
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for an enolate mechanism in the asymmetric Michael reaction of α,β-unsaturated aldehydes and ketones via a hybrid system of two secondary amine catalysts†
Nariyoshi
Umekubo
a,
Takahiro
Terunuma
a,
Eunsang
Kwon
b and
Yujiro
Hayashi
 *a
*a
aDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aramaki Aza Aoba, Aoba-ku, Sendai 980-8578, Japan. E-mail: yujiro.hayashi.b7@tohoku.ac.jp
bResearch and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan
First published on 21st September 2020
Abstract
The key nucleophile was found to be neither an enamine nor an enol, but an enolate in the direct Michael reaction of α,β-unsaturated aldehydes and non-activated ketones catalyzed by two amine catalysts namely diphenylprolinol silyl ether and pyrrolidine. This is a rare example of an enolate from a ketone serving as a key intermediate in the asymmetric organocatalytic reaction involving secondary amine catalysts because the ketone enolates are generally generated using a strong base, and the enamine is a common nucleophile in this type of reaction.
Introduction
Enolates are versatile and important synthetic intermediates for the formation of α-substituted carbonyl compounds, which are widely used in the synthesis of organic molecules. In order to retard nucleophilic attack on the carbonyl group, and to match the weak acidity of the proton, sterically hindered strong amide bases such as lithium diisopropylamide (LDA) and lithium hexamethyldisilyl-amide (LHMDS) are generally employed for the generation of enolates from ketones.1,2 In the field of organocatalysis3 organic super bases such as phosphazene4 and proazaphosphatrane5 have been developed, which have similar or much stronger basicity than LDA and LHMDS.Contrary to organic strong bases, weak secondary and primary amines can be successfully employed for the α-functionalization of non-activated ketones in which the key intermediate is not an enolate but an enamine. Stork was a pioneer in this field of enamine chemistry where an equimolar amount of the amine was employed.6 However, in recent asymmetric reactions, catalytically generated enamines are essentially the key intermediates. In this context, chiral secondary and primary amines act as catalysts for the α-functionalization of ketones in bond forming reactions such as the aldol reaction,3,7 Mannich reaction,3,8 Michael reaction,3 α-aminoxylation3 and α-aminations.3 In the Mannich reaction of acetone catalysed by a primary amine-thiourea organocatalyst, an enol mechanism was proposed,9 while an enamine mechanism is proved by calculation and 13C kinetic isotope effects in the Michael reaction of acetone catalysed by a similar catalyst.10 In the Michael reaction of propanal and methyl vinyl ketone catalysed by pyrrolidine, both enamine and enol mechanisms were computationally investigated, and an enamine is concluded to be a key nucleophile.11 The discrimination of the reaction mechanism of enamine and enol is difficult, and the asymmetric reaction involving an enol is rare. Moreover, the reaction is very rare in which an enolate (not enol) is a key intermediate in the secondary amine mediated asymmetric catalytic reactions of non-activated ketones with excellent enantioselectivity. Although there is one report involving an enolate as an intermediate, as far as we are aware, the enantioselectvitiy is not sufficient.12 In this study, we will demonstrate that the key nucleophile in the Michael reaction of cyclohexanone and α,β-unsaturated aldehyde catalyzed by a secondary amine was neither an enamine nor an enol but a rather unexpected enolate.
Results and discussion
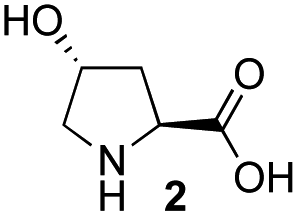
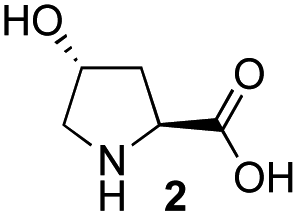
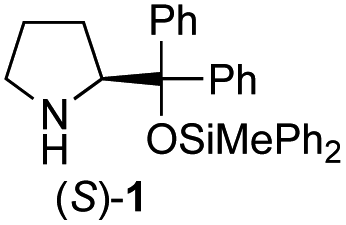
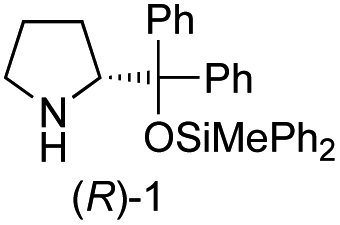
Recently, we reported the first direct asymmetric Michael reaction of non-activated ketones and α,β-unsaturated aldehydes catalyzed by a combination of two organocatalysts namely diphenylprolinol silyl ether 113 and pyrrolidine or 4-hydroxyproline (2) (eqn (1), Table 1, entries 1, 2 and 3).14 Although two similar pyrrolidine-type catalysts were involved in the reaction, an iminium ion generated from the α,β-unsaturated aldehyde was the reactive Michael acceptor based on Mayr's electrophilicity principle.15 This implies that the enantio face-selectivity of the α,β-unsaturated aldehyde was controlled by diphenylprolinol silyl ether. We hypothesized that the nucleophile would be an enamine generated from the reaction of the ketone and either pyrrolidine or 4-hydroxyproline. It has been well established that the enamine generated from the ketone and proline is a reactive nucleophile.3,16 We reasoned that if the second amine can control the enantio-face selectivity of the ketone, both syn and anti-isomers can be selectively synthesized.17 This hypothesis led us to study the effect of the second chiral amine catalyst in more detail with the intention of enhancing the selective control of relative stereochemistry.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst A | Catalyst B | Time [h] | drb | Yieldc [%] | eed [%] |
a Unless otherwise shown, the reaction was performed by employing cinnamaldehyde (0.5 mmol), cyclohexanone (1.5 mmol), catalyst A (0.075 mmol), catalyst B (0.0375 mmol), p-nitrophenol (0.15 mmol), and water (1.5 mmol), in EtOH (0.4 mL) and toluene (0.1 mL) at room temperature. After the reaction, Wittig reagent (0.75 mmol) was added. See the ESI for details.
