
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular assemblers: molecular machines performing chemical synthesis
Rainer
Herges
 *
*
Otto-Diels-Institute of Organic Chemistry, Kiel University, 24118 Kiel, Germany. E-mail: rherges@oc.uni-kiel.de
First published on 15th July 2020
Abstract
Molecular assemblers were proposed by K. Eric Drexler in 1986, based on the ideas of R. Feynman. In his (quite lurid) book “Engines of Creation: The Coming Era of Nanotechnology” and follow-up publications Drexler proposes molecular machines capable of positioning reactive molecules with atomic precision and to build larger, more sophisticated structures via mechanosynthesis. These imaginative visions started a hot controversy. The debate culminated in a cover story of Chemical & Engineering News in 2003 (ref. 1) with the key question: “Are molecular assemblers – devices capable of positioning atoms and molecules for precisely defined reactions – possible?” with Drexler as the proponent and Nobelist Richard E. Smalley being the opponent. Smalley raised two major objections: the “fat fingers” and the “sticky fingers” problem. To grab and guide each individual atom the assembler must have many nano-fingers. Smalley argued that there is just not enough room in the nanometer-sized reaction region to accommodate all the fingers of all the manipulators necessary to have complete control of the chemistry. The sticky finger issue arises from the problem that …“the atoms of the manipulator hands will adhere to the atom that is being moved. So it will often be impossible to release the building block in precisely the right spot.” Smalley concludes that the fat and the sticky finger problems are fundamental and cannot be avoided. While some of the statements of E. Drexler are bold and probably not very realistic, his ideas are inspiring and might be a good starting point to assess on how far laboratory chemistry has advanced towards real “molecular assemblers” within the last two decades.
Introduction & overview
Chemists have been performing reactions with stochastic ensembles in solution or in the gas phase since the very beginning of our field. Chemical bonds form upon random collisions, if the orientations of reaction partners and kinetic energies are favourable. Hence, incomplete conversion and side products are usually unavoidable. Notwithstanding advances in catalysis, control of reaction parameters, protecting group strategies and solid-phase synthesis, natural product synthesis in our laboratories has never reached the level of molecular assemblers in nature. Life has optimized chemical synthesis in a completely different manner and it would not exist based on conventional stochastic chemistry. Living organisms perform reactions in an extremely selective and efficient manner to survive in a competitive environment with limited resources and energy supply, using molecular machines for synthesis.Natural paragons
Ribosomes, non-ribosomal peptide synthases (NRPSs) and polyketide synthetases (PKSs) are molecular assemblers that perform atomically precise movements along a cascade of events. Ribosomes are the most sophisticated, programmable assemblers, and probably far beyond of what we could ever achieve as chemists. NRPSs are still complex, but their mechanisms are suitable as paragons for artificial systems.NRPSs are among the largest enzyme complexes in nature (up to 1800 kDa).2 They synthesize a large part of bioactive compounds such as antibiotics, siderophores, and immunosuppressants. NRPSs resemble assembly lines, organized in up to 15 subsequent modules. Each module is responsible for performing a specific reaction (condensation, adenylation, thiolation, epimerisation, methylation, Claisen ester condensation, oxidation etc.) and for adding a building block to the growing chain (Fig. 1). Each module consists of at least three domains: the A (adenylation) domain selects and activates the substrate, the T (peptidyl carrier, thiolation) domain is responsible for the transportation and orientation of the substrate and the C (condensation) domain catalyses the reaction. Even though the detailed mechanisms are complex, the general synthetic logic is simple. It seems obvious that bioengineering NRPSs (adding, removing and substituting domains or modules) should allow production of many novel bioactive compounds. There is major interest in the use of semi-synthetic hybrids or bioengineered NRPSs as molecular assemblers.3 Success, however, to date has been limited. Each condensation step in NRPS fabricated synthesis is endergonic and driven by ATP. ATP is produced from ADP and Pi by a large enzyme complex ATP synthase (>500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 daltons). ATP synthase also perfectly complies with the definition of a molecular machine performing chemical synthesis.
000 daltons). ATP synthase also perfectly complies with the definition of a molecular machine performing chemical synthesis.
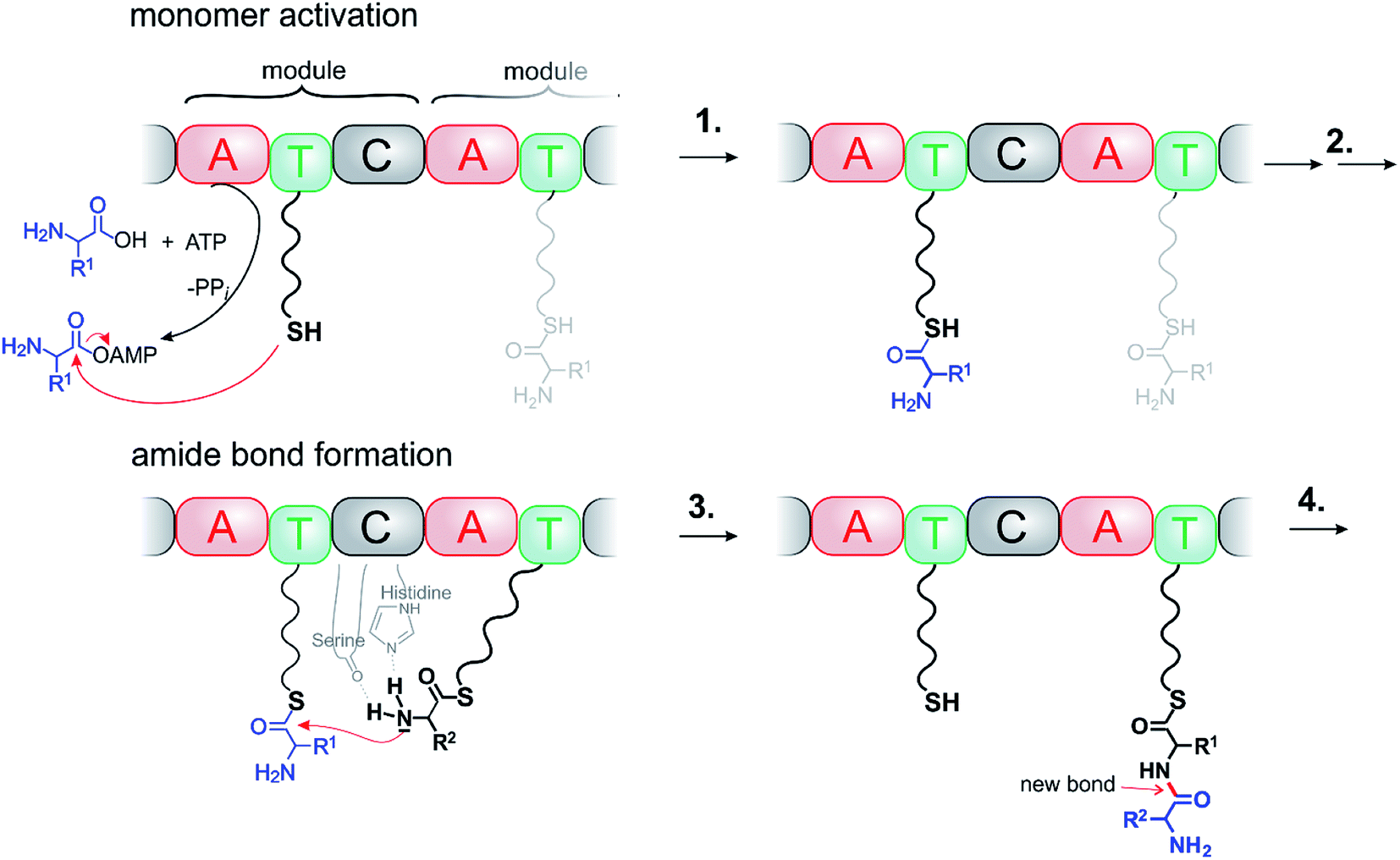 | ||
| Fig. 1 A molecular machine performing chemical synthesis: ATP driven formation of an amide bond in NRPSs. (1) Recognition, activation and binding of a building block, (2) precise orientation of the reactants, (3) catalysed bond formation, and (4) repeating cycles, growing chain and product release. | ||
Surprisingly, structurally diverse and functionally different assemblers such as NRPSs, ATP synthase and chaperones,4 as well as a number of further ATP driven molecular synthetic machines, such as ATP driven proteases,5 and polyketide synthases6 have the following 4-stage assembly sequence in common:
(1) recognition and binding of the reactants.
(2) Orientation of the reactants with atomic precision.
(3) Catalyzed bond formation.
(4) Active (energy consuming) release of the product and repeating the cycle.
The advantages of machine-type syntheses are obvious: the assembler prevents side reactions, and provides chemo-, regio- and stereoselective synthesis without protecting groups or chiral auxiliaries. Notwithstanding the numerous examples in biology and considerable advances in covalent and supramolecular chemistry, artificial, continuously operating assemblers have not been realized so far. Fortunately, there is at least one encouraging argument on Drexler's side. In 1972 the economist Kenneth E. Boulding stated the following:
If something exists, then it must be possible. 7
In terms of molecular assemblers Boulding's law would read: “If nature is using molecular assemblers for synthesis, chemists should (at least in principle) be able to build and operate artificial assemblers in the laboratory”. Biochemical and chemical reactions follow similar principles of chemistry and the same laws of physics. There should not be a fundamental problem about building artificial molecular assemblers; however, the endeavour could still be unrealistically difficult or too expensive. Therefore, it might be expedient to look at molecular assemblers in nature and try to systematically reduce their sophistication and complexity to a level achievable with synthetic chemistry.
Towards artificial molecular assemblers
The first three stages of biological assemblers: (1) recognition and binding of the reactants, (2) orientation of the reactants and (3) bond formation have been realized in a number of artificial systems. Assemblers that operate in a cyclic fashion (stage 4) have not been known until recently.“Mechanical” control of reactions following the chemical potential gradient (downhill)
Hosseini and Lehn8,9 reported a macrocycle catalyzing the formation of pyrophosphate and ATP by hydrolysis of ADP and acetyl phosphate. The latter is an activated phosphate and the reaction is exergonic. Nevertheless, it demonstrates the power of supramolecular catalysis. The first non-continuously operating assemblers were based on DNA architectures and performed multistep organic synthesis by sequential reactions on subsequent stations on a DNA track.10–15 (Fig. 2a) Leigh et al. used rotaxanes as the assembly line. In a landmark paper, they reported the sequence-specific synthesis of a tripeptide16–18 (Fig. 2b).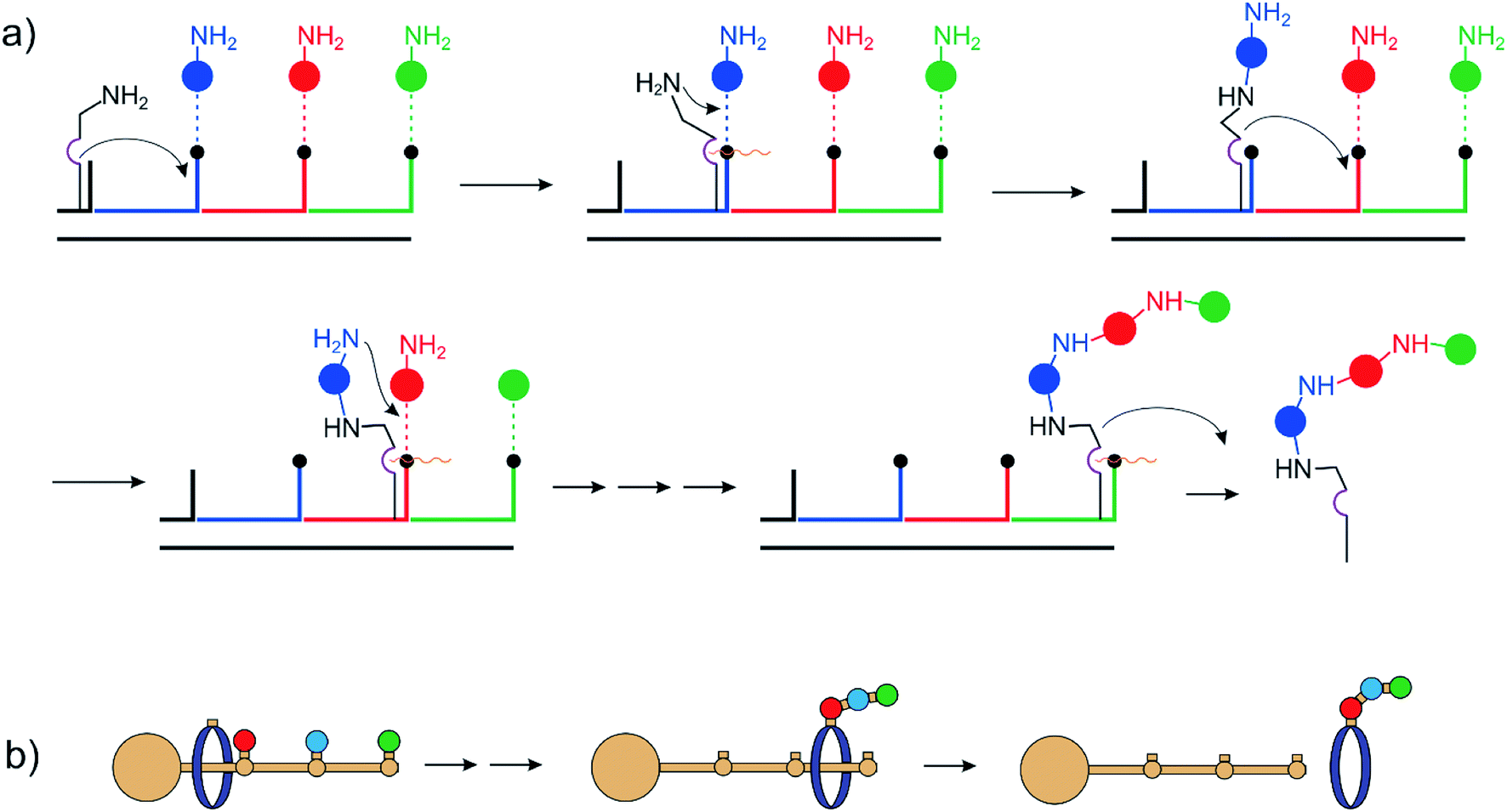 | ||
| Fig. 2 (a) DNA and (b) rotaxane based assembly. | ||
Recently the same group published a programmable molecular machine for stereodivergent synthesis.19 While DNA and rotaxane assembly lines are ingenious designs and impressive examples of the power of modern synthetic and supramolecular chemistry, they suffer from the disadvantage that the information is read in a destructive manner. There is no way to reload the machine for a second run. One equivalent of the machine can only make one equivalent of the product. For practical applications in synthesis, the assembler has to operate thousands of cycles without fatigue to justify the efforts of its synthesis. Precise orientation of the reacting components and a mechanism for the autonomous execution of a protocol alone are not sufficient. A number of additional preconditions have to be met to perform machine-type synthesis similar to ATP synthase or NRPS-type assemblers.
Continuously operating molecular assemblers, similar to macroscopic and molecular machines depend on a continuous energy input. The paragon of artificial molecular machines, the Feringa motor, rotates upon continuous irradiation (light energy), ATP synthase depends on energy supply by a proton gradient (proton motive force) and NRPSs work on incessant supply of ATP (chemical energy).
Driving endergonic processes (directed transport) with external energy sources (uphill)
Early implementations of energy coupled processes include the active transport of ions across membranes building up concentration gradients (active transport, Fig. 3a). Proton, electron and light driven systems have been realized.20 Directed transport or pumping can be performed using two fundamentally different approaches. The energy source is either a steady gradient across a membrane in combination with a responsive carrier, e.g. the combination of a redox gradient and a redox responsive carrier21,22 (Fig. 3a) or an alternating redox potential following the principle of an energy23–25 or information ratchet26,27 (Fig. 3b). Alternatively, directed transport based on a light driven information and energy ratchet has been realized by Credi et al.28 Another category of endergonic processes driven by external energy sources is dissipative assembly of supramolecular structures.29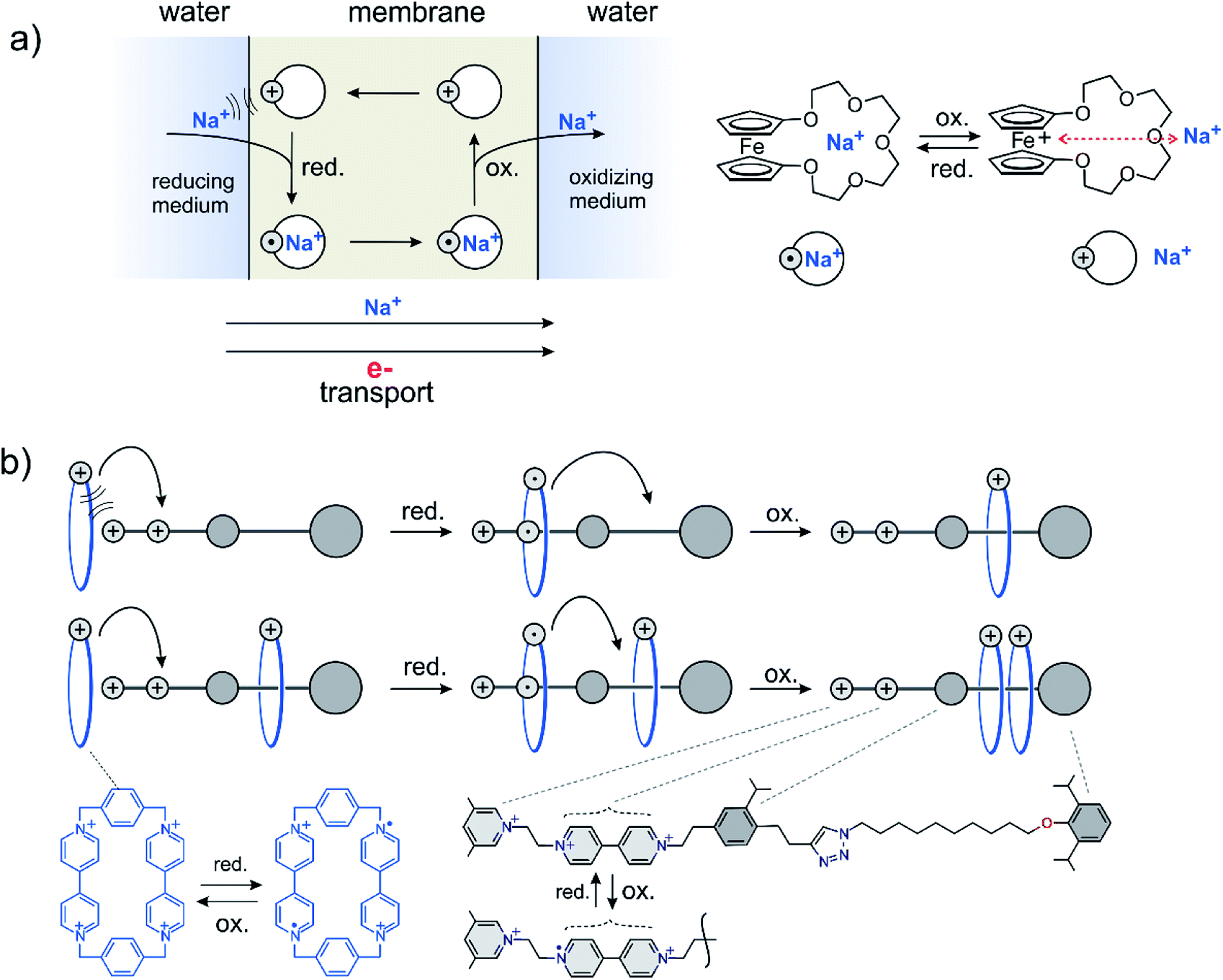 | ||
| Fig. 3 Two different approaches to perform directed transport against a chemical potential using a redox gradient as an energy source. (a) A steady redox gradient (electron transport) drives the active transport of sodium ions across a membrane. (b) An alternating redox potential creates a local concentration of rotaxane rings.30,31 | ||
Continuously operating molecular assemblers (driving endergonic reactions)
Precise mechanical control of chemical reactions and the coupling to external energy sources (as outlined in the previous chapters) are essential functions of molecular assemblers. Molecular assemblers thus comply with the definition of molecular machines, which are molecules or molecular assemblies that use an energy input to perform controlled and directed mechanical movements leading to particular functions.32 Molecular machines performing chemical synthesis have to perform directed motion and hence need a continuous energy input. Early life on earth probably was energized by redox gradients,33 photosynthesis exploits light, ATP synthase is driven by a proton gradient and most biochemical, synthetic processes (e.g. NRPSs, see Fig. 1) use ATP as an ubiquitous energy source. After artificial systems have been realized that use these external energy sources to build up concentration gradients, or to drive self-assembly34 the obvious next step is to develop approaches to make and break covalent chemical bonds, and hence to perform synthesis. Probably the simplest way to couple the reversible reactions in molecular machines to irreversible bond making in chemical synthesis is to use reversible supramolecular binding.For the sake of simplicity, we consider the dimerization of two monomers (blue circles), as a model reaction, which is endergonic in solution (ΔG1 > 0) (Fig. 4). Hence, the equilibrium is on the monomer side. To shift the equilibrium towards dimer formation, a ligand is added, that binds the dimer with a high free enthalpy35 of association (ΔG4). If the overall reaction enthalpy (ΔG1 + ΔG4) is negative, the dimerization would be exergonic. It does not matter whether the dimerization proceeds inside the ligand (ΔG3, ΔG2) or if the ligand “traps” dimer molecules present in small concentrations in equilibrium (ΔG4). However, the dimer now is tightly bound within the ligand, which is equivalent to the “sticky finger problem” raised by Smalley, or what is called product inhibition in catalysis.
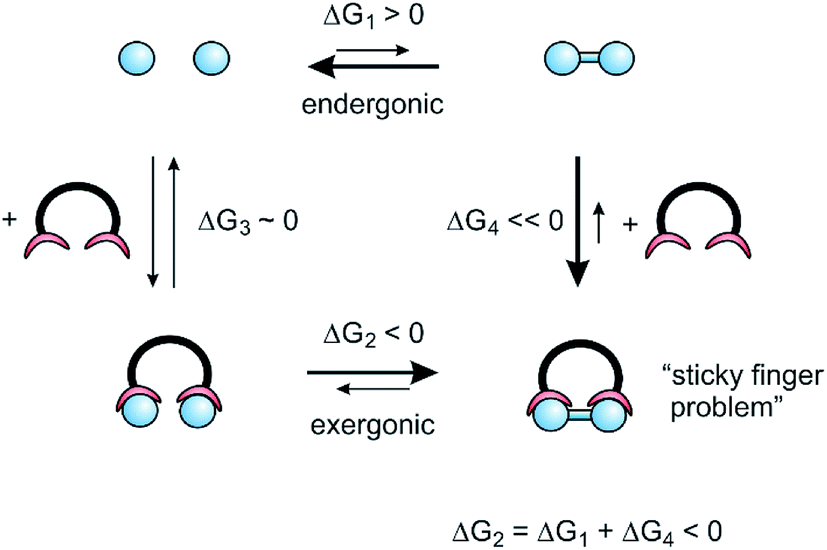 | ||
| Fig. 4 Thermodynamic cycle of the reaction driven by supramolecular binding. | ||
To actively release the product, ditopic switchable ligands are needed (Fig. 5). In response to an external stimulus such as light, the ligand switches between an open and a closed state. The open state preferentially binds to the reactants (monomer) and the closed state strongly binds the product (dimer). The synthetic cycle starts with the open state of the ditopic assembler and two reactants attached to each binding site (1). Upon irradiation the assembler switches to the closed state and brings the two reactants in close proximity (2). A covalent bond between the reactants is formed via catalysis (3). Active release of the product is achieved by photochemical switching to the open state, which subsequently binds the two reactants (1) and thus completes the reaction cycle. Among the four consecutive steps, energy is supplied in the photochemical steps (1) → (2), and most importantly, during active release of the product (3) → (4) It is important to note that it is not the bond forming process, but the product release which needs an energy input. Product release is the energy-consuming step in ATP synthase as well. Without the energy input, ATP synthase generates one molecule of ATP, which remains stuck in the active site and blocks further reaction cycles.
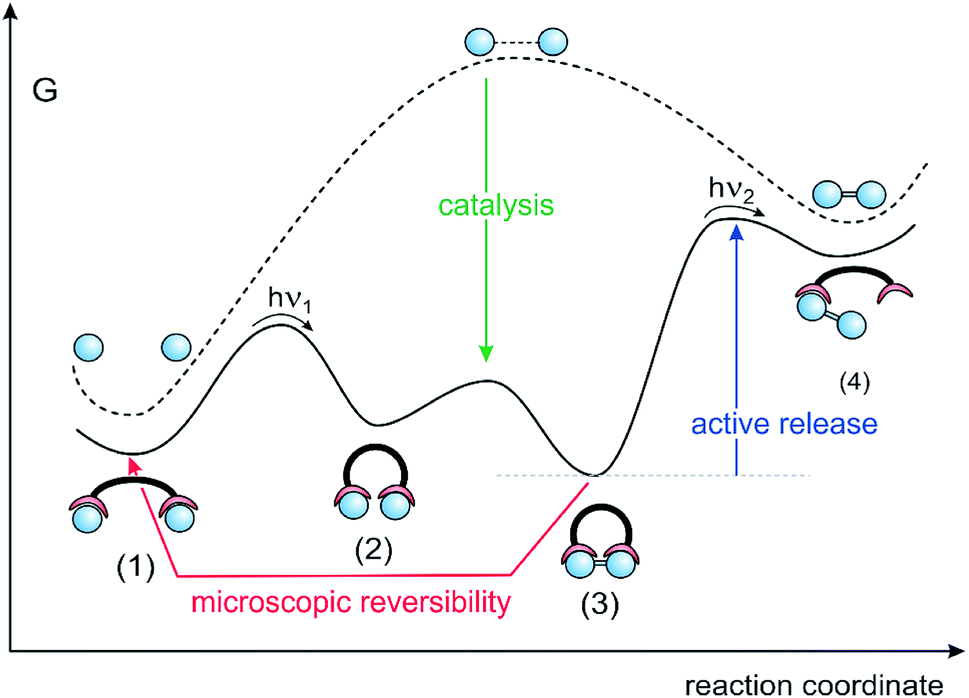 | ||
| Fig. 5 Reaction pathway of a dimerisation reaction in solution (dashed line) driven by a continuously operating molecular assembler, based on a switchable ditopic ligand using light as the energy source (solid line). The three major challenges in realizing a molecular assembler are product destruction upon product release (red arrow), efficient catalysis of the reaction inside the assembler (green arrow) and the energy input for active product release (blue arrow). | ||
The model reaction outside the ditopic assembler (dashed line) is endergonic, and the high-energy product is kinetically stable (high barrier to back-reaction). Inside the closed form of the ditopic receptor, the reaction (2) → (3) is exergonic, driven by the large binding energy of the product. Stage (3) is the lowest point on the reaction pathway. However, the bond forming step (2) → (3) is likely to suffer from a high barrier of activation, because the model reaction in solution must have a high activation to avoid a spontaneous back-reaction. To prevent the assembly stalling at stage (2), the bond forming process has to be catalyzed. A further implication has to be considered, which arises from microscopic reversibility. If the product release is reciprocal to the bond forming process (opening and closing of the assembler), product formation and destruction would be equiprobable and cancel each other on average.
Summing up, the following prerequisites are indispensable for any continuously operating molecular assembler:
• Energy source: light, redox gradient, and concentration gradient.
• Endergonic target reaction generating a kinetically stable product.
• Switchable ligand (molecular assembler) with the following characteristics:
– one state binding the reactants and the other strongly binding the product.
– Catalysis of the bond forming process.
– Active release of the product.
– Avoiding microscopic reversibility of bond making and bond breaking.
ATP-synthase-type assemblers
ATP synthase follows the above outline. The analysis of the operational steps during ATP synthesis provides insight into the needs and problems we will encounter on the way to realize artificial continuously operating assemblers. The energy source of ATP synthase is the proton gradient across mitochondrial membranes driving the endergonic reaction ADP + Pi + H+ → ATP + H2O in an assembler-type mechanosynthesis. ATP synthase uses a three-state ligand, changing the binding affinity for reactants and the product in a defined sequence: (1) binding of the reactants, (2) bringing the reactants in close proximity including bond formation, and (3) active release of the product36,37 (Fig. 5). Equilibrium constants determined for the different steps of ATP synthesis38 are in agreement with the thermodynamic cycle shown in Fig. 4. Product binding of ATP is quite strong with ΔG4 = −18 kcal mol−1 and clearly outmatches the positive free reaction enthalpy of ATP-synthesis of ΔG1 = 7.3 kcal mol−1. The most energy consuming process is active release of the product, which is ΔG1 + ΔG4 = 25.3 kcal mol−1 uphill. Besides these thermodynamic implications, there is a severe kinetic problem to be overcome. If the endergonic model reaction is chosen in such a way that the product (e.g. ATP) is kinetically stable, the barrier to the back-reaction in solution must be high enough to prevent a spontaneous back-reaction. In contrast, bond formation inside the ligand should be spontaneous. Hence, a dramatic lowering of the barrier is necessary to achieve a fast reaction inside the cavity (green arrow in Fig. 5). In ATP synthase the free enthalpy barrier of ATP formation is lowered from ∼40 kcal mol−1 in water to <15 kcal mol−1 in the binding pocket of the catalytic core. The high barrier in water is due to the strong Coulomb repulsion of the negatively charged reactants ADP and Pi. For obvious reasons the strategy to achieve catalysis is to remove charge from one of the two reactants (Fig. 6).9 Positively charged amino acid residues and Mg2+ withdraw charge from the incoming phosphate and convert it to an electrophile in the P–O bond forming reaction. Active removal of water reduces the activation barrier further. Efficient catalysis is probably one of the main challenges in the design of continuously operating assemblers.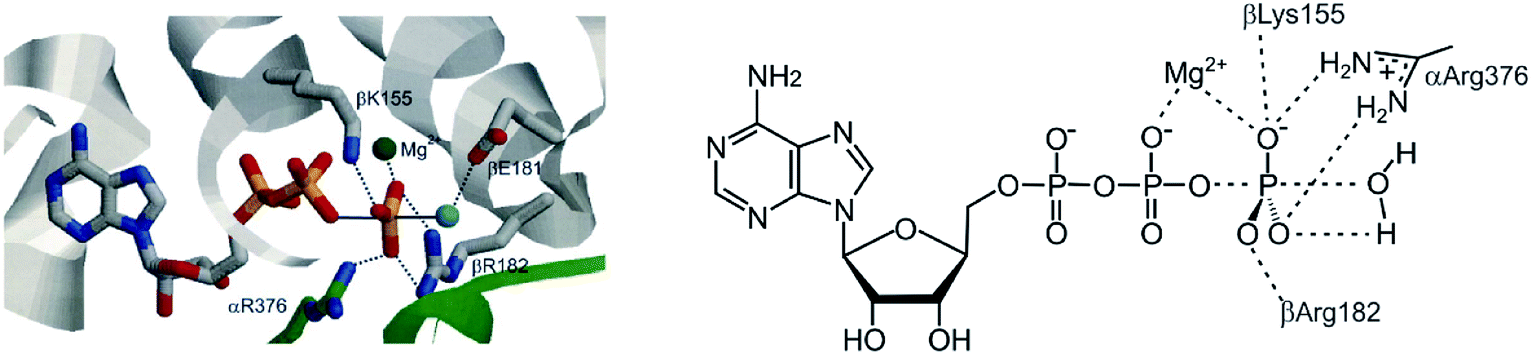 | ||
| Fig. 6 Catalysis of the P–O bond formation in the active site of ATP synthase. | ||
Based on the above principles a simple model assembler was recently developed.39 In view of the high activation barrier of phosphate condensation, vanadate condensation was used as the model reaction, which has a lower barrier of condensation, and therefore is easier to catalyse. A photoswitchable ditopic receptor consisting of a photochromic azobenzene unit and two Zn-cyclene units performs a pincer-type motion upon irradiation with UV and visible light (Fig. 7).
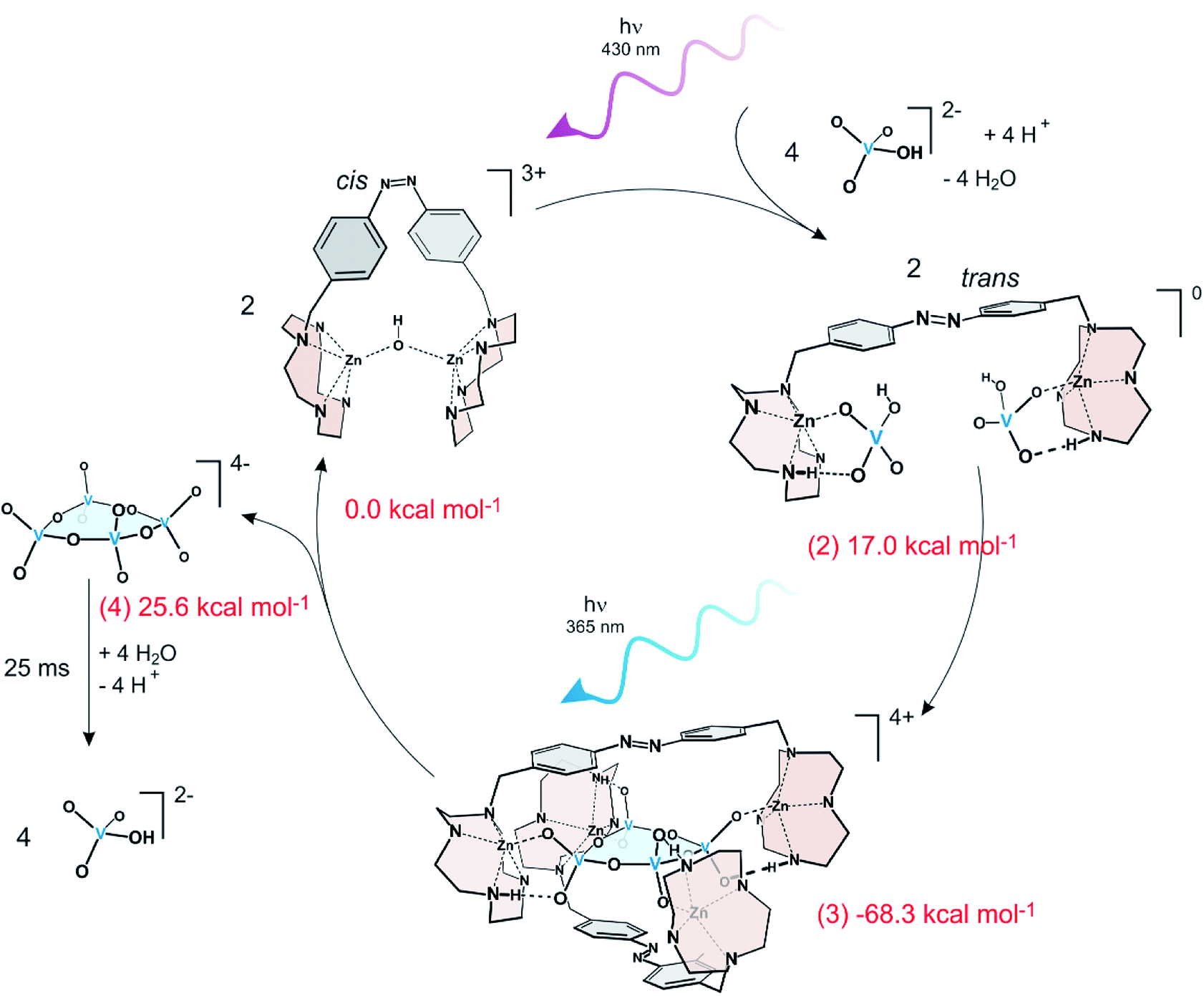 | ||
| Fig. 7 A continuously operating assembler driving the endergonic condensation of monovanadate to cyclic tetravanadate using light energy. ΔG values of the four stages measured by NMR titration are given in red numbers. The numbers in parentheses correspond to the stages in Fig. 5. Formation of cyclic tetravanadate is endergonic by 25.6 kcal mol−1. Cyclic tetravanadate is not stable under the conditions and hydrolyses back to monovanadate. | ||
In the closed form, the two Zn2+ ions in the two cyclene binding units are bridged by an OH− ion and have no affinity towards the reactants. Upon irradiation with violet light the receptor opens and the cyclene units each bind a monovanadate (2). Coordination to Zn2+ catalyzes the condensation of further monovanadates40 and finally a stable tennis ball-type complex is formed consisting of two pincer ligands and a cyclic tetravanadate (3). This stage is the lowest point on the reaction path and corresponds to the product inhibition or “sticky finger” step. Irradiation with UV light isomerizes the ligands to their non-binding cis form and the high energy product cyclic tetravanadate is released (4). Free cyclic tetravanadate is not stable under these conditions. It hydrolyses back to monovanadate. Rebinding of the product in stage (2) thus is avoided. Irradiation with violet light (365 nm) starts the next cycle. The system does not exhibit fatigue over at least several hundred cycles.
The example demonstrates that a well-designed photoswitchable ligand in an appropriate environment can guide reactant molecules through a predefined reaction channel and generate a compound that is not present (in measurable amounts) in the starting solution. Thereby, light energy is converted to chemical energy. Two problems remain: hydrolysis and rebinding of the product. Hydrolysis could be prevented by performing the reaction in a solvent other than water, where the product is stable (e.g. acetonitrile). Active removal of the product either by chemical trapping or diffusion of the product into a different phase or active transport through a membrane could solve both problems.
Conclusion and outlook
Referring to the discussion of Eric Drexler and Richard Smalley about the key question “Are molecular assemblers – devices capable of positioning atoms and molecules for precisely defined reactions – physically possible?” and particularly on Smalley's objections,1 we make the following statements:- the “fat finger problem” can be solved by molecular recognition (product binding).
- The “sticky finger problem” can be solved by active release of the product (coupling to an energy source).
However, programmable fingers (“universal assemblers”) probably are a very distant goal and self-replicating assemblers (“grey goo”) are science fiction. Notwithstanding the challenges of their molecular design and synthesis, continuously operating molecular assemblers are the obvious next step to go in the field of molecular machines and systems chemistry. Molecules that make molecules have the potential to induce a paradigm shift in chemical synthesis (Table 1). Even more challenging than continuously operating assemblers performing single step synthesis (ATP-type assemblers) are multistep (NRPS-type) assemblers.
| Laboratory synthesis | Assembly in nature | |
|---|---|---|
| Reaction environment | Solution | “Machine phase” |
| Bond forming process | Stochastic encounters of well designed, complementary reactive sites | Positioning control including the catalytic step after positioning within the assembler |
| Regio- and stereo control | Implemented within the reactants, catalyst or auxiliary | Information transferred from the assembler |
The advantages of assembler-type synthesis are obvious: prevention of side reactions, selective bond formation without protecting groups, facile stereo control, enforcement of thermodynamically unfavourable reactions, and selection of designated reactants in a mixture of compounds. The latter proficiency is particularly important while operating in more complex environments such as in one pot multistep syntheses and reaction cascades. Even applications in living organisms, fuelled by ubiquitous energy sources such as ATP and using bioavailable building blocks are conceivable. Finally, light driven molecular assemblers provide a new approach to convert light into chemical energy.41
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support by the Deutsche Forschungsgemeinschaft via the SFB 677 function by switching.References
- R. Baum, C&EN, Cover story, 2003, 81, pp. 37–42 Search PubMed.
- S. A. Sieber and M. A. Marahiel, Molecular Mechanisms Underlying Nonribosomal Peptide Synthesis: Approaches to New Antibiotics, Chem. Rev., 2005, 105, 715–738 CrossRef CAS PubMed.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Recent advances in engineering nonribosomal peptide assembly lines, Nat. Prod. Rep., 2016, 33, 317 RSC.
- B. Bakau and A. L. Horwich, The Hsp70 and Hsp60 Chaperone Machines, Cell, 1998, 92, 351–366 CrossRef.
- A. Stolz, W. Hilt, A. Buchberger and D. H. Wolf, Cdc48: A Power Machine in Protein Degradation, Trends Biochem. Sci., 2011, 36, 515–523 CrossRef CAS PubMed.
- C. Khosla, Harnessing the Biosynthetic Potential of Modular Polyketide Synthases, Chem. Rev., 1997, 97, 2577–2590 CrossRef CAS PubMed.
- K. E. Boulding, Stable Peace, 1972 Search PubMed.
- M. W. Hosseini and J.-M. Lehn, Supramolecular catalysis: substrate phosphorylations and adenosine triphosphate synthesis with acetylphosphate catalysed by a macrocycle polyamine, Chem. Commun., 1988, 397–399 RSC.
- M. W. Hosseini, J.-M. Lehn, K. C. Jones, K. E. Plute, K. B. Mertes and M. P. Mertes, Supramolecular catalysis: polyammonium macrocycles as enzyme mimics for phosphoryl transfer in ATP hydrolysis, J. Am. Chem. Soc., 1989, 111, 6330–6335 CrossRef CAS.
- Y. He and D. R. Liu, Autonomous Multistep Organic Synthesis in a Single Isothermal Solution Mediated by a DNA Walker, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed.
- J. Niu, R. Hili and D. R. Liu, Enzyme-free translation of DNA into sequence-defined synthetic polymers structurally unrelated to nucleic acids, Nat. Chem., 2013, 5, 282–292 CrossRef CAS PubMed.
- Z. Chen and D. R. Liu, Nucleic acid-templated synthesis of sequence-defined synthetic polymers, Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley-VCH, 1st edn, 2018, 3, pp. 49–90 Search PubMed.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, R. K. O`Reilly and A. J. Turberfield, Programmable one-pot multistep organic synthesis using DNA junctions, J. Am. Chem. Soc., 2012, 134, 1446–1449 CrossRef CAS PubMed.
- W. Meng, R. A. Muscat, M. L. McKee, P. J. Milnes, A. H. El-Sagheer, J. Bath, B. G. Davis, T. Brown, R. K. O'Reilly and A. J. Turberfield, An Autonomous Molecular Assembler for Programmable Chemical Synthesis, Nat. Chem., 2016, 8, 542–548 CrossRef CAS PubMed.
- For review see: F. C. Simmel, DNA-based Assembly Lines and Nanofactories, Curr. Opin. Biotechnol., 2012, 23, 516–521 CrossRef CAS PubMed.
- B. Lewandowski and D. A. Leigh, et al., Sequence-Specific Peptide Synthesis by an Artificial Small-Molecule Machine, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- G. de Bo, M. A. Y. Gall, S. Kuschel, J. de Winter, P. Gerbaux and D. A. Leigh, Nat. Nanotechnol., 2018, 13, 381–386 CrossRef CAS PubMed.
- A. W. Heard and S. M. Goldup, Simplicity in the design, operation, and applications of mechanically interlocked molecular machines, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- S. Kassem, A. T. L. Lee, D. A. Leigh, V. Marcos, L. I. Palmer and S. Pisano, Stereodivergent synthesis with a programmable molecular machine, Nature, 2017, 549, 374–378 CrossRef CAS PubMed.
- M. Okahara and Y. Nakatsuji, Active transport of ions using synthetic ionophores derived from macrocyclic polyethers and the related compounds, Top. Curr. Chem., 1985, 128, 37–59 CrossRef CAS.
- H. Plenio, H. El-Desoky and J. Heinze, Facile synthesis of ferrocene biscrown ethers and ferrocene cryptands: NMR complexation studies and the redox-switched bonding of H+ and Na+, Chem. Ber., 1993, 126, 2403–2408 CrossRef CAS.
- T. Saji and I. Kinoshita, Electrochemical ion transport with ferrocene functionalized crown ether, Chem. Commun., 1986, 716–717 RSC.
- H. Wang and G. Oster, Ratchets, power strokes, and molecular motors, Appl. Phys. A, 2002, 75, 315–323 CrossRef CAS.
- T. Schneider, Theory of molecular machines II. Energy dissipation form molecular machines, J. Theor. Biol., 1991, 148, 125–137 CrossRef CAS PubMed.
- C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Non-equilibrium supramolecular polymerization, Chem. Soc. Rev., 2017, 46, 5467–5680 RSC.
- D. Astumian, Kinetic asymmetry allows macromolecular catalysts to drive an information ratchet, Nat. Commun., 2019, 10, 3837 CrossRef PubMed.
- V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, A molecular information ratchet, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Light-powered autonomous and directional molecular motion of a dissipative self-assembling system, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Dissipative out-of-equilibrium assembly of man-made supramolecular materials, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, An artificial molecular pump, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- M. Baroncini, S. Silvi and A. Credi, Photo- and Redox-Driven Artificial Molecular Motors, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- M. Baroncini, L. Casimiro, C. de Vet, J. Groppi, S. Silvi and A. Credi, Making and operating molecular machines: a multidisciplinary challenge, ChemistryOpen, 2018, 7, 169–179 CrossRef CAS PubMed.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Hydrothermal vents and the origin of life, Nat. Rev. Microbiol., 2008, 6, 805–814 CrossRef CAS PubMed.
- D. Ragazzon and L. J. Prins, Energy consumption in in chemical fuel-driven self-assembly, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef PubMed.
- R. Kubo, The term free enthalpy is used as a synonym for Gibbs free energy, or Gibbs energy, Thermodynamics: An Advanced Course with Problems and Solutions (Divertissement 8: On the Names of Thermodynamic Functions), North Holland, 1976 Search PubMed.
- T. M. Duncan, V. V. Bulygin, Y. Zhou, M. L. Hutcheon and R. L. Cross, Proc. Natl. Acad. Sci., 1995, 92, 10964–10968 CrossRef CAS PubMed.
- M. J. Gresser, J. A. Myers and P. D. Boyer, J. Biol. Chem., 1982, 257, 12030–12038 CAS.
- H. S. Penefsky, Rate constants and equilibrium constants for the elementary steps of ATP hydrolysis by beef heart mitochondrial ATPase, Methods Enzymol., 1986, 126, 608–618 CAS.
- H. Sell, A. Gehl, D. Plaul, F. D. Sönnichsen, C. Schütt, F. Köhler, K. Steinborn and R. Herges, Towards a light driven molecular assembler, Commun. Chem., 2019, 2, 62, DOI:10.1038/s42004-019-0163-y.
- H. Sell, A. Gehl, F. D. Sönnichsen and R. Herges, Thermodynamic and kinetic stabilization of divanadate in the monovanadate/divanadate equilibrium using a Zn-cyclene derivative: towards a simple ATP synthase model, Beilstein J. Org. Chem., 2012, 8, 81–89 CrossRef CAS PubMed.
- W. Moormann, T. Tellkamp, E. Stadler, F. Röhricht, C. Näther, R. Puttreddy, K. Rissanen, G. Gescheidt and R. Herges, Angew. Chem., Int. Ed., 2020, 59, 1–7 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |