere of H2; Scheme 1). Notably, it appears that the arylimido group engages in stronger π-interactions with the Nb center, as 1,2-addition of H2 occurred solely across the (presumably more polarized) Nb tert-butylimido π-bond. We posit that the asymmetric bis(imido) complex (BDI)Nb(NtBu)(NAr) is less reactive toward H2 than the symmetric bis(imido) complex (BDI)Nb(NtBu)2 for electronic reasons, in particular the increased strength of the arylimido Nb–N bond.
Encouraged by these results, we continued to explore the 1,2-addition reactivity of Nb bis(imido) complexes toward substrates containing heteroatom-hydrogen bonds. We found that both phenylsilane and n-butylsilane readily underwent 1,2-addition across an imido moiety of (BDI)Nb(NtBu)2 to give hydrido-silylamido-imido Nb complexes 3a and 3b, respectively (Scheme 1). These complexes were isolated as pale yellow crystals in 75–85% yield, and displayed broad peaks in their 1H NMR spectra, indicating fluxionality in solution at room temperature. The spectrum of 3a sharpened significantly at −20 °C and clearly exhibited C1 symmetry, with two sharp doublets (2J = 9.1 Hz) observed for the diastereotopic Si–H protons and four septets observed for the isopropyl methine protons. Similar 2J values have been observed in related tantalum phenylsilylamide complexes.66 IR spectra of 3a and 3b both displayed broad absorptions at 1667 cm−1 attributable to Nb–H stretches. Compound 3a was also characterized by X-ray diffraction: in the solid state, 3a exhibits an ideal square based pyramidal structure, analogous to that of 1, with the imido moiety in the apical position (Fig. 1, center, and Table S6, ESI†).
Similarly, treatment of (BDI)Nb(NtBu)2 with pinacolborane gave the hydrido-borylamido-imido Nb complex 4 as a yellow microcrystalline powder in 39% isolated yield (Scheme 1). The 1H NMR spectrum of 4 displayed sharp resonances at room temperature, aside from the broad Nb–H resonance centered at 10.01 ppm. The compound was characterized by X-ray crystallography; unlike 1 and 3a, the solid-state structure of 4 showed a distorted square-based pyramidal geometry with the borylamido group in the apical position (Fig. 1, right, and Table S6, ESI†).
Addition of 1 atmosphere of CO2 to hydrido-amido-imido complexes 3b and 4 in hexane resulted in immediate formation of the corresponding formate complexes 5 and 6, respectively, as yellow crystals in moderate yield (Scheme 1). The 1H NMR spectrum of each complex revealed the disappearance of the broad Nb–H signal and appearance of a sharp singlet corresponding to the formate C–H proton at 7.86 and 8.60 ppm for 5 and 6, respectively. The solid-state structures of both 5 and 6 revealed square-based pyramidal geometries (Fig. S1 and Table S7, ESI†). The Nb–O(1)–C(42) bond angle for the formate ligand of 5 is nearly linear (167.2(2)°), indicating the presence of some π-donation from both lone pairs of the oxygen atom to the Nb center. In contrast, the Nb–O(3)–C(44) angle of 6 is bent (127.8(4)°) and the Nb–O bond distance is 0.027 Å longer than the analogous Nb–O bond in 5, indicating that only one lone pair of the oxygen atom is donating to the metal center.
We continued to explore the 1,2-addition reactivity of the bis(imido) complex (BDI)Nb(NtBu)2, turning our attention toward substrates with relatively acidic S–H and C–H bonds. Correspondingly, we found that the reaction of (BDI)Nb(NtBu)2 with tert-butylacetylene with heating at 60 °C yielded the Nb amido-acetylide complex 7 as pale yellow crystals in 56% yield (Scheme 1); such reactivity patterns are precedented for imido complexes of groups 4 and 5.41,43 The 1H NMR spectrum of 7 contained two sets of broad peaks at room temperature (1.9:1 ratio), likely corresponding to two diastereoisomers. An EXSY correlation was observed between the resonances of the two isomers at room temperature, indicating that the observed fluxionality was likely caused by an interconversion of the two diastereomers. Upon cooling to −20 °C, the spectrum sharpened into two distinct sets of sharp signals, with a 1.4:1 ratio of the two isomers observed. At higher temperatures (60–80 °C), the two sets of peaks began to coalesce into a single set of very broad resonances, consistent with averaging between the two diastereoisomers on the NMR timescale. Only a single isomer was observed in the solid-state structure of 7 (Fig. S2, left, and Table S8, ESI†). Like most of the other 1,2-addition products presented here, 7 has a distorted square-based pyramidal geometry with the imido group in the apical position. It is not clear which solution state isomer the crystal structure corresponds to, nor is it clear what the structure of the second diastereoisomer is, although distorted square-based pyramidal geometries in which one of the ligands other than the imido group occupies the apical position are likely.
Similarly, (BDI)Nb(NtBu)2 reacted with triphenylmethaneylthiol to afford the amido-thiolate complex 8 as pale yellow crystals in 60% yield (Scheme 1). Notably, both 1H NMR spectroscopy and X-ray crystallography were consistent with a structure in which the BDI ligand was bound in a κ1 fashion to the Nb center, presumably to accommodate the sterically demanding triphenylmethanethiolato ligand (Fig. S2, right, and Table S8, ESI†). The κ1 binding of the BDI ligand was apparent from the 1H NMR spectrum, based on the unusually large difference in chemical shift between the two BDI backbone methyl units. While κ1 binding of BDI ligands is relatively unusual, it is not without precedent.72–74 Compound 8 is four-coordinate with a distorted tetrahedral geometry. The Nb–N(1) distance of 2.065(2) Å is 0.1 to 0.2 Å shorter than the distances observed in Nb BDI complexes with κ2-BDI ligands, and is consistent with the localized picture of the bonding in the BDI ligand depicted in Scheme 1, in which the Nb–N(1) interaction is best characterized as an X-type single bond with some dative π-character. Additionally, the Nb–S(1) distance of 2.414(1) Å, which is among the shortest of Nb thiolate bonds reported in the literature, is consistent with considerable degree of Nb–S multiple bond character. The thiolate ligand is bent (Nb–S(1)–C(38) = 121.63(6)°), indicating that, at most, only one of the sulfur p-based orbitals is interacting with the metal center.
Overall, we were able to demonstrate a variety of 1,2-addition reactions across Nb-imido bonds. Addition led to products in which the more electronegative substrate fragment was bound to the electropositive Nb center: in the case of silanes and boranes, Nb-hydrides were formed, while addition of acidic substrates led to protonation of the imido to form Nb-amidos. In contrast to the reactivity patterns presented above, previous reports of related d0 Nb BDI mono(imido) complexes did not display analogous 1,2-addition reactivity; instead, the Nb imido group behaves purely as an ancillary ligand.7,57–64 Specifically, the complexes (BDI)Nb(NtBu)Cl2, (BDI)Nb(NtBu)F2, and (BDI)Nb(NAr)Cl2 were not observed to react with dihydrogen, silanes, boranes, or thiols across the Nb-imido bond.7 Thus, these results support the notion that introduction of a second imido moiety enhances reactivity in these d0 Nb BDI complexes due to a π-loading effect.7
Furthermore, we found the asymmetric bis(imido) complex (BDI)Nb(NtBu)(NAr) to be far less reactive than symmetric (BDI)Nb(NtBu)2. While the latter reacted with a diverse array of substrates, the former was only observed to react reversibly with H2 (preferentially across the tert-butyl imido group) with a smaller equilibrium constant than that of the corresponding reaction with the symmetric bis(imido). We hypothesize that the distinctive reactivity patterns observed between the two complexes can be attributed to electronic factors: the tert-butyl moiety is weakly electron donating, leading to increased electron density at the already electron-rich, π-loaded metal center, while the aromatic 2,6-diisopropylphenyl moiety is weakly electron withdrawing, stabilizing the Nb–NAr bonding interaction. These observations support the idea that increased electron density at the metal center increases the activity of the Nb-imido group and indicate that the choice of imido group substituent plays a significant role in determining the reactivity of these complexes.
Toward Nb oxo imido and sulfido imido complexes: [2 + 2] cycloadditions and retrocyloadditions of Nb bis(imido) complexes
Following our studies involving 1,2-addition reactivity of Nb bis(imido) complexes, we desired to access isolobal terminal oxo imido and sulfido imido Nb complexes and subsequently evaluate the effect of π-loading on their reactivity. Our group recently described the synthesis, electronic structure, and reactivity of the Re(V) oxo imido complexes (BDI)Re(O)(NAr) and (BDI)Re(O)(NtBu): this report demonstrated that both π-loading and the asymmetric π-bonding of an imido ligand and an oxo ligand to the same metal center led to a substantial enhancement of reactivity of the metal-oxo moiety.16 To begin evaluating potential routes to oxo imido and sulfido imido complexes in the Nb system at hand, we began investigating the [2 + 2] cycloaddition reactivity of bis(imido) complexes (BDI)Nb(NtBu)2 and (BDI)Nb(NtBu)(NAr) toward oxygen- and sulfur-containing heteroallenes. Such reactivity patterns are precedented for group 4 complexes, and are known to lead to either isolable carbamate or dithiocarbamate complexes; these products result from further reaction of carbon dioxide with carbamate intermediates, or extrusion of isocyanate or isothiocyanate to ultimately give stable bis-μ-oxo or bis-μ-sulfido dimers.75–80 To date, there has only been one report of group 4 terminal oxo and sulfido complexes accessed via this route.81Accordingly, we found that the bis(imido) complex (BDI)Nb(NtBu)2 reacted with diphenylketene to give the diphenylethyleneformamide imido compound 9 as brown crystals in 56% yield (Scheme 2). The solid-state structure of 9 (Fig. S3 and Table S9, ESI†) showed two distinct molecules in the asymmetric unit, with each exhibiting a distorted square-based pyramidal geometry. Upon heating 9 in solution, we observed a mixture of products including several Nb-containing species and Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNtBu. Previous work from our group has shown that (BDI)Nb(NtBu)(NAr) undergoes analogous [2 + 2] reactivity with tert-butylisocyanate, ultimately degrading to give multiple unidentified Nb species and free carbodiimide.7
CNtBu. Previous work from our group has shown that (BDI)Nb(NtBu)(NAr) undergoes analogous [2 + 2] reactivity with tert-butylisocyanate, ultimately degrading to give multiple unidentified Nb species and free carbodiimide.7
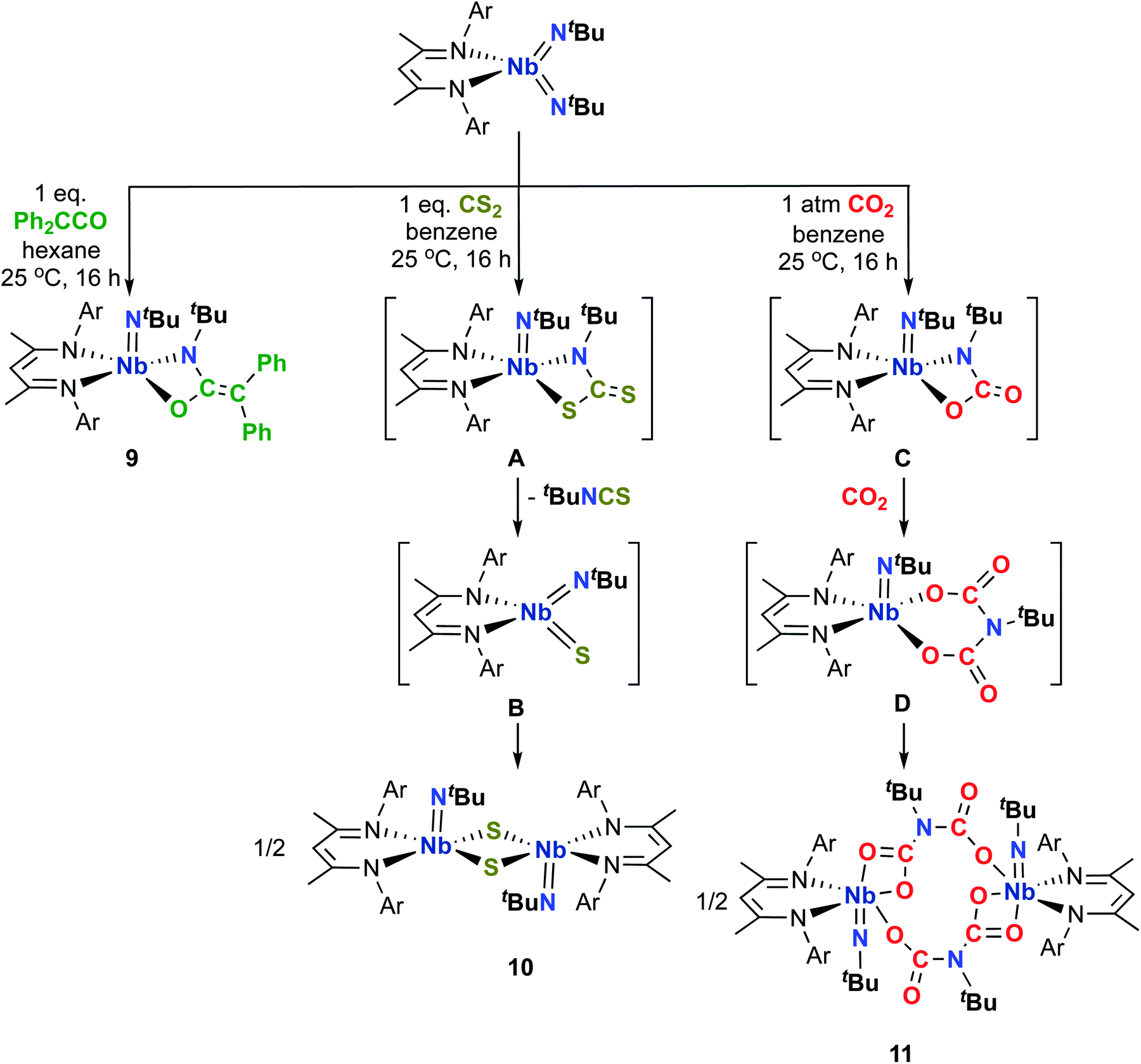 | ||
| Scheme 2 [2 + 2] Cycloaddition of heteroallenes across Nb-imido π-bonds to yield complexes 9, 10, and 11. | ||
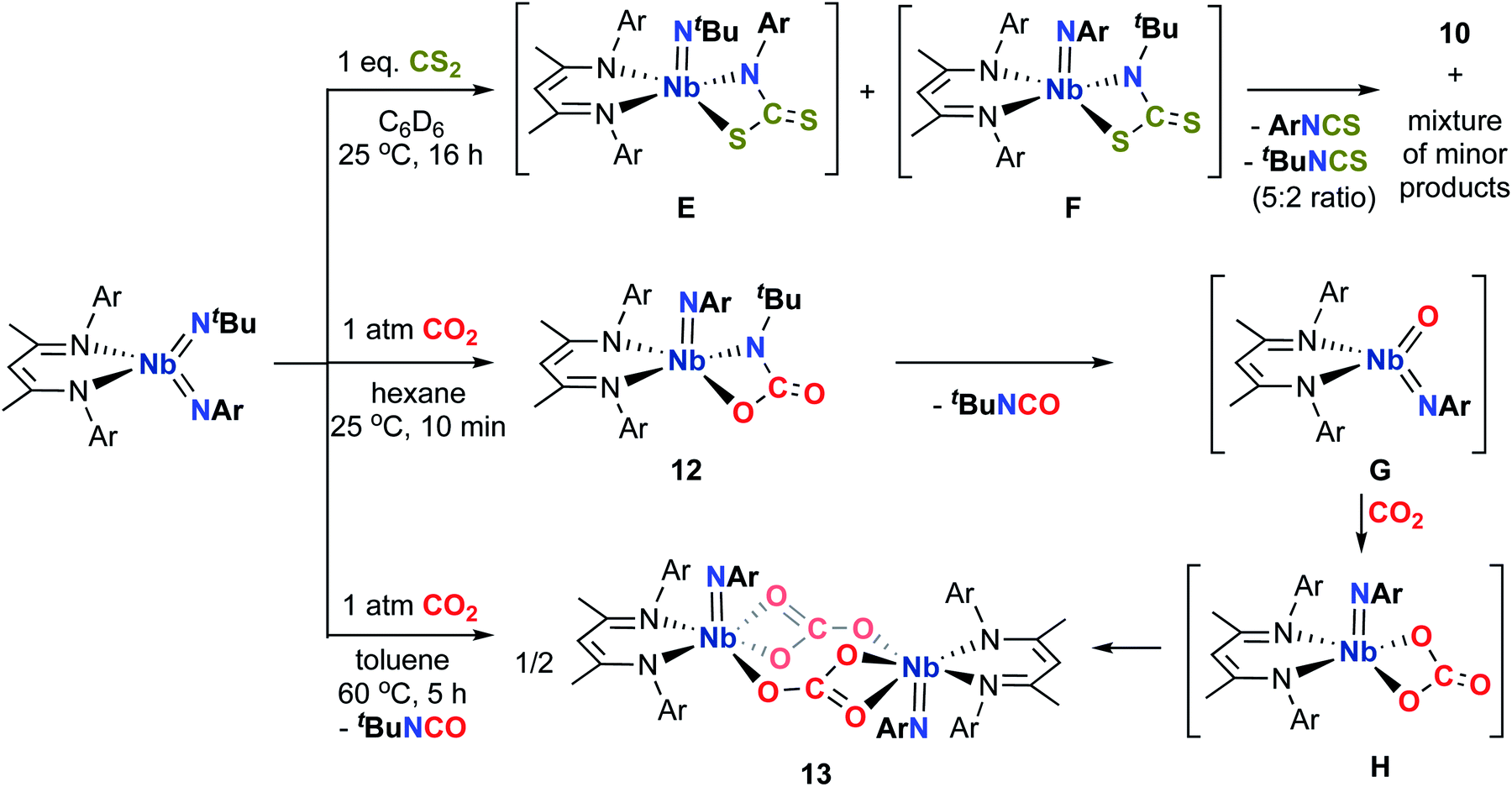
Comparably, the reaction of (BDI)Nb(NtBu)2 and carbon disulfide in benzene resulted in the formation of the dinuclear bis-μ-sulfido complex 10 as large red crystals (Scheme 2). Formation of compound 10 likely proceeds via [2 + 2] cycloaddition of carbon disulfide across a Nb-imido π-bond to give intermediate A, followed by [2 + 2] retrocycloaddition to release tert-butylisothiocyanate (as observed by 1H NMR spectroscopy) to yield terminal Nb sulfido intermediate B, which readily dimerizes. We previously reported an analogous Nb bis-μ-oxo complex, which formed from reaction of a Nb(III) complex with oxygen atom transfer reagents.59 Like the bis-μ-oxo complex, 10 is only sparingly soluble in organic solvents and rapidly crystallizes from solution upon forming. When we carried out the reaction of (BDI)Nb(NtBu)2 and excess carbon disulfide, complex 10 was formed in similar yield, and we observed no evidence of products resulting from insertion of more than one equivalent of carbon disulfide into the Nb-imido bond. Due to the low solubility, complex 10 could not be characterized by NMR spectroscopy. Therefore, confirmation of bulk purity was carried out by combustion analysis. We were also able to obtain a crystal structure of 10, which revealed C2h symmetry in the solid state (Fig. 2, left, and Table S10, ESI†). The Nb–S distances in the dimer were consistent with those of related Nb complexes bearing bridging sulfide ligands.82,83
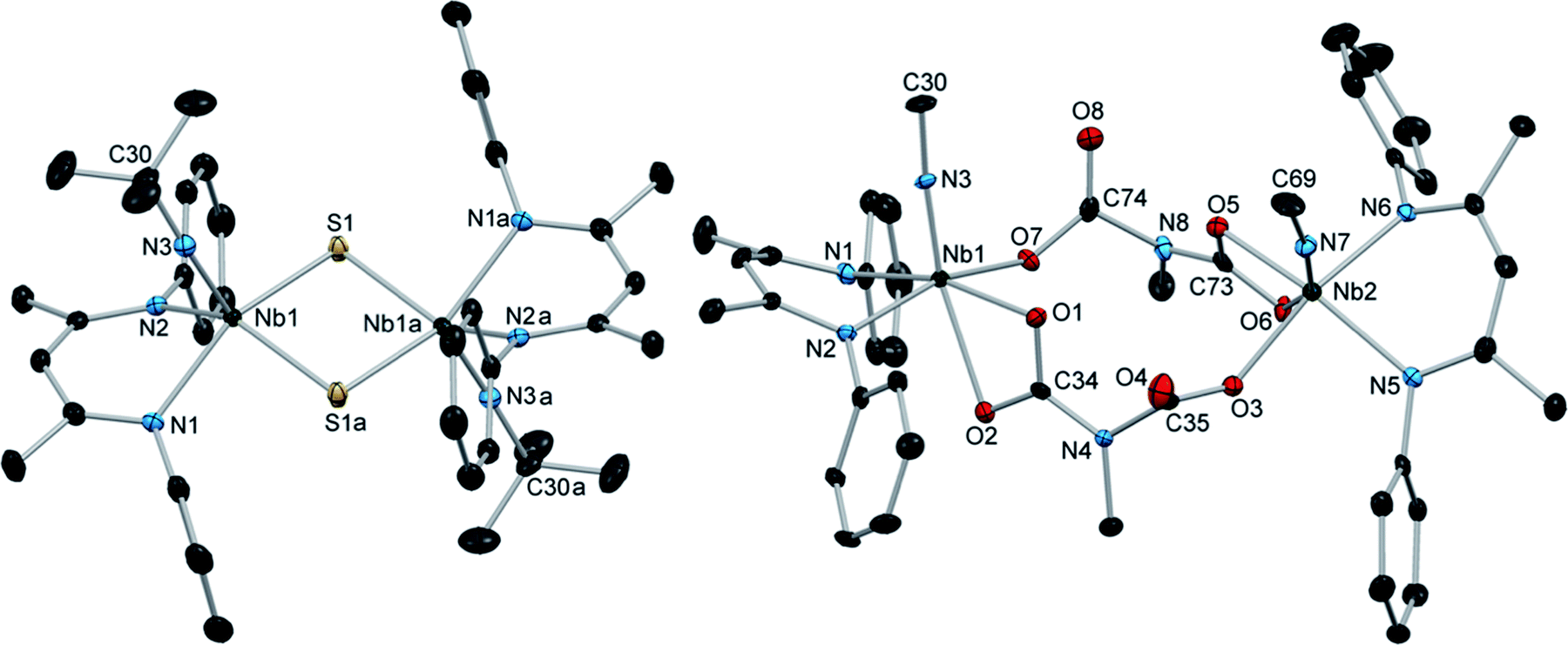 | ||
| Fig. 2 Crystal structures of 10 (left) and 11 (right) with 50% probability thermal ellipsoids. H atoms, aryl iPr groups, and lattice solvent are excluded and selected tBu groups are truncated to the α-carbon. | ||
Surprisingly, rather than reacting with carbon disulfide exclusively at the more basic and reactive tert-butylimido group, the asymmetric Nb bis(imido) complex (BDI)Nb(NtBu)(NAr) reacted at the arylimido group to once again produce 10 as a precipitate, as well as a 5:2 mixture of arylisothiocyanate and tert-butylisothiocyanate (Scheme 3). This reaction differs from all others presented in this work in that we observe products resulting from preferential reactivity across the arylimido group instead of the tert-butylimido group. A complex mixture of other Nb-containing compounds was observed in solution, originating from a putative terminal sulfido intermediate, which in turn results from release of diisopropylphenylisothiocyanate or tert-butylisothiocyanate from intermediates E and F, respectively. The sterically encumbering nature of the arylimido group likely disfavors dimerization of the aforementioned terminal sulfido complex to form a dimeric product analogous to complex 10, bearing arylimido and κ2-BDI ligands. Hence, the highly reactive terminal sulfido intermediate instead either reacts reversibly with tert-butyl- or diisopropylphenylisothiocyanate to regenerate the starting material, which can then react across the arylimido group, or degrades to a mixture of products. The low solubility of product 10, which rapidly crystallizes from solution upon forming, drives this reaction forward.
 | ||
| Scheme 3 [2 + 2] Cycloaddition of heteroallenes across Nb-imido π-bonds to yield complexes 10, 12, and 13. | ||
Introduction of carbon dioxide to a solution of (BDI)Nb(NtBu)2 resulted in conversion to the iminodicarboxylate-bridged dinuclear complex 11, which was isolated as yellow-orange crystals in 76% yield (Scheme 2). Monomeric, chelating iminodicarboxylate complexes resulting from related reactions of titanium and scandium imido complexes with carbon dioxide have been reported.76,77,79,84 When exposed to a single equivalent of carbon dioxide, (BDI)Nb(NtBu)2 reacted to give a mixture of 11 and starting material, indicating that insertion of a second equivalent of carbon dioxide into the Nb–N bond of carbamate intermediate C to give iminodicarboxylate intermediate D occurs faster than [2 + 2] retrocycloaddition to release tert-butylisocyanate. Compound 11 was characterized by 1H and 13C NMR spectroscopy, as well as X-ray crystallography (Fig. 2, right, and Table S10, ESI†).
Exposing the Nb arylimido species (BDI)Nb(NtBu)(NAr) to carbon dioxide resulted in fast, relatively clean conversion to the carbamate complex 12 (Scheme 3). While 12 continued to react further with carbon dioxide in aromatic solvents (vide infra), when the reaction was instead carried out in hexane, this product could be readily isolated as a red powder that precipitated within seconds of introducing carbon dioxide. While the 1H NMR spectrum of 12 displayed broad resonances at room temperature, the resonances sharpened into a spectrum consistent with C1 solution symmetry at 40 °C. The X-ray crystal structure of 12 exhibited distorted square-based pyramidal geometry with the arylimido group in the apical position (Fig. 3, left, and Table S11, ESI†).
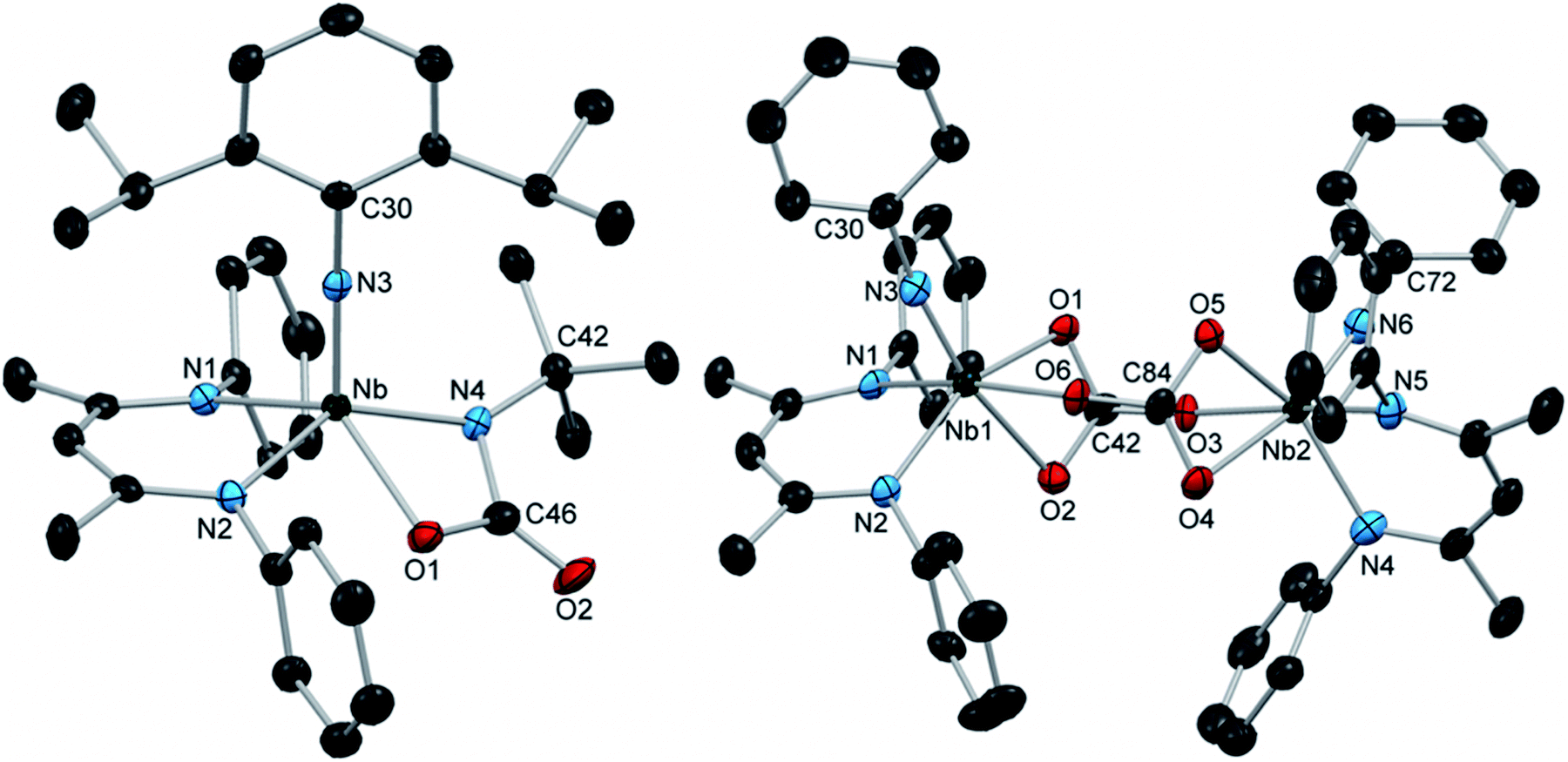 | ||
| Fig. 3 Crystal structures of 12 (left) and 13 (right) with 50% probability thermal ellipsoids. H atoms, aryl iPr groups, and lattice solvent are excluded. | ||
Leaving solutions of (BDI)Nb(NtBu)(NAr) under an atmosphere of carbon dioxide for extended periods resulted in partial conversion of 12 to a second major product. Heating a solution of either (BDI)Nb(NtBu)(NAr) or 12 under an atmosphere of carbon dioxide resulted in a greater degree of conversion to the carbonate-bridged complex 13, which was isolated as red crystalline material in moderate yield, with concomitant release of tert-butylisocyanate, as observed by 1H NMR spectroscopy (Scheme 3). Unlike the related iminodicarboxylate-bridged dimer 11, compound 13 exhibited sharp signals in the 1H NMR spectrum consistent with C1 symmetry in solution. The solution symmetry was also reflected in the solid-state structure (Fig. 3, right, and Table S11, ESI†). Formation of 13 indicates that the putative four-coordinate terminal oxo-arylimido intermediate G is generated during the course of the reaction. Intermediate G reacts with carbon dioxide via [2 + 2] cycloaddition to give Nb carbonate intermediate H, which readily dimerizes to yield 13. We have observed similar [2 + 2] cycloaddition reactivity of CO2 with the terminal oxo moiety of (BDI)Re(O)(NAr); however, the isolable carbonate complex formed in this system remains mononuclear in the solid and solution states, and readily releases CO2 upon standing in solution under an N2 atmosphere.15 Thus, while the reactivity patterns of (BDI)Nb(O)(NAr) and (BDI)Re(O)(NAr) with carbon dioxide are similar, key structural and thermodynamic differences prevail in the resulting carbonate products.
Reactions of early transition metal imido complexes with carbon dioxide are legion: Mountford and co-workers reported reaction of carbon dioxide across the imido functionality in Cp*Ti(NR)[MeC(NiPr)2] (R = tert-butyl or 2,6-diisopropylphenyl).76 Both complexes reacted with one equivalent of carbon dioxide to form a carbamate complex (akin to intermediate C and complex 12 in this work), but, as was also seen in this work, reaction with further equivalents of carbon dioxide led to different products. The arylimido Ti complex reacted cleanly with additional carbon dioxide to form a double insertion product (similar to proposed intermediate D in this work), while the Ti derivative bearing the tert-butylimido group did not react with additional carbon dioxide; instead, it underwent retrocycloaddition to release tert-butylisocyanate to form a Ti-oxo species (akin to proposed intermediate G in this work), which dimerized to form a bis-μ-oxo complex. A similar pattern has been reported for late transition metal monoimidos.85,86 Mountford attributed the different reactivity patterns to electronic differences (i.e. electron-withdrawing versus electron-donating) of the substituent attached to the reacting imido group; in the present work, the substituent effect is instead due to the spectating second imido group, as the reaction occurs across a tert-butyl imido in both Nb bis(imido)complexes. While Mountford observed double insertion only when the electron-withdrawing arylimido group was present (presumably stabilizing the build-up of negative charge on the carbamate nitrogen during in the transition state leading to the double insertion product), we observed double insertion when the (spectating) tert-butyl imido group was present, which serves to stabilize the build-up of positive charge on the metal center during the second insertion. In our work, when the spectating imido group bears an aryl substituent, we did not observe a second insertion and instead the complex released tert-butylisocyanate to yield the proposed oxo-imido intermediate G.
The reactivity of early transition metal terminal oxo complexes often leads to dimerization to form inert bis-μ-oxo complexes.59,76 In the present system, it appears that the combination of a κ2-BDI ligand and an arylimido group provides enough steric encumbrance to prevent dimer formation, based on the following: first, reaction of (BDI)Nb(NtBu)(NAr) with carbon disulfide gave 10 as the major product and did not produce the analogous arylimido-supported bis-μ-sulfido complex and second, oxo imido intermediate G reacted with carbon dioxide to form 13 without generating a bis-μ-oxo complex. Spurred by these observations, we set out to trap terminal oxo-arylimido intermediate G and further study its reactivity.
Accordingly, we found that heating a solution of the carbamate complex 12 with 20 equivalents of pyridine resulted in release of an equivalent of tert-butylisocyanate and relatively clean conversion to a new compound, which was characterized by 1H NMR spectroscopy as the 5-coordinate pyridine adduct of proposed oxo imido intermediate G (Scheme 3), compound 14 (Scheme 4). Compound 14 was isolated as orange-yellow crystals in 22% yield. While 14 exhibited broad 1H NMR signals at room temperature, the signals sharpened in the presence of excess pyridine, indicating the fluxional process responsible for the broad signals involved reversible dissociation of pyridine. The low yield of 14 can be attributed to thermal decomposition; 14 was only stable in solution at low temperature or in the presence of excess pyridine.
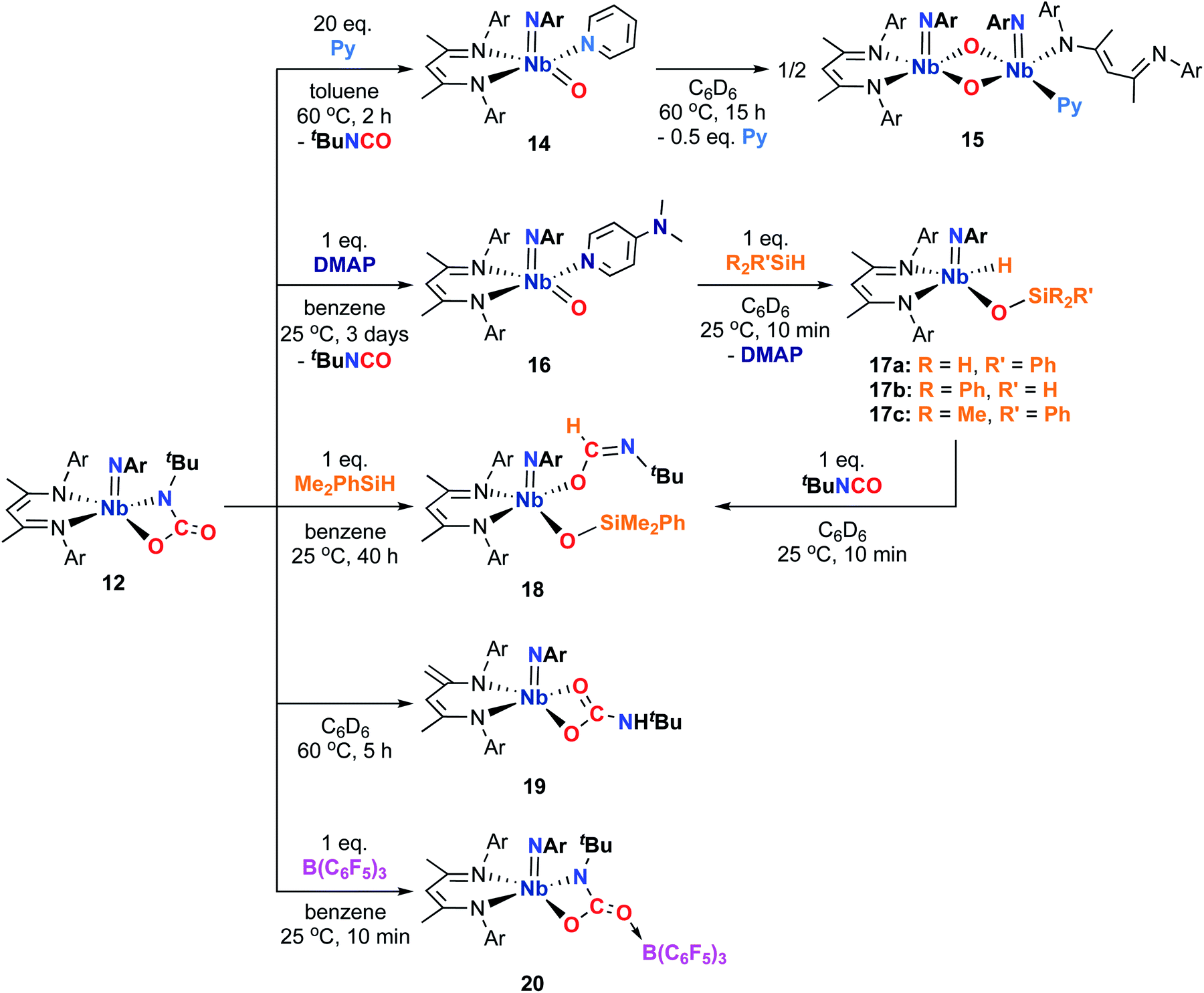 | ||
| Scheme 4 Syntheses of complexes 14, 15, 16, 17a–c, 18, 19, and 20 from Nb imido carbamate complex 12. | ||
When a solution of 14 in C6D6 was subjected to elevated temperatures, the complex decomposed cleanly to give the highly asymmetric bis-μ-oxo dimer 15, which could be isolated as yellow crystals (Scheme 4). The thermal decomposition of 14 proceeded much more rapidly in the absence of excess pyridine; based on this observation, we hypothesize that the transformation is likely initiated by the dissociation of pyridine to generate reactive four-coordinate intermediate G, which subsequently dimerizes by reaction with another equivalent of 14. The solid-state structure of 15 (Fig. 4, left, and Table S12, ESI†) revealed that one of the BDI ligands coordinated in an unusual κ1 binding mode, likely to accommodate the demanding steric environment around the two Nb centers.72–74 This provided further confirmation that the κ2-BDI and arylimido ligands provided sufficient steric support in order to prohibit formation of bis-μ-oxo complexes.
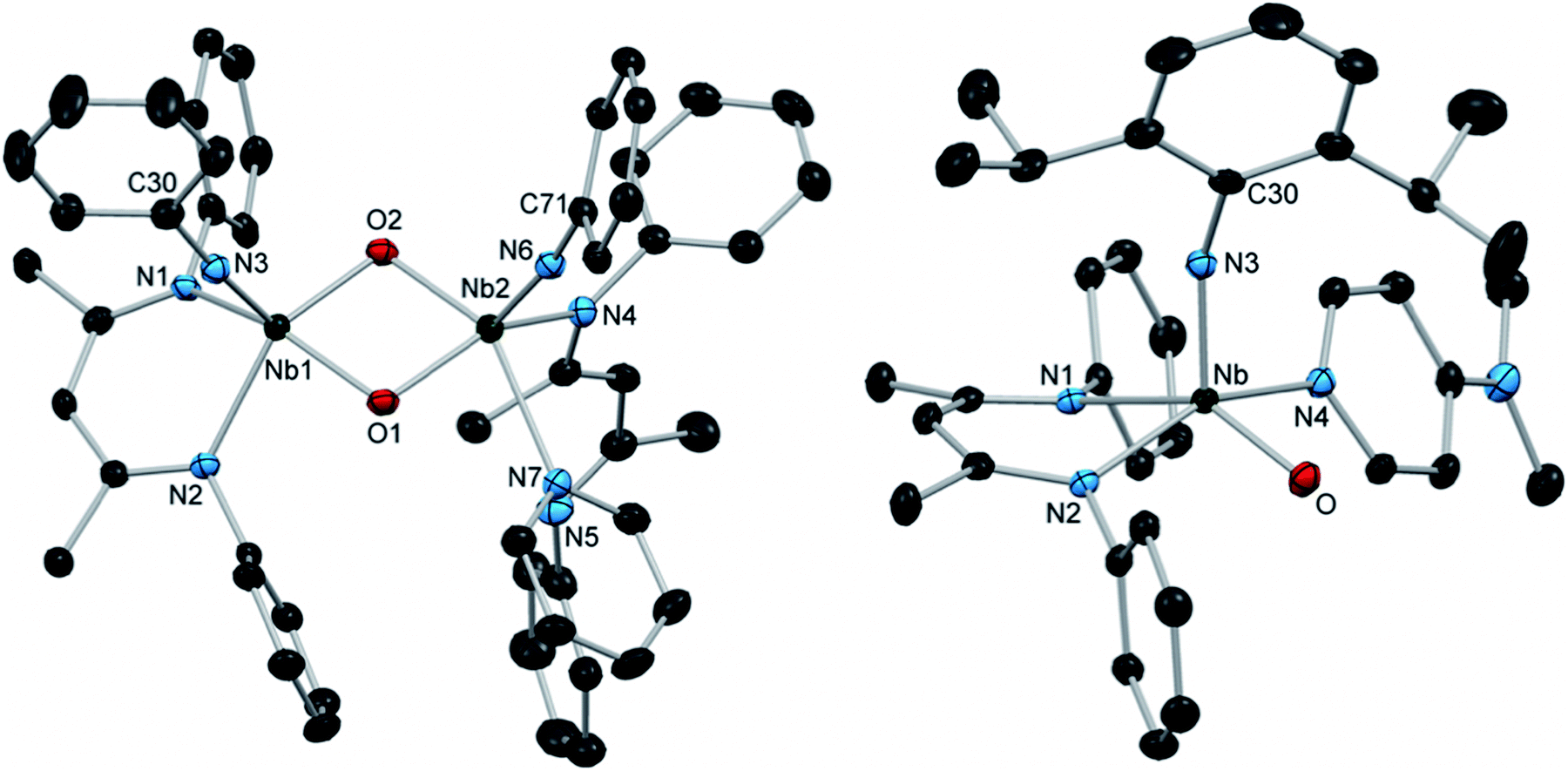 | ||
| Fig. 4 Crystal structures of 15 (left) and 16 (right) with 50% probability thermal ellipsoids. H atoms, selected aryl iPr groups, and lattice solvent are excluded. | ||
In order to access a more thermally stable analog of oxo imido pyridine adduct 14, we investigated the reactivity of arylimido carbamate complex 12 toward more strongly donating ligands. We found that the addition of 4-dimethylaminopyridine (DMAP) to a solution of 12 resulted in a slow color change from red to yellow and precipitation of a yellow powder. Upon workup, the yellow powder was isolated in 61% yield and characterized as the five-coordinate terminal oxo arylimido Nb complex 16 (Scheme 4). To the best of our knowledge, 16 represents the first structurally characterized example of a group 5 transition metal complex bearing both a terminal imido and a terminal oxo group (although many examples of related group 6, 7 and 8 complexes have been reported).15,16,87–94 In contrast to the 1H NMR spectrum of 14, that of 16 displayed sharp signals at room temperature, consistent with a more strongly bound DMAP ligand. The X-ray crystal structure of 16 (Fig. 4, right, and Table S12, ESI†) showed a distorted square-based pyramidal geometry with an apical imido group and an equatorial oxo group. The Nb–O distance of 1.750(1) Å is within the typical range for terminal Nb oxo complexes (ca. 1.7–1.8 Å), while the imido bond distance of 1.816(1) Å is among the longest distances reported for terminal Nb imido bonds and is similar to the distances observed in related bis(imido) complexes.21 Similarly, the Nb–N(3)–C(30) bond angle of 156.1(1)° is among the most bent observed in Nb terminal imido complexes. Thus, the effect of π-loading in this system primarily manifests in the metrical parameters of the imido group; however, this is not an indication that the imido group is expected to be more reactive than the oxo group. In fact, preliminary studies indicate that 16 reacts with substrates exclusively across the Nb oxo moiety.
Group 4 early transition metal oxo complexes are known to engage in 1,2-additions and [2 + 2] cycloadditions across their very polarized oxo groups.47–56 However, while many Nb terminal oxo compounds have been reported, nearly all contain relatively inert NbO bonds and to our knowledge, none have been shown to undergo 1,2-addition reactions to generate Nb hydrides.95 Despite this lack of precedent, we found that compound 16 reacted with silane reagents selectively and relatively cleanly across the oxo group to generate the yellow siloxide compounds 17a–c (with concomitant generation of free DMAP; Scheme 4). While the reactivity of the bis(imido) complex (BDI)Nb(NtBu)2 was limited to primary silanes, 16 reacted with both secondary and tertiary silanes within seconds at room temperature. In solution, 17a and 17b completely degraded to mixtures of products within 12 h at room temperature; 17c was slightly more stable, decomposing after about 24 h at room temperature. Unfortunately, isolation of 17a–c was complicated by the presence of free DMAP, which proved difficult to separate from the Nb hydride products. Hence, our attempts to crystallize 17a–c were not successful, and selective addition of the Si–H bonds across the Nb-oxo group could not be confirmed crystallographically. Nevertheless, the large geminal coupling constant for the Si–H protons observed in the 1H NMR spectrum of 17a (2J = 16 Hz; 1,2-addition across the Nb-oxo moiety) contrasted with the relatively small coupling constant observed for 3a (2J = 9.1 Hz; 1,2-addition across the Nb-imido moiety) is indicative of different electronic environments for these diastereotopic silyl groups.96
While our attempts to thoroughly characterize compounds 17a–c were met with limited success, we were able to provide further support for our connectivity assignment by studying their reactivity. We found that siloxide compound 17c reacted with tert-butylisocyanate via insertion into the Nb–H bond to give the Nb κ1-imidate 18 (Scheme 4). Compound 18 could also be generated readily from the reaction of carbamate complex 12 with dimethylphenylsilane (thus avoiding problematic generation of free DMAP) and isolated as yellow X-ray quality crystals in 41% yield. The solid-state structure of 18 confirmed that, in the original reaction of the oxo imido complex 16 with silanes, the silane reagent indeed added across the Nb-oxo group (Fig. S4, left, and Table S13, ESI†). The formation of complex 18 from 12 likely occurs via two steps: initially, the silane adds across the C–O bond of the four-membered niobacycle to give a niobium dimethylphenylsiloxide tert-butylformamide complex, which is followed by rearrangement from an N-bound formamide to an O-bound imidate, likely through a κ2-[N,O] formamide intermediate. The driving force for this transformation is twofold: first, addition across one side of the four-membered metallacycle relieves ring strain and second, rearrangement from an N-bound formamide to an O-bound imidate is highly favored by the oxophilic early transition metal center and, in addition, relieves steric congestion at the metal center.
While the strong Nb–O and Si–O bonds in 18 rendered the compound quite thermally stable, and no catalytic hydrosilation of isocyanates was observed, the reactivity of 17c with isocyanate notably differed from that of silylamido Nb hydride complexes 3a and 3b, which did not undergo any reaction with isocyanates under similar conditions. We believe this can be attributed to more sterically accessible hydride ligand in 17c relative to those of 3a and 3b. We hope to expand upon this chemistry in order to use reactivity across oxo groups in catalytic hydrosilations and other hydrofunctionalizations of unsaturated substrates.
Having isolated the first examples of group 5 transition metal terminal oxo imido complexes, we next targeted isolable four-coordinate analogs. Our initial attempts to remove the coordinating DMAP ligand from 16 (by treatment with Lewis acids) to generate a four-coordinate complex resulted in degradation to mixtures of products. Next, we hypothesized that heating carbamate complex 12 might result in [2 + 2] retrocycloaddition to release tert-butylisocyanate and generate a four-coordinate terminal oxo complex. However, upon heating 12 in an NMR tube, we instead observed unproductive isomerization to the protonated κ2-[O,O] carbamate complex 19, in which a proton from one of the BDI methyl groups has migrated to the carbamate nitrogen; this product was characterized by 1H NMR spectroscopy (Scheme 4). As a final attempt, we aimed to trap a four-coordinate terminal oxo imido complex generated by loss of tert-butylisocyanate from 12 by capping the oxo moiety with a triarylborane Lewis acid. This approach has been used to trap complexes containing reactive basic functionalities in related systems.97–100 In this case, however, the unbound oxygen of the carbamate functionality simply engaged in a Lewis acid–base interaction with the triarylborane to generate 20, which was isolated as purple crystals in 45% yield (Scheme 4). The X-ray crystal structure of 20 (Fig. S4, right, and Table S13, ESI†) is quite similar to that of 12, although coordination of the borane resulted in lengthening of the C(46)–O(2) distance by ca. 0.9 Å. Rather than leading to release of isocyanate and formation of the desired borane-capped oxo complex, prolonged heating of 20 led to mixtures of unidentified products. While we were ultimately unable to satisfactorily access isolable, four-coordinate oxo imido and sulfido imido complexes, we were able to generate complexes that could act as a source of reactive monomeric oxo imido intermediates, as well as trap these intermediates with strongly σ-donating ligands.
Conclusions
Nb bis(imido) and oxo imido complexes were shown to react with a variety of substrates across their Nb–N and Nb–O π-bonds. First, we demonstrated stoichiometric 1,2-addition reactivity with dihydrogen, silanes, and boranes to generate Nb hydrido-amido-imido complexes 1–4, as well as with substrates containing relatively acidic S–H or C–H bonds to generate products 7 and 8. We observed the symmetric bis(imido) complex (BDI)Nb(NtBu)2 to be considerably more reactive toward 1,2-addition than the asymmetric bis(imido) complex BDI(Nb)(NtBu)(NAr), revealing a significant substituent effect in these complexes. This finding suggests that merely adding a second imido group is not enough to enhance reactivity of a Nb-imido group: both substituents must be electron-donating to make one of the imido groups highly reactive. Second, bis(imido) complexes also exhibited [2 + 2] cycloaddition and retrocycloaddition reactivity with sulfur- and oxygen-containing heteroallenes. While reactions with (BDI)Nb(NtBu)2 generated unreactive dimeric species, the reaction between (BDI)Nb(NtBu)(NAr) and carbon dioxide generated the monomeric Nb carbamate complex 12. Again, the choice of imido substituent played a major role in the reactivity of these complexes toward cycloaddition with carbon dioxide: the substituent attached to the spectating imido group ultimately determined the outcome of the reaction. Under appropriate conditions, compound 12 was shown to behave as a source of the reactive four-coordinate, monomeric Nb oxo imido intermediate G, which could be trapped as the pyridine and DMAP adducts 14 and 16, respectively. These represent the first isolated examples of group 5 transition metal complexes containing both a terminal imido and a terminal oxo group. Likely as a consequence of π-loading effects, compound 16 was reactive toward 1,2-addition of silane substrates across its oxo group. While related reactions across oxo groups have been observed in complexes of groups 4, 6, and 7, this represents a new reaction pathway in group 5 chemistry. The full scope of stoichiometric and catalytic reactions that this reactivity can be applied to remains to be explored, and work in this direction is ongoing. In addition to pursuing the reactivity of 16, we are continuing to target four-coordinate terminal oxo imido complexes of Nb, as well as other isolobal π-loaded Nb complexes with reactive multiply-bonded ligands, such as terminal sulfides, selenides, phosphinidenes, and alkylidenes.Experimental
General considerations
Unless otherwise noted, all reactions were performed using standard Schlenk line techniques or in an MBraun inert atmosphere glove box under an atmosphere of nitrogen (<1 ppm O2/H2O). Glassware and Celite were stored in an oven at ca. 140 °C. Molecular sieves (4 Å) were activated by heating to 300 °C overnight under vacuum prior to storage in a glovebox. n-Hexane, n-pentane, diethyl ether, benzene, toluene, and pyridine were purified by passage through columns of activated alumina and degassed by sparging with nitrogen. HMDSO was vacuum distilled from sodium/benzophenone, degassed by sparging with nitrogen, and stored over molecular sieves. Deuterated solvents were vacuum-transferred from sodium/benzophenone, degassed with three freeze–pump–thaw cycles, and stored over molecular sieves. NMR spectra were recorded on Bruker AV-600, AVB-400, AVQ-400, AV-500, and DRX-500 spectrometers. 1H and 13C{1H} chemical shifts are given relative to residual solvent peaks. 1H and 13C{1H} NMR assignments were routinely confirmed by 1H–1H (COSY and NOESY) and 1H–13C (HSQC and HMBC) experiments. FT-IR samples were prepared as Nujol mulls and were taken between KBr disks using a Nicolet iS10 FT-IR spectrometer. Melting points were determined using an OptiMelt automated melting point system. (BDI)Nb(NtBu)2 and (BDI)Nb(NtBu)(NAr) (BDI = ArNC(Me)CHC(Me)NAr, Ar = 2,6-diisopropylphenyl) were prepared using the literature procedures.21 All other reagents were acquired from commercial sources and used as received. Elemental analyses were determined either at the College of Chemistry, University of California, Berkeley or at the School of Human Sciences, Science Center, London Metropolitan University. X-ray structural determinations were performed at CHEXRAY, University of California, Berkeley on SMART APEX I and SMART APEX II QUAZAR diffractometers.(BDI)NbH(NHtBu)(NtBu) (1)
(BDI)Nb(NtBu)2 (200 mg, 0.300 mmol) was added to a 25 mL Schlenk tube and dissolved in 2 mL HMDSO to give a nearly saturated orange solution. The flask was evacuated under reduced pressure for 5 s, and the headspace was backfilled with H2. The solution was left at room temperature for 1 day, resulting in a slight lightening in color. Storage at −40 °C for 5 days yielded yellow-orange crystals of 1. The crystals were isolated and residual solvent was removed under vacuum. Yield: 91.0 mg, 41%. Generation in solution: (BDI)Nb(NtBu)2 (10.0 mg, 0.015 mmol) was dissolved in C6D6 (0.4 mL) in a 4 mL vial to give an orange-yellow solution and then transferred to a J. Young NMR tube and sealed. The solution was degassed with two freeze–pump–thaw cycles. With ca. 95% of the length of the tube submerged in liquid N2, the tube was evacuated under reduced pressure for 5 s, and the headspace was backfilled with 1 atm H2. The tube was sealed, and then allowed to warm to room temperature. Upon thawing, the solution rapidly changed color from orange-yellow to a lighter yellow-orange. 1H NMR (500 MHz, C6D6, 293 K): δ = 9.09 (br s, 1H, Nb–H) 7.80 (s, 1H, N–H) 7.24–7.07 (m, 6H, Ar), 5.02 (s, 1H, HC(C(Me)NAr)2), 3.56 (sep, 2H, CHMe2), 3.46 (sep, 2H, CHMe2), 1.62 (s, 6H, HC(C(Me)NAr)2), 1.50 (d, 6H, CHMe2), 1.37 (d, 6H, CHMe2), 1.25 (s, 9H, NbNtBu), 1.23 (d, 6H, CHMe2), 1.21 (d, 6H, CHMe2), 1.06 (s, 9H, NbN(H)tBu). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 166.2 (HC(C(Me)NAr)2), 152.0 (Ar), 145.3 (Ar), 142.1 (Ar), 125.9 (Ar), 124.7 (Ar), 124.3 (Ar), 101.0 (HC(C(Me)NAr)2), 67.2 (Cα, NbN(H)tBu), 56.5 (Cα, NbNtBu), 33.3 (Cβ, NbN(H)tBu), 33.1 (Cβ, NbNtBu), 28.7 (CHMe2), 28.6 (CHMe2), 25.3 (CHMe2), 25.2 (CHMe2), 25.1 (CHMe2), 25.0 (CHMe2), 24.9 (HC(C(Me)NAr)2). FT-IR (KBr, Nujol, cm−1): 3344 (sharp, w, N–H stretch), 3323 (sharp, w, N–H stretch), 1652 (broad, m, Nb–H stretch). Anal. calcd (%) for Nb1N4C37H61: C, 67.87; H, 9.39; N, 8.56. Found: C, 67.69; H, 9.33; N, 8.47.(BDI)NbH(NHtBu)(NAr) (2)
(BDI)Nb(NtBu)(NAr) (10.0 mg, 0.013 mmol) was dissolved in C6D6 (0.4 mL) in a 4 mL vial to give a red solution and then transferred to a J. Young NMR tube and sealed. The solution was degassed with two freeze–pump–thaw cycles. With ca. 95% of the length of the tube submerged in liquid N2, the tube was evacuated under reduced pressure for 5 s, and the headspace was backfilled with 1 atm H2. The tube was sealed, and then allowed to warm to room temperature. Upon thawing, the solution lightened in color. 1H NMR (400 MHz, C6D6, 293 K): δ = 9.43 (br s, 1H, Nb–H) 8.66 (s, 1H, N–H) 7.19–6.95 (m, 9H, Ar), 5.24 (s, 1H, HC(C(Me)NAr)2), 4.26 (br s, 1H, CHMe2), 4.08 (br s, 1H, CHMe2), 3.83 (sep, 1H, CHMe2), 3.38 (sep, 1H, CHMe2), 3.25 (sep, 1H, CHMe2), 3.21 (sep, 1H, CHMe2), 1.71 (s, 3H, HC(C(Me)NAr)2), 1.69 (s, 3H, HC(C(Me)NAr)2), 1.37–1.30 (m, 18H, CHMe2), 1.25 (d, 3H, CHMe2), 1.17–1.09 (m, 12H, CHMe2), 1.02 (d, 3H, CHMe2), 0.82 (s, 9H, tBu). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 166.8 (HC(C(Me)NAr)2), 166.1 (HC(C(Me)NAr)2), 153.3 (Ar), 149.0 (Ar), 144.4 (Ar), 142.5 (Ar), 141.5 (Ar), 140.6 (Ar), 140.4 (Ar), 126.6 (Ar), 125.7 (Ar), 125.6 (Ar), 125.1 (Ar), 124.3 (Ar), 124.0 (Ar), 123.8 (Ar), 123.7 (Ar), 122.5 (Ar), 101.8 (HC(C(Me)NAr)2), 57.1 (Cα, tBu), 32.1 (Cβ, tBu), 29.9 (CHMe2), 29.2 (CHMe2), 28.4 (CHMe2), 28.2 (CHMe2), 27.9 (CHMe2), 27.9 (CHMe2), 26.1 (CHMe2), 25.9 (CHMe2), 25.8 (CHMe2), 25.5 (CHMe2), 25.0 (CHMe2), 25.0 (HC(C(Me)NAr)2), 24.8 (HC(C(Me)NAr)2), 24.6 (CHMe2), 24.5 (CHMe2), 24.2 (CHMe2), 24.1 (CHMe2), 24.0 (CHMe2), 23.9 (CHMe2) 23.8 (CHMe2).(BDI)NbH(N[SiH2Ph]tBu)(NtBu) (3a)
(BDI)Nb(NtBu)2 (220 mg, 0.340 mmol) was dissolved in 10 mL hexane in a 20 mL vial to give an orange solution. Phenylsilane (42.0 μL, 0.340 mmol) was added in one portion using a micropipette, resulting in an immediate color change to yellow. The solution was left at room temperature for 15 min, and then the volatile materials were removed under vacuum, leaving a yellow powder. The residue was extracted with hexane and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding 3a as a pale yellow microcrystalline powder. The powder was isolated and residual solvent was removed under vacuum. Yield: 220 mg, 85% over 2 crops. X-ray suitable crystals were obtained by recrystallization from a concentrated HMDSO solution at −40 °C. 1H NMR (600 MHz, C6D6, 293 K): δ = 9.15 (br s, 1H, Nb–H), 8.19 (br d, 2H, SiH2Ph), 7.37–7.08 (br m, 9H, Ar/SiH2Ph), 6.06 (br d, 1H, SiH2Ph), 5.56 (br d, 1H, SiH2Ph), 4.94 (s, 1H, HC(C(Me)NAr)2), 4.42 (sep, 1H, CHMe2), 4.29 (br sep, 1H, CHMe2), 3.22 (br sep, 1H, CHMe2), 3.17 (br sep, 1H, CHMe2), 1.86 (br d, 3H, CHMe2), 1.65–0.80 (br m, 45H, CHMe2/HC(C(Me)NAr)2/tBu). 1H NMR (500 MHz, C7D8, 253 K): δ = 9.05 (br s, 1H, Nb–H) 8.16 (d, 2H, SiH2Ph), 7.26 (t, 3H, SiH2Ph), 7.19 (m, 2H, Ar), 7.15–7.03 (m, 4H, Ar/SiH2Ph), 6.02 (d, 1H, SiH2Ph, 2J = 9.1 Hz), 5.53 (d, 1H, SiH2Ph, 2J = 9.1 Hz), 4.88 (s, 1H, HC(C(Me)NAr)2), 4.40 (sep, 1H, CHMe2), 4.26 (sep, 1H, CHMe2), 3.16 (sep, 1H, CHMe2), 3.11 (sep, 1H, CHMe2), 1.83 (d, 3H, CHMe2), 1.59 (d, 3H, CHMe2), 1.57 (s, 3H, HC(C(Me)NAr)2), 1.53 (s, 3H, HC(C(Me)NAr)2), 1.42 (d, 3H, CHMe2), 1.40 (s, 9H, NbNtBu), 1.29–1.17 (m, 9H, CHMe2), 1.09 (d, 3H, CHMe2), 1.08 (d, 3H, CHMe2), 0.78 (s, 9H, NbN(Si)tBu). 13C{1H} NMR (126 MHz, C7D8, 253 K): δ = 166.2 (HC(C(Me)NAr)2), 165.9 (HC(C(Me)NAr)2), 152.1 (Ar), 147.1 (Ar), 145.3 (Ar), 143.4 (Ar), 142.1 (Ar), 137.9 (Ar), 135.6 (Ar), 129.4 (Ar), 128.5 (Ar), 127.5 (Ar), 126.8 (Ar), 126.1 (Ar), 126.0 (Ar), 124.5 (Ar), 123.5 (Ar), 101.3 (HC(C(Me)NAr)2), 71.0 (Cα, NbN(Si)tBu), 58.6 (Cα, NbNtBu), 32.8 (Cβ, NbtBu), 29.8 (Cβ, NbN(Si)tBu), 28.8 (CHMe2), 28.4 (CHMe2), 28.1 (CHMe2), 27.9 (CHMe2), 26.4 (CHMe2), 25.9 (CHMe2), 25.9 (CHMe2), 25.3 (CHMe2), 25.2 (HC(C(Me)NAr)2), 25.1 (CHMe2), 25.0 (CHMe2), 24.9 (CHMe2), 24.5 (HC(C(Me)NAr)2), 24.4 (CHMe2). FT-IR (KBr, Nujol, cm−1): 2204 (sharp, s, Si–H stretch), 2130 (sharp, s, Si–H stretch), 2103 (sharp, s, Si–H stretch), 1667 (broad, m, Nb–H stretch). Anal. calcd (%) for Nb1Si1N4C43H67: C, 67.87; H, 8.87; N, 7.36. Found: C, 68.02; H, 8.77; N, 7.41. Mp: 120 °C (dec).(BDI)NbH(N[SiH2nBu]tBu)(NtBu) (3b)
(BDI)Nb(NtBu)2 (180 mg, 0.280 mmol) was added to a 50 mL Schlenk flask and dissolved in 5 mL toluene to give an orange solution. N-Butylsilane (36.0 μL, 0.280 mmol) was added using a micropipette, resulting in an immediate color change to orange-yellow. The solution was left at room temperature overnight, then the volatile materials were removed under vacuum leaving an orange-yellow residue. The residue was extracted with HMDSO and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding pale yellow crystals of 3b. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 160 mg, 75% over two crops. 1H NMR (600 MHz, C6D6, 293 K): δ = 8.65 (br s, 1H, Nb–H) 8.19, 7.28–7.07 (br m, 6H, Ar), 5.34 (br s, 1H, SiH2nBu), 5.01 (br s, 1H, SiH2nBu), 4.92 (s, 1H, HC(C(Me)NAr)2), 4.30 (br m, 1H, CHMe2), 4.22 (br m, 1H, CHMe2), 3.19 (br m, 2H, CHMe2), 1.80–0.80 (br m, 57H, CHMe2/HC(C(Me)NAr)2/tBu/nBu). FT-IR (KBr, Nujol, cm−1): 2178 (sharp, m, Si–H stretch), 2092 (sharp, s, Si–H stretch), 1667 (broad, m, Nb–H stretch). Mp: 148–168 °C (melting with dec).(BDI)NbH(N[BO2C6H12]tBu)(NtBu) (4)
(BDI)Nb(NtBu)2 (210 mg, 0.320 mmol) was dissolved in 5 mL hexane in a 20 mL vial to give an orange solution. Pinacolborane (49.0 μL, 0.320 mmol) was added using a micropipette, resulting in an immediate color change to yellow. The solution was left at room temperature for 3 h, and then filtered through a pad of Celite. The volatile materials were removed under vacuum, leaving a yellow residue. The residue was extracted with a hexane/HMDSO mixture and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding 4 as a yellow microcrystalline powder. The powder was isolated and residual solvent was removed under vacuum. Yield: 97.0 mg, 39%. X-ray suitable crystals were obtained by recrystallization from a concentrated HMDSO solution at −40 °C. 1H NMR (600 MHz, C6D6, 293 K): δ = 10.01 (s, 1H, Nb–H), 7.26–7.13 (m, 6H, Ar) 5.11 (s, 1H, HC(C(Me)NAr)2), 3.63 (sep, 1H, CHMe2), 3.52 (sep, 1H, CHMe2), 3.32 (sep, 1H, CHMe2), 3.25 (sep, 1H, CHMe2), 1.69 (s, 3H, HC(C(Me)NAr)2), 1.65 (s, 3H, HC(C(Me)NAr)2), 1.63 (d, 3H, CHMe2), 1.61 (d, 3H, CHMe2), 1.54 (s, 9H, NbNtBu), 1.45 (d, 3H, CHMe2), 1.44 (d, 3H, CHMe2), 1.29 (d, 3H, CHMe2), 1.23 (d, 3H, CHMe2), 1.21 (d, 3H, CHMe2), 1.16 (d, 3H, CHMe2), 1.14 (s, 6H, BPin CH3), 1.13 (s, 6H, BPin CH3), 1.11 (s, 9H, NbN(B)tBu). 13C{1H} NMR (101 MHz, C6D6, 293 K): δ = 165.5 (CH(C(Me)NAr)2), 163.8 (CH(C(Me)NAr)2), 153.2 (Ar), 150.0 (Ar), 144.0 (Ar), 142.5 (Ar), 142.2 (Ar), 142.0 (Ar), 126.3 (Ar), 125.8 (Ar), 125.3 (Ar), 125.1 (Ar), 123.9 (Ar), 123.6 (Ar), 103.7 (CH(C(Me)NAr)2), 82.4 (Cα, NbNtBu), 56.2 (Cα, NbN(B)tBu), 35.4 (Cβ, NbNtBu), 33.3 (Cβ, NbN(B)tBu), 29.1 (CHMe2), 28.9 (CHMe2), 28.0 (CHMe2), 27.4 (CHMe2), 26.8 (CHMe2), 26.4 (CHMe2), 26.0 (HC(C(Me)NAr)2), 25.8 (HC(C(Me)NAr)2), 25.7 (CHMe2), 25.6 (CHMe2), 25.4 (CHMe2), 25.0 (BPin CH3), 24.8 (CHMe2), 24.6 (CHMe2), 24.4 (CHMe2), 24.2 (BPin CH3). FT-IR (KBr, Nujol, cm−1): 1662 (broad, m, Nb–H stretch). Anal. calcd (%) for NbO2N4BC43H72: C, 66.15; H, 9.30; N, 7.18. Found: C, 65.97; H, 9.42; N, 7.08. Mp: 142 °C (dec).(BDI)Nb(OC(H)O)(N[SiH2nBu]tBu)(NtBu) (5)
Compound 3b (190 mg, 0.260 mmol) was added to a 100 mL Schlenk flask and dissolved in 10 mL toluene to give a pale yellow solution. The flask was evacuated under reduced pressure for 5 s, and the headspace was backfilled with CO2, resulting in a slight lightening of the color of the solution. The solution was stirred at room temperature overnight, and then the volatile materials were removed under vacuum, leaving a yellow residue. The residue was extracted with pentane and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding pale yellow crystals of 5. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 95.0 mg, 48% over two crops. 1H NMR (400 MHz, C6D6, 293 K): δ = 7.86 (s, 1H, NbOCHO), 7.22–7.11 (m, 5H, Ar), 7.07 (dd, 1H, Ar), 5.51 (m, 1H, SiH2nBu), 5.13 (s, 1H, HC(C(Me)NAr)2), 5.06 (m, 1H, SiH2nBu), 4.11 (sep, 1H, CHMe2), 3.67 (sep, 1H, CHMe2), 2.96 (sep, 1H, CHMe2), 2.95 (sep, 1H, CHMe2), 1.65–0.99 (m, 6H, nBu CH2), 1.64 (d, 3H, CHMe2), 1.61 (s, 3H, HC(C(Me)NAr)2), 1.61 (s, 3H, HC(C(Me)NAr)2), 1.58 (d, 3H, CHMe2), 1.56 (d, 3H, CHMe2), 1.48 (s, 9H, NbNtBu), 1.37 (d, 3H, CHMe2), 1.25 (d, 3H, CHMe2), 1.23 (d, 3H, CHMe2), 1.08 (br s, 9H, NbN(Si)tBu), 1.04 (d, 3H, CHMe2), 1.04 (d, 3H, CHMe2), 0.92 (t, 3H, nBu CH2). FT-IR (KBr, Nujol, cm−1): 2182 (s, Si–H stretch), 2098 (s, Si–H stretch), 1649 (s, CO stretch). Anal. calcd (%) for NbSiO2N4C42H71: C, 64.26; H, 9.12; N, 7.14. Found: C, 64.10; H 9.30; N, 7.03. Mp: 123 °C (dec).
(BDI)Nb(OC(H)O)(N[BO2C6H12]tBu)(NtBu) (6)
Compound 4 (160 mg, 0.200 mmol) was added to a 100 mL Schlenk flask and dissolved in 10 mL hexane to give a yellow solution. The flask was evacuated under reduced pressure for 5 s, and the headspace was backfilled with CO2, resulting in a slight lightening of the color of the solution. The solution was stirred at room temperature overnight, and then the volatile materials were removed under vacuum, leaving a yellow residue. The residue was extracted with HMDSO and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding yellow crystals of 6. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 57.0 mg, 35% over three crops. 1H NMR (400 MHz, C6D6, 293 K): δ = 8.60 (s, 1H, NbOCHO), 7.28–7.11 (m, 5H, Ar), 7.06 (dd, 1H, Ar), 5.16 (s, 1H, HC(C(Me)NAr)2), 4.15 (sep, 1H, CHMe2), 3.78 (sep, 1H, CHMe2), 2.95 (sep, 1H, CHMe2), 2.90 (sep, 1H, CHMe2), 1.67 (s, 3H, HC(C(Me)NAr)2), 1.60 (s, 3H, HC(C(Me)NAr)2), 1.59 (d, 3H, CHMe2), 1.57 (s, 9H, NbNtBu), 1.53 (d, 3H, CHMe2), 1.29 (s, 9H, NbN(B)tBu), 1.26 (d, 3H, CHMe2), 1.24 (d, 3H, CHMe2), 1.21 (s, 6H, BPin CH3), 1.20 (d, 3H, CHMe2), 1.19 (d, 3H, CHMe2), 1.13 (s, 6H, BPin CH3), 1.06 (d, 3H, CHMe2), 1.04 (d, 3H, CHMe2). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 169.0 (CH(C(Me)NAr)2), 166.2 (CH(C(Me)NAr)2), 164.9 (NbOCHO), 149.1 (Ar), 148.6 (Ar), 145.8 (Ar), 144.4 (Ar), 143.7 (Ar), 140.5 (Ar), 127.3 (Ar), 126.4 (Ar), 125.7 (Ar), 125.0 (Ar), 124.6 (Ar), 123.8 (Ar), 103.9 (CH(C(Me)NAr)2), 82.4 (Cα, NbNtBu), 58.2 (Cα, NbN(B)tBu), 32.6 (Cβ, NbNtBu), 28.9 (CHMe2), 28.8 (CHMe2), 27.9 (CHMe2), 27.8 (CHMe2), 26.9 (CHMe2), 26.6 (HC(C(Me)NAr)2), 26.5 (Cα, NbN(B)tBu), 26.3 (HC(C(Me)NAr)2), 26.3 (CHMe2), 25.9 (CHMe2), 25.9 (CHMe2), 25.8 (CHMe2), 25.5 (CHMe2), 25.4 (CHMe2), 25.0 (BPin CH3), 24.8 (BPin CH3), 24.5 (CHMe2). FT-IR (KBr, Nujol, cm−1): 1661 (s, CO stretch). Anal. calcd (%) for NbO4N4BC44H72: C, 64.26; H, 9.12; N, 7.14. Found: C, 63.89; H 8.87; N, 6.71. Mp: 113–138 °C (melting with dec).
(BDI)Nb(NtBu)(NHtBu)(CCtBu) (7)
(BDI)Nb(NtBu)2 (130 mg, 0.200 mmol) was added to a 100 mL Schlenk flask and dissolved in 10 mL toluene to give an orange solution. tert-Butylacetylene (25.0 μL, 0.200 mmol) was added using a microsyringe. The solution was heated at 60 °C for 15 h, resulting in a color change from orange to pale yellow. The volatile materials were removed under vacuum, leaving a pale yellow powder. The residue was extracted with hexane and the resulting solution was filtered through a pad of Celite and concentrated under vacuum. The solution was stored at −40 °C overnight, yielding 7 as a pale yellow crystalline solid. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 85.0 mg, 56% over 2 crops. X-ray suitable crystals were obtained by recrystallization from a concentrated hexane solution at −40 °C. A 1.9:1 ratio of 7a to 7b (diastereomers) was observed by 1H NMR spectroscopy at 293 K, while a 1.4:1 ratio was observed at 253 K. 1H NMR (600 MHz, C6D6, 293 K): compound 7aδ = 7.67 (br s, 1H, N–H), 7.30–7.05 (br m, 6H, Ar), 5.06 (br s, 1H, HC(C(Me)NAr)2), 3.66 (br m, 2H, CHMe2), 3.06 (br m, 1H, CHMe2), 2.88 (br m, 1H, CHMe2), 1.69–0.94 (br m, 57H, CHMe2/HC(C(Me)NAr)2/tBu). Compound 7bδ = 8.65 (br s, 1H, N–H), 7.30–7.05 (br m, 6H, Ar), 5.03 (br s, 1H, HC(C(Me)NAr)2), 4.19 (br m, 1H, CHMe2), 3.93 (br m, 1H, CHMe2), 3.14 (br m, 2H, CHMe2), 1.83 (br m, 3H, CHMe2), 1.69–0.94 (br m, 54H, CHMe2/HC(C(Me)NAr)2/tBu). 1H NMR (500 MHz, C7D8, 253 K): compound 7aδ = 7.57 (s, 1H, N–H), 7.22–7.04 (m, 6H, Ar), 4.98 (s, 1H, HC(C(Me)NAr)2), 3.61 (sep, 1H, CHMe2), 3.60 (sep, 1H, CHMe2), 3.01 (sep, 1H, CHMe2), 2.80 (sep, 1H, CHMe2), 1.59–0.95 (m, 57H, CHMe2/HC(C(Me)NAr)2/tBu). Compound 7bδ = 8.67 (s, 1H, N–H), 7.30–7.05 (m, 6H, Ar), 4.95 (s, 1H, HC(C(Me)NAr)2), 4.19 (sep, 1H, CHMe2), 3.89 (sep, 1H, CHMe2), 3.10 (sep, 2H, CHMe2), 1.83 (d, 3H, CHMe2), 1.69–0.94 (m, 54H, CHMe2/HC(C(Me)NAr)2/tBu). 1H NMR (500 MHz, C7D8, 333 K): δ = 7.85 (br s, 1H, N–H), 7.19–6.70 (br m, 6H, Ar), 5.09 (s, 1H, HC(C(Me)NAr)2), 3.88–2.60 (br m, 4H, CHMe2), 1.65–0.85 (br m, 57H, CHMe2/HC(C(Me)NAr)2/tBu). 13C{1H} NMR (126 MHz, C7D8, 253 K): δ = 167.7 (HC(C(Me)NAr)2), 166.8 (HC(C(Me)NAr)2), 165.9 (HC(C(Me)NAr)2), 165.5 (HC(C(Me)NAr)2), 154.5 (Ar), 152.5 (Ar), 151.9 (Ar), 150.9 (Ar), 143.2 (Ar), 142.6 (Ar), 141.6 (Ar), 141.3 (Ar), 141.1 (Ar), 140.7 (Ar), 140.4 (Ar), 128.5 (Ar), 127.6 (Ar), 126.2 (Ar), 126.1 (Ar), 125.9 (Ar), 124.7 (Ar), 124.0 (Ar), 123.8 (Ar), 123.6 (Ar), 123.0 (Ar), 122.8 (Ar), 101.2 (HC(C(Me)NAr)2), 100.9 (HC(C(Me)NAr)2), 71.5 (Cα, tBu), 66.1 (Cα, tBu), 57.9 (Cα, tBu), 57.1 (Cα, tBu), 33.7 (Cβ, tBu), 32.3 (Cβ, tBu), 31.7 (Cβ, tBu), 31.5 (Cβ, tBu), 31.3 (Cβ, tBu), 30.0 (Cβ, tBu), 29.5 (CHMe2), 29.2 (CHMe2), 28.8 (CHMe2), 28.5 (CHMe2), 28.4 (CHMe2), 28.0 (CHMe2), 27.4 (CHMe2), 26.8 (CH3), 26.7 (CH3), 26.4 (CH3), 26.2 (CH3), 26.1 (CH3), 25.8 (CH3), 25.7 (CH3), 25.6 (CH3), 25.5 (CH3), 25.4 (CH3), 25.2 (CH3), 25.1 (CH3), 25.1 (CH3), 24.9 (CH3), 24.8 (CH3), 24.5 (CH3), 24.3 (CH3), 23.9 (CH3), 23.7 (CH3). FT-IR (KBr, Nujol, cm−1): 3318 (sharp, w, N–H), 2080 (s, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch). Anal. calcd (%) for NbN4C42H66: C, 70.07; H, 9.24; N, 7.78. Found: C, 70.34; H 8.88; N, 7.80. Mp: 190 °C (dec).
C stretch). Anal. calcd (%) for NbN4C42H66: C, 70.07; H, 9.24; N, 7.78. Found: C, 70.34; H 8.88; N, 7.80. Mp: 190 °C (dec).
(κ1-BDI)Nb(NtBu)(NHtBu)(SCPh3) (8)
(BDI)Nb(NtBu)2 (140 mg, 0.220 mmol) was dissolved in 5 mL toluene in a 20 mL vial to give an orange solution. In a separate vial, triphenylmethanethiol (61.0 mg, 0.220 mmol) was dissolved in 3 mL toluene. The triphenylmethanethiol solution was added to the solution of (BDI)Nb(NtBu)2, resulting in a slight lightening of the solution color. The solution was left at room temperature for 16 h, and then the volatile materials were removed under vacuum, leaving a yellow residue. The residue was extracted with HMDSO and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding 8 as a pale yellow microcrystalline powder. The powder was isolated and residual solvent was removed under vacuum. Yield: 120 mg, 60% over two crops. X-ray suitable crystals were obtained by recrystallization from a concentrated HMDSO solution at −40 °C. 1H NMR (400 MHz, C6D6, 293 K): δ = 7.62 (m, 6H, SCPh3), 7.29–7.05 (m, 6H, Ar), 7.09 (t, 6H, SCPh3), 6.99 (t, 3H, SCPh3), 6.24 (s, 1H, N–H), 4.66 (s, 1H, HC(C(Me)NAr)2), 3.84 (sep, 1H, CHMe2), 3.31 (sep, 1H, CHMe2), 2.94 (sep, 1H, CHMe2), 2.85 (sep, 1H, CHMe2), 2.51 (s, 3H, HC(C(Me)NAr)2), 1.69 (d, 3H, CHMe2), 1.49 (d, 3H, CHMe2), 1.38 (d, 3H, CHMe2), 1.31 (s, 3H, HC(C(Me)NAr)2), 1.29 (d, 3H, CHMe2), 1.25 (d, 3H, CHMe2), 1.20 (s, 9H, NbNtBu), 1.16 (d, 3H, CHMe2), 1.13 (d, 3H, CHMe2), 1.11 (d, 3H, CHMe2), 1.03 (s, 9H, NbN(H)tBu). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 165.0 (CH(C(Me)NAr)2), 153.1 (Ar), 150.5 (Ar), 149.7 (Ar), 148.6 (Ar), 148.0 (Ar), 144.0 (Ar), 142.7 (Ar), 137.0 (Ar), 136.1 (Ar), 131.0 (SCPh3), 130.4 (Ar), 128.7 (Ar), 127.9 (SCPh3), 126.8 (SCPh3), 126.7 (Ar), 124.5 (Ar), 123.3 (Ar), 123.2 (Ar), 123.0 (Ar), 102.5 (CH(C(Me)NAr)2), 73.5 (SCPh3), 69.0 (Cα, NbN(H)tBu), 57.5 (Cα, NbNtBu), 34.5 (Cβ, NbNtBu), 31.3 (Cβ, NbN(H)tBu), 28.7 (CHMe2), 28.6 (CHMe2), 28.5 (CHMe2), 28.4 (CHMe2), 26.3 (CHMe2), 26.1 (CHMe2), 25.3 (CHMe2), 24.4 (CHMe2), 23.9 (CHMe2), 23.4 (CHMe2), 23.2 (CHMe2), 23.0 (CH(C(Me)NAr)2), 22.9 (CHMe2), 22.1 (CH(C(Me)NAr)2). Anal. calcd (%) for NbSN4C56H75: C, 72.39, H, 8.14; N, 6.03. Found: C, 72.28; H, 7.91; N, 5.95. Mp: 169 °C (dec).(BDI)Nb(NAr)(N(tBu)C(CPh2)O) (9)
(BDI)Nb(NtBu)2 (200 mg, 0.310 mmol) was added to a 20 mL vial and dissolved in 10 mL hexane to give an orange solution. In a separate vial, diphenylketene (120 mg, 0.610 mmol) was dissolved in 4 mL hexane. The ketene solution was added to the solution of (BDI)Nb(NtBu)2. The solution was left at room temperature for 16 h, resulting in a color change from orange to brown. The volatile materials were removed under vacuum, leaving a brown residue. The residue was extracted with hexane and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding brown crystals of 9. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 150 mg, 56% over three crops. 1H NMR (600 MHz, C6D6, 293 K): δ = 7.28 (t, 2H, Ar), 7.21 (d, 2H, Ar), 7.19–7.13 (m, 4H, Ar), 7.01 (t, 2H, Ar), 7.00–6.96 (m, 3H, Ar), 6.94 (t, 1H, Ar), 6.79 (br s, 2H, Ar), 5.28 (s, 1H, HC(C(Me)NAr)2), 3.86 (sep, 2H, CHMe2), 2.69 (sep, 2H, CHMe2), 1.63 (s, 6H, HC(C(Me)NAr)2), 1.44 (d, 6H, CHMe2), 1.33 (s, 9H, tBu), 1.26 (s, 9H, tBu), 1.25 (d, 6H, CHMe2), 0.98 (d, 12H, CHMe2). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 169.8 (HC(C(Me)NAr)2), 160.3 (OCN), 144.6 (Ar), 144.2 (Ar), 143.9 (Ar), 143.8 (Ar), 142.8 (Ar), 134.6 (Ar), 130.0 (Ar), 128.0 (Ar), 127.5 (Ar), 127.1 (Ar), 127.0 (Ar), 125.3 (Ar), 125.1 (Ar), 124.5 (Ar), 122.5 (Ar), 105.1 (HC(C(Me)NAr)2), 103.7 (CC(Ph)2), 69.6 (Cα, tBu), 59.8 (Cα, tBu), 34.2 (Cβ, tBu), 32.4 (Cβ, tBu), 28.7 (CHMe2), 27.7 (CHMe2), 25.8 (CHMe2), 25.5 (HC(C(Me)NAr)2), 25.2 (CHMe2), 25.1 (CHMe2), 24.9 (CHMe2). Anal. calcd (%) for Nb1O1N4C51H69: C, 72.32; H, 8.21; N, 6.61. Found: C, 72.14; H, 8.01; N, 6.36. Mp: 150 °C (dec).
[(BDI)Nb(NtBu)(μ-S)]2 (10)
(BDI)Nb(NtBu)2 (100 mg, 0.150 mmol) was dissolved in 6 mL benzene in a 20 mL vial to give an orange solution. Carbon disulfide (9.20 μL, 0.150 mmol) was added using a microsyringe, resulting in an immediate color change from orange to red. The solution was left at room temperature for 16 h, resulting in precipitation of 10 as red crystalline blocks. Yield: 65.0 mg, 69%. Compound 10 was not sufficiently soluble in common NMR solvents to obtain solution 1H or 13C NMR data. Anal. calcd (%) for Nb2S2N6C66H100: C, 64.58; H, 8.21; N, 6.85. Found: C, 64.42; H, 8.53; N, 6.62.[(BDI)Nb(NtBu)(μ-O2CN(tBu)CO2)]2 (11)
(BDI)Nb(NtBu)2 (200 mg, 0.310 mmol) was added to a 100 mL Schlenk flask and dissolved in 25 mL benzene to give an orange solution. The flask was evacuated under reduced pressure for 5 s, and the headspace was backfilled with CO2, resulting in an immediate color change from orange to yellow. The solution was stirred at room temperature overnight. The volatile materials were removed under vacuum, leaving a yellow-orange powder. The residue was extracted with toluene and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding yellow-orange crystals of 11. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 180 mg, 76% over three crops. 1H NMR (400 MHz, C6D6, 293 K): δ = 7.22–7.14 (m, 6H, Ar), 7.07–7.03 (m, 6H, Ar), 5.36 (s, 2H, HC(C(Me)NAr)2), 3.99 (br sep, 4H, CHMe2), 3.00 (br s, 4H, CHMe2), 1.77 (br s, 12H, CHMe2), 1.64 (br s, 12H, HC(C(Me)NAr)2), 1.61 (s, 18H, NbNtBu), 1.40 (d, 12H, CHMe2), 1.27 (br d, 12H, CHMe2), 1.07 (d, 12H, CHMe2), 0.85 (s, 18H O2CNtBu). 13C{1H} NMR (101 MHz, C6D6, 293 K): δ = 168.3 (CH(C(Me)NAr)2), 147.6 (Ar), 144.0 (Ar), 143.3 (Ar), 126.2 (Ar), 125.1 (Ar), 123.6 (Ar), 104.7 (CH(C(Me)NAr)2), 70.3 (Cα, NbNtBu), 54.3 (Cα, O2CNtBu), 31.5 (Cβ, NbNtBu), 28.2 (CHMe2), 28.2 (Cβ, O2CNtBu), 28.1 (CHMe2), 26.4 (CH(C(Me)NAr)2), 26.2 (CHMe2), 25.5 (CHMe2), 25.4 (CHMe2), 24.5 (CHMe2). FT-IR (KBr, Nujol, cm−1): 1720 (s, CO stretch). Anal. calcd (%) for Nb2O8N8C78H118: C, 63.23; H, 8.03; N, 7.56. Found: C, 63.26; H, 7.92; N, 7.60. Mp: 178 °C (dec).
(BDI)Nb(NAr)(N(tBu)CO2) (12)
(BDI)Nb(NtBu)(NAr) (1.80 g, 2.40 mmol) was added to a 1000 mL Schlenk flask and dissolved in 100 mL hexane to give a red solution. The flask was evacuated under reduced pressure for 10 s, and the headspace was backfilled with CO2. The solution was stirred at room temperature for 10 min, resulting in a lightening of the solution color and precipitation of a red powder. The powder was collected on a fritted funnel and washed with hexane (3 × 25 mL), and then extracted with diethyl ether. Upon dissolution of the residue in diethyl ether, red crystalline material began forming at room temperature. The solution was stored at −40 °C overnight, yielding additional red crystals of 12. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 1.01 g, 53% over three crops. 1H NMR (600 MHz, C6D6, 293 K): δ = 7.19–6.87 (br m, 9H, Ar), 5.51 (s, 1H, HC(C(Me)NAr)2), 4.46 (br s, 1H, CHMe2), 4.14 (br s, 1H, CHMe2), 3.23 (br s, 1H, CHMe2), 2.87 (br s, 1H, CHMe2), 2.83 (br s, 1H, CHMe2), 2.73 (br s, 1H, CHMe2), 1.73 (br s, 3H, HC(C(Me)NAr)2), 1.63–1.53 (br m, 6H, CHMe2/HC(C(Me)NAr)2), 1.39 (br s, 3H, CHMe2), 1.29–1.03 (br m, 30H, CHMe2/tBu), 0.95 (br s, 3H, CHMe2), 0.85 (br s, 3H, CHMe2), 0.64 (br s, 3H, CHMe2). 1H NMR (500 MHz, C7D8, 233 K): δ = 7.17–6.82 (m, 9H, Ar), 5.51 (s, 1H, HC(C(Me)NAr)2), 4.46 (br sep, 1H, CHMe2), 4.08 (br sep, 1H, CHMe2), 3.09 (br sep, 1H, CHMe2), 2.82 (br sep, 1H, CHMe2), 2.75 (br sep, 1H, CHMe2), 2.65 (br sep, 1H, CHMe2), 1.68 (s, 3H, HC(C(Me)NAr)2), 1.57 (br d, 3H, CHMe2), 1.47 (s, 3H, HC(C(Me)NAr)2), 1.36 (br d, 3H, CHMe2), 1.33–1.08 (br m, 21H, CHMe2/tBu), 1.02 (br d, 3H, CHMe2), 1.01 (br d, 3H, CHMe2), 0.93 (br d, 3H, CHMe2), 0.77 (br d, 3H, CHMe2) 0.56 (br d, 3H, CHMe2). 13C{1H} NMR (126 MHz, C7D8, 233 K): δ = 170.1 (HC(C(Me)NAr)2), 165.8 (HC(C(Me)NAr)2), 160.6 (NCO2), 154.4 (Ar), 148.8 (Ar), 145.1 (Ar), 144.9 (Ar), 143.1 (Ar), 141.4 (Ar), 141.3 (Ar), 140.5 (Ar), 132.5 (Ar), 129.4 (Ar), 126.9 (Ar), 126.1 (Ar), 125.7 (Ar), 124.2 (Ar), 123.7 (Ar), 123.0 (Ar), 122.3 (Ar), 104.5 (HC(C(Me)NAr)2), 57.7 (Cα, tBu), 31.2 (CHMe2), 30.7 (Cβ, NbtBu), 29.2 (CHMe2), 28.2 (HC(C(Me)NAr)2), 28.2 (CHMe2), 28.0 (CHMe2), 27.7 (CHMe2), 27.2 (CHMe2), 26.9 (CHMe2), 26.6 (CHMe2), 26.0 (CHMe2), 25.7 (CHMe2), 25.2 (CHMe2), 25.0 (CHMe2), 24.9 (CHMe2), 24.4 (CHMe2), 23.9 (CHMe2), 23.7 (HC(C(Me)NAr)2), 22.4 (CHMe2), 21.3 (CHMe2). Anal. calcd (%) for NbO2N4C46H67·C4H10O: C, 68.63; H, 8.87; N, 6.40. Found: C, 68.59; H, 8.72; N, 6.79. Mp: 135 °C (dec).[(BDI)Nb(NAr)(μ-CO3)]2 (13)
(BDI)Nb(NtBu)(NAr) (250 mg, 0.330 mmol) was added to a 100 mL Schlenk flask and dissolved in 10 mL toluene to give a red solution. The flask was evacuated under reduced pressure for 5 s, and the headspace was backfilled with CO2. The solution was stirred at 60 °C under N2 atmosphere for 5 h, resulting in a lightening of the solution color. The volatile materials were removed under vacuum, leaving a red residue. The residue was extracted with HMDSO and the resulting solution was concentrated under vacuum. The solution was stored at −40 °C overnight, yielding red crystals of 13. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 98.0 mg, 40% over three crops. 1H NMR (400 MHz, C6D6, 293 K): δ = 7.19 (d, 2H, Ar), 7.13 (d, 2H, Ar), 7.12–7.04 (m, 10H, Ar), 7.00 (d, 2H, Ar), 6.92 (t, 2H, Ar), 5.56 (s, 2H, HC(C(Me)NAr)2), 4.50 (sep, 2H, CHMe2), 4.19 (sep, 2H, CHMe2), 3.68 (sep, 2H, CHMe2), 3.39 (sep, 2H, CHMe2), 2.92 (sep, 2H, CHMe2), 2.83 (sep, 2H, CHMe2), 1.78 (s, 6H, HC(C(Me)NAr)2), 1.75 (s, 6H, HC(C(Me)NAr)2), 1.44 (d, 6H, CHMe2), 1.25–1.15 (m, 15H, CHMe2), 1.09 (d, 6H, CHMe2), 1.06–1.00 (m, 24H, CHMe2), 0.98 (d, 6H, CHMe2). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 168.6 (HC(C(Me)NAr)2), 168.2 (HC(C(Me)NAr)2), 166.5 (CO3), 152.2 (Ar), 149.6 (Ar), 146.9 (Ar), 144.8 (Ar), 144.7 (Ar), 144.4 (Ar), 143.0 (Ar), 142.2 (Ar), 142.2 (Ar), 127.4 (Ar), 127.0 (Ar), 126.7 (Ar), 125.2 (Ar), 124.8 (Ar), 124.7 (Ar), 124.0 (Ar), 123.2 (Ar), 121.7 (Ar), 105.9 (CH(C(Me)NAr)2), 28.8 (CHMe2), 28.7 (CHMe2), 28.5 (CHMe2), 28.4 (CHMe2), 28.3 (CHMe2), 28.1 (CHMe2), 27.3 (CHMe2), 27.2 (CHMe2), 26.6 (CHMe2), 26.5 (CH(C(Me)NAr)2), 26.5 (CHMe2), 26.1 (CHMe2), 26.0 (CHMe2), 25.8 (CHMe2), 25.7 (CH(C(Me)NAr)2), 25.5 (CHMe2), 25.0 (CHMe2), 24.7 (CHMe2), 24.4 (CHMe2), 24.2 (CHMe2). Anal. calcd (%) for Nb2O6N6C84H116·2C6H18OSi2: C, 63.48; H, 8.43; N, 4.63. Found: C, 63.45; H, 8.68; N, 4.63. Mp: 171 °C (dec).(BDI)Nb(NAr)(O)(Py) (14)
Compound 12 (300 mg, 0.340 mmol) was added to a 50 mL Schlenk flask and dissolved in 10 mL toluene to give a red solution. Pyridine (0.55 mL, 6.90 mmol) was added to the solution of 12. The solution was stirred at 60 °C for 2 h, resulting in a color change from red to orange. The volatile materials were removed under vacuum, leaving an orange residue. The residue was triturated with hexane, and then extracted with toluene. The resulting solution was concentrated under vacuum and then stored at −40 °C overnight, yielding orange crystals of 14. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 59.0 mg, 22% over two crops. 1H NMR (600 MHz, C6D6, 293 K): δ = 8.33 (br s, 2H, Py), 7.32–6.82 (m, 9H, Ar), 6.56 (br s, 1H, Py), 6.17 (br s, 2H, Py), 5.19 (s, 1H, HC(C(Me)NAr)2), 4.68 (br s, 1H, CHMe2), 4.26 (br s, 1H, CHMe2), 3.52 (br sep, 4H, CHMe2), 1.70 (s, 6H, HC(C(Me)NAr)2), 1.65–0.83 (m, 36H, CHMe2). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 167.7 (HC(C(Me)NAr)2), 157.8 (Ar), 153.4 (Ar), 152.0 (Py), 149.5 (Ar), 144.3 (Ar), 141.9 (Ar), 137.9 (Ar), 136.9 (Py), 129.3 (Ar), 128.6 (Ar), 126.1 (Ar), 125.7 (Ar), 124.6 (Ar), 123.5 (Py), 122.7 (Ar), 99.6 (CH(C(Me)NAr)2), 29.4 (CHMe2), 28.1 (CHMe2), 25.9 (CH(C(Me)NAr)2), 25.3 (CHMe2), 25.0 (CHMe2), 24.8 (CHMe2). Anal. calcd (%) for NbON4C46H63: C, 70.75; H, 8.13; N, 7.17. Found: C, 70.18; H, 7.77; N, 6.72. Mp: 143 °C (dec).(κ1-BDI)Nb(Py)(NAr)-(μ-O)2-(NAr)Nb(BDI) (15)
Compound 12 (10.0 mg, 0.013 mmol) was dissolved in C6D6 (0.4 mL) in a 4 mL vial and transferred to a J. Young NMR tube. The solution was heated at 60 °C for 24 h, resulting in a color change from orange to yellow. A 1H NMR spectrum indicated clean conversion to 15. 1H NMR (600 MHz, C6D6, 293 K): δ = 8.86 (br s, 2H, Py), 7.31–6.87 (m, 18H, Ar/Py), 6.84 (t, 1H, Ar), 6.60 (br d, 2H, Py), 5.50 (s, 1H, κ2-BDI HC(C(Me)NAr)2), 4.50 (s, 1H, κ1-BDI HC(C(Me)NAr)2), 4.35 (sep, 1H, CHMe2), 4.00 (br sep, 2H, CHMe2), 3.92 (br sep, 1H, CHMe2), 3.76 (sep, 1H, CHMe2), 3.27 (br sep, 1H, CHMe2), 3.04 (br sep, 1H, CHMe2), 3.03 (br sep, 1H, CHMe2), 2.82 (br sep, 1H, CHMe2), 2.78 (br sep, 1H, CHMe2), 2.71 (br sep, 2H, CHMe2), 2.66 (s, 3H, κ1-BDI HC(C(Me)NAr)2), 1.64 (s, 3H, κ2-BDI HC(C(Me)NAr)2), 1.61 (s, 3H, κ2-BDI HC(C(Me)NAr)2), 1.54 (br d, 3H, CHMe2), 1.51 (br d, 3H, CHMe2), 1.48 (br d, 3H, CHMe2), 1.39 (br d, 3H, CHMe2), 1.32 (br d, 3H, CHMe2), 1.31–1.11 (br m, 30H, CHMe2), 1.29 (s, 3H, κ1-BDI HC(C(Me)NAr)2), 1.06 (br d, 3H, CHMe2), 1.05 (br d, 3H, CHMe2), 0.93 (br d, 3H, CHMe2), 0.89 (br d, 3H, CHMe2), 0.87 (br d, 3H, CHMe2), 0.67 (br d, 3H, CHMe2), 0.64 (br d, 3H, CHMe2), 0.59 (br d, 3H, CHMe2), 0.20 (br d, 3H, CHMe2). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 169.5 (κ2-BDI HC(C(Me)NAr)2), 169.0 (κ2-BDI HC(C(Me)NAr)2), 165.3 (κ1-BDI HC(C(Me)NAr)2), 157.8 (κ1-BDI HC(C(Me)NAr)2), 154.3 (Ar), 154.1 (Ar), 151.0 (Py), 149.4 (Ar), 149.0 (Ar), 146.6 (Ar), 145.2 (Ar), 144.7 (Ar), 144.5 (Ar), 144.3 (Ar), 143.6 (Ar), 143.3 (Ar), 141.9 (Ar), 141.6 (Ar), 139.9 (Ar), 137.9 (Ar), 137.1 (Ar), 136.6 (Ar), 129.3 (Ar), 128.6 (Ar), 128.0 (Ar), 127.6 (Ar), 127.4 (Ar), 126.9 (Ar), 126.4 (Ar), 126.0 (Ar), 125.7 (Ar), 124.5 (Py), 124.1 (Ar), 123.7 (Ar), 123.6 (Ar), 123.5 (Py), 123.4 (Ar), 123.0 (Ar), 122.9 (Ar), 122.5 (Ar), 122.4 (Ar), 122.0 (Ar), 105.6 (κ2-BDI CH(C(Me)NAr)2), 104.0 (κ1-BDI CH(C(Me)NAr)2), 29.8 (CHMe2), 29.6 (CHMe2), 28.8 (CHMe2), 28.7 (CHMe2), 28.5 (CHMe2), 28.4 (CHMe2), 28.3 (CHMe2), 28.3 (CHMe2), 28.1 (CHMe2), 28.0 (CHMe2), 28.0 (CHMe2), 27.7 (CHMe2), 27.1 (κ2-BDI CH(C(Me)NAr)2), 27.0 (κ2-BDI CH(C(Me)NAr)2), 26.9 (CHMe2), 26.5 (CHMe2), 26.2 (CHMe2), 25.9 (CHMe2), 25.8 (CHMe2), 25.7 (CHMe2), 25.5 (CH(C(Me)NAr)2), 25.3 (CHMe2), 25.1 (CHMe2), 24.9 (CHMe2), 24.7 (CHMe2), 24.3 (CHMe2), 23.9 (CHMe2), 23.8 (κ1-BDI CH(C(Me)NAr)2), 23.6 (CHMe2), 23.5 (CHMe2), 23.1 (κ1-BDI CH(C(Me)NAr)2), 22.3 (CHMe2).(BDI)Nb(NAr)(O)(DMAP) (16)
Compound 12 (200 mg, 0.250 mmol) was dissolved in benzene (3 mL) in a 20 mL vial to give a red solution. In a separate vial, 4-dimethylaminopyridine (DMAP) (33.0 mg, 0.270 mmol) was dissolved in 2 mL benzene. The DMAP solution was added to the solution of 12. The solution was left at room temperature for 3 days, resulting in a slow color change from red to orange-yellow and precipitation of a yellow powder. The volatile materials were removed under vacuum, leaving an orange-yellow residue. The residue was washed with hexane (3 × 10 mL), and residual solvent was removed under vacuum to give 16 as a yellow powder. Yield: 126 mg, 61%. X-ray suitable crystals were obtained by recrystallization via slow cooling of a concentrated solution in benzene from 60 °C to room temperature. 1H NMR (600 MHz, CDCl3, 293 K): δ = 7.90 (d, 2H, DMAP), 7.17 (t, 1H, Ar), 7.13 (d, 1H, Ar), 7.08 (d, 1H, Ar), 7.07 (d, 1H, Ar), 6.96 (d, 1H, Ar), 6.92 (t, 1H, Ar), 6.89 (d, 1H, Ar), 6.74 (t, 1H, Ar), 6.63 (d, 1H, Ar), 5.92 (d, 2H, DMAP), 5.35 (s, 1H, HC(C(Me)NAr)2), 4.22 (sep, 1H, CHMe2), 3.98 (sep, 1H, CHMe2), 3.54 (sep, 1H, CHMe2), 3.34 (sep, 1H, CHMe2), 3.14 (sep, 1H, CHMe2), 2.99 (sep, 1H, CHMe2), 2.93 (s, 6H, DMAP CH3), 1.92 (s, 3H, HC(C(Me)NAr)2), 1.73 (s, 3H, HC(C(Me)NAr)2), 1.48 (d, 3H, CHMe2), 1.28 (d, 3H, CHMe2), 1.25 (d, 3H, CHMe2), 1.19 (d, 3H, CHMe2), 1.18 (d, 3H, CHMe2), 1.17 (d, 3H, CHMe2), 1.03 (d, 3H, CHMe2), 0.99 (d, 3H, CHMe2), 0.98 (d, 3H, CHMe2), 0.97 (d, 3H, CHMe2), 0.88 (d, 3H, CHMe2), 0.51 (d, 3H, CHMe2). 13C{1H} NMR (151 MHz, C6D5Br, 293 K): δ = 167.2 (HC(C(Me)NAr)2), 166.2 (HC(C(Me)NAr)2), 153.4 (DMAP), 153.3 (Ar), 152.8 (Ar), 151.5 (DMAP), 147.1 (Ar), 144.7 (Ar), 143.8 (Ar), 142.5 (Ar), 141.4 (Ar), 140.9 (Ar), 137.1 (Ar), 125.7 (Ar), 125.4 (Ar), 125.2 (Ar), 124.6 (Ar), 123.8 (Ar), 123.0 (Ar), 122.8 (Ar), 121.4 (Ar), 121.3 (Ar), 104.8 (DMAP), 99.3 (CH(C(Me)NAr)2), 38.3 (DMAP CH3), 29.8 (CHMe2), 28.6 (CHMe2), 27.7 (CHMe2), 27.6 (CHMe2), 27.5 (CHMe2), 26.9 (CHMe2), 25.9 (CH(C(Me)NAr)2), 25.6 (CHMe2), 25.4 (CH(C(Me)NAr)2), 25.4 (CHMe2), 25.3 (CHMe2), 25.2 (CHMe2), 25.0 (CHMe2), 24.9 (CHMe2), 24.8 (CHMe2), 24.2 (CHMe2), 24.1 (CHMe2), 23.5 (CHMe2), 22.5 (CHMe2), 15.6 (CHMe2). Anal. calcd (%) for NbON5C48H68: C, 69.97; H, 8.32; N, 8.50. Found: C, 70.35; H, 8.26; N, 8.26. Mp: 219 °C (dec).(BDI)NbH(O[SiH2Ph])(NAr) (17a)
Compound 16 (10.0 mg, 0.012 mmol) was suspended in C6D6 (0.4 mL) in a 4 mL vial. Phenylsilane (1.60 μL, 0.013 mmol) was added by microsyringe, resulting in immediate dissolution of the suspended solid and a slight lightening of the solution color. The resulting solution was transferred to a J. Young NMR tube. A 1H NMR spectrum indicated clean conversion to 17a and free DMAP. 1H NMR (400 MHz, C6D6, 293 K): δ = 11.20 (br s, 1H, Nb–H), 7.34 (d, 2H, SiH2Ph), 7.18–6.95 (m, 12H, Ar/SiH2Ph), 5.19 (s, 1H, HC(C(Me)NAr)2), 5.14 (d, 1H, SiH2Ph, 2J = 16 Hz), 4.99 (d, 1H, SiH2Ph, 2J = 16 Hz), 3.98 (sep, 2H, CHMe2), 3.59 (sep, 1H, CHMe2), 3.56 (sep, 1H, CHMe2), 3.17 (br sep, 1H, CHMe2), 3.14 (sep, 1H, CHMe2), 1.63 (s, 3H, HC(C(Me)NAr)2), 1.60 (s, 3H, HC(C(Me)NAr)2), 1.33 (d, 3H, CHMe2), 1.29–1.11 (m, 30H, CHMe2), 1.07 (d, 3H, CHMe2).(BDI)NbH(O[SiHPh2])(NAr) (17b)
Compound 16 (10.0 mg, 0.012 mmol) was suspended in C6D6 (0.4 mL) in a 4 mL vial. Diphenylsilane (2.50 μL, 0.013 mmol) was added by microsyringe, resulting in immediate dissolution of the suspended solid and a slight lightening of the solution color. The resulting solution was transferred to a J. Young NMR tube. A 1H NMR spectrum indicated clean conversion to 17b and free DMAP. 1H NMR (600 MHz, C6D6, 293 K): δ = 11.32 (br s, 1H, Nb–H), 7.45 (d, 2H, SiHPh2), 7.30 (d, 2H, SiHPh2), 7.21–6.95 (m, 15H, Ar/SiHPh2), 5.55 (s, 1H, SiHPh2), 5.20 (s, 1H, HC(C(Me)NAr)2), 3.96 (br s, 2H, CHMe2), 3.60 (br sep, 1H, CHMe2), 3.57 (br sep, 1H, CHMe2), 3.09 (br sep, 1H, CHMe2), 3.08 (br sep, 1H, CHMe2), 1.64 (s, 3H, HC(C(Me)NAr)2), 1.58 (s, 3H, HC(C(Me)NAr)2), 1.39–0.93 (m, 36H, CHMe2).(BDI)NbH(O[SiMe2Ph])(NAr) (17c)
Compound 16 (10.0 mg, 0.012 mmol) was suspended in C6D6 (0.4 mL) in a 4 mL vial. Dimethylphenylsilane (2.00 μL, 0.013 mmol) was added by microsyringe, resulting in dissolution of the suspended solid and a slight lightening of the solution color within 5 min. The resulting solution was transferred to a J. Young NMR tube. A 1H NMR spectrum indicated clean conversion to 17c and free DMAP. 1H NMR (600 MHz, C6D6, 293 K): δ = 11.02 (br s, 1H, Nb–H), 7.33 (d, 2H, SiMe2Ph), 7.21–6.97 (m, 12H, Ar/SiMe2Ph), 5.21 (s, 1H, HC(C(Me)NAr)2), 4.01 (br sep, 1H, CHMe2), 3.92 (br sep, 1H, CHMe2), 3.61 (sep, 1H, CHMe2), 3.55 (sep, 1H, CHMe2), 3.08 (br sep, 1H, CHMe2), 3.07 (br sep, 1H, CHMe2), 1.64 (s, 3H, HC(C(Me)NAr)2), 1.60 (s, 3H, HC(C(Me)NAr)2), 1.36–1.02 (m, 36H, CHMe2), 0.15 (s, 3H, SiMe2Ph), 0.12 (s, 3H, SiMe2Ph).(BDI)Nb(OCHNtBu)(O[SiMe2Ph])(NAr) (18)
Compound 12 (200 mg, 0.250 mmol) was dissolved in 4 mL benzene in a 20 mL vial to give a red solution. In a separate vial, dimethylphenylsilane (29.0 mg, 0.250 mmol) was dissolved in 2 mL benzene. The silane solution was added to the solution of 12. The solution was left at room temperature for 40 h, resulting in a slow color change from red to yellow. The volatile materials were removed under vacuum, leaving a yellow residue. The residue was extracted with hexane and the resulting solution was concentrated under vacuum and then stored at −40 °C overnight, yielding yellow crystals of 18. The crystalline material was isolated and residual solvent was removed under vacuum. Yield: 94.0 mg, 41%. 1H NMR (600 MHz, C6D6, 293 K): δ = 11.02 (br s, 1H, Nb–H), 7.88 (d, 2H, SiMe2Ph), 7.29 (t, 2H, SiHPh2), 7.24 (tt, 1H, Ar), 7.13 (t, 1H, Ar), 7.09 (dd, 1H, Ar), 7.07–7.00 (m, 5H, Ar), 6.98 (dd, 1H, Ar), 6.93 (t, 1H, SiMe2Ph), 6.33 (s, 1H, OCHNtBu), 5.33 (s, 1H, HC(C(Me)NAr)2), 4.16 (sep, 1H, CHMe2), 3.98 (sep, 1H, CHMe2), 3.65 (sep, 1H, CHMe2), 3.36 (sep, 1H, CHMe2), 2.93 (sep, 1H, CHMe2), 2.81 (sep, 1H, CHMe2), 1.60 (s, 6H, HC(C(Me)NAr)2), 1.33 (d, 3H, CHMe2), 1.31 (d, 3H, CHMe2), 1.27 (d, 3H, CHMe2), 1.26 (d, 3H, CHMe2), 1.23 (d, 3H, CHMe2), 1.14 (d, 3H, CHMe2), 1.15 (s, 9H, tBu), 1.13 (d, 3H, CHMe2), 1.08 (d, 3H, CHMe2), 1.03 (d, 3H, CHMe2), 0.99 (d, 3H, CHMe2), 0.98 (d, 3H, CHMe2), 0.87 (d, 3H, CHMe2), 0.34 (s, 3H, SiMe2Ph), 0.25 (s, 3H, SiMe2Ph). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 167.8 (HC(C(Me)NAr)2), 167.7 (HC(C(Me)NAr)2), 154.8 (OCHNtBu), 153.0 (Ar), 151.5 (Ar), 150.7 (Ar), 145.0 (Ar), 143.3 (Ar), 142.7 (Ar), 141.5 (Ar), 141.1 (Ar), 141.1 (Ar), 139.6 (Ar), 135.3 (SiMe2Ph), 129.1 (Ar), 127.6 (SiMe2Ph), 127.5 (Ar), 126.7 (Ar), 126.4 (Ar), 126.0 (Ar), 125.4 (Ar), 124.8 (Ar), 124.8 (SiMe2Ph), 123.9 (Ar), 123.2 (Ar), 122.3 (Ar), 102.8 (HC(C(Me)NAr)2), 52.8 (Cα, tBu), 31.0 (Cβ, tBu), 29.7 (CHMe2), 29.4 (CHMe2), 28.3 (CHMe2), 28.1 (CHMe2), 28.0 (CHMe2), 27.8 (CHMe2), 26.4 (HC(C(Me)NAr)2), 26.2 (HC(C(Me)NAr)2), 25.6 (CHMe2), 25.5 (CHMe2), 25.2 (CHMe2), 25.1 (CHMe2), 25.0 (CHMe2), 25.0 (CHMe2), 24.9 (CHMe2), 24.8 (CHMe2), 24.6 (CHMe2), 24.2 (CHMe2), 24.1 (CHMe2), 23.7 (CHMe2), 1.1 (SiMe2Ph), 0.6 (SiMe2Ph). Anal. calcd (%) for NbO2N4C54H79: C, 69.20; H, 8.50; N, 5.98. Found: C, 69.28; H, 8.30; N, 5.90. Mp: 205–210 °C (melting with dec).(BDI#)Nb(NAr)(O2CNH(tBu)) (19)
Compound 12 (10.0 mg, 0.012 mmol) was dissolved in C6D6 (0.4 mL) in a J. Young NMR tube. The solution was heated at 60 °C for 5 h. A 1H NMR spectrum indicated conversion to 19. 1H NMR (400 MHz, C6D6, 293 K): δ = 7.33 (dd, 1H, Ar), 7.27–7.03 (m, 5H, Ar), 6.91–6.80 (m, 3H, Ar), 5.46 (s, 1H, HC(C(Me)NAr)), 4.51 (s, 1H, N–H), 3.95 (sep, 1H, CHMe2), 3.88 (sep, 1H, CHMe2), 3.75 (sep, 1H, CHMe2), 3.74 (sep, 1H, CHMe2), 3.69 (s, 1H, CH2), 3.53 (s, 1H, CH2), 3.36 (sep, 1H, CHMe2), 3.33 (sep, 1H, CHMe2), 1.70 (s, 3H, HC(C(Me)NAr)), 1.64 (d, 3H, CHMe2), 1.53 (d, 3H, CHMe2), 1.47 (d, 3H, CHMe2), 1.44 (d, 3H, CHMe2), 1.35 (d, 3H, CHMe2), 1.30 (d, 3H, CHMe2), 1.16 (d, 3H, CHMe2), 1.01 (d, 6H, CHMe2), 0.98 (s, 9H, tBu), 0.87 (d, 6H, CHMe2), 0.77 (d, 3H, CHMe2).(BDI)Nb(NAr)(N(tBu)CO2B(C6F5)3) (20)
Compound 12 (200 mg, 0.250 mmol) was dissolved in 5 mL benzene in a 20 mL vial to give a red solution. In a separate vial, tris(perfluorophenyl)borane (130 mg, 0.250 mmol) was dissolved in 5 mL benzene. The borane solution was added to the solution of 12, resulting in an immediate color change from red to purple. The solution was left at room temperature for 5 min, and then the volatile materials were removed under vacuum, leaving a purple powder. The residue was extracted with diethyl ether and the resulting solution was concentrated under vacuum and then stored at −40 °C overnight, yielding purple crystals of 20. Yield: 150 mg, 45%. 1H NMR (600 MHz, C6D6, 293 K): δ = 7.33 (t, 1H, Ar), 7.11–7.06 (m, 2H, Ar), 6.97 (d, 1H, Ar), 6.91–6.81 (m, 5H, Ar), 5.59 (s, 1H, HC(C(Me)NAr)2), 4.09 (sep, 1H, CHMe2), 3.73 (sep, 1H, CHMe2), 2.91 (sep, 1H, CHMe2), 2.48 (br m, 2H, CHMe2), 2.34 (sep, 1H, CHMe2), 1.58 (d, 3H, CHMe2), 1.50 (s, 3H, HC(C(Me)NAr)2), 1.41 (s, 3H, HC(C(Me)NAr)2), 1.35 (d, 3H, CHMe2), 1.08 (d, 3H, CHMe2), 1.06–1.02 (m, 12H, CHMe2/tBu), 0.99 (d, 3H, CHMe2), 0.95 (d, 3H, CHMe2), 0.82 (d, 3H, CHMe2) 0.80 (d, 3H, CHMe2), 0.74 (d, 3H, CHMe2), 0.70 (d, 3H, CHMe2) 0.31 (d, 3H, CHMe2). 13C{1H} NMR (151 MHz, C6D6, 293 K): δ = 172.3 (NCO2), 168.2 (HC(C(Me)NAr)2), 167.4 (HC(C(Me)NAr)2), 156.0 (Ar), 151.6 (Ar), 149.4 (C6F5), 147.8 (C6F5), 145.6 (Ar), 144.4 (Ar), 144.0 (Ar), 142.2 (Ar), 142.1 (Ar), 140.9 (Ar), 138.1 (C6F5), 136.4 (C6F5), 131.4 (Ar), 129.0 (Ar), 128.5 (Ar), 127.9 (Ar), 125.9 (Ar), 124.4 (Ar), 123.9 (Ar), 123.5 (Ar), 122.7 (Ar), 107.4 (HC(C(Me)NAr)2), 57.8 (Cα, tBu), 32.5 (CHMe2), 30.5 (Cβ, NbtBu), 29.4 (CHMe2), 29.0 (HC(C(Me)NAr)2), 28.8 (CHMe2), 27.9 (CHMe2), 27.9 (CHMe2), 27.4 (CHMe2), 27.0 (CHMe2), 26.7 (CHMe2), 26.3 (CHMe2), 25.9 (CHMe2), 25.5 (CHMe2), 25.0 (CHMe2), 24.9 (HC(C(Me)NAr)2), 24.5 (CHMe2), 24.3 (CHMe2), 23.7 (CHMe2), 23.7 (CHMe2), 22.9 (CHMe2), 22.3 (CHMe2). Anal. calcd (%) for NbF15O2N4C64H67: C, 58.55; H, 5.14; N, 4.27. Found: C, 58.41; H, 5.18; N, 4.37. Mp: 185–195 °C (melting with dec).X-ray crystallographic studies
Single crystals of 1, 3a, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16, 18, and 20 were coated in Paratone-N oil, mounted on a Kaptan loop, transferred to a Bruker APEX CCD area detector,101 centered in the beam, and cooled by a nitrogen flow low-temperature apparatus. Preliminary orientation matrices and cell constants were determined by collection of 36 10s frames, followed by spot integration and least-squares refinement. An arbitrary hemisphere of data was collected, and the raw data were integrated using SAINT.102 An empirical absorption correction based on comparison of redundant and equivalent reflections was applied using SADABS.103 Structures were solved by direct methods with the aid of successive difference Fourier maps and were refined against all data using the SHELXTL 5.0 software package.104 Thermal parameters for all non-hydrogen atoms were refined anisotropically. The PLATON/SQUEEZE procedure105 was used to remove extraneous electron density due to highly disordered lattice solvent in the structures of compounds 3 and 7. ORTEP diagrams were created using the ORTEP-3 software package106 and Mercury.107 All structures were deposited to the Cambridge Crystallographic Data Centre (CCDC) with deposition numbers: CSD 2004310 (1), 2004311 (3a), 2004312 (4), 2004313 (5), 2004314 (6), 2004315 (7), 2004316 (8), 2004177 (9), 2004133 (10), 2004175 (11), 2004134 (12), 2004135 (13), 2004176 (15), 2004136 (16), 2004137 (18), and 2004138 (20).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSF (Grant No. CHE-1465188 and 1954612) and the AFOSR (Grant No. FA9550-11-1-0008) for financial support. J. I. F. acknowledges support from a NSF graduate research fellowship (Grant No. DGE 1752814). T. D. L thanks the U.S. DOE Integrated University Program for a graduate research fellowship. The CHEXRAY X-ray crystallographic facility is supported by the NIH (Grant No. S10-RR027172). We thank Dr M. A. Boreen, E. T. Ouellette, Prof. C. Camp, Dr J. A. Ziegler, Dr T. Saito, and Prof. T. L. Gianetti for helpful discussions.Notes and references
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, UK, 2nd edn, 1997 Search PubMed.
- W. A. Nugent and J. M. Mayer, Metal-Ligand Multiple Bonds, Wiley-VCH Verlag, 1988 Search PubMed.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS.
- W. A. Nugent and B. L. Haymore, Coord. Chem. Rev., 1980, 31, 123–175 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- K. Kawakita, B. F. Parker, Y. Kakiuchi, H. Tsurugi, K. Mashima, J. Arnold and I. A. Tonks, Coord. Chem. Rev., 2020, 407, 213118 CrossRef CAS.
- N. C. Tomson, J. Arnold and R. G. Bergman, Organometallics, 2010, 29, 2926–2942 CrossRef CAS.
- H. S. La Pierre, J. Arnold and F. D. Toste, Angew. Chem., Int. Ed., 2011, 50, 3900–3903 CrossRef CAS.
- A. H. Obenhuber, T. L. Gianetti, R. G. Bergman and J. Arnold, Chem. Commun., 2015, 51, 1278–1281 RSC.
- J. de With, A. D. Horton and A. G. Orpen, Organometallics, 1993, 12, 1493–1496 CrossRef CAS.
- T. R. Cundari, Organometallics, 1994, 13, 2987–2994 CrossRef CAS.
- G. Liang, T. K. Hollis and C. E. Webster, Organometallics, 2018, 37, 1671–1681 CrossRef CAS.
- T. R. Helgert, X. Zhang, H. K. Box, J. A. Denny, H. U. Valle, A. G. Oliver, G. Akurathi, C. E. Webster and T. K. Hollis, Organometallics, 2016, 35, 3452–3460 CrossRef CAS.
- K. Kawakita, E. P. Beaumier, Y. Kakiuchi, H. Tsurugi, I. A. Tonks and K. Mashima, J. Am. Chem. Soc., 2019, 141, 4194–4198 CrossRef CAS.
- T. D. Lohrey, E. A. Cortes, J. I. Fostvedt, A. K. Oanta, A. Jain, R. G. Bergman and J. Arnold, Inorg. Chem., 2020, 59, 11096–11107 CrossRef CAS.
- T. D. Lohrey, E. A. Cortes, R. G. Bergman and J. Arnold, Inorg. Chem., 2020, 59, 7216–7226 CrossRef CAS.
- C. C. Cummins, S. M. Baxter and P. T. Wolczanski, J. Am. Chem. Soc., 1988, 110, 8731–8733 CrossRef CAS.
- P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729–8731 CrossRef CAS.
- A. P. Duncan and R. G. Bergman, Chem. Rec., 2002, 2, 431–445 CrossRef CAS.
- N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS.
- B. M. Kriegel, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2016, 138, 52–55 CrossRef CAS.
- S. W. Krska, R. L. Zuckerman and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 11828–11829 CrossRef CAS.
- R. L. Zuckerman, S. W. Krska and R. G. Bergman, J. Am. Chem. Soc., 2000, 122, 751–761 CrossRef CAS.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 2669–2670 CrossRef CAS.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 974–985 CrossRef CAS.
- P. J. Walsh, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1992, 114, 1708–1719 CrossRef CAS.
- Y. Li, Y. Shi and A. L. Odom, J. Am. Chem. Soc., 2004, 126, 1794–1803 CrossRef CAS.
- A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 CrossRef CAS.
- A. M. Baranger, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 2753–2763 CrossRef CAS.
- L. L. Anderson, J. Arnold and R. G. Bergman, Org. Lett., 2004, 6, 2519–2522 CrossRef CAS.
- F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104–114 RSC.
- R. T. Ruck, R. L. Zuckerman, S. W. Krska and R. G. Bergman, Angew. Chem., Int. Ed., 2004, 43, 5372–5374 CrossRef CAS.
- F. Basuli, H. Aneetha, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2005, 127, 17992–17993 CrossRef CAS.
- Z. W. Davis-Gilbert, L. J. Yao and I. A. Tonks, J. Am. Chem. Soc., 2016, 138, 14570–14573 CrossRef CAS.
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS.
- E. P. Beaumier, M. E. McGreal, A. R. Pancoast, R. H. Wilson, J. T. Moore, B. J. Graziano, J. D. Goodpaster and I. A. Tonks, ACS Catal., 2019, 9, 11753–11762 CrossRef CAS.
- A. J. Pearce, R. P. Harkins, B. R. Reiner, A. C. Wotal, R. J. Dunscomb and I. A. Tonks, J. Am. Chem. Soc., 2020, 142, 4390–4399 CrossRef CAS.
- A. I. Nguyen, R. A. Zarkesh, D. C. Lacy, M. K. Thorson and A. F. Heyduk, Chem. Sci., 2011, 2, 166–189 RSC.
- J. Chu, E. Lu, Y. Chen and X. Leng, Organometallics, 2013, 32, 1137–1140 CrossRef CAS.
- J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894–10897 CrossRef CAS.
- J. L. Polse, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 13405–13414 CrossRef CAS.
- T. E. Hanna, I. Keresztes, E. Lobkovsky, W. H. Bernskoetter and P. J. Chirik, Organometallics, 2004, 23, 3448–3458 CrossRef CAS.
- R. E. Blake, D. M. Antonelli, L. M. Henling, W. P. Schaefer, K. I. Hardcastle and J. E. Bercaw, Organometallics, 1998, 17, 718–725 CrossRef CAS.
- H. E. Toomey, D. Pun, L. F. Veiros and P. J. Chirik, Organometallics, 2008, 27, 872–879 CrossRef CAS.
- W. Ren, E. Zhou, B. Fang, G. Zi, D. C. Fang and M. D. Walter, Chem. Sci., 2014, 5, 3165–3172 RSC.
- E. Zhou, W. Ren, G. Hou, G. Zi, D. C. Fang and M. D. Walter, Organometallics, 2015, 34, 3637–3647 CrossRef CAS.
- M. J. Carney, P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 8751–8753 CrossRef CAS.
- M. J. Carney, P. J. Walsh, F. J. Hollander and R. G. Bergman, Organometallics, 1992, 11, 761–777 CrossRef CAS.
- J. L. Polse, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 5393–5394 CrossRef CAS.
- M. J. Carney, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1990, 112, 6426–6428 CrossRef CAS.
- W. A. Howard, T. M. Trnka, M. Waters and G. Parkin, J. Organomet. Chem., 1997, 528, 95–121 CrossRef CAS.
- G. D. Kortman, M. J. Orr and K. L. Hull, Organometallics, 2015, 34, 1013–1016 CrossRef CAS.
- T. T. Nguyen, G. D. Kortman and K. L. Hull, Organometallics, 2016, 35, 1713–1725 CrossRef CAS.
- W. A. Howard, M. Waters and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4917–4918 CrossRef CAS.
- Z. K. Sweeney, J. L. Polse, R. G. Bergman and R. A. Andersen, Organometallics, 1999, 18, 5502–5510 CrossRef CAS.
- T. E. Hanna, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2007, 46, 2359–2361 CrossRef CAS.
- S. Hohloch, B. M. Kriegel, R. G. Bergman and J. Arnold, Dalton Trans., 2016, 45, 15725–15745 RSC.
- N. C. Tomson, A. Yan, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2008, 130, 11262–11263 CrossRef CAS.
- N. C. Tomson, J. Arnold and R. G. Bergman, Organometallics, 2010, 29, 5010–5025 CrossRef CAS.
- N. C. Tomson, J. Arnold and R. G. Bergman, Dalton Trans., 2011, 40, 7718–7729 RSC.
- T. L. Gianetti, N. C. Tomson, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2011, 133, 14904–14907 CrossRef CAS.
- T. L. Gianetti, G. Nocton, S. G. Minasian, N. C. Tomson, A. L. D. Kilcoyne, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2013, 135, 3224–3236 CrossRef CAS.
- T. L. Gianetti, R. G. Bergman and J. Arnold, Chem. Sci., 2014, 5, 2517–2524 RSC.
- T. L. Gianetti, G. Nocton, S. G. Minasian, N. Kaltsoyannis, A. L. D. Kilcoyne, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, R. G. Bergman and J. Arnold, Chem. Sci., 2015, 6, 993–1003 RSC.
- T. L. Gianetti, H. S. La Pierre and J. Arnold, Eur. J. Inorg. Chem., 2013, 3771–3783 CrossRef CAS.
- T. I. Gountchev and T. D. Tilley, J. Am. Chem. Soc., 1997, 119, 12831–12841 CrossRef CAS.
- A. H. Obenhuber, T. L. Gianetti, X. Berrebi, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2014, 136, 2994–2997 CrossRef CAS.
- While [2 + 2] cycloadditions are formally symmetry-forbidden, the Woodward–Hoffmann rule for the general [2 + 2] cycloaddition process does not necessarily apply to metal complexes. For an in-depth theoretical discussion of [2 + 2] cycloadditions across early transition metal-imido bonds, see N. Ochi, Y. Nakao, H. Sato and S. Sakaki, J. Phys. Chem., 2010, 114, 659–665 CrossRef CAS.
- B. J. Burger, B. D. Santarsiero, M. S. Trimmer and J. E. Bercaw, J. Am. Chem. Soc., 1988, 110, 3134–3146 CrossRef CAS.
- J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2003, 125, 4020–4021 CrossRef CAS.
- K. Y. Dorogov, A. V. Churakov, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Eur. J. Inorg. Chem., 2004, 771–775 CrossRef CAS.
- F. Basuli, L. A. Watson, J. C. Hu and D. J. Mindiola, Dalton Trans., 2003, 4228–4229 RSC.
- N. Carrera, N. Savjani, J. Simpson, D. L. Hughes and M. Bochmann, Dalton Trans., 2011, 40, 1016–1019 RSC.
- J. A. Ziegler, R. G. Bergman and J. Arnold, Dalton Trans., 2016, 45, 12661–12668 RSC.
- A. J. Blake, J. M. Mcinnes, P. Mountford, G. I. Nikonov, D. Swallow and D. J. Watkin, Dalton Trans., 1999, 1, 379–391 RSC.
- A. E. Guiducci, A. R. Cowley, M. E. G. Skinner and P. Mountford, J. Chem. Soc. Dalton Trans., 2001, 1392–1394 RSC.
- A. E. Guiducci, C. L. Boyd and P. Mountford, Organometallics, 2006, 25, 1167–1187 CrossRef CAS.
- C. L. Boyd, E. Clot, A. E. Guiducci and P. Mountford, Organometallics, 2005, 24, 2347–2367 CrossRef CAS.
- P. J. Tiong, A. Nova, L. R. Groom, A. D. Schwarz, J. D. Selby, A. D. Schofield, E. Clot and P. Mountford, Organometallics, 2011, 30, 1182–1201 CrossRef CAS.
- C. L. Boyd, T. Toupance, B. R. Tyrrell, B. D. Ward, C. R. Wilson, A. R. Cowley and P. Mountford, Organometallics, 2005, 24, 309–330 CrossRef CAS.
- S. H. Hsu, J. C. Chang, C. L. Lai, C. H. Hu, H. M. Lee, G. H. Lee, S. M. Peng and J. H. Huang, Inorg. Chem., 2004, 43, 6786–6792 CrossRef CAS.
- M. G. B. Drew, D. A. Rice and D. M. Williams, J. Chem. Soc., Dalton Trans., 1985, 417–421 RSC.
- Y. V. Skripkin, I. L. Eremenko, A. A. Pasynskii, Y. T. Struchkov and V. E. Shklover, J. Organomet. Chem., 1984, 267, 285–292 CrossRef CAS.
- J. Chu, E. Lu, Z. Liu, Y. Chen, X. Leng and H. Song, Angew. Chem., Int. Ed., 2011, 50, 7677–7680 CrossRef CAS.
- D. J. Mindiola, R. Waterman, V. M. Iluc, T. R. Cundari and G. L. Hillhouse, Inorg. Chem., 2014, 53, 13227–13238 CrossRef CAS.
- V. M. Iluc, A. J. M. Miller, J. S. Anderson, M. J. Monreal, M. P. Mehn and G. L. Hillhouse, J. Am. Chem. Soc., 2011, 133, 13055–13063 CrossRef CAS.
- M. Baltrun, F. A. Watt, R. Schoch and S. Hohloch, Organometallics, 2019, 38, 3719–3729 CrossRef CAS.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Inorg. Chim. Acta, 2011, 369, 103–119 CrossRef CAS.
- G. Du and M. Abu-Omar, Curr. Org. Chem., 2008, 12, 1185–1198 CrossRef CAS.
- D. E. Wigley, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1994, pp. 239–482 Search PubMed.
- K. Muñiz, Chem. Soc. Rev., 2004, 33, 166–174 RSC.
- K. Muñiz, A. Iesato and M. Nieger, Chem.–Eur J., 2003, 9, 5581–5596 CrossRef.
- N. E. Travia, Z. Xu, J. M. Keith, E. A. Ison, P. E. Fanwick, M. B. Hall and M. M. Abu-Omar, Inorg. Chem., 2011, 50, 10505–10514 CrossRef CAS.
- W. M. Vaughan, K. A. Abboud and J. M. Boncella, J. Organomet. Chem., 1995, 485, 37–43 CrossRef CAS.
- G. Parkin, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1998, pp. 1–165 Search PubMed.
- R. Cahill, R. C. Cookson and T. A. Crabb, Tetrahedron, 1969, 25, 4711–4735 CrossRef CAS.
- U. J. Kilgore, F. Basuli, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2006, 45, 487–489 CrossRef CAS.
- A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS.
- C. Camp, L. N. Grant, R. G. Bergman and J. Arnold, Chem. Commun., 2016, 52, 5538–5541 RSC.
- A. M. Fuller, D. L. Hughes, G. A. Jones and S. J. Lancaster, Dalton Trans., 2012, 41, 5599–5609 RSC.
- SMART, Area-Detector Software Package, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2001-2003 Search PubMed.
- SAINT, SAX Area-Detector Integration Program, V6.40, Bruker Analytical X-ray Systems Inc.: Madison, WI, 2003 Search PubMed.
- SADABS, Bruker-Nonius Area Detector Scaling and Absorption v. 2.05, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. A, 2008, 64, 112 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2004310 (1), 2004311 (3a), 2004312 (4), 2004313 (5), 2004314 (6), 2004315 (7), 2004316 (8), 2004177 (9), 2004133 (10), 2004175 (11), 2004134 (12), 2004135 (13), 2004176 (15), 2004136 (16), 2004137 (18), and 2004138 (20). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03489d |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |