
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeeing luminescence appear as crystals crumble. Isolation and subsequent self-association of individual [(C6H11NC)2Au]+ ions in crystals†
Lucy M. C.
Luong
 ,
Christopher D.
Lowe
,
Alexandria V.
Adams
,
Venoos
Moshayedi
,
Marilyn M.
Olmstead
and
Alan L.
Balch
*
,
Christopher D.
Lowe
,
Alexandria V.
Adams
,
Venoos
Moshayedi
,
Marilyn M.
Olmstead
and
Alan L.
Balch
*
Department of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA. E-mail: albalch@ucdavis.edu
First published on 10th September 2020
Abstract
Non-luminescent, isostructural crystals of [(C6H11NC)2Au](EF6)·C6H6 (E = As, Sb) lose benzene upon standing in air to produce green luminescent (E = As) or blue luminescent (E = Sb) powders. Previous studies have shown that the two-coordinate cation, [(C6H11NC)2Au]+, self-associates to form luminescent crystals that contain linear or nearly linear chains of cations and display unusual polymorphic, vapochromic, and/or thermochromic properties. Here, we report the formation of non-luminescent crystalline salts in which individual [(C6H11NC)2Au]+ ions are isolated from one another. In [(C6H11NC)2Au](BArF24) ((BArF24)− is tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) each cation is surrounded by two anions that prohibit any close approach of the gold ions. Crystallization of [(C6H11NC)2Au](EF6) (E = As or Sb, but not P) from benzene solution produces colorless, non-emissive crystals of the solvates [(C6H11NC)2Au](EF6)·C6H6. These two solvates are isostructural and contain columns in which cations and benzene molecules alternate. With the benzene molecules separating the cations, the shortest distances between gold ions are 6.936(2) Å for E = As and 6.9717(19) Å for E = Sb. Upon removal from the mother liquor, these crystals crack due to the loss of benzene from the crystal and form luminescent powders. Crystals of [(C6H11NC)2Au](SbF6)·C6H6 that powder out form a pale yellow powder with a blue luminescence with emission spectra and powder X-ray diffraction data that show that the previously characterized [(C6H11NC)2Au](SbF6) is formed. In the process, the distances between the gold(I) ions decrease to ∼3 Å and half of the cyclohexyl groups move from an axial orientation to an equatorial one. Remarkably, when crystals of [(C6H11NC)2Au](AsF6)·C6H6 stand in air, they lose benzene and are converted into the yellow, green-luminescent polymorph of [(C6H11NC)2Au](AsF6) rather than the colorless, blue-luminescent polymorph. Paradoxically, the yellow, green-luminescent powder that forms as well as authentic crystals of the yellow, green-luminescent polymorph of [(C6H11NC)2Au](AsF6) are sensitive to benzene vapor and are converted by exposure to benzene vapor into the colorless, blue-luminescent polymorph.
Introduction
There is significant current interest in the development of new, environmentally responsive, luminescent metal–organic compounds.1–3 These compounds show an array of properties that allow them to be useful in data recording,4 electrooptical device development,5 and biological imaging.6,7 Such materials are also valuable for sensing temperature,8–10 pressure,11–13 and the vapors of volatile organic compounds.14–16 For example, molecular cages involving platinum(II) centers and bridging ligands based on the emissive tetraphenylethylene core have been shown to display tunability in the emissive behavior and white light generation.17 A molecular box based on connecting six gold(I) centers with a non-emissive triphosphine ligand has produced a pressure sensitive salt whose structure and luminescence changes upon grinding.18Materials involving gold(I) complexes are frequently luminescent19–21 and particularly prone to sensitivity to environmental factors including responses to mechanical pressure or organic vapors.6,10,14,22 Linear, two-coordinate gold(I) complexes frequently self-associate in the solid state to form dimers, trimers, and extended, nearly linear chains.23–27 In these assemblies, the individual gold(I) ions are connected through attractive aurophilic interactions, which result from a combination of relativistic effects and dispersion forces.28,29 The strength of these aurophilic interactions ranges from 6 to 11 kcal mol−1.22–30 While most two-coordinate gold(I) complexes are non-luminescent, self-association into dimers, trimers and chains frequently produces luminescent materials and luminescent crystals due to the new chromophores produced by the aurophilic interactions. In this sense, such gold(I) complexes exhibit aggregation-induced emission (AIE).31 With organic luminophores, AIE occurs generally because of restrictions in molecular motion that can cause quenching in more mobile environments.32 With gold(I) complexes and other metal complexes, AIE results from the creation of new luminophores such as those formed by the association of gold centers. Environmental factors can cause rearrangements of these aurophilic interactions and cause changes in the spectroscopic properties of the compounds involved.
For example, several two-coordinate gold(I) complexes involving one aryl isocyanide ligand and one aryl group have been found to be sensitive to the application of mechanical pressure.33–38 Phenyl(phenyl isocyanide)gold(I) crystallizes as two polymorphs, which are crystals of identical composition that differ in the way the contents pack.39,40 A pressure sensitive form is distinguished by a blue emission with the closest Au⋯Au contact of 5.733 Å, which indicates that there is no aurophilic interaction in this form. Upon application of pressure this blue-emitting form is converted into a form with yellow emission. In the process two of the gold complexes come together to form a dimer with a short Au⋯Au contact of 3.177 Å.
The gold(I) complex [(C6H11NC)2Au](PF6) crystallizes in two polymorphic forms: yellow [(C6H11NC)2Au](PF6) (1), which exhibits green luminescence, and colorless [(C6H11NC)2Au](PF6) (2), which displays blue luminescence.41 Similarly, [(C6H11NC)2Au](AsF6) forms two polymorphs: yellow [(C6H11NC)2Au](AsF6) (3), which exhibits green luminescence, and colorless [(C6H11NC)2Au](AsF6) (4), which displays blue luminescence.42 Colorless [(C6H11NC)2Au](PF6) (2) and colorless [(C6H11NC)2Au](AsF6) (4) are isostructural, while yellow [(C6H11NC)2Au](PF6) (1), and yellow [(C6H11NC)2Au](AsF6) (3) crystallize in two different space groups. The cations in these salts self-associate through aurophilic interactions to form extended chains. The chains found in the two polymorphs of [(C6H11NC)2Au](AsF6) are illustrated in Fig. 1. The yellow polymorphs of [(C6H11NC)2Au](PF6) (1) and [(C6H11NC)2Au](AsF6) (3) are converted into the respective colorless polymorphs (2) and (4) upon exposure to organic vapors of benzene, methanol, or dichloromethane without any uptake of vapor molecules. However, pale yellow [(C6H11NC)2Au](SbF6) (5), which has a blue luminescence, does not form polymorphs but crystallizes with two different chains of cations in the crystal as shown in Fig. 1.43 Crystals of pale yellow [(C6H11NC)2Au](SbF6) (5) are not sensitive to vapors of volatile organic compounds. Once dissolved in a solvent, these salts are no longer emissive. The excitation and emission originate in the chromophores produced by the chains of cations that form in the crystals. Here, we examine the hypothesis that if the [(C6H11NC)2Au]+ ions are separated from one another in crystals, the crystals will be non-luminescent and examine the remarkable reactivity of the new crystals containing isolated [(C6H11NC)2Au]+ ions.
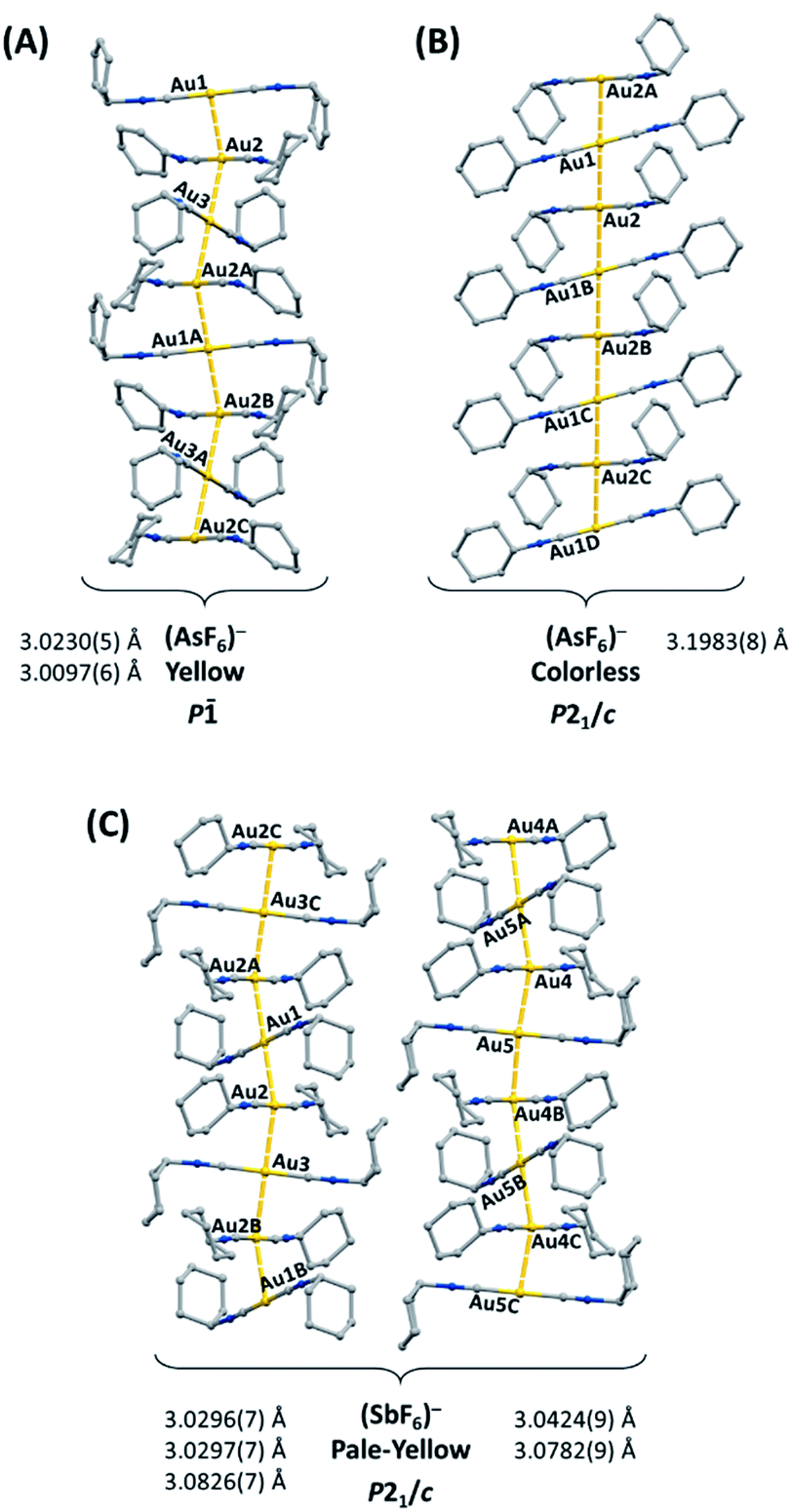 | ||
| Fig. 1 The columnar structures of the yellow [(C6H11NC)2Au](AsF6) (3) and colorless [(C6H11NC)2Au](AsF6) (4) and of [(C6H11NC)2Au](SbF6) (5), which does not form polymorphs but has two different chains in each pale-yellow crystal.42,43 For clarity, anions and hydrogen atoms are omitted. | ||
Results
Isolating the [(C6H11NC)2Au]+ cation to form colorless, non-luminescent salts
Two strategies have been employed to form solids in which the [(C6H11NC)2Au]+ cation is isolated from other cations. One involves the use of a large anion to surround the cation, while the other uses solvate molecules to separate the cations.Using the first method, colorless [(C6H11NC)2Au](BArF24) (6) ((BArF24)− is tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) was prepared by the route used for the formation of [(C6H11NC)2Au]-(PF6) but with sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)-borate replacing ammonium hexafluorophosphate. The colorless, non-luminescent salt was crystallized from methanol by evaporation. The structure of the anion and cation are shown in Fig. 2. The asymmetric unit consists of a half of a cation and a half of an anion. The gold ion resides on a center of symmetry, while the boron atom resides on a two-fold axis. The C–Au–C portion is required crystallographically to be linear. The two cyclohexyl groups occupy an equatorial position. As shown in the drawing in Fig. 3, two anions completely surround the linear cation and prevent self-association through aurophilic interactions. This colorless salt shows no luminescence at room temperature or at 77 K.
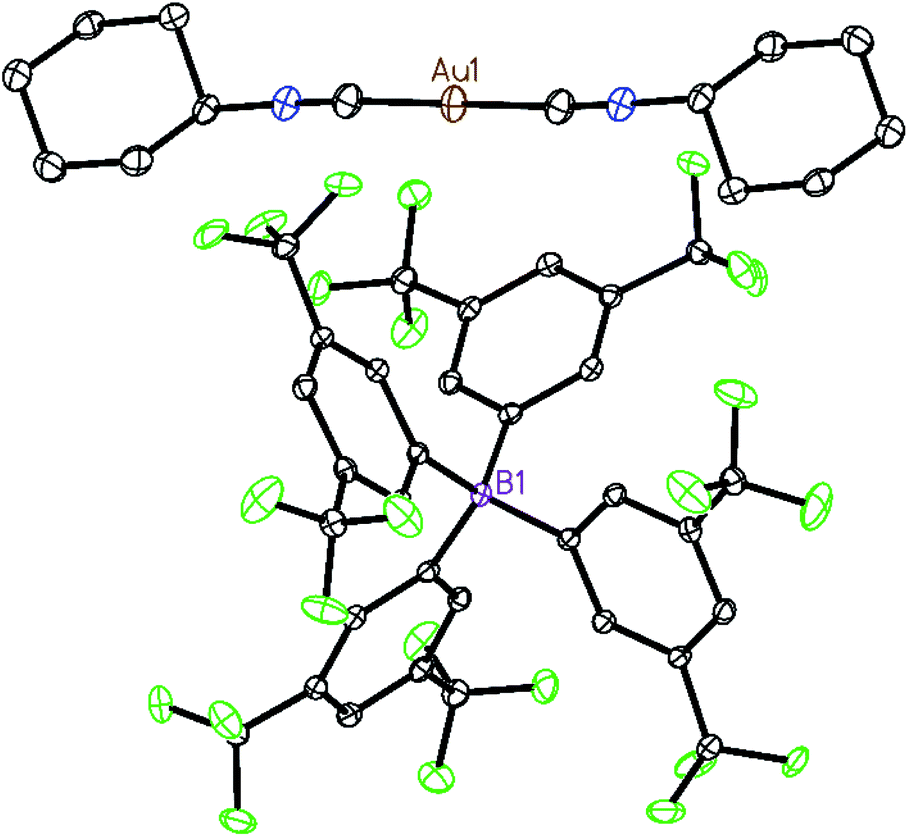 | ||
| Fig. 2 The structure of [(C6H11NC)2Au](BArF24) (6) with 50% thermal contours. Hydrogen atoms are omitted for clarity. Atom colors are: Au, orange; C, grey; N, blue; B, pink; F, green. | ||
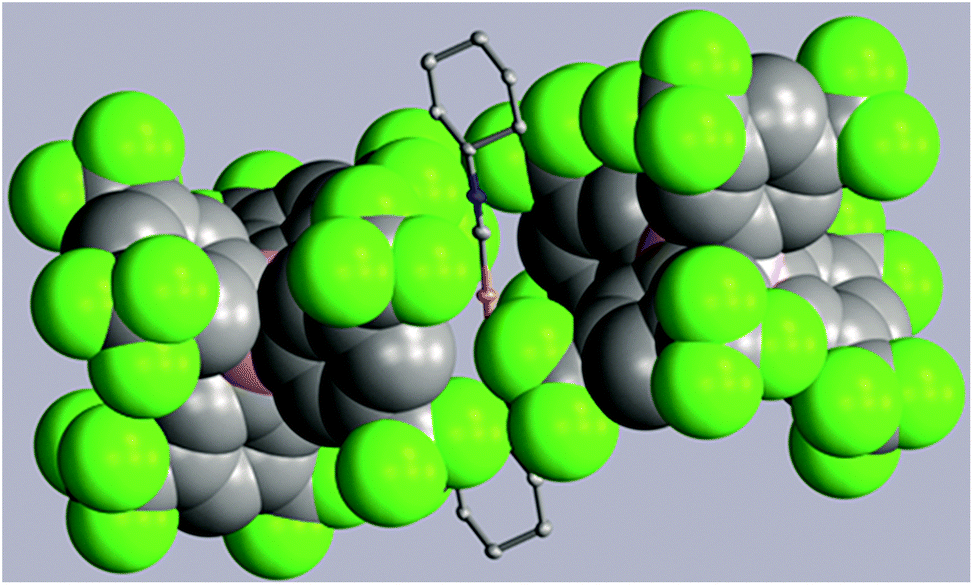 | ||
| Fig. 3 Two (BArF24)− anions (drawn with space-filling contours) surrounding a [(C6H11NC)2Au]+ cation in (6). Atom colors are: Au, orange; C, grey; N, blue; B, pink; F, green. | ||
The second method of separation of these cations involves the formation of benzene solvates, [(C6H11NC)2Au](SbF6)·C6H6 (7) and [(C6H11NC)2Au](AsF6)·C6H6 (8). These solvates were obtained by crystallization of samples of [(C6H11NC)2Au](SbF6) (5) and [(C6H11NC)2Au](AsF6) (3) or (4) from a solution of the complex in benzene with an overlayer of diethyl ether at 5 °C. The process produces colorless crystals of non-luminescent [(C6H11NC)2Au](SbF6)·C6H6 (7) exclusively. However, as shown in Fig. 4, crystallization of [(C6H11NC)2Au](AsF6)·C6H6 (8) is accompanied by the concomitant crystallization of both yellow [(C6H11NC)2Au](AsF6) (3) and colorless [(C6H11NC)2Au](AsF6) (4). The colorless non-luminescent solvate crystals (8) form in the lower sections of the tubes. This is the region in the tube where the benzene concentration is greatest, while the non-solvated polymorphs form in the upper region of the tubes where the ether concentration is high. Such spatially separated formation of crystals of the polymorphs of [(C6H11NC)2Au](AsF6) has been reported previously.35 However, extensive efforts to obtain a benzene solvate of [(C6H11NC)2Au](PF6) have not been successful.
 | ||
| Fig. 4 Spatially separated formation of crystals from a benzene solution of [(C6H11NC)2Au](AsF6) with diethyl ether layered on top in 5 mm outside diameter glass tubes. The photograph was taken under UV irradiation. The non-luminescent crystals of [(C6H11NC)2Au](AsF6)·C6H6 (8) are seen at the lower part of each tube. Above that the pink-tinged crystals are the colorless [(C6H11NC)2Au](AsF6) (4), but the camera captures the emission color as pink rather than the blue seen by eye. Further up the tubes are crystals of the yellow [(C6H11NC)2Au](AsF6) (3), which glow green. | ||
Crystals of the benzene solvates are isostructural and form in the tetragonal space group P4/mnc (128). Gold ions are separated by 6.9717(19) Å in [(C6H11NC)2Au](SbF6)·C6H6 (7) and by 6.936(2) Å in [(C6H11NC)2Au](AsF6)·C6H6 (8). The structure of [(C6H11NC)2Au](SbF6)·C6H6 (7) is shown in Fig. 5. The gold atom resides on a site (c) of 2/m symmetry while the hexafluoroantimonate ion resides on a site (e) with 4-fold symmetry. Benzene molecules are disordered with respect to crystallographic 2-fold axes (d). The cations are stacked along the crystallographic c axis with benzene molecules between cations so that there is no close contact between the cations. The distance between Au1 and the centroid of the benzene ring is 3.486 Å. The distances between Au1 and the carbon atoms of the benzene ring are all greater than 3.7 Å and there appears to be no coordination occurring. Cases where bonding interactions between gold and an arene ring are proposed usually involve a phosphine ligand that positions a gold ion close to one or two carbon atoms of an aryl group of the ligand with Au to C distances in the 3.12–3.20 Å range.44–46 The cyclohexyl rings are in axial positions. A view down the c axis is given in Fig. 6.
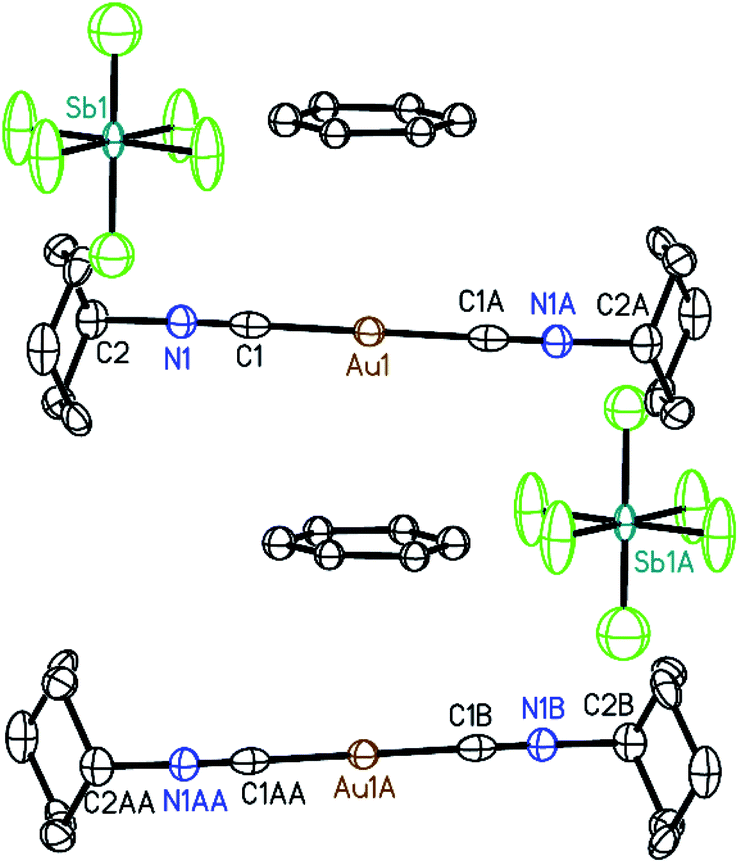 | ||
| Fig. 5 A portion of the structure of [(C6H11NC)2Au](SbF6)·C6H6 (7) that depicts the sandwich of benzene between two of the gold(I) cations with 50% thermal contours. Hydrogen atoms and the alternate position for the benzene molecules are omitted for clarity. | ||
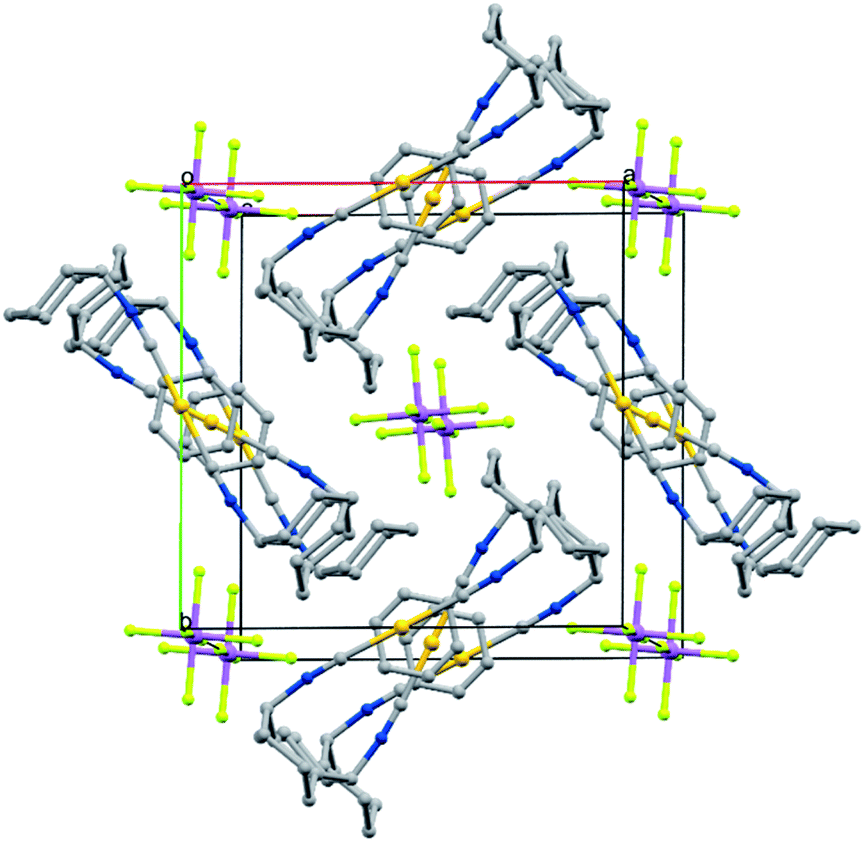 | ||
| Fig. 6 The structure of [(C6H11NC)2Au](SbF6)·C6H6 (7) looking down the c axis with hydrogen atoms and disorder in the benzene molecules omitted. Atom colors are as follows: Au, orange; C, grey; N, blue; F, yellow-green; Sb, purple. | ||
Turning on luminescence through solvate loss: [(C6H11NC)2Au](SbF6)·C6H6 (7)
When removed from the mother liquor and exposed to air, the colorless, non-luminescent crystals of [(C6H11NC)2Au](SbF6)·C6H6 (7) lose their crystallinity and become a pale yellow powder with blue luminescence. The process takes only few hours to accomplish. Fig. 7 shows relevant emission and excitation spectra for the sample after standing in air and compares those spectra to spectra obtained from a sample of pale yellow [(C6H11NC)2Au](SbF6) (5) that had been crystallized from solution. Fig. 8 compares the powder X-ray diffraction from an air dried sample of [(C6H11NC)2Au](SbF6)·C6H6 (7) with a polycrystalline sample of [(C6H11NC)2Au](SbF6) (5) that was grown from a dichloromethane solution layered with diethyl ether. These data indicate the non-emissive [(C6H11NC)2Au](SbF6)·C6H6 (7) is transformed into pale yellow, [(C6H11NC)2Au](SbF6) (5) through simple air drying. Additionally, comparison of the infrared spectrum of colorless [(C6H11NC)2Au](SbF6)·C6H6 (7) with that of the air-dried yellow powder show that the C–H stretching band from the benzene solvate at 3000 cm−1 is lost upon air drying.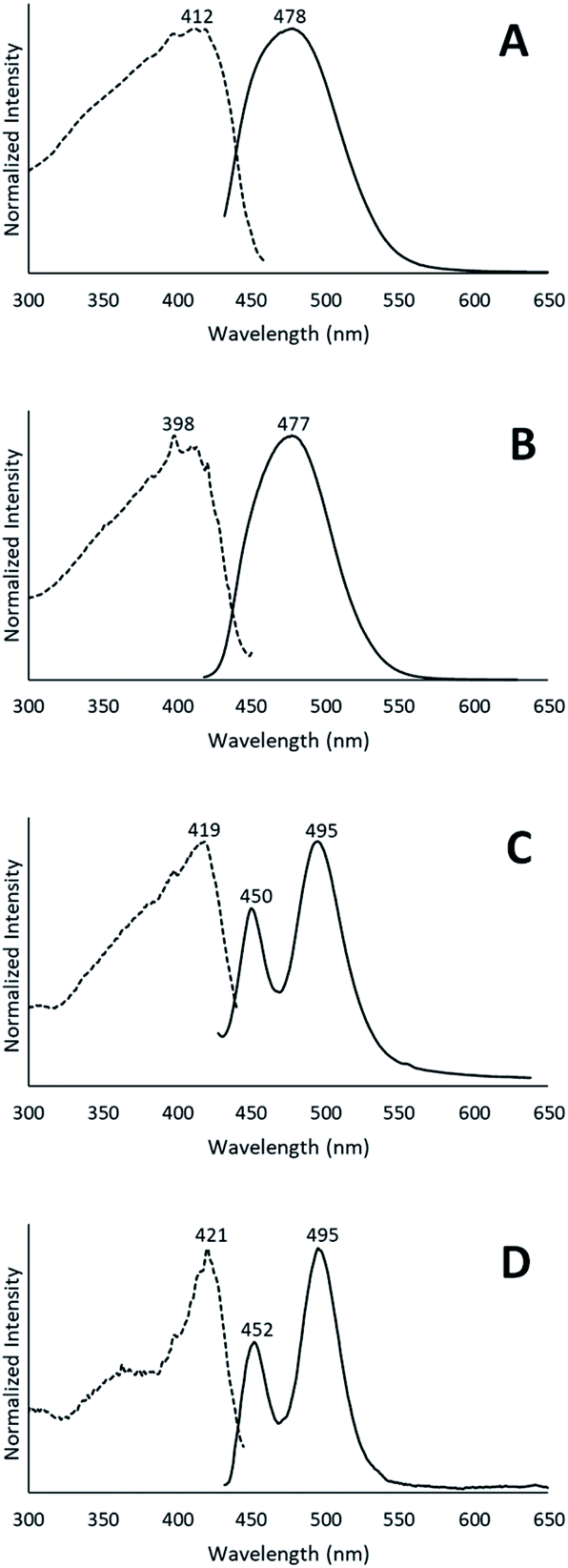 | ||
| Fig. 7 The emission and excitation spectra of (A) [(C6H11NC)2Au](SbF6)·C6H6 (7) after air drying at 298 K; (B) crystalline [(C6H11NC)2Au](SbF6) (5) grown from solution at 298 K; (C) [(C6H11NC)2Au](SbF6)·C6H6 (7) after air drying at 77 K; (D) crystalline [(C6H11NC)2Au](SbF6) (5) grown from solution at 77 K. | ||
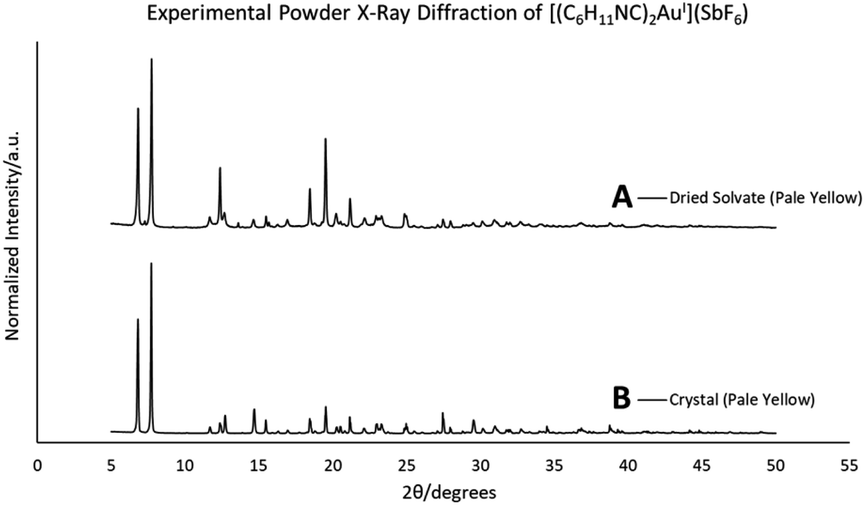 | ||
| Fig. 8 A powder X-ray diffraction comparison: top, air-dried sample of [(C6H11NC)2Au](SbF6)·C6H6 (7); bottom, polycrystalline sample of pale yellow [(C6H11NC)2Au](SbF6) crystals (5). Data taken at 298 K. | ||
Turning on luminescence through solvate loss: [(C6H11NC)2Au](AsF6)·C6H6 (8). Which polymorph forms?
Colorless non-luminescent crystals of [(C6H11NC)2Au](AsF6)·C6H6 (8) also crumble and become green-luminescent upon drying in air. Indeed, crystals of [(C6H11NC)2Au](AsF6)·C6H6 (8) are quite fragile and begin to turn luminescent once they are removed from solution. In an hour they are completely converted into a green-luminescent powder. Fig. 9 compares the excitation and emission spectra of the air dried powder with the corresponding spectra of the yellow and colorless polymorphs of unsolvated [(C6H11NC)2Au](AsF6). The spectra show that the yellow [(C6H11NC)2Au](AsF6) (3) is selectively formed when [(C6H11NC)2Au](AsF6)·C6H6 (8) is allowed to air dry. This conclusion is reinforced by the X-ray powder diffraction data shown in Fig. 10. Trace A shows the X-ray powder diffraction data for the air dried sample of [(C6H11NC)2Au](AsF6)·C6H6 (8), while Trace B shows the X-ray powder diffraction data for the yellow [(C6H11NC)2Au](AsF6) (3). The two powder patterns are similar, but distinct from that of the colorless [(C6H11NC)2Au](AsF6) (4), which is shown in Trace D. The small peak at 7.7° in Trace A suggests that a small amount of the colorless polymorph of [(C6H11NC)2Au](AsF6) (4) also forms during the air drying of [(C6H11NC)2Au](AsF6)·C6H6 (8).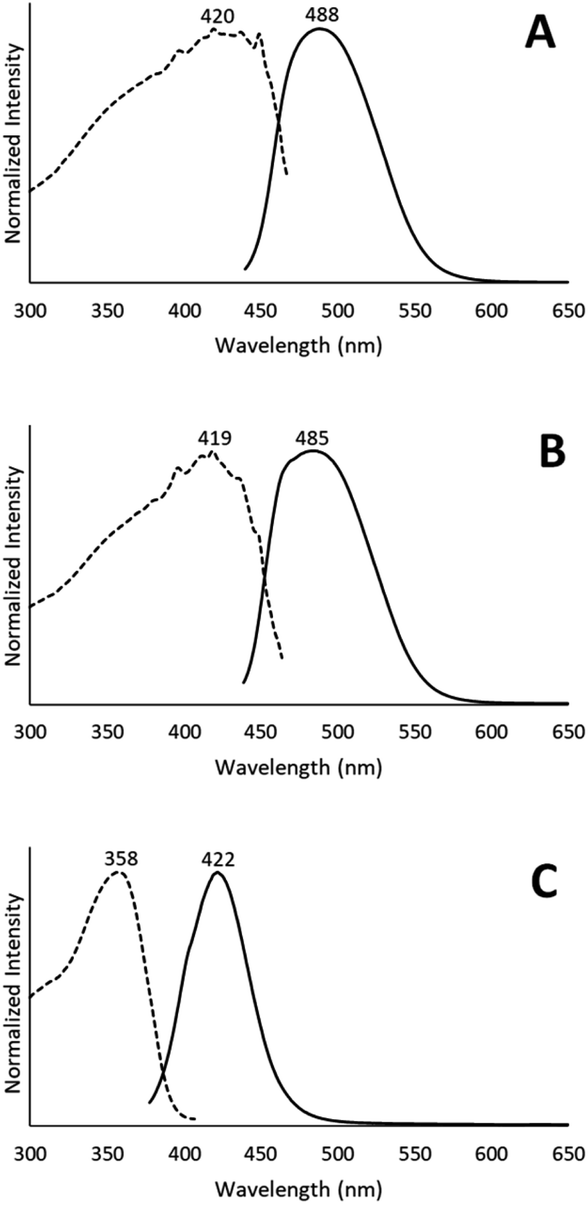 | ||
| Fig. 9 The emission and excitation spectra of (A) [(C6H11NC)2Au](AsF6)·C6H6 (8) after air drying at 298 K; (B) yellow [(C6H11NC)2Au](AsF6) (3) at 298 K; (C) colorless [(C6H11NC)2Au](AsF6) (4) at 298 K. | ||
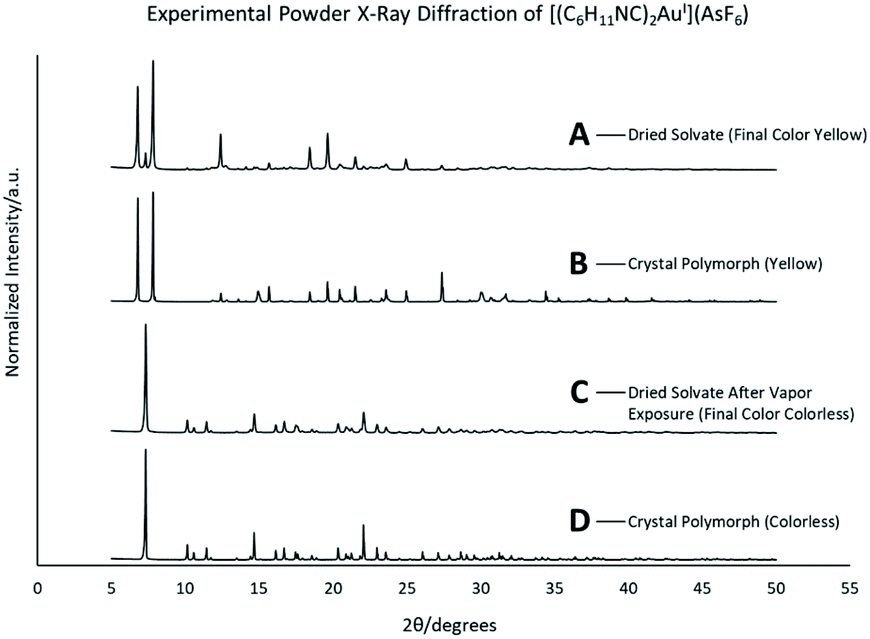 | ||
| Fig. 10 The X-ray powder diffraction data for (A) [(C6H11NC)2Au](AsF6)·C6H6 (8) after air drying; (B) yellow [(C6H11NC)2Au](AsF6) (3); (C) [(C6H11NC)2Au](AsF6)·C6H6 (8) after air drying and exposure to benzene vapor; (D) colorless [(C6H11NC)2Au](AsF6) (4). Data taken at 298 K. | ||
The conditions in which [(C6H11NC)2Au](AsF6)·C6H6 (8) is handled can alter the turn on of the luminescence. When [(C6H11NC)2Au](AsF6)·C6H6 (8) is vacuum dried or benzene is removed by vigorous washing with pentane, the sample shows both green luminescence and blue luminescence and both polymorphs of [(C6H11NC)2Au](AsF6) form.
The conversion of [(C6H11NC)2Au](AsF6)·C6H6 (8) through the loss of benzene into the yellow [(C6H11NC)2Au](AsF6) (3) is surprising, since it was previously shown that crystals of yellow [(C6H11NC)2Au](AsF6) (3) were sensitive to benzene vapor and the vapors of other volatile organic compounds.35 Consequently, we examined the behavior of an air dried sample of [(C6H11NC)2Au](AsF6)·C6H6 (8) in the presence of benzene vapor at 5 °C. The sample does respond to the presence of benzene vapor with the emission color changing from green to blue. Relevant excitation and emission spectra are shown in Fig. 11. Exposure to benzene vapor causes the luminescence of an air dried sample of [(C6H11NC)2Au](AsF6)·C6H6 (8) to change from green to blue. The excitation and emission spectra of the sample at 298 K are shown in Trace A of Fig. 11 where they are compared to the corresponding spectra of crystals of the colorless, [(C6H11NC)2Au](AsF6) (4) in Trace B. The spectra are similar but there is a slight shift of the emission to lower energy. We suspect that this shift is caused by the presence of a second, unidentified material in the sample. Further evidence for this conclusion is seen in the excitation and emission spectra taken at 77 K. As seen in Trace C, there is a shoulder in the emission spectrum in the 500 nm region that is absent in the corresponding spectrum of the colorless [(C6H11NC)2Au](AsF6) (4) shown in Trace D. In previous studies, we have observed similar features whose intensities vary from sample to sample.36 Thus, this shoulder probably comes from a second unidentified product.
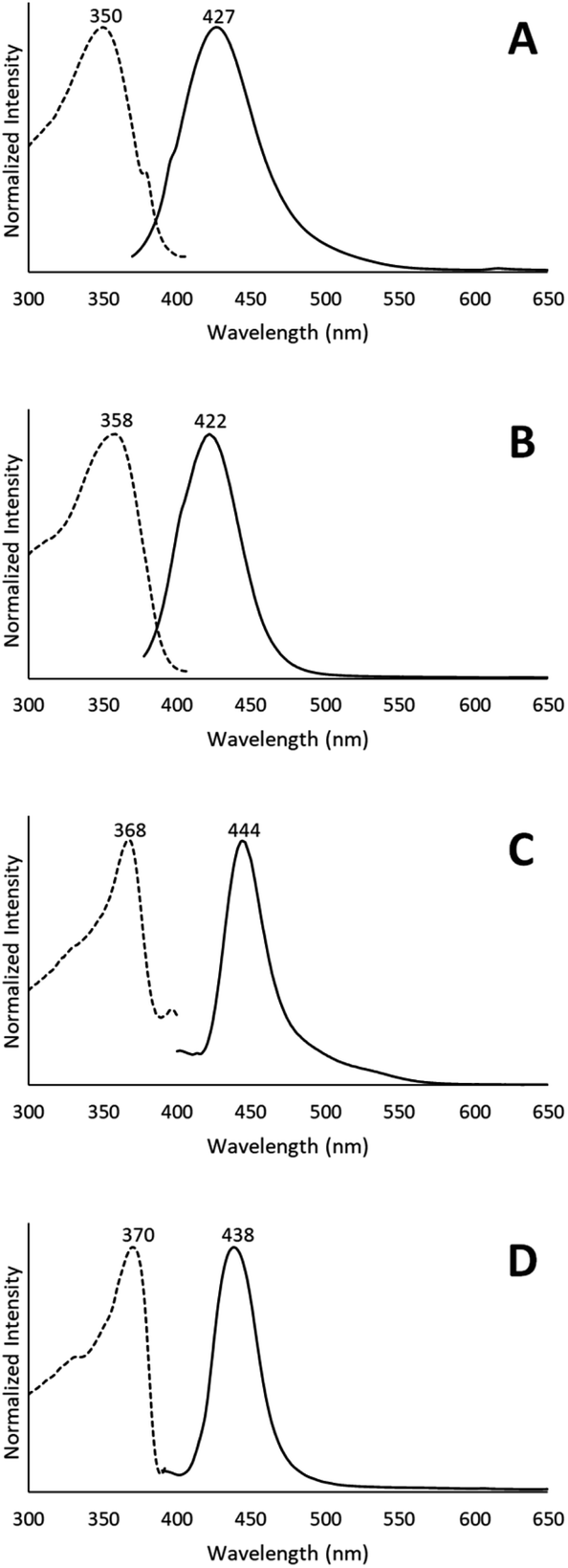 | ||
| Fig. 11 The emission and excitation spectra of (A) [(C6H11NC)2Au](AsF6)·C6H6 (8) after air drying and exposure to benzene vapor at 298 K; (B) yellow [(C6H11NC)2Au](AsF6) (3) grown from solution at 298 K; (C) [(C6H11NC)2Au](AsF6)·C6H6 (8) after air drying and exposure to benzene vapor at 77 K; (D) colorless [(C6H11NC)2Au](AsF6) (4) grown from solution at 77 K. | ||
This conversion of an air dried sample of [(C6H11NC)2Au](AsF6)·C6H6 (8) into the yellow [(C6H11NC)2Au](AsF6) (3) has also been examined by powder X-ray diffraction. Relevant data are shown in Fig. 10. The lower two traces (C and D) compare the powder patterns for air dried sample of [(C6H11NC)2Au](AsF6)·C6H6 (8) and yellow [(C6H11NC)2Au](AsF6) (3). There is good agreement between the two powder patterns.
Discussion
We have shown that the two-coordinate cation, [(C6H11NC)2Au]+ can be kept from self-association by crystallization with a large anion, (BArF24)−, or by formation of benzene solvates [(C6H11NC)2Au](EF6)·C6H6 with E = Sb (7) or As (8) in which the columns of alternating benzene molecules and cations form. With the cations isolated, these colorless crystals are not emissive at 298 or 77 K. However, these benzene solvates [(C6H11NC)2Au](EF6)·C6H6 with E = Sb (7) or As (8) become luminescent simply upon drying in air. Although the two solvates are isostructural, each converts into a unique product upon standing in air. Thus, the solvate [(C6H11NC)2Au](SbF6)·C6H6 (7) converts into pale yellow [(C6H11NC)2Au](SbF6) (5), which contains two different chains of cations within the crystal as shown in Fig. 12.36 In contrast, [(C6H11NC)2Au](AsF6)·C6H6 (8) selectively converts into yellow [(C6H11NC)2Au](AsF6) (3), whose columnar structure is shown in Fig. 13.35 In order to transform into the unsolvated forms, the Au⋯Au separations in the solvated crystals must decrease significantly. Indeed, the ∼3.8 Å difference between the separation between gold ions in the solvate and the Au⋯Au distances in the solvate free products is remarkably large, particularly when compared to the comparable change in separations seen in the mechanochromic PhAuCNPh.32,33 The conversions of these solvated crystals into the respective unsolvated forms are dynamic examples of aggregation-induced emission (AIE) in which the creation of aurophillic interactions produces new chromophores.31 Additionally, in this process half of the cyclohexyl groups need to move into equatorial positions to yield an array in which the positions of the cyclohexyl groups alternate between axial and equatorial along the chain.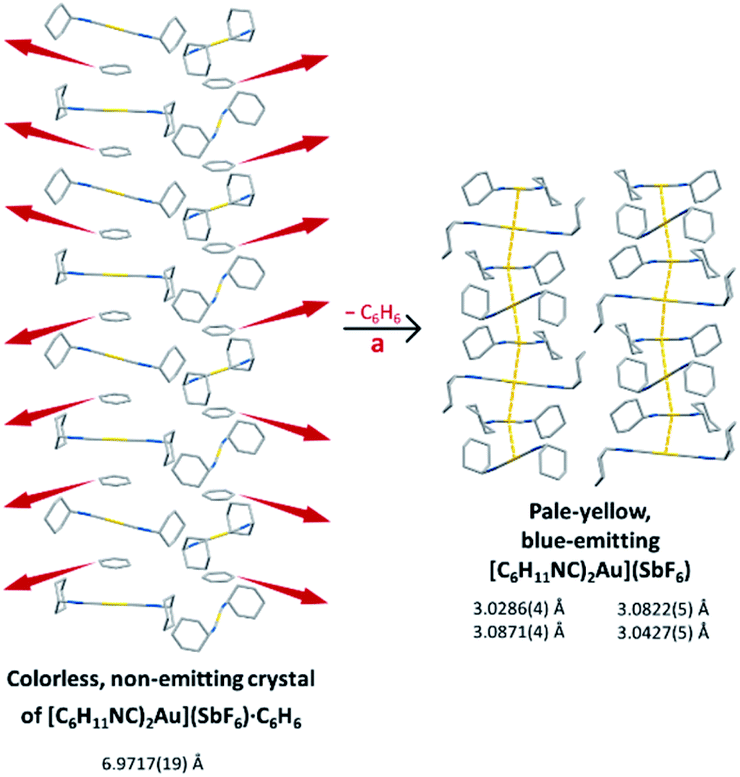 | ||
| Fig. 12 Process of benzene loss from crystalline [(C6H11NC)2Au](SbF6)·C6H6 (7). The distances are the closest spacings between gold(I) ions. | ||
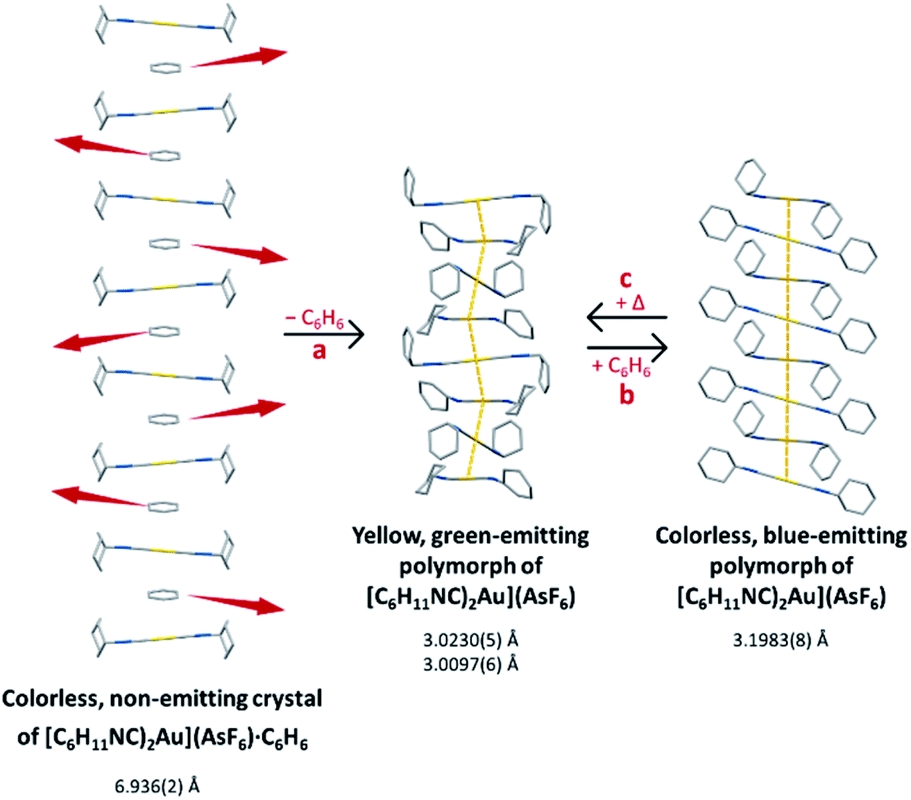 | ||
| Fig. 13 Process of benzene loss from crystalline [(C6H11NC)2Au](AsF6)·C6H6 (8). The distances are the closest spacings between gold(I) ions. | ||
Fig. 12 and 13 are drawn showing the process beginning with the loss of benzene molecules. As the benzene molecules leave, shortening of the Au⋯Au distances occurs. It seems likely that the process proceeds gradually in a local fashion where a few benzene molecules leave and then the cations in that region move close together in segments along the chain. Additionally, mobile solution-like pockets may form in which the cations are free to rearrange into the solvate-free polycrystals.
Additionally, the non-coordinating anions play a role in determining the final structure formed. Thus, with hexafluoroantimonate, a structure with two independent chains forms. However, with hexafluoroarsenate, yellow [(C6H11NC)2Au](AsF6) (3) forms selectively if the crystals are simply separated from the mother liquor and allowed to stand in air. The formation of this particular polymorph is remarkable. This is the polymorph with the shortest Au⋯Au distances and the polymorph that is sensitive to the presence of organic vapors including benzene. Apparently, the partial pressure of benzene during the process of solvate loss is not high enough to alter the structure of the microcrystalline powder of yellow [(C6H11NC)2Au](AsF6) (3) that desolvation produces.
Conclusions
Crystalline salts containing individual [(C6H11NC)2Au]+ ions that are not involved in any aurophilic interactions have been prepared and demonstrated to be non-luminescent. The solvated non-luminescent crystals [(C6H11NC)2Au](SbF6)·C6H6 (7) and [(C6H11NC)2Au](AsF6)·C6H6 (8), which are isostructural, are sensitive to the loss of benzene and decompose to form luminescent polycrystalline solids with quite different structures. Remarkably, [(C6H11NC)2Au](AsF6)·C6H6 (8) selectively forms yellow [(C6H11NC)2Au](AsF6) (3) rather than its polymorph, colorless [(C6H11NC)2Au](AsF6) (4). These results give unusual insight into the structural changes that occur as a crystal decomposes due to solvate loss, a common occurrence that frequently results in the formation of a poorly characterized powder.Experimental
Materials
Cyclohexyl isocyanide was purchased from Acros Organics and used as received. This foul-smelling compound is toxic and must be handled in a well-ventilated hood. Chloro(tetrahydrothiophene)gold(I) was prepared by an known procedure.47 Samples of [(C6H11NC)2Au](AsF6)35 and of [(C6H11NC)2Au](SbF6)36 were prepared as described previously. NaBArF24 was prepared by a standard route.48Preparation of [(C6H11NC)2Au](BArF24) (6)
In a vial, 0.0494 g (0.154 mmol) of chloro(tetrahydrothiophene)gold(I), 0.0307 g (0.281 mmol) of cyclohexyl isocyanide, and 4 mL of acetonitrile were added and stirred to form a homogenous, clear solution. A solution containing 0.1377 g (0.155 mmol) of sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate and 5 mL of acetonitrile was then added to the vial. A white precipitate immediately formed. The mixture was stirred for 30 seconds and subsequently filtered. Rotary evaporation was used to remove the solvent. A colorless oil resulted and was then dissolved in 5 mL of dichloromethane. This solution was filtered through a small pad of Celite. Rotary evaporation was used to remove the solvent. A hazy white film resulted and was then dissolved in 5 mL of methanol. Rotary evaporation was used to remove the solvent. White crystals resulted, were washed with pentane, and filtered. The dried product yielded 0.1548 g (0.121 mmol, 78.6%) of colorless blocks of [(C6H11NC)2Au](BArF24) (6). The non-luminescent, colorless crystals melted from 122–125 °C.Crystallization of [(C6H11NC)2Au](EF6)·C6H6
A 0.0550 g sample of [(C6H11NC)2Au](EF6) (E = As or Sb) was dissolved in 1 mL of benzene. Then, 0.5 mL of this solution was then filtered into a 5 mm od glass tube. A layer of 0.25 mL of benzene was placed over it. The solution in the crystal tube was chilled in an ice bath for a few minutes. Following this step, 1.5 mL of diethyl ether, previously chilled in an ice bath, was filtered into the tube. The crystal tube was stored at approximately 5 °C. Within one week, colorless crystals with no luminescence grew. For the crystallization of [(C6H11NC)2Au](AsF6)·C6H6 (8), concomitant growth of yellow [(C6H11NC)2Au](AsF6) (3) and colorless [(C6H11NC)2Au](AsF6) (4) occurred, as shown in Fig. 3. With the concomitant growth, the crystals were manually separated. With crystallization of [(C6H11NC)2Au](SbF6), the only crystals that grew were those of colorless [(C6H11NC)2Au](SbF6)·C6H6 (7). Attempts to use toluene, o-xylene or p-xylene rather than benzene for crystallization have not produced any solvated crystals.X-ray crystallography and data collection
All crystals were transferred with a small amount of mother liquor to a microscope slide and immediately coated with a hydrocarbon oil. A suitable crystal of [(C6H11NC)2Au](AsF6)·C6H6 (8) (or [(C6H11NC)2Au](SbF6)·C6H6 (7)) was mounted in the 100 K nitrogen cold stream provided by an Oxford Cryostream low temperature apparatus on the goniometer head of a Bruker D8 Venture Kappa DUO diffractometer equipped with Bruker Photon 100 CMOS detector. A suitable crystal of [(C6H11NC)2Au](BArF24) (6) was mounted in the 90 K nitrogen cold stream provided by a Cryo Industries low-temperature apparatus on the goniometer head of a Bruker APEX II sealed-tube diffractometer and CCD detector. All data were collected with the use of MoKα (λ = 0.71073 Å) radiation. A multi-scan absorption correction was applied with the program SADABS.49 The structure was solved by a dual space method, (SHELXT)50 and refined by full-matrix least-squares on F2 (SHELXL-2018).51Powder X-ray diffraction patterns were taken with Cu Kα radiation (λ = 1.5405 Å) on a Bruker D8 Eco Advance diffractometer operated at 40 kV and 40 mA at room temperature from 5° to 50° 2θ. The crystals were ground and sieved (200 mesh) to produce a fine, uniform powder. A thin layer of grease was applied to the PXRD sample holder, and the powder was scattered on to the greased area to form a single layer of the powder for data collection.
Physical measurements
IR spectra were recorded on a Bruker Alpha FT-IR spectrometer using attenuated total reflectance (ATR). Fluorescence excitation and emission spectra were recorded on a PerkinElmer LS50B luminescence spectrophotometer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ARCS Foundation for two awards to LMCL, the National Science Foundation (Grant CHE-1807637) for partial support of this project, and Grant CHE-1531193 for the Dual source X-ray diffractometer.References
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 Search PubMed
.
- X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276–15286 Search PubMed
- M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem.–Eur. J., 2019, 25, 5105–5112 Search PubMed
- K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 Search PubMed
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 Search PubMed
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W. H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 Search PubMed
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 Search PubMed
- S. Perruchas, X. F. Le Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin and J.-P. Boilot, J. Am. Chem. Soc., 2010, 132, 10967–10969 Search PubMed
- X.-D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 Search PubMed
- B. Li, H.-T. Fan, S.-Q. Zang, H. Y. Li and L.-Y. Wang, Coord. Chem. Rev., 2018, 377, 307–329 Search PubMed
- A. L. Balch, Angew. Chem., Int. Ed., 2009, 38, 2641–2644 Search PubMed
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 Search PubMed
- X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 Search PubMed
- X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 Search PubMed
- O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 Search PubMed
- X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 Search PubMed
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 Search PubMed
- D. T. Walters, R. B. Aghakhanpour, X. B. Powers, K. B. Ghiassi, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2018, 140, 7533–7542 Search PubMed
- N. Mirzadeh, S. H. Privér, A. J. Blake, H. Schmidbaur and S. K. Bhargava, Chem. Rev., 2020, 120, 7551–7591 Search PubMed
- X.-Y. Wang, Y.-X. Hu, X.-F. Yang, J. Yin, Z. Chen and S. H. Liu, Org. Lett., 2019, 21, 9945–9949 Search PubMed
- Z. Chen, J. Zhang, M. Song, J. Yin, G.-A. Yu and S. H. Liu, Chem. Commun., 2015, 51, 326–329 Search PubMed
- S. H. Lim, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 10229–10238 Search PubMed
- D. V. Toronto, B. D. S. Weissbart, D. S. Tinti and A. L. Balch, Inorg. Chem., 1996, 35, 2484–2489 Search PubMed
- J. C. Vickery, M. M. Olmstead, E. Y. Fung and A. L. Balch, Angew. Chem., Int. Ed., 2013, 36, 1179–1181 Search PubMed
- A. L. Balch, Struct. Bonding, 2007, 123, 1–40 Search PubMed
- V. W.-W. Yam, V. K.-A. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 Search PubMed
- M. Głodek, S. Pawledzio, A. Makal and D. Plazuk, Chem.–Eur. J., 2019, 25, 13131–13145 Search PubMed
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 Search PubMed
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 Search PubMed
- D. E. Harwell, M. D. Mortimer, C. B. Knobler, F. A. L. Anet and M. F. Hawthorne, J. Am. Chem. Soc., 1996, 118, 2679–2685 Search PubMed
- J. Mei, N. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 Search PubMed
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. T. Tang, Chem.–Eur. J., 2014, 20, 15349–15353 Search PubMed
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 1–5 Search PubMed
- T. Seki, K. Sakurada, M. Muromoto, S. Seki and H. Ito, Chem.–Eur. J., 2016, 22, 1968–1978 Search PubMed
- T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 Search PubMed
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 Search PubMed
- S. Aono, T. Seki, H. Ito and S. Sakaki, J. Phys. Chem. C, 2019, 123, 4773–4794 Search PubMed
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 Search PubMed
-
J. Bernstein, Polymorphism in Molecular Crystals, Clarendon Press, Oxford, 2002 Search PubMed
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 Search PubMed
- R. L. White-Morris, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 1033–1040 Search PubMed
- M. A. Malwitz, S. H. Lim, R. L. White-Morris, D. M. Pham, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 10885–10893 Search PubMed
- L. M. C. Luong, M. A. Malwitz, V. Moshayedi, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2020, 142, 5689–5701 Search PubMed
- D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2008, 27, 28–32 Search PubMed
- Q.-S. Li, C.-Q. Wan, R.-Y. Zou, F.-B. Xu, H.-B. Song, X.-J. Wan and Z.-Z. Zhang, Inorg. Chem., 2006, 45, 1888–1890 Search PubMed
- Q.-L. Ni, X.-F. Jiang, T.-H. Huang, X.-J. Wang, L.-C. Gui and K.-G. Yang, Organometallics, 2012, 31, 2343–2348 Search PubMed
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler Jr, Inorg. Synth., 1989, 26, 85–86 Search PubMed
- N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 Search PubMed
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 Search PubMed
- G. M. Sheldrick, SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information and infrared spectra. X-ray crystallographic files in CIF format for [(C6H11NC)2Au](BArF24) (6), [(C6H11NC)2Au](SbF6)·C6H6 (7) and [(C6H11NC)2Au](AsF6)·C6H6 (8). CCDC 2009608–2009610. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03299a |
| This journal is © The Royal Society of Chemistry 2020 |