
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering of chiral nanomaterials for biomimetic catalysis
Hongyu
Zhang
,
Si
Li
,
Aihua
Qu
,
Changlong
Hao
,
Maozhong
Sun
,
Liguang
Xu
,
Chuanlai
Xu
 and
Hua
Kuang
*
and
Hua
Kuang
*
International Joint Research Laboratory for Biointerface and Biodetection, State Key Lab of Food Science and Technology, School of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu 214122, P. R. China. E-mail: kuangh@jiangnan.edu.cn
First published on 21st October 2020
Abstract
Chiral nanomaterial-based biomimetic catalysts can trigger a similar biological effect to natural catalysts and exhibit high performance in biological applications. Especially, their active center similarity and substrate selectivity promoted their superior biocatalytic activity. Here, modification of critical elements, such as size, morphology, nanocrystal facets, chiral surface and active sites, for controlling the catalytic efficiency of individual chiral nanoparticles (NPs) and chiral nanoassemblies has been demonstrated, which had a synergistic effect on overcoming the defects of pre-existing nanocatalysts. Noticeably, application of external forces (light or magnetism) has resulted in obvious enhancement in biocatalytic efficiency. Chiral nanomaterials served as preferable biomimetic nanocatalysts due to their special structural configuration and chemical constitution advantages. Furthermore, the current challenges and future research directions of the preparation of high-performance bioinspired chiral nanomaterials for biological applications are discussed.
1. Introduction
Chirality, a fundamental aspect of nature, was first identified by Louis Pasteur during the separation of tartaric acid crystals in the early nineteenth century. Chiral biomolecules, for example, amino acids and nucleic acids, are the basic elements of the origin of life and are essential in biological organisms. However, chirality is not only restricted to the molecular level, but also appears on a large scale, especially in chiral nanocrystals and nanoassemblies.1 The chiral ligand-induced method or the seed growth method is widely used for fabricating chiral nanocrystals. However, DNA, antigen–antibody and polymer additives are used to build chiral nanostructures. These chiral nanostructures can be of different lengths ranging from the nanoscale to the macroscale.Chiral nanomaterial-based biomimetic catalysts, a concept derived from simulating natural enzymes, possess a great number of artificial nanomaterials with superior catalytic activity.2 The actual enzymes are often limited due to their high cost, instability, difficult storage and purification, susceptible devitalized catalytically active sites and difficult mass production, which has led to their limited widespread utilization.3–5 Since high-performance peroxidase-like metal oxide NPs were found by Yan's group, diverse types of nanozymes have emerged which mimic natural enzymes with different catalytic properties.6–9 To date, the intractable problems of fabrication are still an issue and include precise structural design of bioinspired nanocatalysts, good stereoselectivity between the nanocatalysts and corresponding substrates, and high reactivity. Thus, well-defined biomimetic nanomaterials which can successfully mimic the active sites, specifically recognize the substrate binding center and cope with strict reactive environments are constantly desired by researchers. It is worth noting that chiral nanomaterial-based biomimetic catalysts as up-coming materials not only possess the general properties of many other nanocatalysts, but also have preferable selectivity and biocompatibility attributed to their specific chemical composition as they are enriched with natural amino acids or peptides during initial fabrication.
Along with a further systematic understanding of the structure–function effect of natural catalysts, some initiatives have been reported for enhancing the catalytic efficiency of nanomaterials. In particular, recent progress has shown that applicable size, suitable morphology and specific crystal facet exposure of biomimetic nanomaterials were prerequisites for potent catalytic interaction. Moreover, dimensional matching, surface modification and enzyme-like active sites have been adopted to promote selectivity to corresponding substrates. The assistance of external driving forces (light or magnetism) enables high performance. Collectively, these reformation principles depend on advanced techniques to provide easily tunable structures and flexible interaction properties to cope with complex biological environments.
Benefiting from the previous rapid development of the synthetic methods of nanocatalysts and tailoring of the reactive properties of chiral nanomaterial-based biomimetic catalysts to mimic natural active materials, many researchers have paid more attention to the biological utilization of these advanced materials. To date, research has demonstrated their potential applications in biological catalysis, such as cutting, theranostics, and peptide synthesis, and in many other biomedical fields.10
This study aimed to provide a comprehensive overview of enzyme-like chiral nanomaterial synthesis and structure mediated catalysis in biological applications (Fig. 1). First, we briefly introduce fabrication methodologies for chiral NPs and chiral nanoassemblies. Then, we illustrate many well-designed strategies to regulate the reactivity of the prepared bioinspired nanocatalysts with superior catalytic performance. Moreover, we summarize the processes of chiral nanomaterial induced biological catalysis and focus on the detailed interaction mechanism, for example, substrate selective bonding, electron transfer, transition state formation and final production output, and many biological applications such as peptide synthesis, theranostics, protein or DNA cleavage, tumor treatment and potential biomedical features are also discussed.11–14 A comprehensive review of the current challenges and future perspective of chiral nanomaterials for biomimetic catalysis is also included.
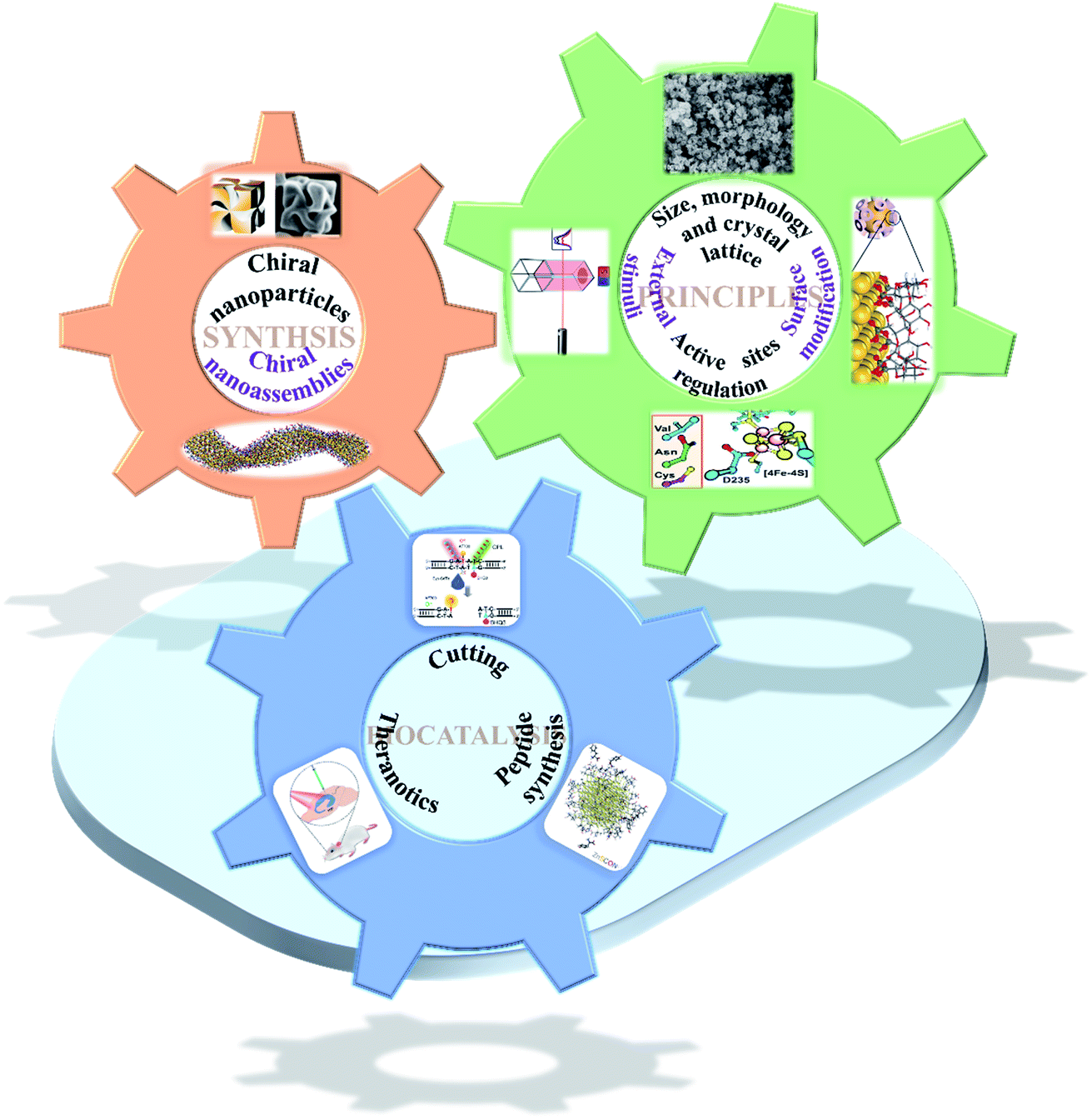 | ||
| Fig. 1 Scheme of chiral nanomaterial fabrication: (1) chiral nanoparticles.31 Adapted with permission from ref. 31. Copyright 2018 Springer Nature. (2) Chiral nanoassemblies.48 Adapted from ref. 48. Copyright© Copyright 2020 The American Association for the Advancement of Science. Smart strategies for preparing various species basis bioinspired catalysts of high performance: (3) size, morphology and crystal lattice.62 Adapted from ref. 62. Copyright© 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (4) Surface modification.72 Adapted from ref. 72. Copyright© 2016 American Chemical Society. (5) Active sites regulation.77 Adapted from ref. 77. Copyright© 2018 The American Association for the Advancement of Science. (6) External stimuli.78 Adapted from ref. 78. Copyright© 2017 Springer Nature. Biocatalytic applications of chiral nanomaterials. (7) Cutting.95 Adapted with permission from ref. 95. Copyright© 2018 Springer Nature. (8) Theranostics.98 Adapted with permission from ref. 98. Copyright© 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (9) Peptide synthesis.101 Adapted with permission from ref. 101. Copyright© 2019 Springer Nature. | ||
2. Synthesis of chiral nanomaterials
In recent years, various methodologies have been reported regarding the synthesis of chiral nanomaterials, and many researchers have summarized these methodologies.15 Chiral nanomaterials reported to date include: (1) individual chiral NPs and (2) complex chiral nanostructures/nanoassemblies.16–19 However, individual chiral nanomaterials can also be divided into two categories: chiral NPs which have a chiral shape, and those that acquired chirality by the interaction between a chiral ligand and an achiral core.20 Chiral nanostructures or nanoassemblies consist of individual NPs assembled into larger structures with external forces, such as DNA strands, antigens, antibodies and polymers, or internal driving forces, such as hydrophobic interactions, electrostatic interactions and hydrogen bonding, which can achieve a collective chirality by bringing asymmetry factors into their spatial geometry.21,22 Therefore, many chiral superstructures (helix, nanowires, etc.) can be acquired through the rational arrangement of basic building blocks. According to the differentiation of the main element composition, chiral nanomaterials can also be divided into chiral novel NPs, chiral semiconductor NPs, chiral metal oxides, chiral carbon nanomaterials and others.23–282.1 Chiral nanoparticles
Individual chiral NPs have distinct optical activity and bio-specific interaction provided many more opportunities for catalysis in the biological field. Their tunable chemical properties and tailored stereo configuration have provided them with superior catalytic activity.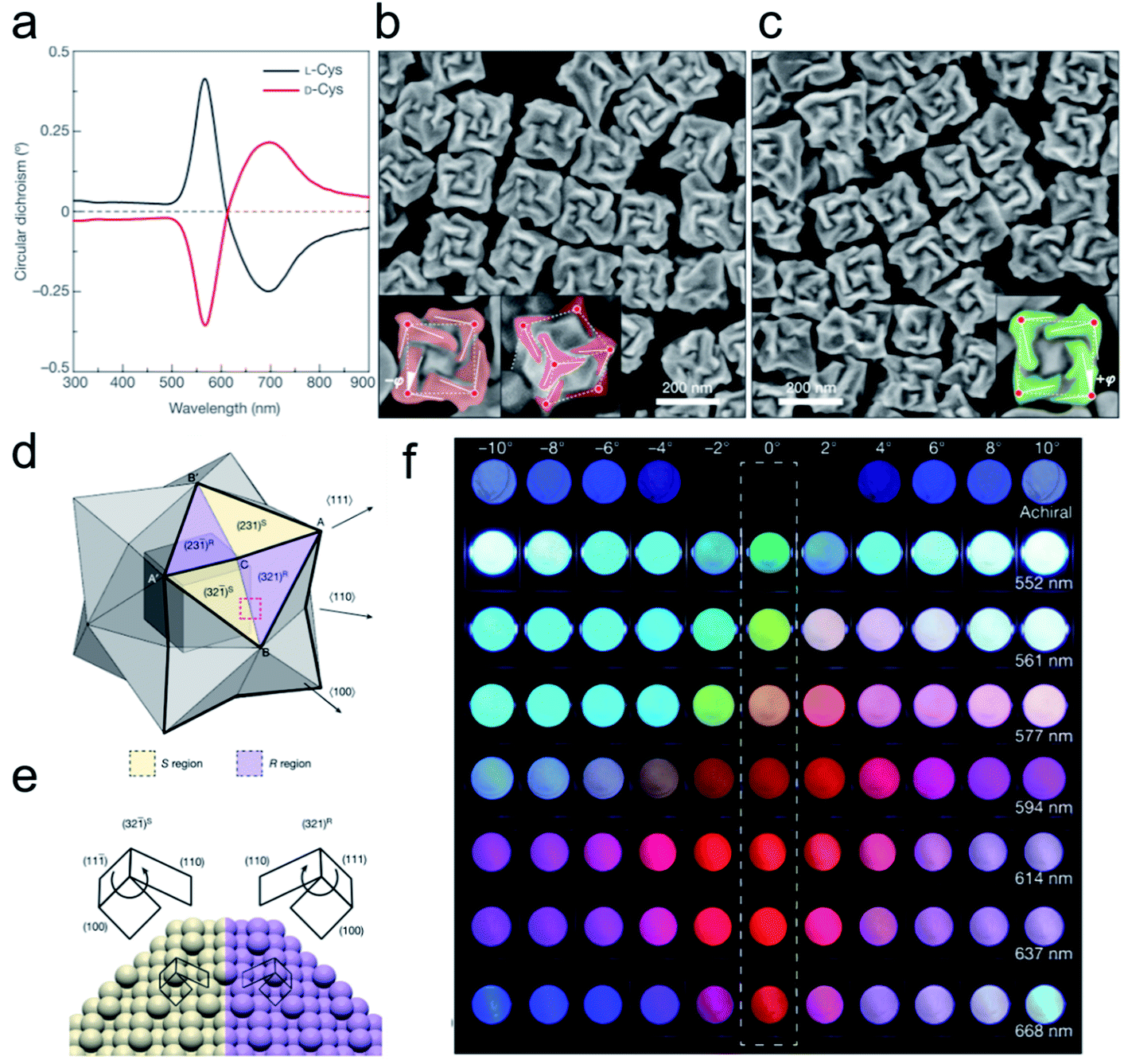 | ||
Fig. 2 Opposite handedness of three-dimensional plasmonic helicoids controlled by cysteine chirality transfer.31 (a) Circular dichroism spectra of chiral nanoparticles synthesized using L-Cys (black) and D-Cys (red). (b) SEM image of L-Cys nanoparticles. The highlighting in the insets illustrates the fact that the edges (solid lines) are tilted by an angle −ϕ with respect to the vertices (red dots) and cubic outline (dashed lines), as viewed along the [100] (left) and [111] (right) directions. (c) SEM image of D-Cys nanoparticles. The inset highlights the tilted edges (solid lines), cubic outline (dashed lines) and tilt angle (+ϕ). Mechanism of chirality evolution through the interplay between the enantioselective binding of molecules and the asymmetric growth of high-index facets. (d) Schematic of a stellated octahedron, differentiated by high-index facets consisting of [321]S, (S region, yellow) and [321]R (R region, purple) configurations. The vertices of the [111], [100] and [110] directions are indicated as A, B and C, respectively; A′ and B′ refer to the symmetric points of A and B, respectively. (e) Comparison of the atomic arrangement of the (321)R and (32![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) )S gold surfaces for the region indicated by the red dotted box in a. The conformation at ‘kink’ sites is defined by the rotational direction of low-index microfacets in the sequence (111) → (100) → (110): clockwise, R region; anticlockwise, S region. (f) Polarization-resolved colors of light transmitted through seven different 432 helicoid III solutions with different λmax values (shown on the right) and an achiral nanoparticle solution (top row). The rotational angle of the analyzer was increased from −10° (left-most) to 10° (right-most) (see Methods). An angle of 0°, indicated by the dashed box, represents cross-polarized conditions. Adapted with permission from ref. 31. Copyright 2018 Springer Nature. )S gold surfaces for the region indicated by the red dotted box in a. The conformation at ‘kink’ sites is defined by the rotational direction of low-index microfacets in the sequence (111) → (100) → (110): clockwise, R region; anticlockwise, S region. (f) Polarization-resolved colors of light transmitted through seven different 432 helicoid III solutions with different λmax values (shown on the right) and an achiral nanoparticle solution (top row). The rotational angle of the analyzer was increased from −10° (left-most) to 10° (right-most) (see Methods). An angle of 0°, indicated by the dashed box, represents cross-polarized conditions. Adapted with permission from ref. 31. Copyright 2018 Springer Nature. | ||
2.2 Chiral nanoassemblies
Chiral nanoassemblies, composed of individual NPs orderly arranged into superstructures, showed hierarchically collective conformations and special chiroptical properties.35–37 The nanoassemblies with intrinsic chiral morphology did not just depend on whether the building blocks possessed chiral units. In this section, we mainly present two common methods for constructing chiral nanoscale assemblies. First, exogenous additives, such as DNA or polymers, play an essential role in chiral nanostructure assembly.38,39 Benefiting from their specific bridging effect, individual NPs orderly assemble into different geometries.40 In the past decades, many asymmetric configurations of chiral assemblies have been reported based on DNA techniques, such as dimers, trimers, pyramidal assemblies and helix structures.41–44 The obtained chiroptical activity is mainly derived from the dissymmetry and free rotation of the nanoassemblies in their spatial arrangement. Recently, chirally engineered Co3O4 supraparticles (SPs) were prepared by assembling 2–3 nm cysteine stabilized cobalt oxide NPs in the presence of dithiol poly(ethylene glycol) (PEG) and PEG methyl ether, which efficiently drive particles to assemble into superstructures.45 Noncovalent forces mediated the assembly process, electrostatic interactions, hydrophobic interactions, hydrogen bond formation, coulombic forces and van der Waals forces, and were always manipulated by the polarity of the solvent, pH changes and ion strength. Chiral cysteine ligand-modified cadmium telluride nanoparticles (CdTe NPs) can be homogeneously assembled into same handedness semiconductor helices by changing the solvent from water to methanol.46 The electrostatic attraction forces between chiral CdTe NPs drive the orderly arrangement of semiconductor subunits into specific chiral dissymmetric collections. Critical performance indicators, such as pitch length, diameter, and thickness, of the prepared chiral helices can efficiently regulate their optical properties. Therefore, the solvent-mediated chiral controllable assembly promotes the topography-dependent optical activity. In another representative study, Kotov and co-workers elaborately explained the cooperative effect of different solvent proportions, pH changes, precursor addition and coordination bonding interactions in controlling the formation of chiral assemblies.47 The mixture solvent, water and methanol, with different ratios directly affects the final output and conformation of the nanostructures. In addition, the acid environment facilitated the increased ionization of surface chiral cysteine leading to the generation of uniform twisted fibrous bundles. Moreover, precise tuning of the amount of Cd and Cys can result in different stacked morphologies and achieve highly purified enantiopure isomers. And also, the coordination effect induced by the interstitial lattice possibly produces hierarchical chiral mesoscale assemblies with excellent chiroptical activity. This benefits their flexible structure tunability and specific interspace, and the chiral nanoassemblies provide more possibilities for high performance biological catalysis. Recently, a series of well-designed hierarchically programmed chiral nanoassemblies were reported (Fig. 3).48 The twisted spike configurations were built up through orderly stacking of chiral amino acid stabilized nanoplatelets. In particular, cetyltrimethylammonium bromide (CTAB) surfactant incorporation efficiently decreased the electrostatic repulsion among the particles by altering their negative charge to a positive charge, which facilitated their staggered pattern to edge to edge not face to face. Moreover, the mutually competitive interaction of anisotropic electrostatic and elastic forces determined their complex chiral architecture formation. Thus, the advanced optical activity and hierarchical structure ensured their superior physicochemical properties in biomimetic catalysis. Therefore, encouraged by the flexible controlled methods and diverse conformations, chiral nanoassemblies may enable more biotransformation processes.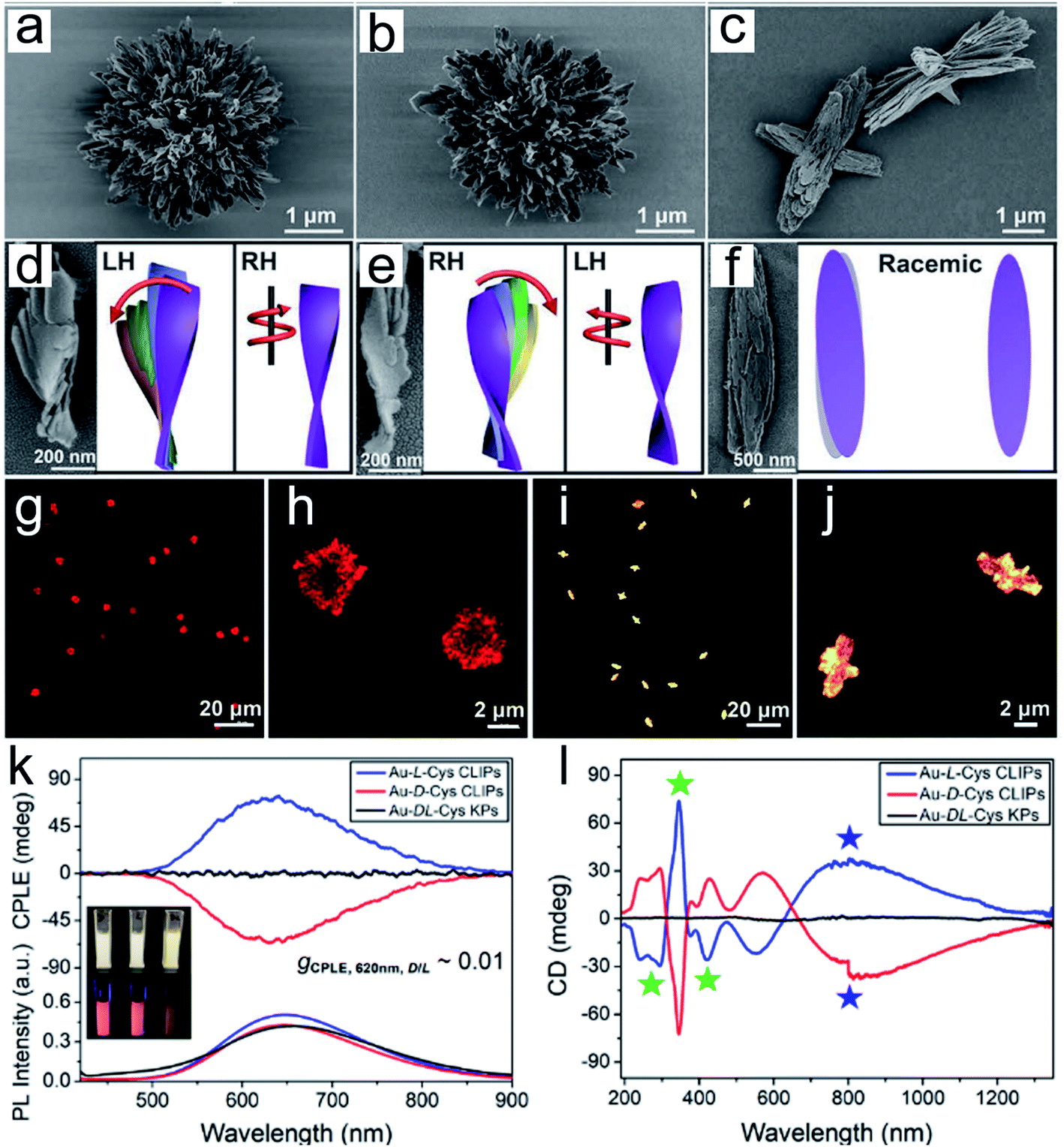 | ||
| Fig. 3 Gold thiolate hierarchically organized particles (HOPs) with cysteine surface ligands.48 (a to c) SEM images of Au-L-Cys (a), Au-D-Cys (b) coccolith-like particles (CLIPs) and Au-DL-Cys (c) kayak particles with a low magnification. (a to c) Enlarged SEM images of Au-L-Cys (a), Au-D-Cys (b) CLIPs and Au-DL-Cys (c) kayak particles. (d to f) SEM images and corresponding schematic illustrations of segments of Au-L-Cys (d), Au-D-Cys (e) and Au-DL-Cys (f). Statistical analysis of SEM images for 100 assembly segments indicates that the pitch of individual nanoribbons in the stacks is 1300 ± 123 nm and their average width is 16 ± 1.8 nm, while the average angle between two neighboring nanoribbons in the stack is 7 ± 0.7° with the pitch of nanoribbon stacks being 820 ± 10 nm. (g to j) Confocal microscopy images of Au-L-Cys (g and h) CLIPs, and Au-DL-Cys (i and j) kayak particles. Chemical properties of Au-Cys dispersions in dimethylformamide and optical CLIPs. (k and l) CPLE (k) and CD (l) spectra of Au-L-Cys (blue), Au-D-Cys (red) CLIPs and Au-DL-Cys (black) kayak particles. Inset in (k): photos of Au-L-Cys, Au-D-Cys and Au-DL-Cys dispersions under daylight and UV light illumination. Adapted from ref. 48. Copyright© Copyright 2020 The American Association for the Advancement of Science. | ||
3. General strategies for promoting biomimetic chiral nanocatalysts with high performance
To our knowledge, natural active enzymes, apoprotein and active catalytic sites, have exclusive selectivity and high catalytic activity to their corresponding substrates. However, the fabrication of enzyme-like artificial chiral nanocatalysts to achieve a greater biological transition has always been a problem which needs to be resolved. To construct high performance nanocatalysts to mimic actual active materials, a deep understanding of the basic interaction mechanisms between natural enzymes and their substrates is necessary. To date, the most appropriate theoretical foundation is the induced-fit hypothesis which suggests that the active site of enzymes can change their conformation to fit the substrates like a well-fitting glove. Recent progress has displayed many advanced methodologies for tailoring the building blocks and elevating the matching domain between artificial chiral nanocatalysts and substrates. Emerging research has since elaborated many biomimetic nanoenzymes, such as metal organic frameworks (MOFs), single atom catalysts (SACs), metal or metal oxide/clusters and carbonic nanomaterials, which exhibit their unique catalytic advantages to their corresponding substrates.49–58 Moreover, the multiple constitution and tunable physicochemical properties of the prepared biocatalytic chiral nanomaterials enable the requirements of complex environments and substrate diversity.59 Therefore, inspired by the structure–function effect of the enzyme, we provided versatile strategies for more precise and efficient sculpturing of artificial nanomaterials to obtain high catalytic ability in this section.3.1 Size, morphology and crystal facets
It is widely acknowledged that the size of nanomaterials has an effect on their catalytic ability. To confirm this, Colvin et al. illustrated the antioxidant activity of monodisperse cerium oxide (CeO2) nanocrystals of different sizes.60 Two different diameters of NPs, 3.8 nm and 8.2 nm, were chosen to examine the scavenging effect of reactive oxygen species (ROS). Based on gas chromatography-mass spectrometry (GC-MS) and luminol assay experimental results, they found that the smaller size nanoceria coated with a thinner surface polymer exhibited higher reduction capacity towards hydrogen peroxide (H2O2) than that of the 8.2 nm CeO2 NPs. Moreover, the morphology of nanocatalysts played an essential role in the catalytic processes. In particular, space confinement synthesis of functional nanocatalysts was performed by Klajn's group to study their specific physicochemical properties.61 They used ultraviolet or visible light to regulate molecule diffusion among confined nanoflakes through their surface light-response ligands, which provided more possibilities for the fabrication of specific geometrical nanomaterials. Recently, five different shaped manganese oxide (Mn3O4) nanomaterials, flowers, cubes, polyhedrons, hexagonal plates and flakes, were fabricated by Mugesh and co-workers to explore the relationship between multi-enzyme like activity and their intrinsic morphologies.62 The Mn3O4 nanoflowers displayed a superior multi-catalytic activity compared with the other four configurations due to their larger size, porosity and higher surface area. Chen, Zhou and colleagues discovered that palladium (Pd) nanocrystals with octahedron morphology exhibited higher ROS removal ability than the cube conformation and showed better antibacterial behavior.63 The crystal facets of nanocatalysts also had a crucial role in bioconversion.64 A series of vanadium pentoxide (V2O5) nanomaterials with redox and glutathione peroxidase (GPx)-like activity were used to investigate the facet effects.65 Four different morphologies of V2O5 nanomaterials, nanowires, nanosheets, nanoflowers and nanospheres, with the same orthorhombic nanocrystals but dissimilar crystal facet exposure displayed distinct redox activity. Surprisingly, the researchers further confirmed that the surface area was of little relevance to their catalytic ability, but the different exposed crystal facets of each shaped V2O5 nanomaterial led to distinct H2O2 reduction capacities. The TEM and density functional theory (DFT) calculations showed that 100 and 010 facets of V2O5 exhibited greater catalytic ability than the 001 facet attributed to their high H2O2 molecule conjunction property and generated a vanadium-peroxide intermediate on their surface.3.2 Surface functionalization
Surface modification of engineered nanocatalysts is considered to be an efficient technique for manipulating the biocatalytic activity.66–71 For example, Xia's group used cyclodextrin (CD) molecules as a stabilizer and reducing agent to construct a multifunctional gold nanoplatform.72 The β-CD functionalized Au NPs displayed a host–guest interaction pathway for the fluorescence sensing, controllable self-assembly and glucose oxidase and horseradish peroxidase mimicking activity. The DFT analysis demonstrated that the CD coated Au NPs accelerated electron transfer from the gold atom surface to ligands which efficiently improved their catalytic efficiency. It is well known that natural enzymes intrinsically possess selectivity and recognize their substrates benefiting from the secondary structures constituted by a series of amino acids or metallic active sites. It is noteworthy that chiral recognition, as a branch of molecular recognition, has attracted the attention of many researchers due to their superior selectivity. Chiral ligands, amino acids or peptides, capped on the surface of NPs or inherently changed morphology of the nanostructure can provide specific physiological functions to the prepared nanomaterials compared to those without modifications. Therefore, recent studies examined the dynamic process between chiral penicillamine functionalized Au NPs and transferrin protein (Fig. 4).73 The chiral ligands predominantly determined their interaction orientation and changed their original conformation in order to match the absorbed transferrin. The D-penicillamine modified Au NPs tended to bind to the a2-side face of transferrin, and had a more negative charge, which was different from the L-type and racemic penicillamine functionalized Au NPs tended to interact with the a1-side. However, the Au NPs(D) and transferrin nanocomplex provided a relative outside binding site which was preferable for capturing more receptors than that of the Au NPs(L) and Au NPs(D/L) conformations.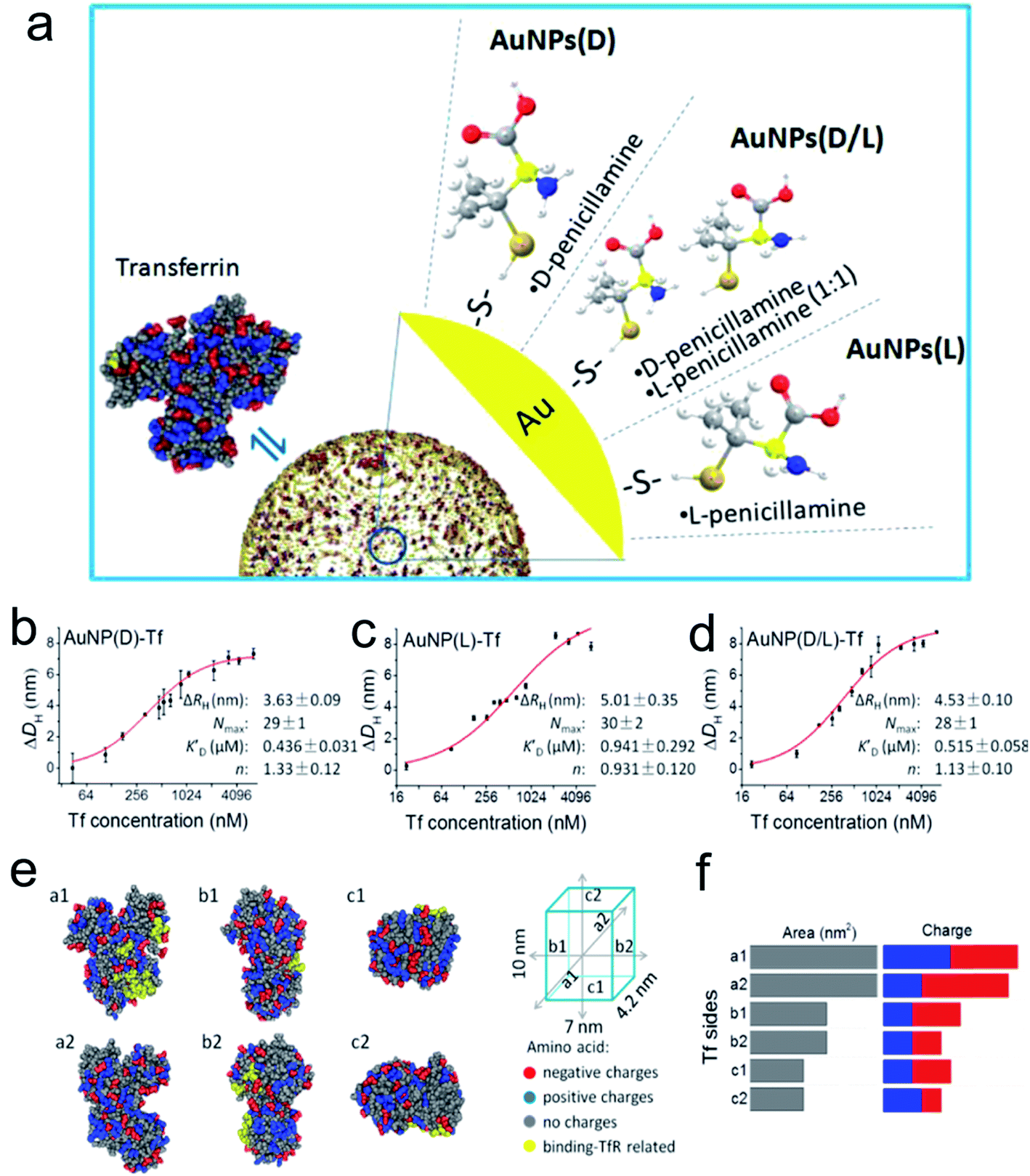 | ||
| Fig. 4 (a) Schematic diagram of modification of the AuNP surface with chiral molecules and their interaction with Tf. Quantitative analysis of Tf corona formation on Pen-AuNPs. (b–d) The hydrodynamic radius measurements of AuNP(D)-Tf (b), AuNP(L)-Tf (c), and AuNP(D/L)-Tf (d), plotted as a function of the concentration of Tf. The final concentrations of all 14 nm AuNP solutions were 150 μg mL−1, and the DLS was measured after mixing AuNPs with the desired concentrations of Tf for 2 h at room temperature. The curves (red solid lines) were fitted according to the Hill equation, and the best-fit parameters are listed on the right. (e) The distribution of amino acids with charges and binding-TfR-related residues on the six side faces of Tf. The 3D structure of Tf was described using a space-filling model of Tf (PDB ID: 4X1B) with color-coded types of charge (red, negative charge; blue, positive charge; gray, no charge). Residues marked by yellow represent the binding-TfR-related residues in the C-lobe and N-lobe of Tf. (f) Statistical distribution of charged amino acids on each side of the virtual cube of Tf. All data are the mean ± SD of three replicates.73 *P < 0.05. Adapted with permission from ref. 73. Copyright© 2017 American Chemical Society. | ||
3.3 Precise regulation of the intrinsic active sites within biomimetic nanocatalysts
It is widely known that the natural reactive center of enzymes has remarkable conversion efficiency in organisms. Recently, many artificial nanomaterials were synthesized to mimic natural enzymes using well-designed organic synthesis techniques.74 Zhang et al. fabricated a Mn4Ca-cluster to mimic the oxygen-evolving center (OEC) of the photosynthesis process (Fig. 5a–f).75 The as-prepared artificial Mn4Ca-cluster involved a metal–oxygen core and water binding sites, which highly resembled the cubane domain of natural OEC. In addition, the clusters identically complied with the four electron redox transformation principle that was similar to the native asymmetric structure. Similarly, Wu and colleagues also constructed a bioinspired Cu4O4 cubane to mimic the conformation of Mn4CaO5.76 Taking advantage of the good coordination effect between the (3-methoxy-salicylidene)-amino acid (glycine, glutamic acid) chelates and Cu(OAc)2, they obtained a series of high yield cuboidal [(LGly-Cu)4] and [(LGlu-Cu)4] artificial catalysts. Their turnover frequency of the two products was 267 s−1 at 1.70 V and 105 s−1 at 1.56 V, respectively. The outstanding conversion efficiency of the Cu4O4 moieties was mainly attributed to their multielectron transfer, which provided enough driving force for water oxidation. As another significant natural catalytic site paradigm, Lu and co-workers reported a heme-[4Fe–4S] cofactor in cytochrome c peroxidase, which served as an artificial sulfite reductase for multielectron redox reactions (Fig. 5g).77 For the requirements of the six-electron and seven-proton reaction, they adjusted the spatial interaction environment of [4Fe–4S] and its corresponding binding sites for efficient targeting. The selective mutation of D235V, D235N and D235C points significantly enhanced their bonding opportunities to the [4Fe–4S] cavity core through hydrogen bond interactions. The similar structure and function of the artificial nanocomplex achieved efficient conversion comparable to the natural sulfite reductase, and it is expected to be a strong potential candidate for catalysis.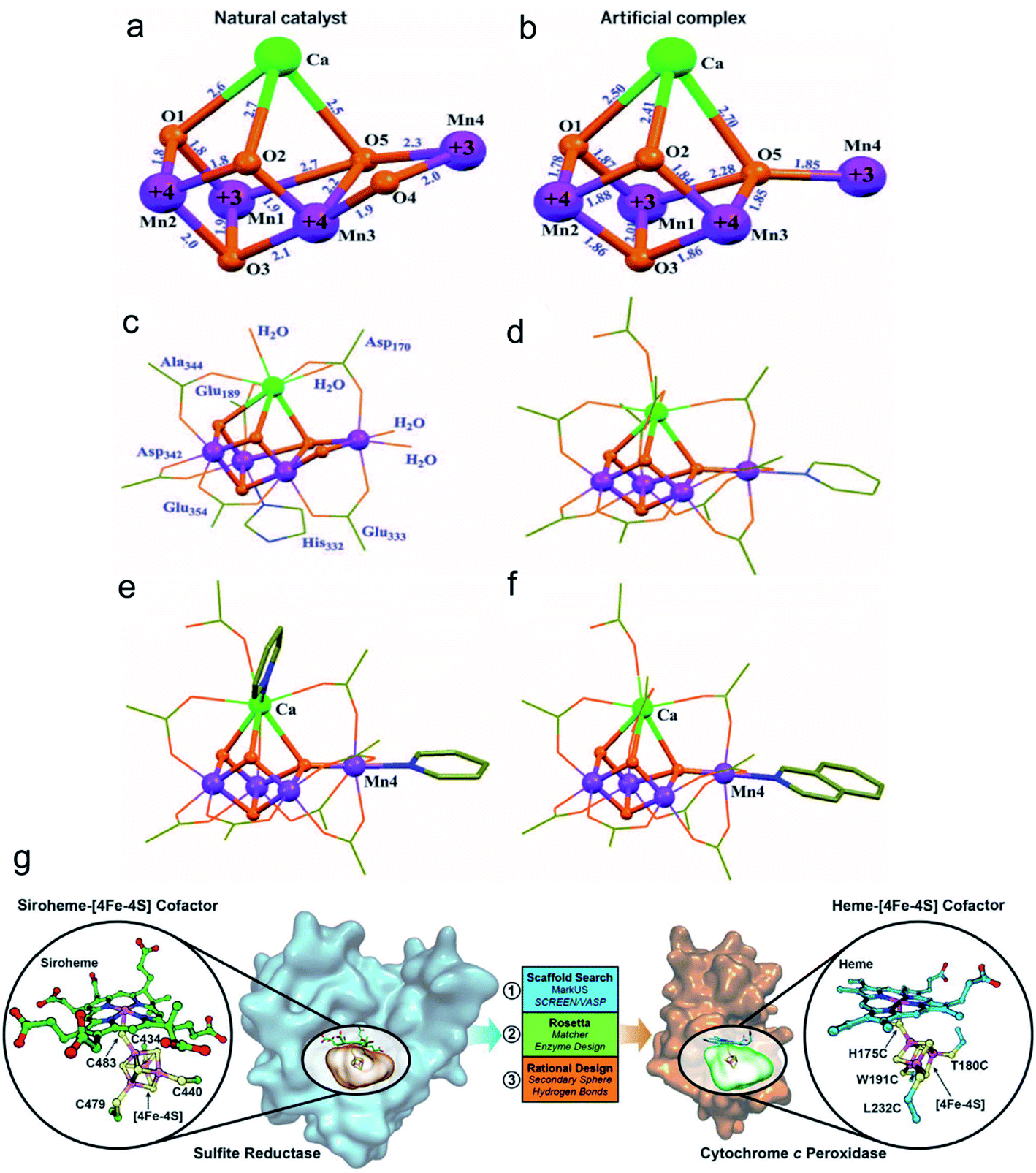 | ||
| Fig. 5 Crystal structures of the native OEC and the synthetic Mn4Ca complex I.75 (a) Mn4CaO5 core of the native OEC (8). (b) Mn4CaO4 core of I. (c) Structure of the native OEC, including ligating protein sidechains and water molecules. (d) Structure of I, including all ligand groups. Structures of complexes with exchanged terminal ligands. Adapted from ref. 75. Copyright© 2015 The American Association for the Advancement of Science. The complexes differ from I by ligand exchange either on (e) Ca(II) or (f) Mn4(III). (g) Design of a [4Fe–4S] binding site in CcP to mimic the heme[4Fe–4S] center in native SiR.77 Adapted from ref. 77. Copyright© 2018 The American Association for the Advancement of Science. | ||
3.4 External stimuli forces
As a noninvasive method, a biosystem allows precise regulation of spatio-temporal dimensions, the magnetic field and light providing more potent catalytic ability. The magnetic field as a universal resource has a wide distribution worldwide. Many species, such as pigeons, butterflies and migratory birds, can adjust their flight by perceiving magnetic field changes. Moreover, the physicochemical behavior of magnetic NPs can also be regulated by altering the magnetic intensity, frequency and orientation. Recently, Minko et al. used magnetic NPs to establish a nanoplatform to remotely regulate a biocatalysis process under a specified magnetic intensity intervention (Fig. 6a and b).78 The enzyme encapsulated magnetic nanocompartment and substrate embedded magnetic cargo can be quickly merged leading to their degradation and the release of therapeutic drugs attributed to external magnetic stimuli forces between the two individual magnetic units. Kim and co-workers observed that magnetized Au NPs can accelerate the conversion efficiency of somatic fibroblasts to dopamine neurons when exposed to a specific magnetic intensity and frequency in vitro and in vivo.79 Furthermore, magnetic iron oxide nanocubes coated with tissue plasminogen activator molecules and serum albumin as a thrombolytic scaffold displayed a superior advantage in thrombolysis therapy by controlling the magnetic field.80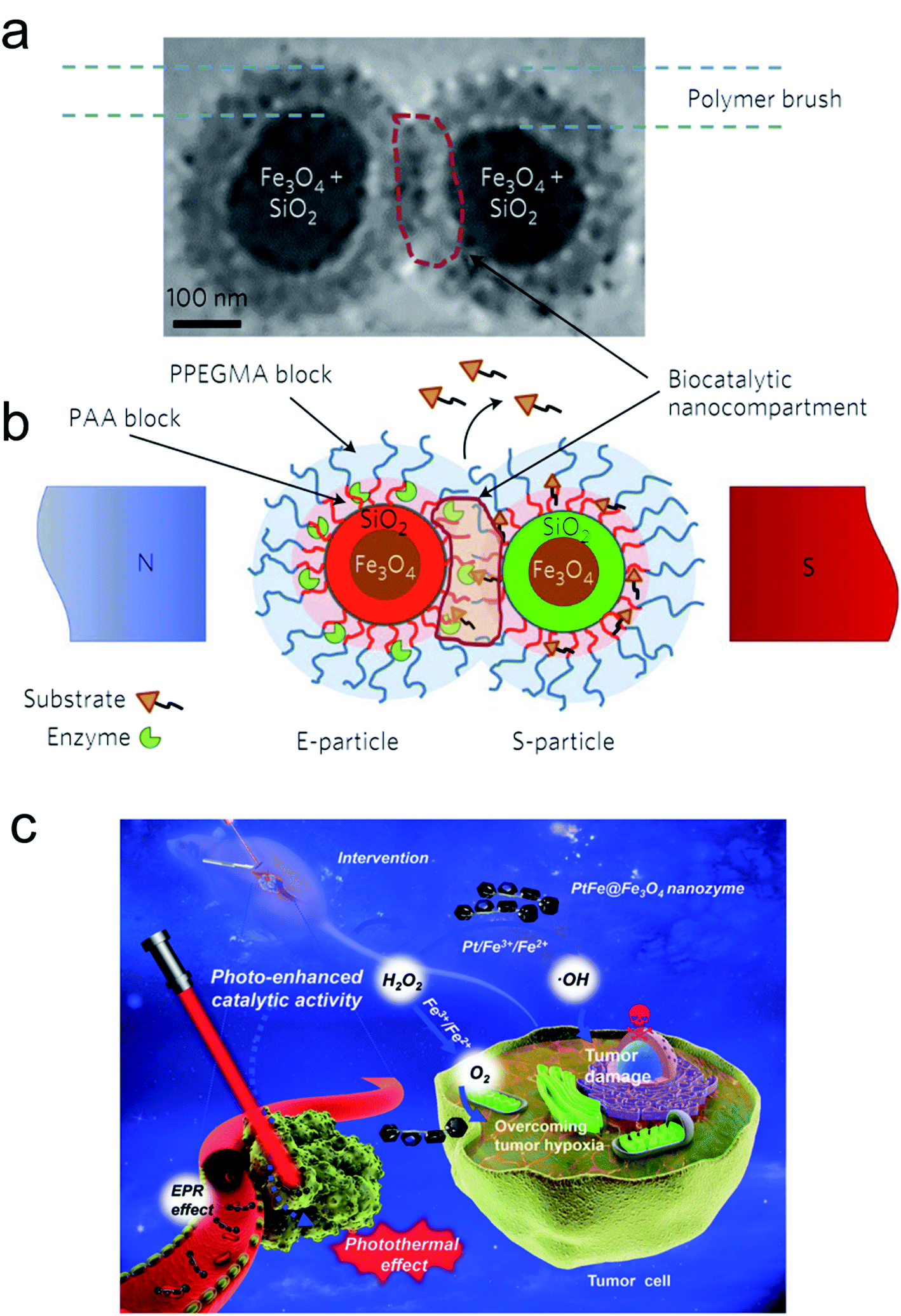 | ||
| Fig. 6 E- and S-type superparamagnetic nanoparticles carrying the enzyme and the substrate.78 (a and b) Cryo-transmission electron microscopy (cryo-TEM) image (a) and schematic (b) explaining the concept of the magnetic-field-triggered biocatalysis. The particle superparamagnetic core is made of Fe3O4 nanoparticles enveloped by silica. The silica envelope is labelled with covalently bound fluorescent dyes (red for E-particles and green for S-particles). In the magnetic field, due to dipole–dipole interactions, the particles are brought into contact, so that the brush-like double-layer shells merge and intertwine, enabling interactions between the enzyme and substrate. The inner layers of the brush-shell are made of polyacrylic acid (PAA), which carries conjugated molecules of enzymes and substrates and provides the acidic environment for hydrolytic reactions. The external shell of the poly(ethylene glycol methyl ether acrylate) polymer (PPEGMA) secures a barrier function to block ‘unauthorized’ or premature reactions of the enzyme and the substrate. The biocatalytic reaction is localized within the biocatalytic nanocompartment, which is generated in the magnetic field. The reaction is monitored by detecting the released cargo molecules. Adapted with permission from ref. 78. Copyright© 2017 Springer Nature. (c) Schematic of the photo-enhanced tumor catalytic therapy based on the PtFe@Fe3O4 nanozyme.84 Adapted with permission from ref. 84. Copyright© 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Light irradiation is another effective tool for bio-nano interactions.81 A large number of metallic nanomaterials have good light absorbance, especially near infrared (NIR) light, which has great potential in biological applications due to its deep penetration in vivo.82 Our group fabricated optical shell-satellite assemblies based on DNA techniques.83 The chiral Au–Ag shell-satellite nanostructures could generate a high yield of ROS under 532 nm right circular polarized light, which served as a photodynamic inducer for tumor treatment. In addition, considering its deep penetration and the accelerated electron transfer effect of NIR light, Liu's group designed a NIR light-assisted multi-metal incorporated nanoplatform for deep pancreatic cancer therapy (Fig. 6c).84 The as-prepared PtFe@Fe3O4 nanostructures integrated peroxidase and catalase activity which enabled tumor treatment in a hypoxia environment. The NIR light not only triggered high-energy electron generation but also facilitated the electron–hole separation which obviously promoted catalytic activity. The Fe3O4 NPs can easily capture electrons in the presence of H2O2 and constantly transfer them to the surface of Pt atoms leading to more production of •OH and further inducing subsequent cascade reactions.
3.5 Biocatalytic applications of chiral nanomaterials
Chiral nanomaterials as bioinspired nanozymes with simple and diverse synthetic methods, abundant precursor sources, and extreme pH and temperature tolerance, which provide more opportunities for tuning their sizes and morphologies with chiral ligands, promoted the atomic spatial arrangement of nanocrystalline structure for matching the domain of biomolecules, facilitated the apoprotein-like selectivity, which enabled corresponding substrate recognition like the function of apoenzymes, enhanced their catalytic performance and increased their biocompatibility.85 Encouraged by their superior characteristics, artificial chiral nanomaterials were widely utilized to investigate the interactions with DNA, protein, small molecules and other biological media.44,86–94 In the past few years, our group has consistently focused on the fabrication of chiral bionanomaterials to mimic native active materials with the aim of achieving great catalytic outcomes.3.6 Cutting
Inspired by the well-defined dimensional structure and exclusive selectivity, chiral nanocatalysts can provide high performance for biomacromolecules, such as DNA, protein and others. It is well known that bioenzymic guided degradation for DNA cutting has attracted much attention due to its genetic significance to human evolution. For instance, we first reported endonuclease-like cystine stabilized cadmium telluride (CdTe) NPs for shearing specific sites of salmon sperm DNA, while another six chiral ligand-modified CdTe NPs had no effect under the same treatment (Fig. 7).95 In particular, the G and C bases tended to anchor on the CdTe crystal surface, while the “ATAT” sequences were adsorbed on the marginal edges, which generated a specific “pocket” and enhanced DNA cleavage. The phosphodiester bond between the A and T bases was obviously stretched when the CdTe NPs embedded in the DNA pocket. Therefore, the 1839 base pairs of DNAs can be selectively cut at specific segments of GATATC between the T and A bases due to in situ accumulation of large amounts of hydroxyl radicals under 405 nm circular polarized light (left- or right-handed) irradiation when pretreated with D-Cys or L-Cys CdTe semiconductor QDs. Furthermore, a simplified 90 bp DNA model with different mutations was used to screen the necessary fragment lengths for DNA cutting and further verify that the GATATC sequence was an essential section for DNA scission. As a result, the suitable size, high affinity and special truncated tetrahedron-shape of the chiral CdTe NPs enabled a preferable match to certain DNA strand pockets and achieved precise cutting. Recently, chiral cysteine modified carbon dots (CDs) showed enantioselectivity to supercoiled DNA and successfully catalyzed the DNA into a nicked open-circular conformation through an intercalative interaction (Fig. 8).96 The D-CD configuration displayed higher affinity than the L-CD configuration, which was attributed to their stronger hydrophobic interaction and hydrogen bond generation at the CDs-DNA interfaces. In addition, in situ production of hydroxyl radicals efficiently broke the phosphodiester bond leading to the stereochemical structure transformation but retained their initial genetic properties, which was similar to the topological rearrangement of topoisomerase I. Another study by our group focused on protein cutting performance with the assistance of a protease-like chiral semiconductor (Fig. 9a–f).97 Chiral Cu2−xS NPs were first prepared using a simple one-pot synthetic method with a g-factor value up to 0.01. Penicillamine stabilized Cu2−xS NPs showed good selectivity for bovine serum albumin (BSA) and cut it into two parts under 660 nm circular polarized light irradiation for 12 hours. Higher affinity of the L-Pen functionalized Cu2−xS than that of D-Pen modified NPs to BSA promoted more ROS production under light which efficiently accelerated BSA hydrolysis. Thus, chiral nanocatalysts have a higher efficient route for the cleavage of biomacromolecules.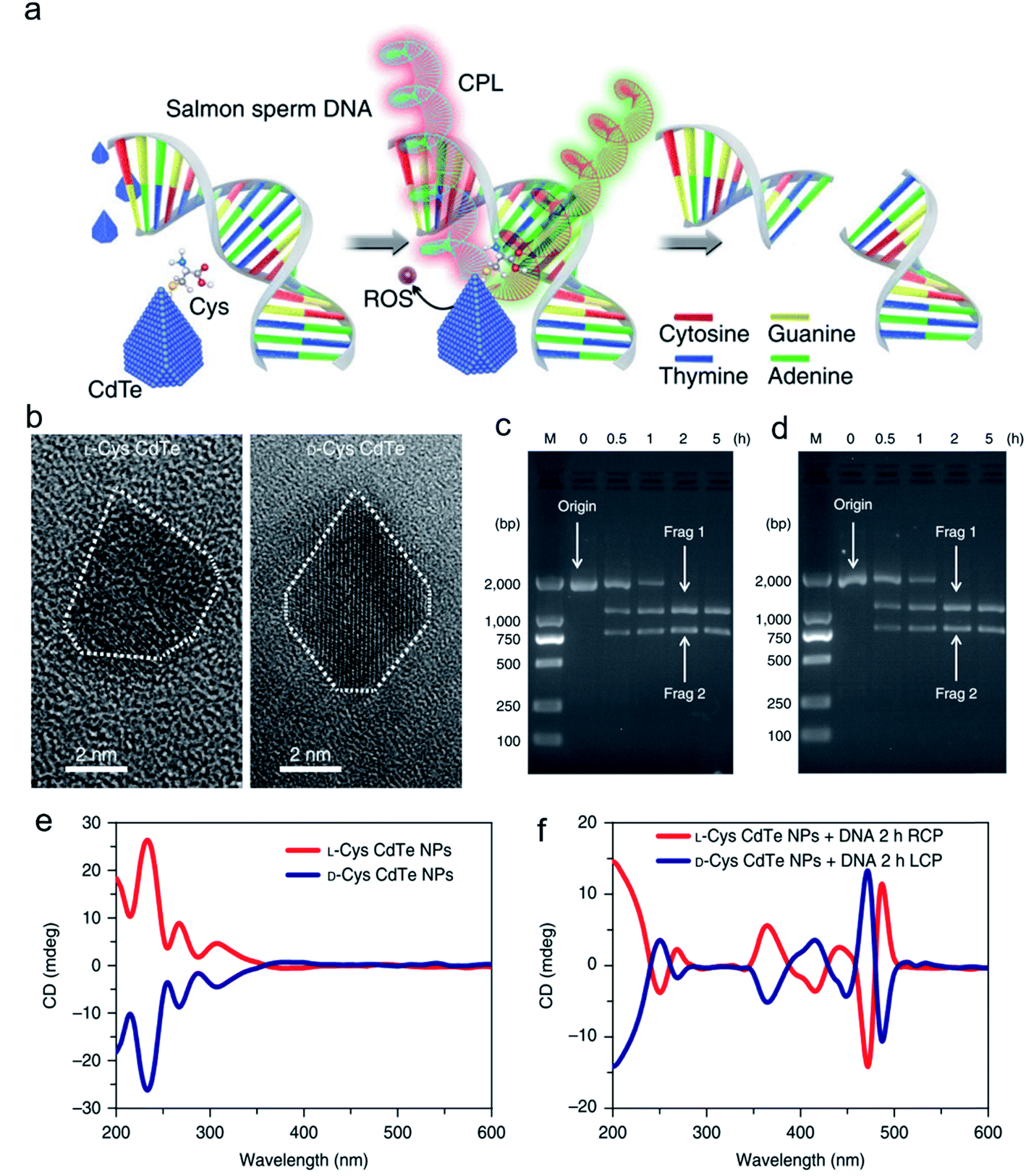 | ||
| Fig. 7 Chiral CdTe nanoparticle preparation and characterization. (a) Schematic illustration of chiral CdTe-based specific DNA cleavage under CPL irradiation. Site-selective DNA cleavage. (b) High-resolution TEM images of truncated tetrahedron-shaped (white dashed areas) L-Cys and D-Cys CdTe nanoparticles. (c and d) Electrophoresis images of L-Cys CdTe (c) and D-Cys CdTe (d) nanoparticles with 1839 bp DNA illuminated with 405 nm RCP/LCP for 2 h. The two DNA fragments after cleavage are denoted as frag 1 and frag 2. (e) CD spectra of L-Cys and D-Cys CdTe nanoparticles (NPs). (f) CD spectra of L-Cys and D-Cys CdTe nanoparticles mixed with 1839 bp DNA and illuminated under 405 nm RCP/LCP for 2 h. Strategy for site-selective DNA cleavage.95 Adapted with permission from ref. 95. Copyright© 2018 Springer Nature. | ||
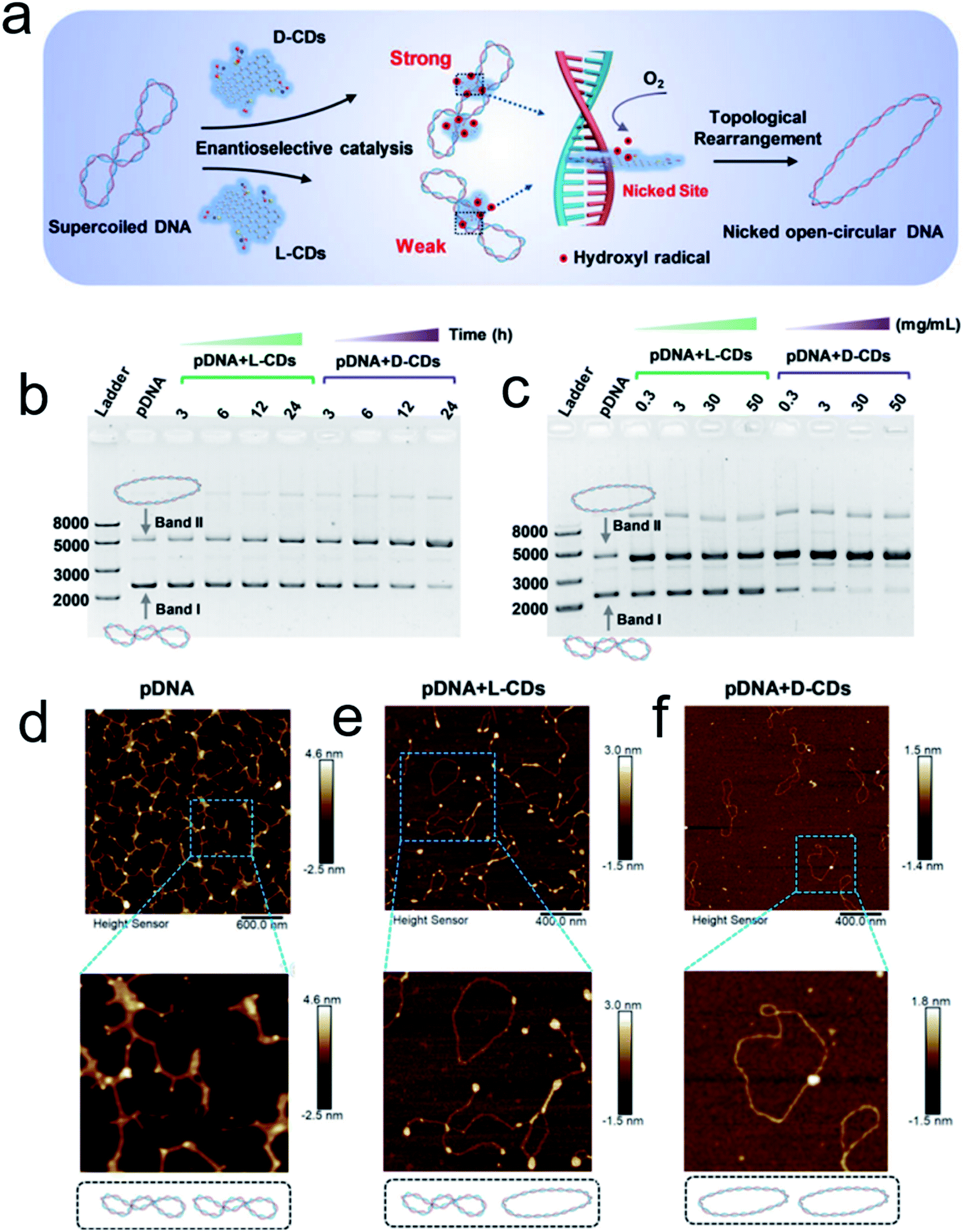 | ||
| Fig. 8 (a) Schematic illustration of chiral CDs mediated topological rearrangement of supercoiled DNA with enantioselectivity. D-CDs can intercalatively bind with the DNA double helix more strongly than L-CDs. The intercalative CDs catalyzed the production of hydroxyl radicals and cleaved the phosphate backbone in one strand of the double helix, leading to a topological transition of supercoiled DNA. Agarose gel electrophoresis and AFM analysis of the topological rearrangement of pDNA treated with chiral CDs. (b) Agarose gel electrophoresis analysis of pDNA treated with L-CDs and D-CDs for 3, 6, 12, and 24 h, respectively. The mass concentration of CDs was 30 mg mL−1, and the concentration of pDNA was 60 ng μL−1. (c) Agarose gel electrophoresis analysis of pDNA treated with L-CDs and D-CDs with concentrations ranging from 0.3 to 50 mg mL−1 for 24 h. (C–E) AFM image of pristine pDNA (d), pDNA treated with L-CDs (30 mg mL−1) for 24 h (e), and pDNA treated with D-CDs (30 mg mL−1) for 24 h (f).96 Adapted with permission from ref. 96. Copyright© 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
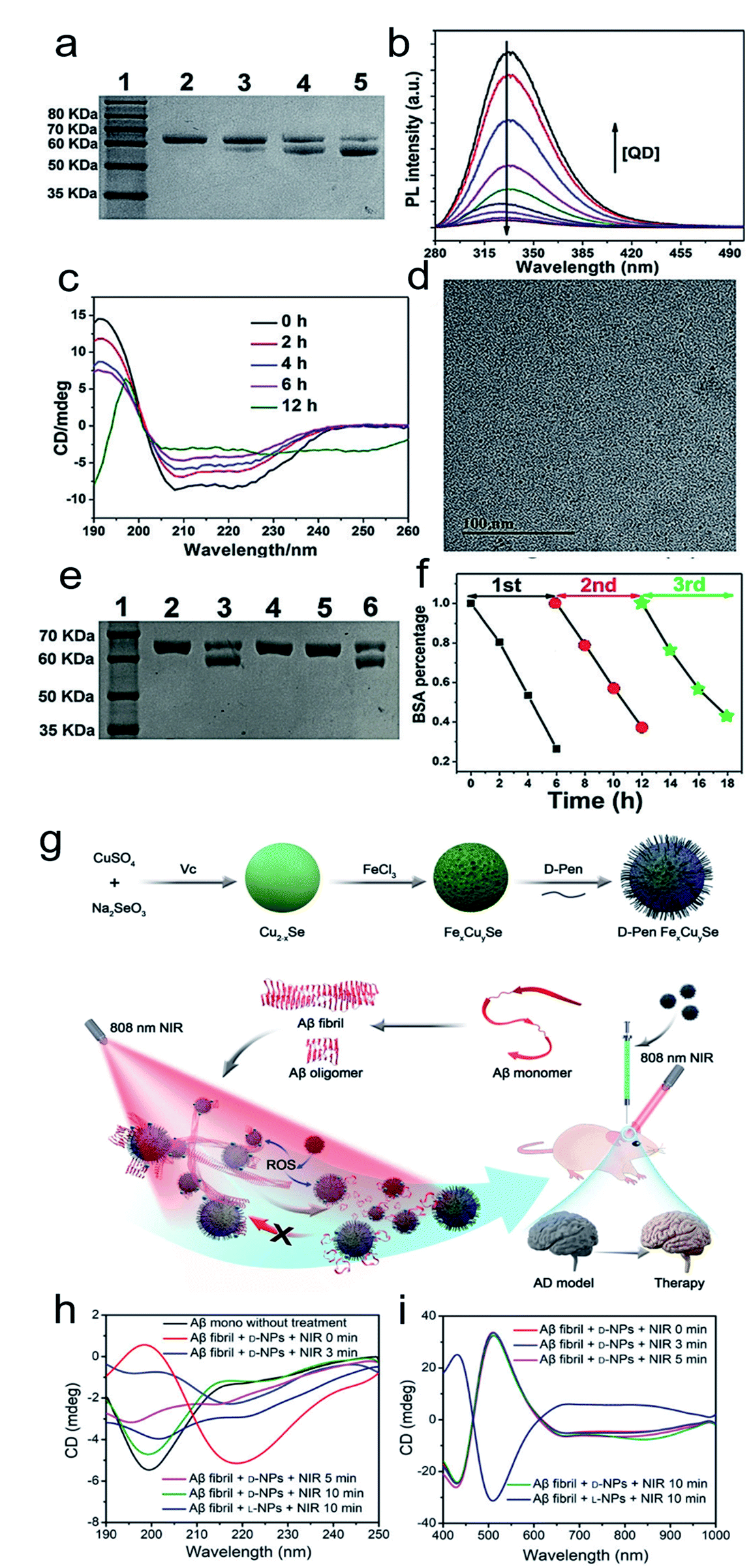 | ||
| Fig. 9 (a) SDS-PAGE analysis shows the light dependence of the cleavage of BSA by L-QDs with exposure to different kinds of light for 12 h. Lane 1, molecular marker; lane 2, BSA; lane 3, BSA + RCP; lane 4, BSA + LP; lane 5, BSA + LCP. (b) PL spectra of BSA in the absence and presence of different concentrations of L-QDs. (c) CD spectra of BSA photocleavage at different reaction times. (d) TEM image of L-QDs after the cutting of BSA. (e) SDS-PAGE analysis shows the results for the cleavage of BSA by L-QDs under LCP irradiation for 6 h: lane 1, molecular marker; lane 2, BSA control; lane 3, BSA + QD; lane 4, BSA + QD + KI; lane 5, BSA + QD + ethanol; lane 6, BSA + QD + NaN3. (f) The recyclability of L-QDs for BSA cutting.97 Adapted with permission from ref. 97. Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (g) Synthesis of penicillamine-modified FexCuySe nanoparticles and illustration of the inhibition and disassembly effects of D-Pen FexCuySe on Aβ42 aggregation and mitigation of potential neurotoxicity in an AD mice model. (h–i) CD spectra of Aβ fibrils with L/D-NPs and NIR at 190–250 nm (h) and 400–1000 nm (i).98 Adapted with permission from ref. 98. Copyright 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
3.7 Theranostics
The good biological selectivity and remarkable activity of chiral nanomaterials have prompted research on their extensive theranostics. Chiral multi-metal incorporated nanomaterials provide an intriguing alternative for biological catalysis. In particular, they exhibit advantages in misfolded protein regulation. Recently, our group fabricated chiral iron copper selenium (FexCuySe) NPs to prevent beta-amyloid (Aβ) fibril-induced neurodegenerative diseases (Fig. 9g–i).98 The D-penicillamine (Pen) stabilized FexCuySe NPs had marked stereoselectivity and higher affinity than the L-type enantiomer to Aβ fibrils. The superior affinity of D-FexCuySe NPs was attributed to their higher conformation matching between the dimensional structures of chiral NPs and Aβ fibrils. The light-induced ROS production of NPs promoted the dissociation of the dense structure of fibrils into short protein fragments. The chiral NPs successfully alleviated fibril-induced neural damage in MN9D neurons and in vivo, and recovered cognitive ability in AD model mice. Moreover, a well-defined chiral AuCuAu heterostructure was used for tumor treatment with the assistance of circular polarized light.99 The copper side growth of the gold NR and dipeptide (cysteine–phenylalanine, Cys–Phe) modification led to an obvious CD signal in the NIR region with a high anisotropy of 0.57 × 10−2. The L-Cys-Phe stabilized AuCuAu heteronanorods resulted in a 20-fold increase in ROS production ability, which was more than that of bare gold NRs under 808 nm LCP laser irradiation, and efficiently promoted cancer therapy. Recently, our group fabricated multi-metallic chiral CuxCoyS NPs for senescent cell elimination (Fig. 10).100D-CuxCoyS NPs have a better cell internalization efficiency than the L-CuxCoyS NPs. Beta 2 macroglobulin functionalized chiral NPs can in situ generate a large number of ROS under NIR light illumination for accelerating senescent cell apoptosis. In addition, the magnetic chiral NPs mechanically damaged the cellular skeleton with the assistance of an external magnetic field. Encouraged by their preferable catalytic properties, the effects of chiral nanomaterials have been widened to medical fields.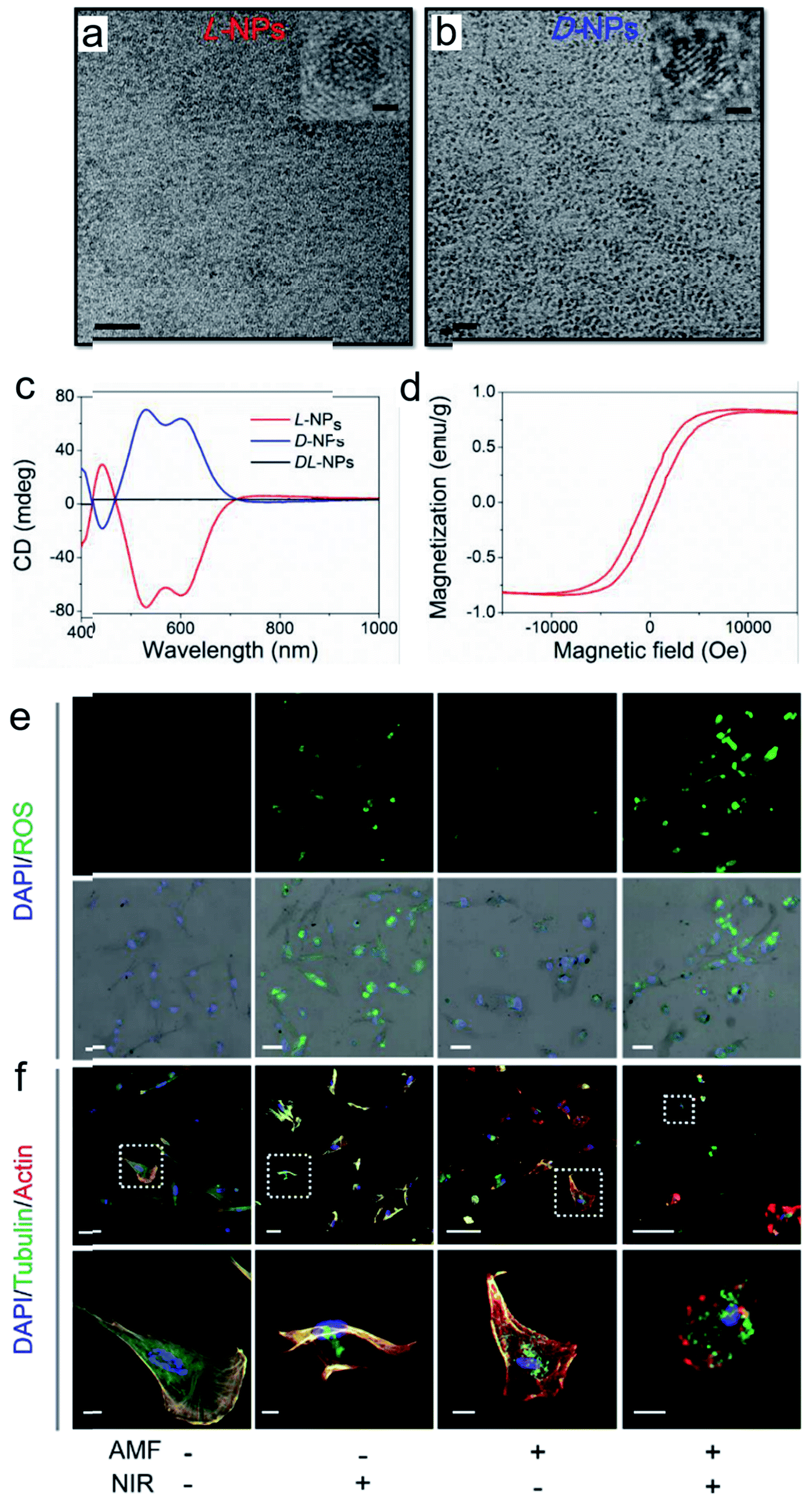 | ||
| Fig. 10 TEM images of (a) L- and (b) D-Pen stabilized CuxCoyS NPs, scale bar = 20 nm. The right-upper images were high resolution images of corresponding chiral NPs, scale bar = 1 nm. (c) CD spectra of L-, D-, and DL-CuxCoyS NPs. (d) Equilibrium magnetization measurements of L-CuxCoyS NPs. (e) ROS levels with D-NPs, scale bar = 50 μm. (f) Cellular skeleton change with D-NPs. Scale bar = 100 μm, and the scale bar of magnified images was 20 μm.100 Adapted with permission from ref. 100. Copyright 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
3.8 Peptide synthesis
Chiral nanomaterial-based biomimetic catalysts have high performance, superior selectivity and good stability for peptide fabrication, similar to natural catalysts. In addition, chiral SPs are used in biocatalytic systems. Hybrid chiral SPs were constructed by assembling penicillamine-modified ZnS semiconductors and glutathione-stabilized Au NPs into a nanostructure with homogeneous sub-nanoscale porosity of the nanoassembly (Fig. 11a–e).101 The as-prepared chiral SPs exhibited high stereoselectivity in photocatalysis which allowed the transformation of tyrosine into dityrosine like the photosynthetic bacteria. A molecular dynamic simulation analysis between the bioinspired SPs and their substrates showed that the confined porosities of the SPs had sufficient spaces for promoting the coupling of two tyrosine enantiomers. Simultaneously, the inlaid Au NPs significantly accelerated electronic mobility with light irradiation. The chiral biocatalytic SPs with intrinsic metallic active sites were functionalized with amino acids as biological recognition units, which creatively paved a potential biotransformation pattern based on chiral SPs. Chiral tungsten oxide hydrate (WO3−x·H2O) NPs were first fabricated for peptide catalysis (Fig. 11f–i). The chiral WO3−x·H2O NPs modified with the amino acids, aspartic acid (Asp) and proline (Pro) efficiently elevated their substrate retention and decreased the energy barrier of the catalytic system.102 The alcoholic solvent further enhanced peptide production by weakening the potential of bound water. The L-Asp functionalized NP catalysts tended to generate an Asp–Asp ethanol ester product with a molecular weight of 276, while the L-Pro stabilized NPs gained an Asp–Pro dipeptide of 230. In the experiments, the chiral bioinspired nanocatalysts not only exhibited their size and morphology advantages for matching the corresponding substrates but also imparted high performance to the bio-nano interactions.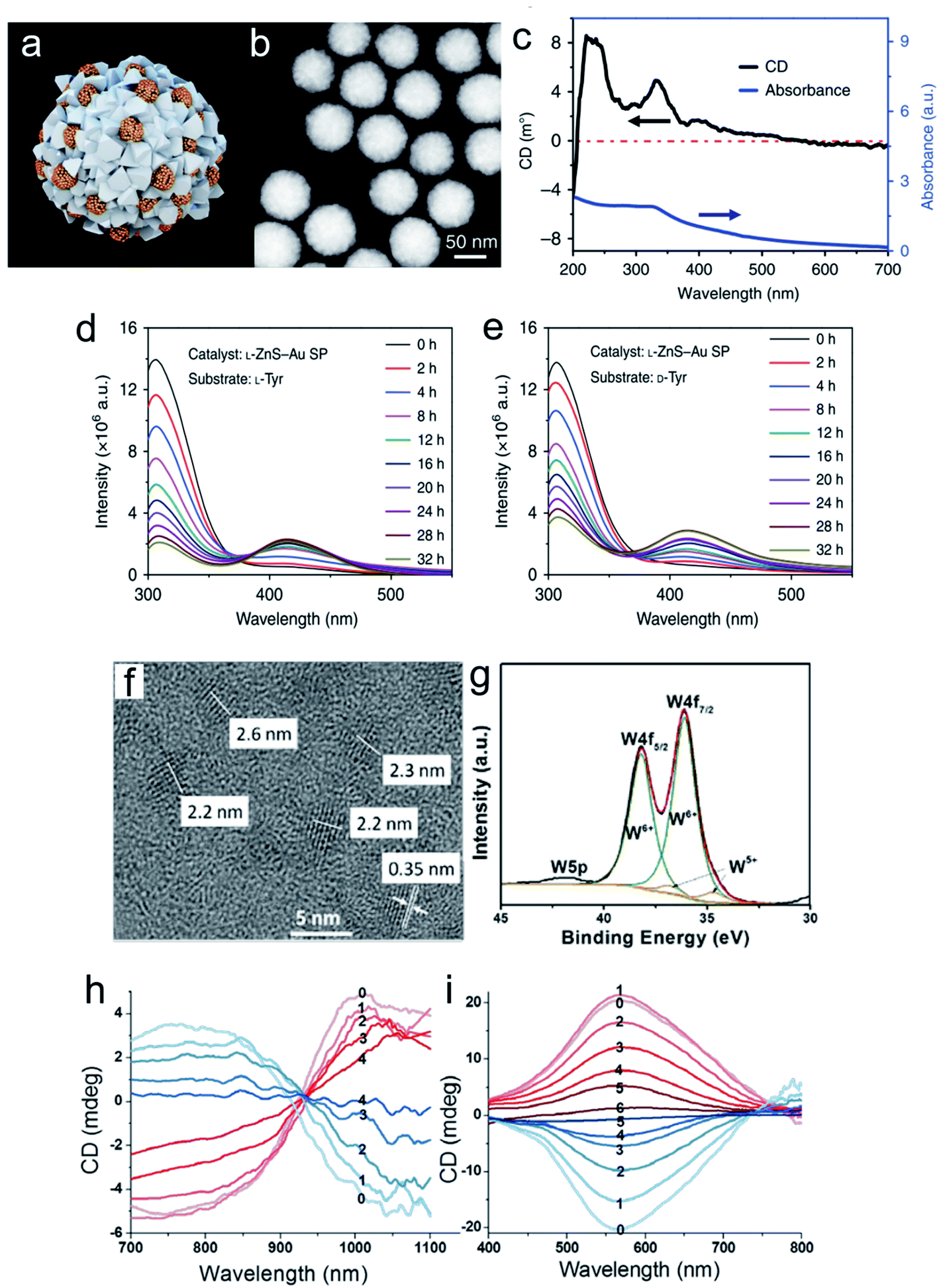 | ||
| Fig. 11 Morphological and spectral characterization of chiral ZnS–Au SPs. (a) Model of multi-component L-ZnS–Au SPs. (b) HAADF-STEM images of multi-component L-ZnS–Au SPs. (c) Elemental maps of L-ZnS–Au SPs of zinc, gold, and sulfur, respectively. (d) CD and UV spectra of multicomponent L-ZnS–Au SPs. Catalysis and stability of ZnS–Au SPs for photoinduced oxidative coupling of L-Tyr and D-Tyr. Fluorescence spectra of L-Tyr or D-Tyr after (e) L-Tyr with L-ZnS–Au SPs and (f) D-Tyr with L-ZnS–Au SPs being illuminated with different periods of time in the presence of ZnS–Au SPs. (g) Extracted ion chromatograms (EIC) of L-Tyr and Tyr-related products of L-ZnS–Au SP L-Tyr samples obtained at 0, 18, and 32 h, m/z = 405.10, 383.12, and 361.14, and m/z = 242.04, 220.06, and 198.08 are attributed to DOPA-Na + Na+, DOPA + Na+, and DOPA + H+ and diTyr-Na + Na+, diTyr + Na+ and diTyr + H+, respectively.101 Adapted with permission from ref. 101. Copyright 2019 Springer Nature. (f) Representative high-resolution TEM image of L-Asp-NPs. (g) XPS spectra with deconvoluted 4f peaks corresponding to two valence states of tungsten. CD spectra of (h) L- and D-Asp-NPs (red and blue, respectively) after addition of H2O2 at different concentrations: 0, 0 mmol L−1; 1, 0.03 mmol L−1; 2, 0.06 mmol L−1; 3, 0.09 mmol L−1; and 4, 0.12 mmol L−1, respectively. (i) L- and D-Pro-NPs (red and blue, respectively) after addition of H2O2 at concentrations of 0, 0 mmol L−1; 1, 0.06 mmol L−1; 2, 0.12 mmol L−1; 3, 0.18 mmol L−1; 4, 0.24 mmol L−1; 5, 0.30 mmol L−1; and 6, 0.36 mmol L−1, respectively.102 Adapted with permission from ref. 102. Copyright 2017 American Chemical Society. | ||
4. Conclusions and perspective
As a substitution for natural enzymes, chiral biomimetic nanocatalysts provided more flexible interaction modes and preferable catalytic efficiency for biocatalysis. Along with the developing nanotechnologies, these nanomaterials have been extended in terms of their fabrication and functionalization. The precise synthesis of the specific structure of chiral nanomaterials for efficient biomimetic catalysis is just in the initial stage which is confronted with many challenges. In this review, we demonstrated some critical points to fabricate chiral nanoparticles and nanoassemblies for biomimetic catalysis, and some principles either internal or external factors for regulating their bioactivity.Based on tremendous progress achieved in fabricating biomimetic nanomaterials, for a further understanding of the principles of designing high-performance nanomaterials and the interaction mechanism between chiral biomimetic nanostructures and substrates, we provided an outlook about the further development of chiral nanomaterials in biomedical catalysis as follows:
(1) Although chiral nanomaterials can be well controlled, present studies still lack precise synthetic strategies to mimic natural active sites for high performance biological interaction. Nevertheless, to screen advanced techniques for enhancing the catalytic activity of chiral nanomaterials is imperative. Especially, chiral nanoassemblies have great potential for high performance biological catalysis by rationally designing their chemical construction.
(2) Allowing for their reactivity in complex biological systems, it is inevitable for researchers to construct chiral nanomaterials with good stability and selectivity to their biomass substrates. Amino acid and peptide involved chiral nanomaterial fabrication provided a platform for good biocompatible and bio-specific interaction.
(3) Single component chiral nanomaterials cannot satisfy the diversified biological applications. Therefore, multi-component incorporated and multi-functional biomimetic nanomaterials for biogenic cascade reactions have gradually risen into a mainstream in the biocatalytic field.
(4) At the same time, the potential applications of chiral biomimetic nanomaterials are not only limited to those previously referred, as many innovative biological applications are being explored. Based on their outstanding photocatalytic properties and chiral ligand induced stereoselectivity to different substrates, they can also be used for virus-related protein cleavage, DNA specific cutting and cellulose degradation.103 Chiral nanomaterials also provide the possibility of enhancing immunological therapy or stimulating neuron cell differentiation due to their specific interaction to biomass.
(5) Inspired by the natural hierarchical chiral configuration, chiral nanoassemblies can provide an extremely similar structure which possessed great potential for efficient biological transformation. Chiral nanomaterials can also create a specific stereochemical environment for enhancing enantioselectivity that is beneficial for their asymmetric catalytic ability.
Nevertheless, chiral biomimetic nanomaterials for further biological applications still have many challenges which need to be overcome, but we anticipate that these barriers can be overcome in the near future.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is financially supported by National Key R&D Program (2017YFA0206902).References
- A. Ben-Moshe, B. M. Maoz, A. O. Govorov and G. Markovich, Chem. Soc. Rev., 2013, 42, 7028–7041 RSC.
- D. Jiang, D. Ni, Z. T. Rosenkrans, P. Huang, X. Yan and W. Cai, Chem. Soc. Rev., 2019, 48, 3683–3704 RSC.
- M. Liang and X. Yan, Acc. Chem. Res., 2019, 52, 2190–2200 CrossRef CAS.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- B. Jiang, D. Duan, L. Gao, M. Zhou, K. Fan, Y. Tang, J. Xi, Y. Bi, Z. Tong, G. F. Gao, N. Xie, A. Tang, G. Nie, M. Liang and X. Yan, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS.
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, e1805368 CrossRef.
- B. Yang, Y. Chen and J. Shi, Adv. Mater., 2019, 31, e1901778 CrossRef.
- P. Zhang, D. Sun, A. Cho, S. Weon, S. Lee, J. Lee, J. W. Han, D. P. Kim and W. Choi, Nat. Commun., 2019, 10, 940 CrossRef.
- C. Hao, L. Xu, H. Kuang and C. Xu, Adv. Mater., 2019, e1802075, DOI:10.1002/adma.201802075.
- J. Huang, J. Li, Y. Lyu, Q. Miao and K. Pu, Nat. Mater., 2019, 18, 1133–1143 CrossRef CAS.
- J. Yu, J. Wang, Y. Zhang, G. Chen, W. Mao, Y. Ye, A. R. Kahkoska, J. B. Buse, R. Langer and Z. Gu, Nat. Biomed. Eng., 2020, 4, 499–506 CrossRef CAS.
- G. Chen, Z. Chen, D. Wen, Z. Wang, H. Li, Y. Zeng, G. Dotti, R. E. Wirz and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3687–3692 CrossRef CAS.
- K. Fan, J. Xi, L. Fan, P. Wang, C. Zhu, Y. Tang, X. Xu, M. Liang, B. Jiang, X. Yan and L. Gao, Nat. Commun., 2018, 9, 1440 CrossRef.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS.
- X. Wu, C. Hao, J. Kumar, H. Kuang, N. A. Kotov, L. M. Liz-Marzan and C. Xu, Chem. Soc. Rev., 2018, 47, 4677–4696 RSC.
- S. Li, L. Xu, W. Ma, X. Wu, M. Sun, H. Kuang, L. Wang, N. A. Kotov and C. Xu, J. Am. Chem. Soc., 2016, 138, 306–312 CrossRef CAS.
- W. Ma, P. Fu, M. Sun, L. Xu, H. Kuang and C. Xu, J. Am. Chem. Soc., 2017, 139, 11752–11759 CrossRef CAS.
- U. Hananel, A. Ben-Moshe, H. Diamant and G. Markovich, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11159–11164 CrossRef CAS.
- U. Hananel, A. Ben-Moshe, D. Tal and G. Markovich, Adv. Mater., 2019, e1905594, DOI:10.1002/adma.201905594.
- L. Xu, Y. Gao, H. Kuang, L. M. Liz-Marzan and C. Xu, Angew. Chem., Int. Ed., 2018, 57, 10544–10548 CrossRef CAS.
- X. Wu, L. Xu, L. Liu, W. Ma, H. Yin, H. Kuang, L. Wang, C. Xu and N. A. Kotov, J. Am. Chem. Soc., 2013, 135, 18629–18636 CrossRef CAS.
- W. Ma, M. Sun, P. Fu, S. Li, L. Xu, H. Kuang and C. Xu, Adv. Mater., 2017, 29, 1703410 CrossRef.
- X. Gao, B. Han, X. Yang and Z. Tang, J. Am. Chem. Soc., 2019, 141, 13700–13707 CrossRef CAS.
- F. Purcell-Milton, R. McKenna, L. J. Brennan, C. P. Cullen, L. Guillemeney, N. V. Tepliakov, A. S. Baimuratov, I. D. Rukhlenko, T. S. Perova, G. S. Duesberg, A. V. Baranov, A. V. Fedorov and Y. K. Gun'ko, ACS Nano, 2018, 12, 954–964 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS.
- T. Kang, Y. G. Kim, D. Kim and T. Hyeon, Coord. Chem. Rev., 2020, 403, 213092 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS.
- A. Ben-Moshe, S. G. Wolf, M. Bar Sadan, L. Houben, Z. Fan, A. O. Govorov and G. Markovich, Nat. Commun., 2014, 5, 4302 CrossRef CAS.
- P. P. Wang, S. J. Yu, A. O. Govorov and M. Ouyang, Nat. Commun., 2017, 8, 14312 CrossRef CAS.
- H. E. Lee, H. Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 CrossRef CAS.
- J. Yeom, U. S. Santos, M. Chekini, M. Cha, A. F. de Moura and N. A. Kotov, Science, 2018, 359, 309–314 CrossRef CAS.
- M. P. Moloney, J. Govan, A. Loudon, M. Mukhina and Y. K. Gun'ko, Nat. Protoc., 2015, 10, 558–573 CrossRef CAS.
- X. Gao, X. Zhang, L. Zhao, P. Huang, B. Han, J. Lv, X. Qiu, S. H. Wei and Z. Tang, Nano Lett., 2018, 18, 6665–6671 CrossRef CAS.
- L. Shi, L. Zhu, J. Guo, L. Zhang, Y. Shi, Y. Zhang, K. Hou, Y. Zheng, Y. Zhu, J. Lv, S. Liu and Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 15397–15401 CrossRef CAS.
- A. Guerrero-Martínez, J. L. Alonso-Gómez, B. Auguié, M. M. Cid and L. M. Liz-Marzán, Nano Today, 2011, 6, 381–400 CrossRef.
- L. Ma, Y. Cao, Y. Duan, L. Han and S. Che, Angew. Chem., Int. Ed., 2017, 56, 8657–8662 CrossRef CAS.
- A. Qu, X. Wu, S. Li, M. Sun, L. Xu, H. Kuang and C. Xu, Adv. Mater., 2020, 32, e2000184 CrossRef.
- X. Wu, L. Xu, W. Ma, L. Liu, H. Kuang, N. A. Kotov and C. Xu, Adv. Mater., 2016, 28, 5907–5915 CrossRef CAS.
- M. Sun, T. Hao, X. Li, A. Qu, L. Xu, C. Hao, C. Xu and H. Kuang, Nat. Commun., 2018, 9, 4494 CrossRef.
- L. Xu, W. Yan, W. Ma, H. Kuang, X. Wu, L. Liu, Y. Zhao, L. Wang and C. Xu, Adv. Mater., 2015, 27, 1706–1711 CrossRef CAS.
- M. Sun, A. Qu, C. Hao, X. Wu, L. Xu, C. Xu and H. Kuang, Adv. Mater., 2018, 30, e1804241 CrossRef.
- C. Li, S. Li, A. Qu, H. Kuang, L. Xu and C. Xu, Adv. Funct. Mater., 2020, 2001451, DOI:10.1002/adfm.202001451.
- R. Gao, L. Xu, C. Hao, C. Xu and H. Kuang, Angew. Chem., Int. Ed., 2019, 58, 3913–3917 CrossRef CAS.
- J. Yeom, P. P. G. Guimaraes, H. M. Ahn, B. K. Jung, Q. Hu, K. McHugh, M. J. Mitchell, C. O. Yun, R. Langer and A. Jaklenec, Adv. Mater., 2020, 32, e1903878 CrossRef.
- W. Feng, J. Y. Kim, X. Wang, H. A. Calcaterra, Z. Qu, L. Meshi and N. A. Kotov, Sci. Adv., 2017, 3, e1601159 CrossRef.
- J. Yan, W. Feng, J.-Y. Kim, J. Lu, P. Kumar, Z. Mu, X. Wu, X. Mao and N. A. Kotov, Chem. Mater., 2019, 32, 476–488 CrossRef.
- W. Jiang, Z.-b. Qu, P. Kumar, D. Vecchio, Y. Wang, Y. Ma, J. H. Bahng, K. Bernardino, W. R. Gomes, F. M. Colombari, A. Lozada-Blanco, M. Veksler, E. Marino, A. Simon, C. Murray, S. R. Muniz, A. F. de Moura and N. A. Kotov, Science, 2020, eaaz7949, DOI:10.1126/science.aaz7949.
- M. Huo, L. Wang, Y. Wang, Y. Chen and J. Shi, ACS Nano, 2019, 13, 2643–2653 CAS.
- L. Jiao, H. Yan, Y. Wu, W. Gu, C. Zhu, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2020, 59, 2565–2576 CrossRef CAS.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497–501 CrossRef CAS.
- H. J. Kwon, D. Kim, K. Seo, Y. G. Kim, S. I. Han, T. Kang, M. Soh and T. Hyeon, Angew. Chem., Int. Ed., 2018, 57, 9408–9412 CrossRef CAS.
- H. Sun, Y. Zhou, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 9224–9237 CrossRef CAS.
- B. Xu, H. Wang, W. Wang, L. Gao, S. Li, X. Pan, H. Wang, H. Yang, X. Meng, Q. Wu, L. Zheng, S. Chen, X. Shi, K. Fan, X. Yan and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 4911–4916 CrossRef CAS.
- I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127–4170 RSC.
- X. Hu, F. Li, F. Xia, X. Guo, N. Wang, L. Liang, B. Yang, K. Fan, X. Yan and D. Ling, J. Am. Chem. Soc., 2020, 142, 1636–1644 CrossRef CAS.
- L. Huang, J. X. Chen, L. F. Gan, J. Wang and S. J. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS.
- Q. Wang, X. Zhang, L. Huang, Z. Zhang and S. Dong, Angew. Chem., Int. Ed., 2017, 56, 16082–16085 CrossRef CAS.
- W. Ma, C. Hao, M. Sun, L. Xu, C. Xu and H. Kuang, Mater. Horiz., 2018, 5, 141–161 RSC.
- S. S. Lee, W. Song, M. Cho, H. L. Puppala, N. Phuc, H. Zhu, L. Segatori and V. L. Colvin, ACS Nano, 2013, 7, 9693–9703 CrossRef CAS.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kucanda, D. Manna, P. K. Kundu, J. W. Lee, P. Kral and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS.
- N. Singh, M. A. Savanur, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2017, 56, 14267–14271 CrossRef CAS.
- G. Fang, W. Li, X. Shen, J. M. Perez-Aguilar, Y. Chong, X. Gao, Z. Chai, C. Chen, C. Ge and R. Zhou, Nat. Commun., 2018, 9, 129 CrossRef.
- C. Ge, G. Fang, X. Shen, Y. Chong, W. G. Wamer, X. Gao, Z. Chai, C. Chen and J. J. Yin, ACS Nano, 2016, 10, 10436–10445 CrossRef CAS.
- S. Ghosh, P. Roy, N. Karmodak, E. D. Jemmis and G. Mugesh, Angew. Chem., Int. Ed., 2018, 57, 4510–4515 CrossRef CAS.
- Q. Zhang, D. Dehaini, Y. Zhang, J. Zhou, X. Chen, L. Zhang, R. H. Fang, W. Gao and L. Zhang, Nat. Nanotechnol., 2018, 13, 1182–1190 CrossRef CAS.
- Q. Zhou, S. Shao, J. Wang, C. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Nat. Nanotechnol., 2019, 14, 799–809 CrossRef CAS.
- Y. Wang, R. Cai and C. Chen, Acc. Chem. Res., 2019, 52, 1507–1518 CrossRef CAS.
- X. Han, S. Shen, Q. Fan, G. Chen, E. Archibong, G. Dotti, Z. Liu, Z. Gu and C. Wang, Sci. Adv., 2019, 5, eaaw6870 CrossRef CAS.
- J. Wang, J. Yu, Y. Zhang, X. Zhang, A. R. Kahkoska, G. Chen, Z. Wang, W. Sun, L. Cai, Z. Chen, C. Qian, Q. Shen, A. Khademhosseini, J. B. Buse and Z. Gu, Sci. Adv., 2019, 5, eaaw4357 CrossRef CAS.
- J. Zhuang, H. Gong, J. Zhou, Q. Zhang, W. Gao, R. H. Fang and L. Zhang, Sci. Adv., 2020, 6, eaaz6108 CrossRef CAS.
- Y. Zhao, Y. Huang, H. Zhu, Q. Zhu and Y. Xia, J. Am. Chem. Soc., 2016, 138, 16645–16654 CrossRef CAS.
- X. Wang, M. Wang, R. Lei, S. F. Zhu, Y. Zhao and C. Chen, ACS Nano, 2017, 11, 4606–4616 CrossRef CAS.
- A. Bhagi-Damodaran, J. H. Reed, Q. Zhu, Y. Shi, P. Hosseinzadeh, B. A. Sandoval, K. A. Harnden, S. Wang, M. R. Sponholtz, E. N. Mirts, S. Dwaraknath, Y. Zhang, P. Moenne-Loccoz and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6195–6200 CrossRef.
- C. Zhang, C. Chen, H. Dong, J. R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS.
- X. Jiang, J. Li, B. Yang, X. Z. Wei, B. W. Dong, Y. Kao, M. Y. Huang, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 7850–7854 CrossRef CAS.
- E. N. Mirts, I. D. Petrik, P. Hosseinzadeh, M. J. Nilges and Y. Lu, Science, 2018, 361, 1098–1101 CrossRef CAS.
- A. Zakharchenko, N. Guz, A. M. Laradji, E. Katz and S. Minko, Nat. Catal., 2017, 1, 73–81 CrossRef.
- J. Yoo, E. Lee, H. Y. Kim, D. H. Youn, J. Jung, H. Kim, Y. Chang, W. Lee, J. Shin, S. Baek, W. Jang, W. Jun, S. Kim, J. Hong, H. J. Park, C. J. Lengner, S. H. Moh, Y. Kwon and J. Kim, Nat. Nanotechnol., 2017, 12, 1006–1014 CrossRef CAS.
- E. Voros, M. Cho, M. Ramirez, A. L. Palange, E. De Rosa, J. Key, Z. Garami, A. B. Lumsden and P. Decuzzi, Adv. Funct. Mater., 2015, 25, 1709–1718 CrossRef CAS.
- S. Liang, X. Deng, Y. Chang, C. Sun, S. Shao, Z. Xie, X. Xiao, P. Ma, H. Zhang, Z. Cheng and J. Lin, Nano Lett., 2019, 19, 4134–4145 CrossRef CAS.
- W. Yin, J. Yu, F. Lv, L. Yan, L. R. Zheng, Z. Gu and Y. Zhao, ACS Nano, 2016, 10, 11000–11011 CrossRef CAS.
- F. Gao, M. Sun, W. Ma, X. Wu, L. Liu, H. Kuang and C. Xu, Adv. Mater., 2017, 29, 1606864 CrossRef.
- S. Li, L. Shang, B. Xu, S. Wang, K. Gu, Q. Wu, Y. Sun, Q. Zhang, H. Yang, F. Zhang, L. Gu, T. Zhang and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 12624–12631 CrossRef CAS.
- F. Neubrech, M. Hentschel and N. Liu, Adv. Mater., 2020, e1905640, DOI:10.1002/adma.201905640.
- J. Kumar and L. M. Liz-Marzán, Bull. Chem. Soc. Jpn., 2019, 92, 30–37 CrossRef CAS.
- X. Zhao, L. Xu, M. Sun, W. Ma, X. Wu, C. Xu and H. Kuang, Nat. Commun., 2017, 8, 2007 CrossRef.
- J. Kumar, H. Erana, E. Lopez-Martinez, N. Claes, V. F. Martin, D. M. Solis, S. Bals, A. L. Cortajarena, J. Castilla and L. M. Liz-Marzan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3225–3230 CrossRef CAS.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS.
- Q. Xin, Q. Liu, L. Geng, Q. Fang and J. R. Gong, Adv. Healthcare Mater., 2017, 6, 1601011 CrossRef.
- G. Qing, S. Zhao, Y. Xiong, Z. Lv, F. Jiang, Y. Liu, H. Chen, M. Zhang and T. Sun, J. Am. Chem. Soc., 2014, 136, 10736–10742 CrossRef CAS.
- A. Yuan, C. Hao, X. Wu, M. Sun, A. Qu, L. Xu, H. Kuang and C. Xu, Adv. Mater., 2020, e1906580, DOI:10.1002/adma.201906580.
- C. Hao, A. Qu, L. Xu, M. Sun, H. Zhang, C. Xu and H. Kuang, J. Am. Chem. Soc., 2019, 141, 1091–1099 CrossRef CAS.
- C. Hao, X. Wu, M. Sun, H. Zhang, A. Yuan, L. Xu, C. Xu and H. Kuang, J. Am. Chem. Soc., 2019, 141, 19373–19378 CrossRef CAS.
- M. Sun, L. Xu, A. Qu, P. Zhao, T. Hao, W. Ma, C. Hao, X. Wen, F. M. Colombari, A. F. de Moura, N. A. Kotov, C. Xu and H. Kuang, Nat. Chem., 2018, 10, 821–830 CrossRef CAS.
- F. Li, S. Li, X. Guo, Y. Dong, C. Yao, Y. Liu, Y. Song, X. Tan, L. Gao and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 11087–11092 CrossRef CAS.
- C. Hao, R. Gao, Y. Li, L. Xu, M. Sun, C. Xu and H. Kuang, Angew. Chem., Int. Ed., 2019, 58, 7371–7374 CrossRef CAS.
- F. Li, S. Li, X. Guo, Y. Dong, C. Yao, Y. Liu, Y. Song, X. Tan, L. Gao and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 11087–11092 CrossRef CAS.
- J. Wang, X. Wu, W. Ma and C. Xu, Adv. Funct. Mater., 2020, 2000670, DOI:10.1002/adfm.202000670.
- S. Li, M. Sun, C. Hao, A. Qu, X. Wu, L. Xu, C. Xu and H. Kuang, Angew. Chem., Int. Ed., 2020, 59, 13915–13922 CrossRef CAS.
- S. Li, J. Liu, N. S. Ramesar, H. Heinz, L. Xu, C. Xu and N. A. Kotov, Nat. Commun., 2019, 10, 4826 CrossRef.
- S. Jiang, M. Chekini, Z. B. Qu, Y. Wang, A. Yeltik, Y. Liu, A. Kotlyar, T. Zhang, B. Li, H. V. Demir and N. A. Kotov, J. Am. Chem. Soc., 2017, 139, 13701–13712 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CAS.
| This journal is © The Royal Society of Chemistry 2020 |