b dr ratio (syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti) was determined by 1H-NMR.
c Isolated yield of the diastereomer mixture.
d Determined by HPLC analysis on a chiral column material. :anti) was determined by 1H-NMR.
c Isolated yield of the diastereomer mixture.
d Determined by HPLC analysis on a chiral column material.
|
||||||
| 1 |
|
|
24 | 5:1 |
74 | 91 |
| 2 |
|
|
40 | 15:1 |
74 | 96 |
| 3 |

|
|
40 | 10:1 |
70 | −95 |
| 4 |
|
|
30 | 5:1 |
68 | 93 |
| 5 |
|
|
30 | 9:1 |
70 | −94 |
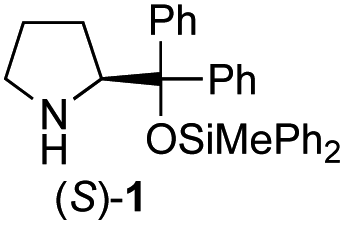
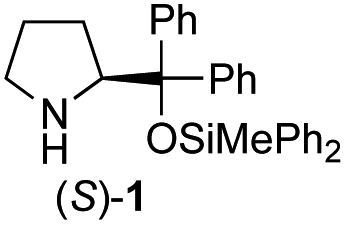
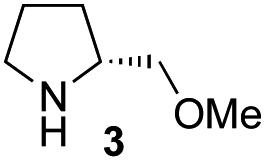
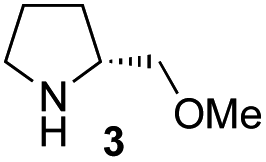
In our previous paper, we reported the results of (S)-diphenylprolinol silyl ether (S)-1 with (2S,4R)-4-hydroxyproline 2 and (R)-1 with (2S,4R)-2 in the Michael reaction of cinnamaldehyde and cyclohexanone (Table 1, entries 2 and 3).14 Although the enantio face-selectivity of an enamine originating from (2S,4R)-4-hydroxyproline 2 is not known, that of an (R)-2-(methoxymethyl)pyrrolidine 3 has been well investigated.18 Thus, the effect of this amine 3 with a combination of (S)-1 and (R)-1 was examined (entries 4 and 5). Although the diastereoselectivity was slightly different according to the relative fractions of the two catalysts, the syn-isomer was predominantly obtained in all cases. As for the enantioselectivity, the absolute configuration was controlled by the chirality of the diphenylprolinol silyl ether 1 (ref. 19) regardless of the chirality of the second amine catalyst 3. These results cast a doubt about the involvement of any enamine as an intermediate in the reaction.
The possible nucleophiles would be the enamine 4, the enol 5 and the enolate 6 (Fig. 1). Hence, we investigated the reactivity of these species. The reactivity of the enamine 7,18 which was generated from (R)-2-(methoxymethyl)pyrrolidine 3, was examined (Scheme 1). The equimolar reaction of enamine 7 and iminium ion (S)-8 (ref. 20) proceeds in the presence of 2,6-lutidine and MS4A to afford the bicyclic compound 11 (ref. 21) in 57% yield. 11 was formed by the Michael reaction, followed by an exchange of the enamine and iminium ion and cyclization.22 The stereochemistry at the α-position of the obtained cyclohexanone was R whereas the observed stereochemistry from the catalytic Michael reaction was the opposite S (Table 1, entry 4).23 This result implies that the enamine 7 was not an intermediate in the catalytic reaction. Moreover, apart from the secondary amine, tertiary amines such as i-Pr2NEt can successfully act as a co-catalyst to afford the Michael product with excellent enantioselectivity (71%, syn:anti = 5:1, 97% ee). This result was also a piece of evidence to support the claim that the enamine was not a key nucleophile in the reaction.
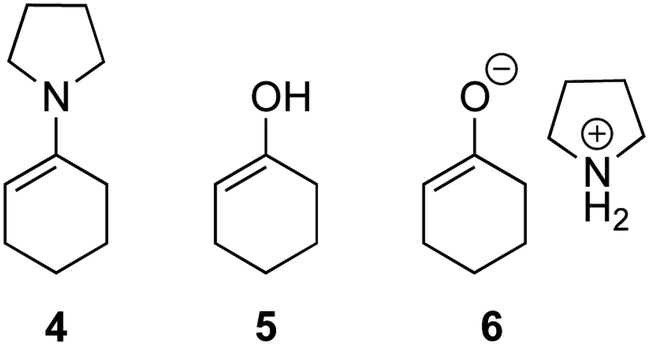 | ||
| Fig. 1 Enamine, enol and enolate. | ||
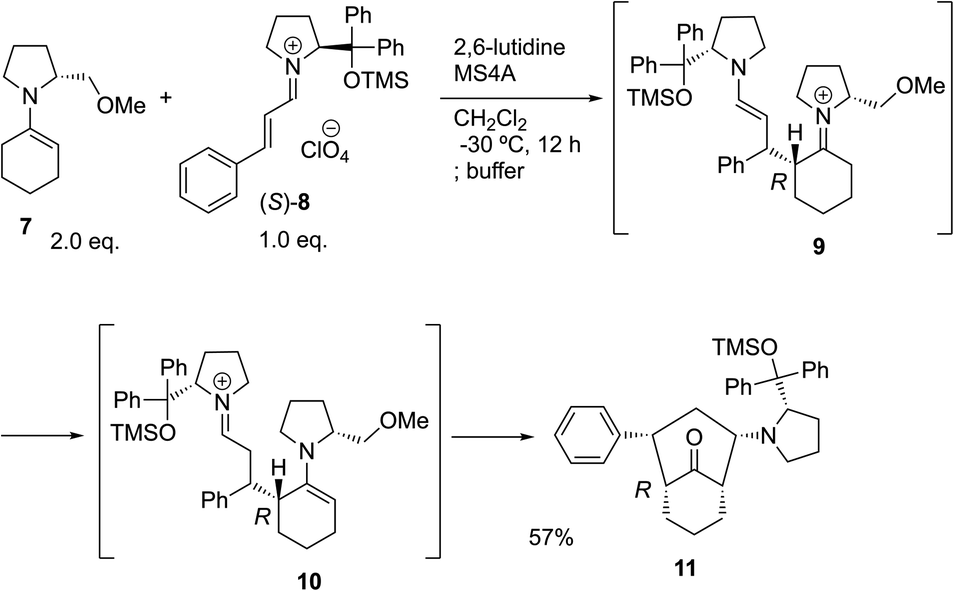 | ||
| Scheme 1 The reaction of enamine 7 and iminium ion (S)-8. | ||
The equimolar reactions of the iminium ion with 1-methoxycyclohex-1-ene and 1-(trimethylsiloxy)cyclohex-1-ene, and a combination of 1-(trimethylsiloxy)cyclohex-1-ene and tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) were investigated. The two former nucleophiles would possess a similar reactivity to 1-hydroxycyclohex-1-ene 5, but they did not react at all even after a longer reaction time (eqn (2)). In the reaction of a combination of silyl enol ether and TASF, which is known to generate an enolate,24 the Michael product was obtained at −30 °C after 1 h with a similar diastereo- and enantio-selectivity as observed in the catalytic reaction (eqn (3)). This result indicates that an enolate was the likely intermediate.
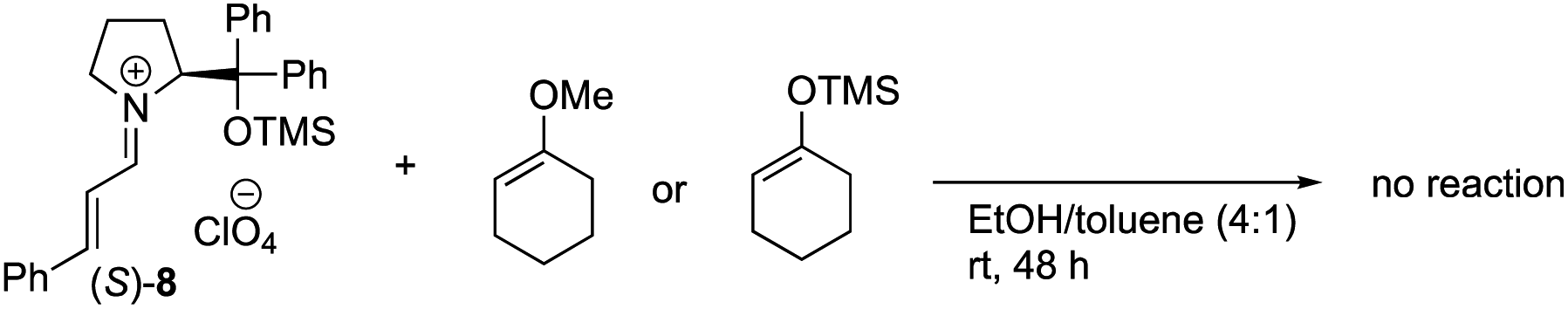 | (2) |
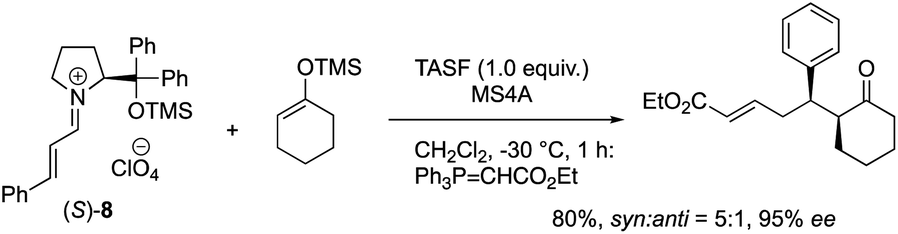 | (3) |
If the enolate was the actual nucleophile, a question arose about how the enolate would be generated from the ketone and pyrrolidine or i-Pr2NEt. The direct deprotonation of the ketone by pyrrolidine would be very difficult considering their relative pKa values. That is to say that the pKa value of an α-proton on cyclohexanone in DMSO is 26.4 (ref. 25) while that of an ammonium ion of Et3N is only 9.00.26 This is why a strong base such as LDA is usually employed. But the enolate could be generated considering an equilibrium between the keto and enol forms, and that the O–H proton of the enol form is rather acidic, although the content of enol is very low.27
Next, we investigated the generation speed of the enolate. The combined generation speed of both the enamine, the enol and the enolate can be monitored by the H/D exchange of the α-proton of the carbonyl group in the reaction with D2O (eqn (4)). The generation speed of the enamine can also be monitored by the 16O/18O exchange in the reaction with H218O (eqn (5)).28 Moreover, as a tertiary amine can also acts as a co-catalyst in which the enamine would not be involved, the generation of only the enol and the enolate can be monitored by the H/D exchange reaction with D2O in the case of the tertiary amine (eqn (4)).
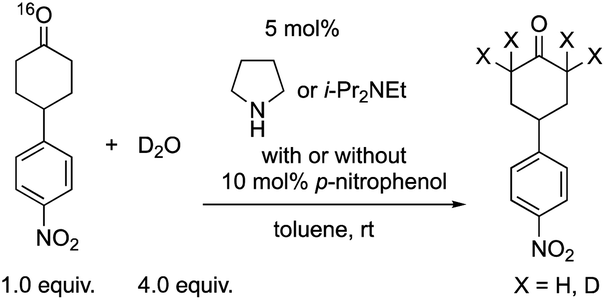 | (4) |
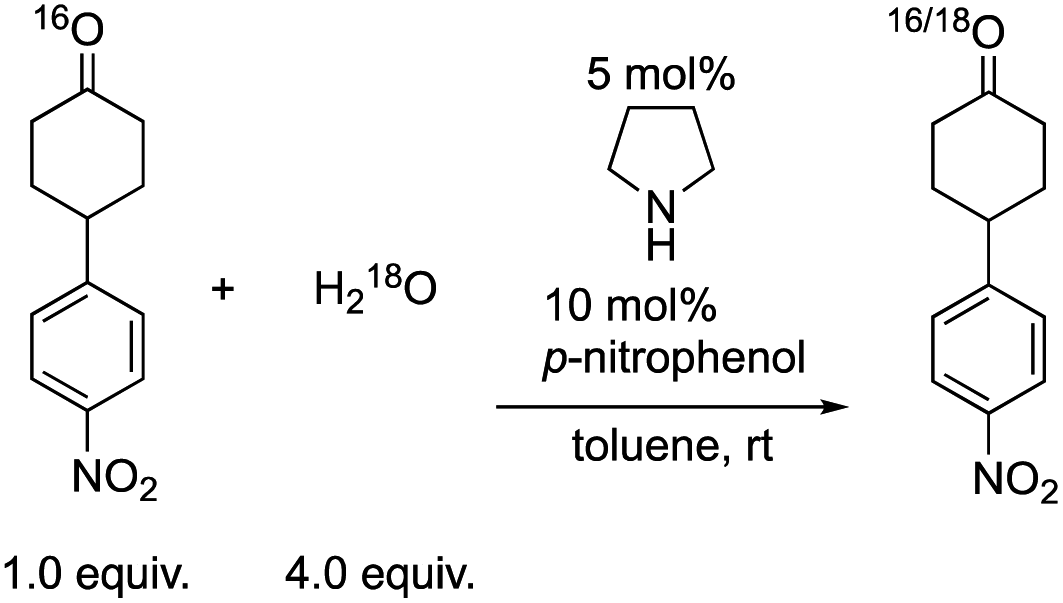 | (5) |
We examined the H/D exchange experiments in the presence of (1) pyrrolidine + p-nitrophenol (red), (2) p-nitrophenol (blue), (3) i-Pr2NEt + p-nitrophenol (green), and (4) i-Pr2NEt (yellow) (Fig. 2). A reaction using H218O was conducted using pyrrolidine and p-nitrophenol (purple) (Fig. 2). Fig. 2 indicates that the generation speed of the enol and the enolate under the reaction conditions is rather fast compared to the Michael reaction. This indicates that the rapid kinetic of deprotonation is fast, although the concentration of the enol and enolate is quite low. The speed of generation of the enolate was faster in the case of i-Pr2NEt and p-nitrophenol compared to pyrrolidine and p-nitrophenol. Although each acid and base are known to accelerate the keto/enol equilibrium,29p-nitrophenol did not promote the H/D exchange under the reaction conditions. Moreover, the tertiary amine did not promote the exchange either. However, it should be noted that a combination of both acid and base facilitates the generation of the enolate. Their cooperative role would be explained as follows (Scheme 2):30p-nitrophenol and i-Pr2NEt partially form an ammonium ion, and all these species such as acids, bases and ammonium ions are present in a mixture under equilibrium based on their pKa values.26,31 A protonation would occur at the carbonyl oxygen, which would increase the acidity of an α-proton of a carbonyl of cyclohexanone. Then, this α-proton of a carbonyl can be deprotonated by i-Pr2NEt. From the generated vinyl alcohol, deprotonation of the O–H proton32 would proceed very fast to afford the enolate.33 If so, the combination of acidity of the acid and basicity of the base should be important. In fact, when we used phenol or p-methoxyphenol34 instead of p-nitrophenol, the reaction became slow (see the ESI†). Moreover, the reaction did not proceed at all in the presence of CF3CO2H35 with a combination of i-Pr2NEt (see the ESI†).
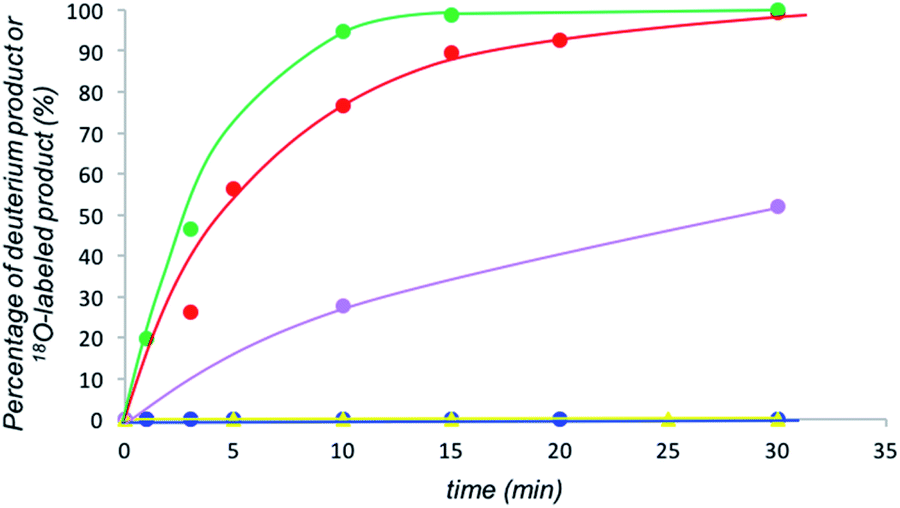 | ||
| Fig. 2 Generation of deuterated substrates in eqn (4), green: i-Pr2NEt and p-nitrophenol, red: pyrrolidine and p-nitrophenol, blue: p-nitrophenol, yellow: i-Pr2NEt, and generation of 18O labelled substrate in eqn (5), purple. | ||
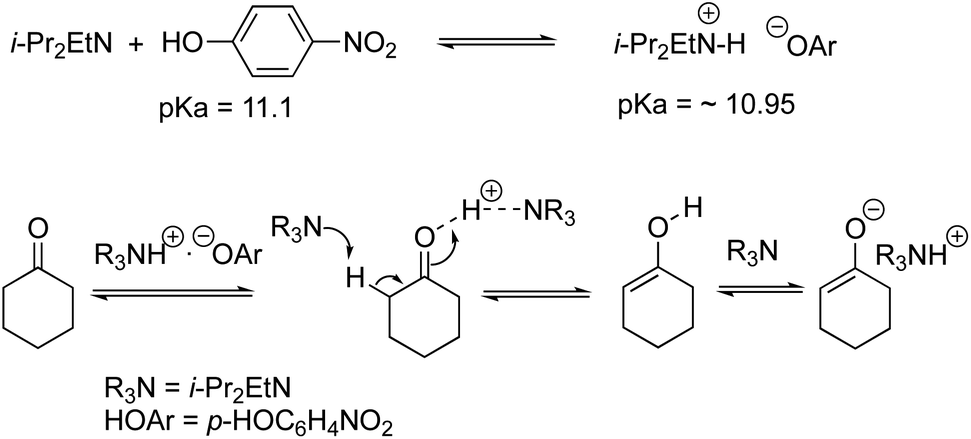 | ||
| Scheme 2 Generation of the enolate. | ||
One of the roles of the acid is to facilitate the generation of iminium ions from the α,β-unsaturated aldehyde and diphenylprolinol silyl ether, but this result indicates that another role of the acid is to accelerate the generation of the enolate in combination with the amine.
Based on the above investigations, the reaction was considered to proceed as follows (Fig. 3): diphenylprolinol silyl ether reacts with cinnamaldehyde to generate an iminium salt. On the other hand, cyclohexanone is in equilibrium with the enol form in the presence of p-nitrophenol and pyrrolidine, and the enol tautomer would then react with another molecule of pyrrolidine to afford the ammonium enolate. Ion exchange occurs between the iminium salt and ammonium enolate, followed by a coupling reaction to provide the enamine, which is hydrolyzed to provide the Michael product with the regeneration of the catalyst. Thus, the role of the second amine was to accelerate the equilibrium of keto and enol with a combination of p-nitrophenol, and to also deprotonate the O–H proton in the enol tautomer of the cyclohexanone.
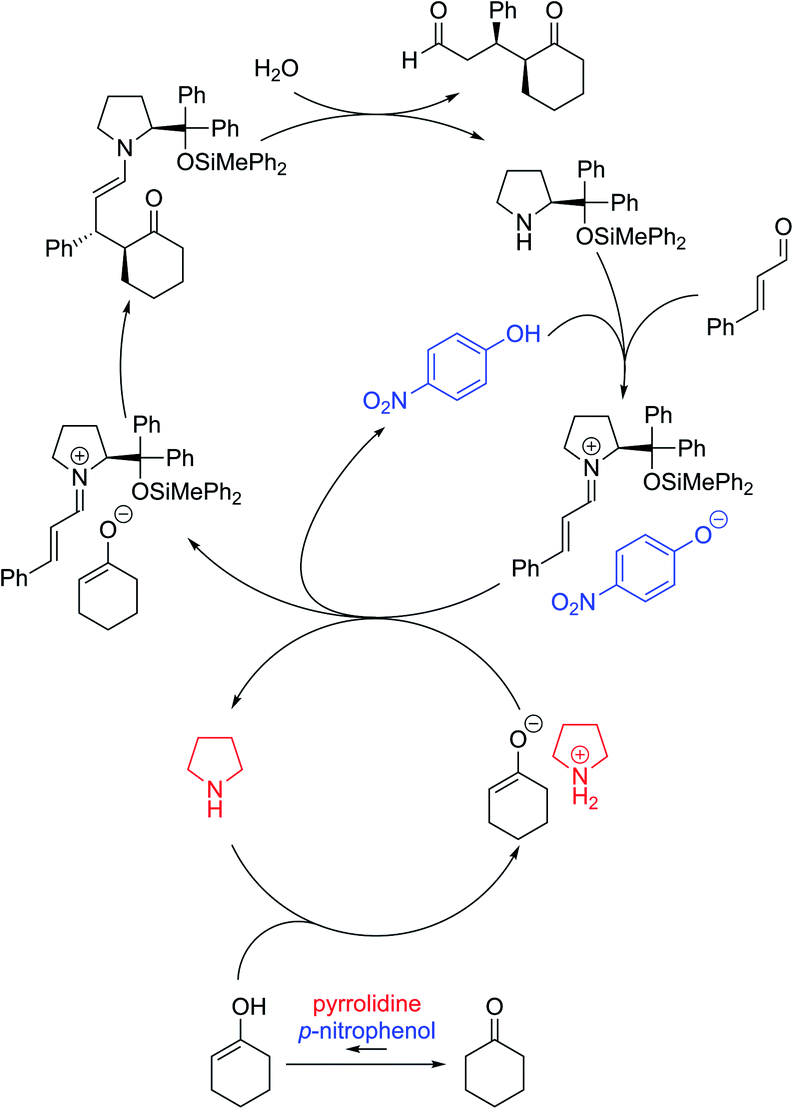 | ||
| Fig. 3 The catalytic cycle of the reaction. | ||
Conclusions
In summary, we have identified the actual nucleophile in the direct Michael reaction of α,β-unsaturated aldehydes and non-activated ketones catalyzed by two amine catalysts. The generation speed of the enamine, enol, and enolate was examined along with the reactivity of these species using both catalytic and equimolar reactions of the isolated iminium ions (S)-8 and (R)-8. The reaction was investigated using chiral (R)-2-(methoxymethyl)pyrrolidine 3 and its corresponding enamine from cyclohexanone with the chiral iminium ions (S)-8 and (R)-8. We also investigated the reactivity of the enamine, the enol and the enolate ion. Based on these experiments, we have concluded that the key nucleophile in the direct Michael reaction was neither an enamine nor an enol, but an enolate. Although the enolate of cyclohexanone is usually generated with a strong base, a secondary amine can generate the enolate by the deprotonation of the O–H proton in the enol form. Even though the concentration of the enol form is very low, there is a rapid keto–enol conversion in the joint presence of an acid and a base compared with the Michael reaction. This is a rare asymmetric catalytic reaction using a secondary amine catalyst, in which the key nucleophile is not the enamine but the enolate of a non-activated ketone.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Tohru Fukuyama for the discussion about the generation of enolates from ketones and secondary amines. We thank reviewers for the useful comments. We thank Mr Amaechi Shedrack Odoh in my group for the English corrections. This work was supported by JSPS KAKENHI Grant Number JP20H04801 in Hybrid Catalysis for Enabling Molecular Synthesis on Demand, and JP19H05630.Notes and references
- (a) H. B. Mekelburger and C. S. Wilcox, Comprehensive Organic Synthesis, Elsevier, Amsterdam, 2nd edn, 2014, vol. 2, pp. 243–272 Search PubMed; (b) B. M. Stoltz, N. B. Bennett, D. C. Duquette, A. F. G. Goldberg, Y. Liu, M. B. Loewinger and C. M. Reeves, Comprehensive Organic Synthesis, Elsevier, Amsterdam, 2nd edn, 2014, vol. 3, pp. 1–55 Search PubMed; (c) H. O. House, Modern Synthetic Reactions, 2nd edn, Benjamin, Philippines, 1972, p. 492 Search PubMed.
- While the pKa of cyclohexanone is 26.4 (in DMSO), those of isopropylamine and TMS2NH are 36 and 30 (in THF), respectively. Evans' pKa Table can be found in http://evans.rc.fas.harvard.edu/pdf/evans_pKa_table.pdf.
- For representative reviews on organocatalysis, see; (a) Asymmetric Organocatalysis 1: Lewis Base and Acid Catalysts, ed. B. List, Thieme, Stuttgart, 2012 Search PubMed; (b) Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2013 Search PubMed.
- R. Schwesinger and H. Schlemper, Angew. Chem., Int. Ed. Engl., 1987, 26, 1167 Search PubMed.
- (a) J. G. Verkade and P. B. Kisanga, Tetrahedron, 2003, 59, 7819 Search PubMed; (b) J. G. Verkade and P. B. Kisanga, Aldrichchimica Acta, 2004, 37, 3 Search PubMed.
- (a) M. B. Smith, in March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, Hoboken, 7th edn, 2013 Search PubMed; (b) G. Stork and S. R. Dowd, J. Am. Chem. Soc., 1963, 85, 2178 Search PubMed.
- (a) Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, vol. 1–2, 2004 Search PubMed; (b) N. Mase and Y. Hayashi, Comprehensive Organic Synthesis, Elsevier, Amsterdam, 2nd edn, 2014, vol. 2, pp. 273–339 Search PubMed.
- Y. Hayashi, J. Synth. Org. Chem., Jpn., 2014, 72, 1228 Search PubMed.
- D. A. Yalalov, S. B. Tsogoeva, T. E. Shubina, I. M. Martynova and T. Clark, Angew. Chem., Int. Ed., 2008, 47, 6624 Search PubMed.
- V. A. Roytman, R. W. Karugu, Y. Hong, J. S. Hirschi and M. J. Vetticatt, Chem.–Eur. J., 2018, 24, 8098 Search PubMed.
- M. P. Patil and R. B. Sunoj, Chem.–Asian J., 2009, 4, 714 Search PubMed.
- J. Muzart, Tetrahedron: Asymmetry, 2014, 25, 697 Search PubMed.
- (a) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 Search PubMed; (b) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 Search PubMed. For reviews, see; (c) C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876 Search PubMed; (d) A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922 Search PubMed; (e) L. W. Xu, L. Li and Z. H. Shi, Adv. Synth. Catal., 2010, 352, 243 Search PubMed; (f) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 Search PubMed; (g) H. Gotoh and Y. Hayashi, Sustainable Catalysis, Wiley, Hoboken, 2013, pp. 287–316 Search PubMed; (h) B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov and K. A. Jørgensen, Angew. Chem. Int. Ed., 2015, 54, 13860 Search PubMed; (i) G. J. Reyes-Rodriguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221 Search PubMed.
- Y. Hayashi and N. Umekubo, Angew. Chem., Int. Ed., 2018, 57, 1958 Search PubMed.
- S. Lakhdar, T. Tokuyasu and H. Mayr, Angew. Chem., Int. Ed., 2008, 47, 8723 Search PubMed.
- (a) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615 Search PubMed; (b) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496 Search PubMed; (c) B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395 Search PubMed.
- For the reactions to control relative and absolute configurations via two catalysts, see; (a) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 Search PubMed; (b) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020 Search PubMed; (c) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093 Search PubMed; (d) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 Search PubMed; (e) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029 Search PubMed; (f) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 Search PubMed.
- (a) S. J. Blarer, W. B. Schweizer and D. Seebach, Helv. Chim. Acta, 1982, 65, 1637 Search PubMed; (b) S. J. Blarer and D. Seebach, Chem. Ber., 1983, 116, 2250 Search PubMed; (c) D. Seebach, M. Missbach, G. Calderari and M. Eberle, J. Am. Chem. Soc., 1990, 112, 7625 Search PubMed; (d) E. Butkus and A. Stončius, Synlett, 1999, 234 Search PubMed; (e) T. Husch, D. Seebach, A. K. Beck and M. Reiher, Helv. Chim. Acta, 2017, 100, e1700182 Search PubMed.
- Y. Hayashi, D. Okamura, T. Yamazaki, Y. Ameda, H. Gotoh, S. Tsuzuki, T. Uchimaru and D. Seebach, Chem.–Eur. J., 2014, 20, 17077 Search PubMed.
- H. Gotoh, T. Uchimaru and Y. Hayashi, Chem.–Eur. J., 2015, 21, 12337 Search PubMed.
- The structure of 11 was determined by converting to the crystalline substrate and X-ray crystallographic analysis was conducted, see the ESI† for details. This reaction was conducted at −30 °C. The reaction also proceeded at rt, but the yield was lower because of the side reactions. On the other hand, a catalytic reaction (Table 1, entry 4) proceeded at room temperature, which was very slow at −30 °C.
- Another pathway for the formation of 10 is the ene reaction of 7 and 8.
- When the reaction was stopped in a short reaction time, similar enantioselectivity has been obtained. Thus, the Michael product was obtained kinetically.
- (a) E. Nakamura, M. Shimizu, I. Kuwajima, J. Sakata, K. Yokoyama and R. Noyori, J. Org. Chem., 1983, 48, 932 Search PubMed; (b) R. Noyori, I. Nishida and J. Sakata, J. Am. Chem. Soc., 1983, 105, 1598 Search PubMed.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1991, 56, 4218 Search PubMed.
- As we cannot find the pKa of ammonium ions of i-Pr2NEt, we will discuss the pKa of ammonium ions of a similar tertiary amine such as Et3N. (a) I. M. Kolthoff, M. K. Chantooni and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23 Search PubMed; (b) M. R. Crampton and I. A. Robotham, J. Chem. Res., 1997, 22 Search PubMed.
- (a) J. P. Guthrie and P. A. Cullimore, Can. J. Chem., 1979, 57, 240 Search PubMed; (b) J. P. Guthrie, Can. J. Chem., 1979, 57, 1177 Search PubMed.
- For the reaction using H218O to determine the reaction mechanism, see; Y. Hayashi, T. Mukaiyama, M. Benohoud, N. R. Gupta, T. Ono and S. Toda, Chem.–Eur. J., 2016, 22, 5868 Search PubMed.
- See the general organic textbook, for instance, J. McMurry, Organic Chemistry, Physical Sciences, Belmont, 7th edn, 2008, p. 842.
- The generation of an enol (not enolate) from an aldehyde (not ketone) using pyrrolidine was observed to proceed through a cyclic replay mechanism, see ref. 11.
- As the solvent of the reaction is EtOH/toluene (4/1), we discuss pKa in MeOH. The pKa of o-nitrophenol and Et3NH+ (in water) is 7.1 and 10.75, respectively, based on Evans' pKa table (ref. 2). The pKa of o-nitrophenol and Et3NH+ in MeOH is estimated to be 11.1 and 10.95, respectively, by the use of the empirical conversion method by Knapp. See, E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653 Search PubMed.
- The enol O–H of the cyclohexanone is acidic enough to be deprotonated by a tertiary amine. The pKa of the enol O–H of the cyclohexanone is 12.1 in water, see; (a) J. P. Guthrie and P. A. Cullimore, Can. J. Chem., 1979, 57, 240 Search PubMed; (b) J. P. Guthrie, Can. J. Chem., 1979, 57, 1177 Search PubMed . The calculated pKa using Advanced Chemistry Development (ACD/Labs) Software V11.02 is 11.5 (gas phase).
- The deprotonation of enol–OH is very fast (often diffusion controlled), see H. O. House, Modern Synthetic Reactions, Benjamin, Philippines, 2nd edn, 1972, p. 495.
- The pKa of phenol and p-methoxyphenol in MeOH is estimated to be 13.95 and 14.2, respectively, see ref. 2 and 31..
- The value of pKa in MeOH is estimated to be 4.75, respectively, see ref. 2 and 31.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03359f |
| This journal is © The Royal Society of Chemistry 2020 |