
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe double life of conductive nanopipette: a nanopore and an electrochemical nanosensor
Rui
Jia
 ab and
Michael V.
Mirkin
*ab
ab and
Michael V.
Mirkin
*ab
aDepartment of Chemistry and Biochemistry, Queens College-CUNY, Flushing, NY 11367, USA. E-mail: mmirkin@qc.cuny.edu
bThe Graduate Center of CUNY, New York, NY 10016, USA
First published on 5th August 2020
Abstract
The continuing interest in nanoscale research has spurred the development of nanosensors for liquid phase measurements. These include nanopore-based sensors typically employed for detecting nanoscale objects, such as nanoparticles, vesicles and biomolecules, and electrochemical nanosensors suitable for identification and quantitative analysis of redox active molecules. In this Perspective, we discuss conductive nanopipettes (CNP) that can combine the advantages of single entity sensitivity of nanopore detection with high selectivity and capacity for quantitative analysis offered by electrochemical sensors. Additionally, the small physical size and needle-like shape of a CNP enables its use as a tip in the scanning electrochemical microscope (SECM), thus, facilitating precise positioning and localized measurements in biological systems.
1. Introduction
Many types of nanometer-scale objects, such as nanoparticles (NP), nanodroplets, vesicles, and nanobubbles, play important roles in nanotechnology and nanomedicine. They are used as building blocks for nanostructured materials and catalysts,1,2 drug delivery vehicles,3 biosensing,4 imaging,5 and diagnostic6 tools. A number of biologically important species are stored and released in biological vesicles, e.g., synaptic vesicles7,8 and lysosomes.9 To deliver, detect, and measure various nanoobjects, one needs comparably sized tools and sensors. Among them are solid-state nanopores and nanopipettes. Nanopore sensing techniques, such as resistive-pulse and current rectification sensing,10–13 are based on measuring the change in ion current flowing through the pore orifice induced by analyte species (Fig. 1). In a resistive pulse experiment, the base current through the pore (i0) is driven by voltage applied between two reference electrodes (Fig. 1A). A nanosized object can enter through the orifice and partially block the ion current (Fig. 1B). In rectification sensors, the analyte species typically affect the surface charge density and, therefore, the extent of ion current rectification (Fig. 1C and D). These techniques found numerous applications in detection and sizing of single NPs, vesicles and large molecules.14–19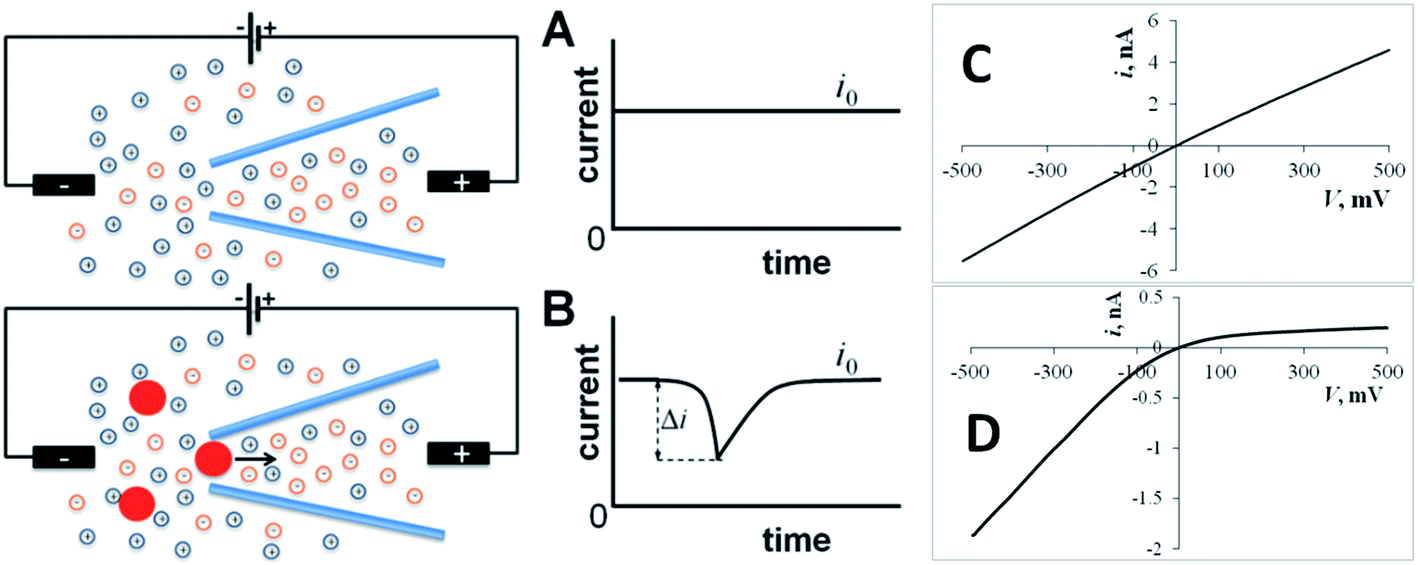 | ||
| Fig. 1 Resistive-pulse and current rectification sensing with a conical nanopore or nanopipette. (A) Constant ion current (i0) flows through the orifice with no particles present in solution. (B) A blockage event (resistive pulse) can be seen in the current vs. time curve when a particle translocates through the pore orifice. (C) The ion current vs. voltage (i–V) curve is essentially linear for a large (200 nm diameter) pipette. (D) A small (24 nm diameter) pipette exhibits strong current rectification. | ||
Although the diameter of the pore orifice can be very small (nm-range), a much larger physical size of the device (Fig. 2A) limits most nanopore applications to amperometric measurements in the bulk solution. While the nanopipette inside geometry is similar to that of a conical nanopore, the nanoscale tip diameter (Fig. 2B) makes pipettes suitable for localized experiments in small spaces.20–22 Nanopipette-based resistive-pulse and electrochemical techniques provided several promising approaches to single-entity measurements.17–19,23,24 A needle-like nanopipette can serve as a scanning ion conductive microscopy (SICM) or scanning electrochemical microscopy (SECM) tip that can be inserted into a living cell or positioned close to its surface.25–28 In this way, nanopipettes can be used for detecting NPs and biological vesicles in living cells with potential applications in different areas of biomedical research ranging from nanotoxicology to neurochemistry, photodynamic therapy, and immunology.4–6,9,29
 | ||
| Fig. 2 SEM (A and C) and TEM (B and D) images of the single-nanopore membrane (A), quartz nanopipette (B), carbon fiber nanoelectrode (C) and disk-type platinized carbon nanoelectrode (D). Adapted with permission from ref. 30; copyright 2006, American Chemical Society (A), ref. 32; copyright 2015, Wiley-VCH (C), and ref. 35; copyright 2017, American Chemical Society (D). | ||
Unlike a nanopore, amperometric nanoelectrode signal is the faradaic current produced by oxidation/reduction of electroactive species at its conductive surface. Nanoelectrode measurements are complementary to nanopore experiments – they are more suitable for determining concentrations of molecular analytes than for detecting nanoscale objects. Amperometric nanoelectrodes, including glass-sealed disk-type Pt tips,31 have been used for electrochemical experiments inside living cells. However, many important analytes, such as dopamine and reactive oxygen and nitrogen species (ROS/RNS), passivate the surface of a nanoelectrode. To determine concentrations of such species, the electrode surface area must be sufficiently large, and no analytically useful response could be recorded at a polished disk nanoelectrode. Carbon nanofibers (Fig. 2C),32–34 and platinized nanodisk-type electrodes (Fig. 2D)35,36 and nanowires37 were employed for measurements of neurotransmitters and ROS/RNS in single cells and biological vesicles.7
Coating the inner wall of a glass (or quartz) nanopipette with a thin layer of conductive material, such as Au38,39 (Fig. 3A), Pt40 (Fig. 3B), Ag41 (Fig. 3C), or conductive polymer,42 yields a conductive nanopipette (CNP) that can function as a nanopore or/and a nanoelectrode sensor. Carbon CNPs with different geometries were produced by chemical vapor deposition of carbon developed by the Bau43 and Gogotsi44 groups into quartz pipettes45 (Fig. 3D). In these devices, the conductive film is deposited onto the inner pipette wall leaving an open pass in the middle. Two examples of sensors different from but closely related to CNPs are also shown Fig. 3. In an “electrochemical nanosampler”46 (Fig. 3E), the interior of the pulled nanocapillary is completely filled with carbon, leaving only a small cavity adjacent to the orifice. The amperometric response of a nanosampler is similar to that of a CNP, but it has no open pass in the middle and, therefore, is not suitable for conventional resistive-pulse experiments (electrochemical resistive-pulse experiments (see Section 4) with a nanosampler are conceptually possible, but have not yet been reported). Metallized glass nanopipettes were prepared by sputtering a thin layer of either Au (Fig. 3F)47 or Pt48 onto the outer wall. Such pipettes have been used for single-molecule dielectrophoretic trapping (shown schematically in Fig. 3F)47 and in a nanokit48 in which the reagents were injected into a living cell through the pipette, and the Pt ring served for electrochemical measurement of hydrogen peroxide.
 | ||
| Fig. 3 SEM (A–C and F) and TEM (D and E) images of CNPs (A–D) and closely related devices (E and F). (A) Side view of a nanopipette with a gold nanotip after FIB splitting along the tip. Adapted with permission from ref. 38; copyright 2018, Wiley-VCH. (B) Pt layer (shiny region) on the inner wall of capillary after FIB splitting along the tip. Adapted with permission from ref. 40; copyright 2018, National Academy of Sciences. (C) Wireless nanopore electrode prepared by depositing Ag on the inner wall of a quartz nanopipette by electron beam evaporation technique. Adapted with permission from ref. 41; copyright 2017, American Chemical Society. (D) Open carbon CNP. Adapted with permission from ref. 28; copyright 2020, American Chemical Society. (E) Carbon nanocavity electrode (“electrochemical nanosampler”). Adapted with permission from ref. 46; copyright 2014, American Chemical Society. (F) Gold-coated nanopipette (scale bar 5 μm) and schematic representation of a dielectrophoretic experiment. Insets: SEM image of the tip visualized parallel to the barrel and intensity line plot; scale bar, 50 nm. Adapted with permission from ref. 47; copyright 2016, Springer Nature. | ||
Important advantages of conductive pipettes for nanopore sensing (tunable surface charge and potential) and nanoelectrode applications (a small physical size and a significant conductive surface area) are discussed below along with a recently developed combination of resistive-pulse sensing with nanoelectrochemistry.49
2. Resistive-pulse and current rectification sensing with conductive pipettes
Analytical applications of nanopore- and nanopipette-based sensors typically rely on control of the surface charge and chemical state of the inner wall that can influence both ion current rectification and resistive-pulse sensing. To optimize the pipette response, its inner wall can be chemically modified and functionalized using different reagents,27,50–52 but these procedures are labor-intense and plagued by various technical issues. An alternative approach—to coat the inner wall with a conductive film—can yield a nanopipette with the tunable surface charge and potential, thus, improving its selectivity and sensitivity.24The potential and surface charge of the conductive layer can be controlled either by electrically connecting it to the potentiostat or under open circuit conditions. The current rectification exhibited by carbon CNPs at open circuit was stronger than that observed with similarly sized quartz pipettes in the same solution pointing to a significant negative charge density on the carbon surface. The extent of ion current rectification was changed systematically by varying the potential of a carbon CNP used as a working electrode.45 It became more pronounced with the increasingly negative potential applied to the carbon layer. When the carbon layer was biased positively (e.g., +500 mV vs. Ag/AgCl), the rectification disappeared completely, and the corresponding i–V curve became linear.45
Although the current rectification can be controlled by varying the applied CNP potential, this approach is not very useful because the conductive surface area exposed to the solution is by many orders of magnitude larger than that of the orifice, and a significant faradaic current flowing at the carbon surface interferes with the ion current through the pipette. This problem can be avoided by keeping the conductive surface at the open circuit potential. One approach is to let the conductive film float and act as a bipolar junction.39,53 It was shown that polarizing an Au film placed at the entrance of a SiN nanopore enables voltage control of local ionic concentrations.53 The surface charge in a nanopore rectifier changed with the transmembrane potential, and the induced surface charge density and ionic concentrations in the pore were regulated by magnitude and direction of the external electric field, leading to ion current rectification.
An alternative approach is to control the rectification properties of a floating carbon CNP through tuning its open circuit potential by low (e.g., pM to nM) concentrations of redox species in the solution.54 The redox mediators with positive standard potentials (E0) tend to decrease the negative charge density on the carbon surface, thus, decreasing the ion current in the high-conductance state and the degree of ion current rectification (Fig. 4A), while redox species with a negative E0 produce the opposite effect (Fig. 4B). At very low concentrations, the faradaic current produced by these redox species is immeasurably small; however, by changing the open circuit potential, they affect the much larger ion current through the CNP (“electron-transfer gated ion transport”54).
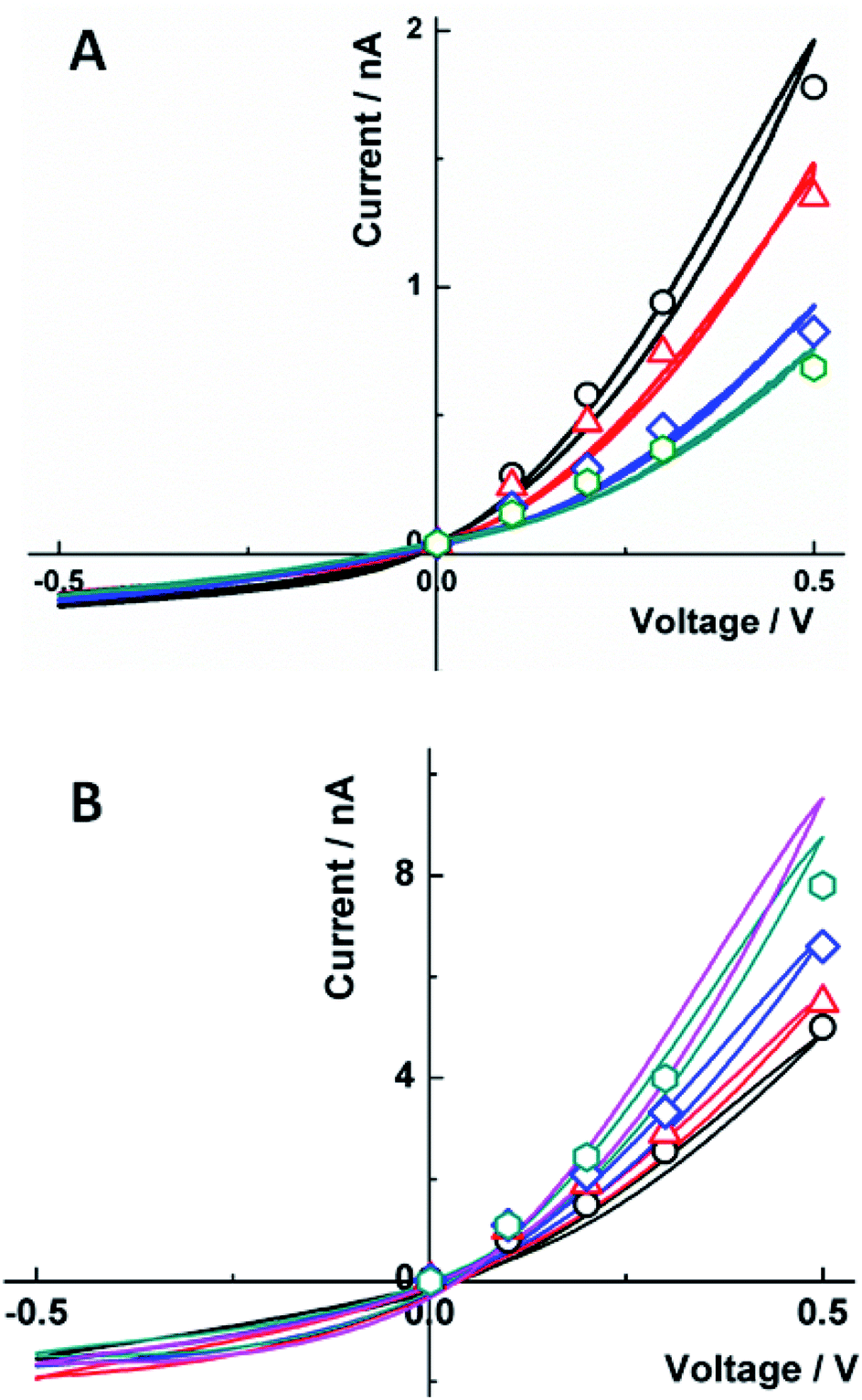 | ||
| Fig. 4 Effect of (A) Fe(CN)63− and (B) Ru(NH3)63+ concentration on the carbon CNP i–V curves in 10 mM KCl. (A) From top to bottom, experimental (solid lines) and simulated (symbols) i–V curves for [K3Fe(CN)6], nM: 0 (black), 100 (red), 200 (blue), and 300 (green); and surface charge density, σ (mC m−2) = −23, −10, −4, and −2. (B) [Ru(NH3)6Cl3], pM: 0 (black), 100 (red), 200 (blue), and 300 (green); and fitted σ (mC m−2) = −5, −10, −30, and −60. The magenta curve was obtained with [Ru(NH3)6Cl3] = 100 nM. The CNP orifice radius was 50 nm. Reprinted with permission from ref. 54; copyright 2017, American Chemical Society. | ||
The effect of Ru(NH3)63+ on current rectification in Fig. 4B was observed at much lower concentrations than that of Fe(CN)63− in Fig. 4A. This difference was attributed to positive ionic charge of Ru(NH3)63+. The multivalent cations (including electrochemically inactive Ca2+) adsorb on the carbon surface and affect the ionic transport processes, in contrast to apparent insensitivity of the ion current to higher (e.g., sub-μM) concentrations of multivalent anions.
Nevertheless, the main mechanism in electron-transfer gating of ion current is bipolar electrochemistry rather than ionic adsorption. This effect was observed with different redox species, including neutral molecules such as aqueous ferrocenemethanol and ferrocene in acetonitrile; both of them exerted a stronger effect on current rectification than Fe(CN)63−. In addition to redox control of ion current rectification in conductive nanopores, these results suggested the possibility of trace level sensing of redox analytes with CNPs.
The possibility of reversibly changing the potential and surface charge of the inner wall suggests that a CNP can be used as a versatile resistive-pulse sensor whose properties can be tuned to detect a specific analyte. The first application of a conductive (Au coated) conical nanopore as a single entity translocation sensor was reported by the Martin group.55 Unlike stochastic resistive-pulse sensors, the detection of protein molecules was based on total current blockage by protein analyte binding to a biochemical molecular-recognition agent immobilized at the smaller opening of the conical pore and did not rely on measurement of transient current pulses.
Carbon CNPs were used for resistive-pulse detection of 10 nm Au NPs with covalently attached monoclonal primary antihuman PSA antibodies and prostate specific antigen, and the recorded current spikes were similar to those obtained with quartz nanopipettes.45 In both cases, current blockages were observed only when a positive potential was applied to the internal reference electrode. However, the voltage applied to record current blockages was only +100 mV for CNPs as opposed to +600 mV that was required to detect the same nanoparticles using quartz nanopipettes.
Metalized (Au-coated) nanopores were used for resistive-pulse sensing of insulin.53 An attempt to detect insulin with unmodified metallic or SiN-based nanopores was not successful presumably due to the small size of the protein species and the high speed of its translocation. The translocation signal was recorded after modifying the Au surface with homocysteine (Fig. 5), and the authors concluded that the residence time of the protein inside the pore was governed by the hydrophobic rather than electrostatic interactions between the protein and the homocysteine layer.
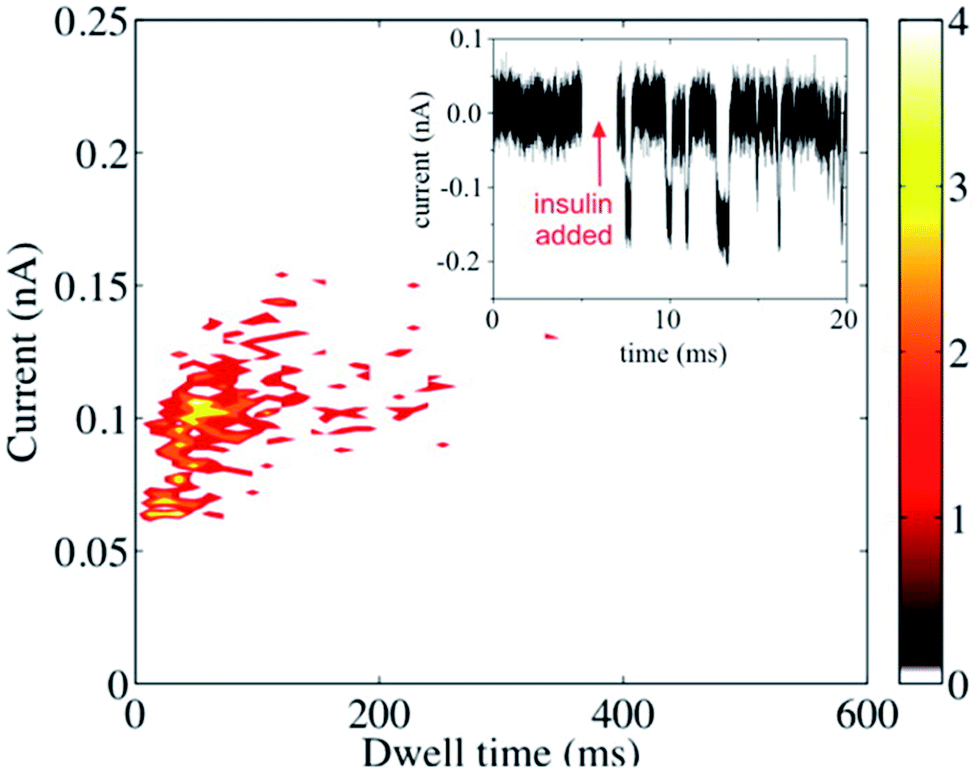 | ||
| Fig. 5 Resistive-pulse sensing of insulin molecules with a metallized nanopore. Scatter plot and the current–time recording (inset) before and after the addition of the 0.1 mg mL−1 insulin to the solution containing 0.1 M KCl and 40 mM HCl. V = −400 mV. Reprinted with permission from ref. 53; copyright 2015, American Chemical Society. | ||
Resistive-pulse sensing of liposomes49 and intracellular biological vesicles28 with carbon CNPs was reported recently. Similar to quartz pipettes, the translocation of negatively charged liposomes through a floating carbon CNP was found to be driven by electroosmosis. In ref. 28, a carbon CNP was inserted into a RAW 264.7 murine macrophage to record ion current blockages produced by cellular vesicles. These experiments showed the possibility of resistive-pulse measurements inside a living cell that was not obvious because of likely plugging of the pipette orifice (either glass or carbon) by lipids, proteins, and other biomolecules. Nevertheless, the base current was relatively stable during long (up to an hour) recordings inside the cell. The current transients recorded with carbon CNPs28,45,49 were qualitatively similar to those obtained with quartz pipettes, and the expected advantages of tunable potential and surface charge for ion-current-based stochastic sensing have yet to be demonstrated.
3. Conductive nanopipettes as amperometric nanosensors
When a CNP is immersed in solution containing redox species, the faradaic current at the conductive film that covers the inner pipette wall near its orifice and serves as a working electrode comprises two components – the steady-state current due to convergent diffusion of redox species to the CNP tip in the external solution and the transient component produced by oxidation/reduction of the sampled redox species inside the pipette (Fig. 6A). If the conductive surface area exposed to solution is small (i.e. either the conductive coating is only applied to the pipette tip38,40 or the solution volume inside the CNP is very small45,46), voltammograms obtained at a slow scan rate are sigmoidal and retraceable (curve 1 in Fig. 6A), suggesting that the non-steady-state current inside the pipette completely vanishes on a relatively long experimental time scale. At a faster scan rate, cyclic voltammograms exhibit a pair of prominent peaks corresponding to oxidation/reduction of the redox species inside the CNP (curve 2 in Fig. 6B). The rapid mass transfer inside the nanocavity results in exhaustive electrolysis of sampled molecules even on a short experimental time scale, and at E ≫ E°′, the anodic current decreases to the steady-state value. This value is similar to the steady-state diffusion limiting current at the inlaid nanodisk electrode of the same radius,46 and therefore it can be used to evaluate the radius of the CNP orifice.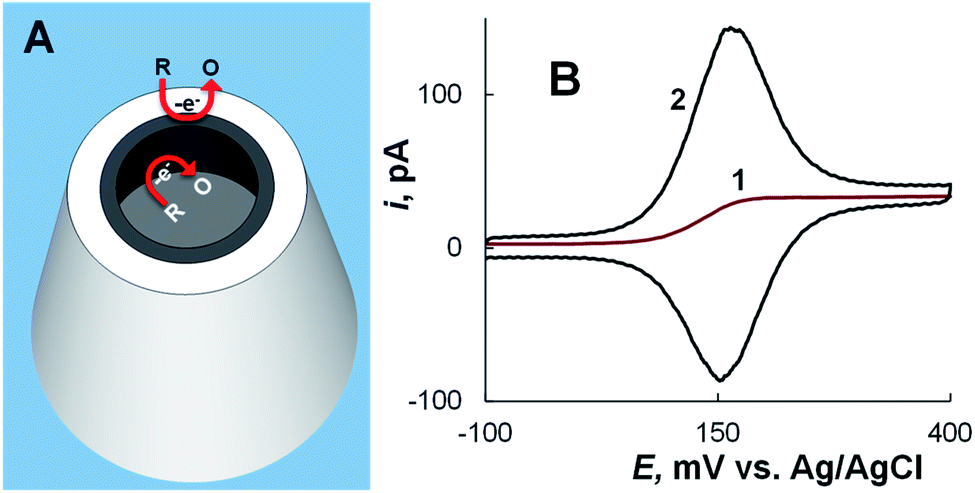 | ||
| Fig. 6 CNP as a nanoelectrode. (A) Schematic representation of oxidation of electroactive species at the conductive nanoring exposed to external solution and at the inner CNP wall. (B) Cyclic voltammograms of 1 mM ferrocenemethanol in 0.2 M KCl solution obtained using an ∼100 nm-radius CNP at different potential sweep rates, ν = 10 mV s−1 (1) and 200 mV s−1 (2). Adapted with permission from ref. 46; copyright 2014, American Chemical Society. | ||
The shapes of steady-state current vs. distance curves obtained with a CNP serving as an SECM tip were in agreement with the theory for a disk-shaped tip.46 suggesting that the SECM can be used for CNP positioning and control of its insertion into biological cells (see Section 4).
The mass-transfer inside the CNP is rapid, whereas the diffusion of species out of the cavity is relatively slow. Thus, a CNP can be used for sampling and electrochemical analysis of molecular species and single entities. In ref. 40, a CNP with a Pt ring inside its tip (Fig. 7) was inserted into a living HeLa cell to sample single lysosomes and characterize glucosidase activity in them. Lysosomes were driven inside the CNP by voltage applied between the inner Ag/AgCl reference and external electrode and lysed by Triton X-100 present inside the pipette to release glucosidase. The CNP also contained β-D-glucopyranoside and glucose oxidase. The released glucosidase converted β-D-glucopyranoside to glucose that reacted with glucose oxidase producing H2O2. The glucosidase activity was evaluated from the oxidation of H2O2 at the Pt surface. The data analysis was challenging because of complicated transport processes and difficulties in ascertaining that the measured signal was produced by a single lysosome.
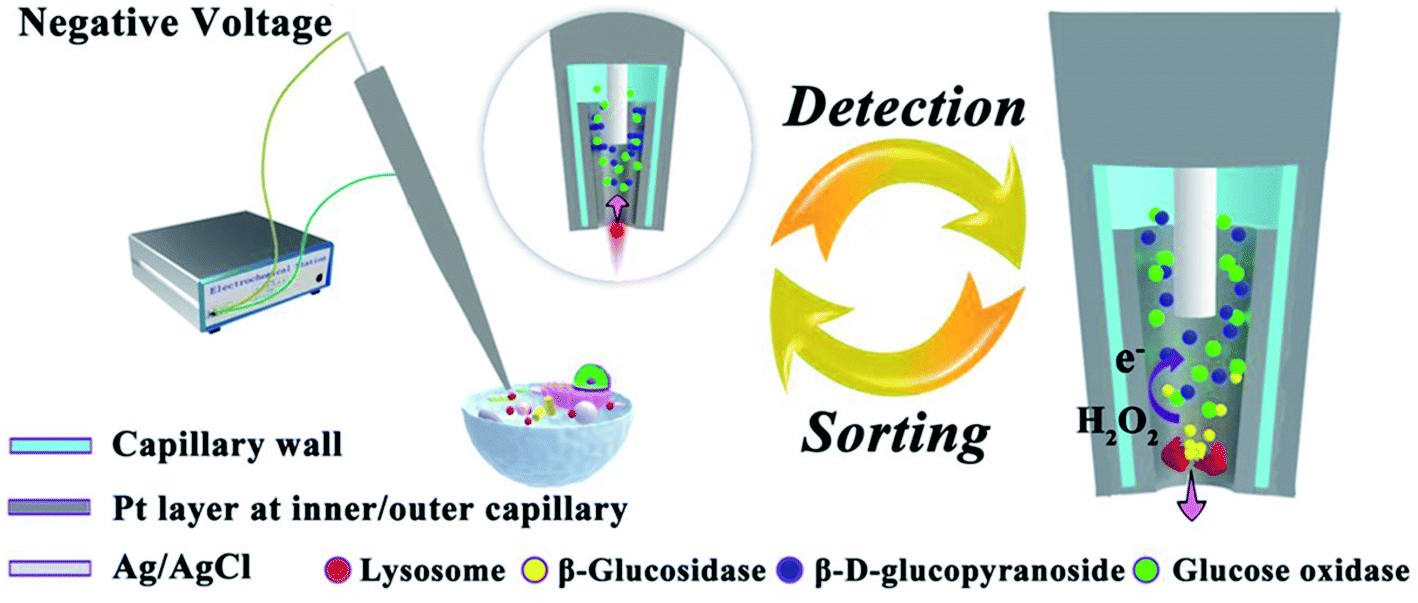 | ||
| Fig. 7 Schematic representation of the electrochemical setup for evaluating glucosidase activity in single lysosomes sampled from a living cell. Inset: a lysosome is driven into the Pt-coated capillary tip by applied voltage. The arrow shows the flow direction of buffer with the lysosome. Right picture: glucosidase is released during the lysis of a lysosome; hydrogen peroxide is oxidized at the Pt layer inside the CNP. Adapted with permission from ref. 40; copyright 2018, National Academy of Sciences. | ||
The charged inner surface of a CNP tip can give rise to permselective behavior. High negative surface charge on the carbon wall54 results in a significant accumulation of cations and depletion of anions inside a CNP. The lower the concentration of supporting electrolyte and smaller the CNP radius the larger the magnitude of these effects that also increase with the ionic charge. The electrostatic effects can be used for improving the CNP sensitivity to ionic redox species and eliminating charged interferences. Thus, the 2000 fold accumulation of Ru(NH3)63+ in 1 mM KCl solution inside a CNP enabled the detection of this species at the level of 10 pM, while the current produced by the 106 times higher concentration of Fe(CN)64− was completely suppressed.56
The sensitivity and selectivity of CNPs can be further enhanced by adsorption of analyte species and suppression of electrochemically irreversible interferences. The adsorption of cationic dopamine on the carbon CNP surface resulted in a sub-nM detection limit.57 High selectivity for dopamine detection over ascorbic acid—a major interferent in dopamine analysis—was attained by redox cycling for dopamine. The electrochemically irreversible signal produced by oxidation of anionic ascorbic acid was diminished after the first voltammetric cycle because of its relatively slow mass transport into the CNP from external solution. The applicability of carbon CNPs to fast scan voltammetry of dopamine (e.g., with the potential sweep rate, ν = 400 V s−1) relevant to analysis in biological systems has recently been demonstrated, and exogenously applied dopamine was detected in mouse-brain slices.58
The conductive film inside a CNP can also work as a wireless bipolar nanoelectrode (Fig. 8). The proof-of-concept experiment (Fig. 8A) was done using a CNP with the carbon layer floating (i.e., not connected to the potentiostat).45 The two parallel pathways for the current flow through a bipolar CNP—the ion current through the pipette orifice and electronic current through the conductive carbon layer—are represented by a simplified equivalent circuit (inset I). The relative magnitudes of the ionic and electronic components of the total current flowing between the two reference electrodes are determined by the resistance associated with each pathway. In ref. 45, the ionic pathway was eliminated by not filling the CNP with liquid phase, so that only the back portion of the carbon layer and its nm-sized tip were exposed to solution (Fig. 8A). The voltage drop along the carbon layer induced two opposite electrochemical reactions, i.e. the oxidation of ferrocenedimethanol at the tip and a cathodic process (presumably, oxygen reduction) at the backside side of the CNP. The steady-state voltammogram of ferrocenedimethanol obtained in this configuration by sweeping the voltage applied between two reference electrodes (red curve in the inset II) and the background voltammogram obtained with no added redox species (black curve) are very similar to the curves that would be measured at a similarly sized carbon disk nanoelectrode. Because the carbon area exposed to solution on the backside of the asymmetric bipolar electrode is large (∼mm2), voltammetric current is fully determined by diffusion of redox species to the microscopic tip.
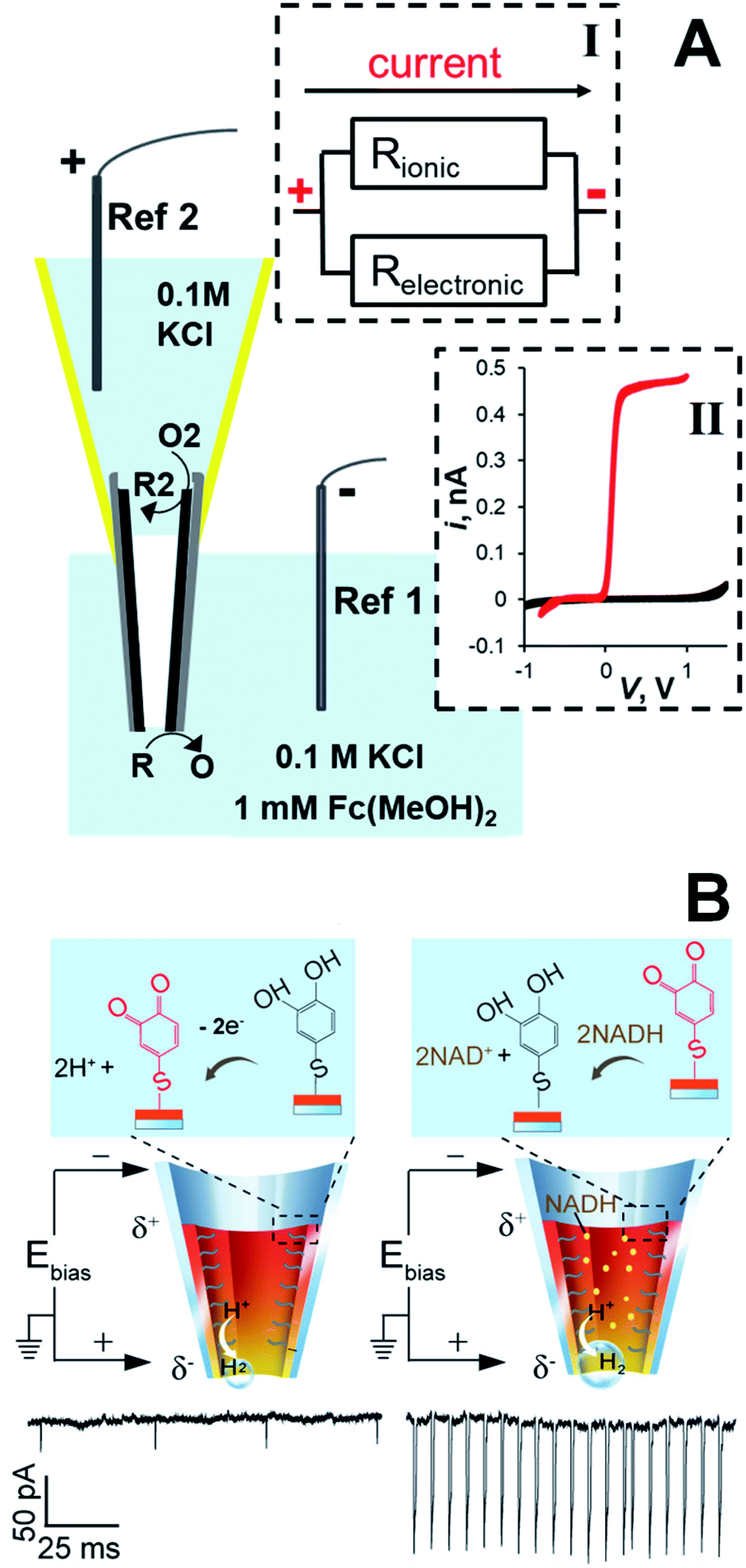 | ||
| Fig. 8 CNP as an asymmetric bipolar nanoelectrode. (A) Schematic representation of a carbon CNP used as a wireless nanoelectrode. Two faradaic processes, i.e. oxidation of R at the nanotip and reduction of O2 species at the macroscopic backside of the CNP are driven by the voltage drop (V) along the carbon film. Inset I: Equivalent circuit showing ionic and electronic pathways for the current flow through the CNP. Inset II: Cyclic voltammograms obtained with a bipolar CNP electrode. External solution contained 0.1 M KCl and 0 (black) or 1 mM (red) of ferrocenedimethanol. Adapted with permission from ref. 45; copyright 2014, American Chemical Society. (B) The 4-thiol-catechol-modified CNP exhibits current spikes due to the generation of H2 nanobubbles. Left: The catechol is oxidized at the anodic pole, whereas H2 is generated at the cathodic pole. Right: The mediation of NADH oxidation by the catechol/o-benzoquinone couple results in increased ion current spikes. V = −0.7 V. Adapted with permission from ref. 59; copyright 2018, American Chemical Society. | ||
The applicability of asymmetric bipolar nanoelectrodes to the detection of single small molecules and electroanalysis in living cells was demonstrated in ref. 41, 59 and 60 where CNPs were prepared by coating the inner wall of a glass nanopipette with a Ag or Au layer and filled with solution to measure the ion current induced by the bias voltage (Fig. 8B). The cathodic reaction occurring at the CNP tip was the reduction of protons that resulted in the formation of hydrogen bubbles. The ion current spikes caused by the bubble translocation through the CNP allowed the detection of just a few hydrogen molecules.41 In ref. 59, the Au surface inside the CNP was modified with 4-thiol-catechol, and its oxidation to o-benzoquinone was coupled with the H2 evolution at the tip (Fig. 8B, left panel). A novel strategy was used to amplify the signal produced by a very low concentration of redox species in solution: when nicotinamide adenine nucleotide (NADH) was added to the solution, catechol mediation of its oxidation to NAD+ resulted in the increased frequency and amplitude of current spikes (Fig. 8B, right panel). In this way, the immeasurably low current of NADH oxidation at the CNP was amplified, enabling NADH monitoring in single living cells and its detection at concentrations as low as 1 pM.
4. Electrochemical resistive-pulse sensing in solution and in biological cells
In electrochemical resistive-pulse experiments, a CNP serves as a working electrode and no reference electrode is placed inside it (Fig. 9A). Only a small (μm-long) part of the CNP shaft adjacent to its orifice is filled with solution, and the measured signal is the faradaic current flowing at the microscopic portion of the conductive film exposed to solution. The blockage of the diffusion current of redox species by a particle (e.g., a liposome49) translocating through the CNP orifice results in a resistive pulse (green peak in the inset; Fig. 9A). Because the conductive carbon surface is essentially equipotential, the voltage drop along the pipette axis inside its shaft is small. The translocation of particles is driven by diffusion rather than electroosmosis or electrophoresis, and current blockages can be observed at either positive or negative CNP bias with respect to the external reference.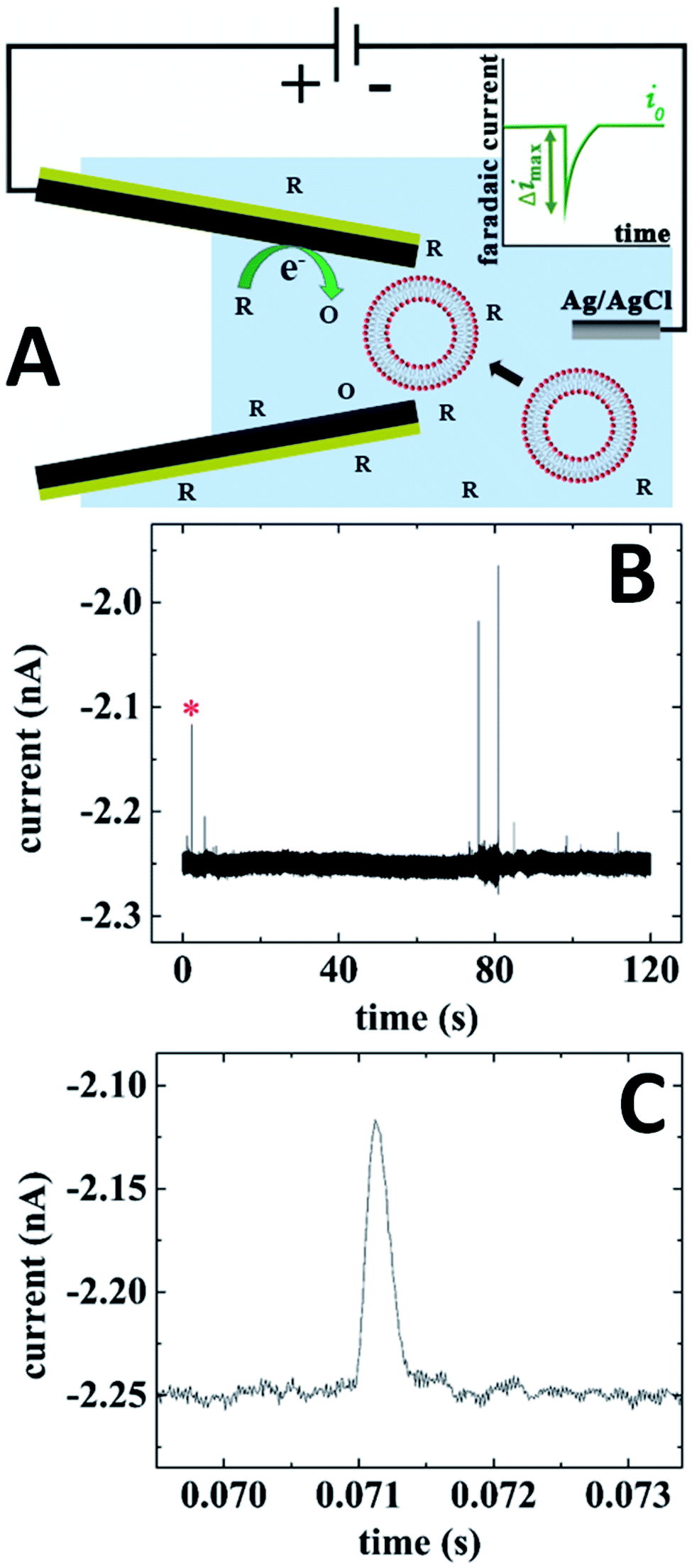 | ||
| Fig. 9 Electrochemical resistive-pulse sensing with a carbon CNP. (A) Schematic representation of the experiment involving translocation of liposomes through a CNP. The inset shows a faradaic current blockage produced by the liposome translocation. (B) Current–time recording obtained with a 250 nm CNP biased at −0.5 V vs. Ag/AgCl reference. (C) Blowup of the transient labeled by red asterisk in (B). 10 mM PBS (pH 7.4) solution contained 10 mM K3[Fe(CN)6] and 15.6 nM of liposomes. Adapted with permission from ref. 49; copyright 2019, American Chemical Society. | ||
In Fig. 9B, the current was recorded in solution containing 10 mM Fe(CN)63− that was reduced at the carbon CNP potential of −0.5 V vs. Ag/AgCl. A relatively small magnitude (Δimax/i0 ≪ 1), shape (Fig. 9C), and the mean half-width (0.29 ms) of the faradaic current blockages by liposomes are comparable to those recorded in conventional resistive-pulse experiments. The electrochemical resistive pulses are produced by liposomes entering the CNP rather just plugging its orifice from the solution side; therefore, no current spikes could be measured using CNPs with the orifice diameter smaller than that of the liposomes.49 Using oxygen reduction as a source of faradaic current, one can carry out electrochemical resistive-pulse experiments in biological systems with no redox mediator added to solution.
The concept of combining electrochemical resistive-pulse sensing with electroanalysis of single entities is shown schematically in Fig. 10A, where liposomes are loaded with the reduced form of redox species (R). Resistive-pulse recordings in this case are expected to include current blockages associated with liposome translocations (green peak in Fig. 10A) and current upsurges caused by oxidation of R during liposome collisions with the CNP inner wall (purple peak).
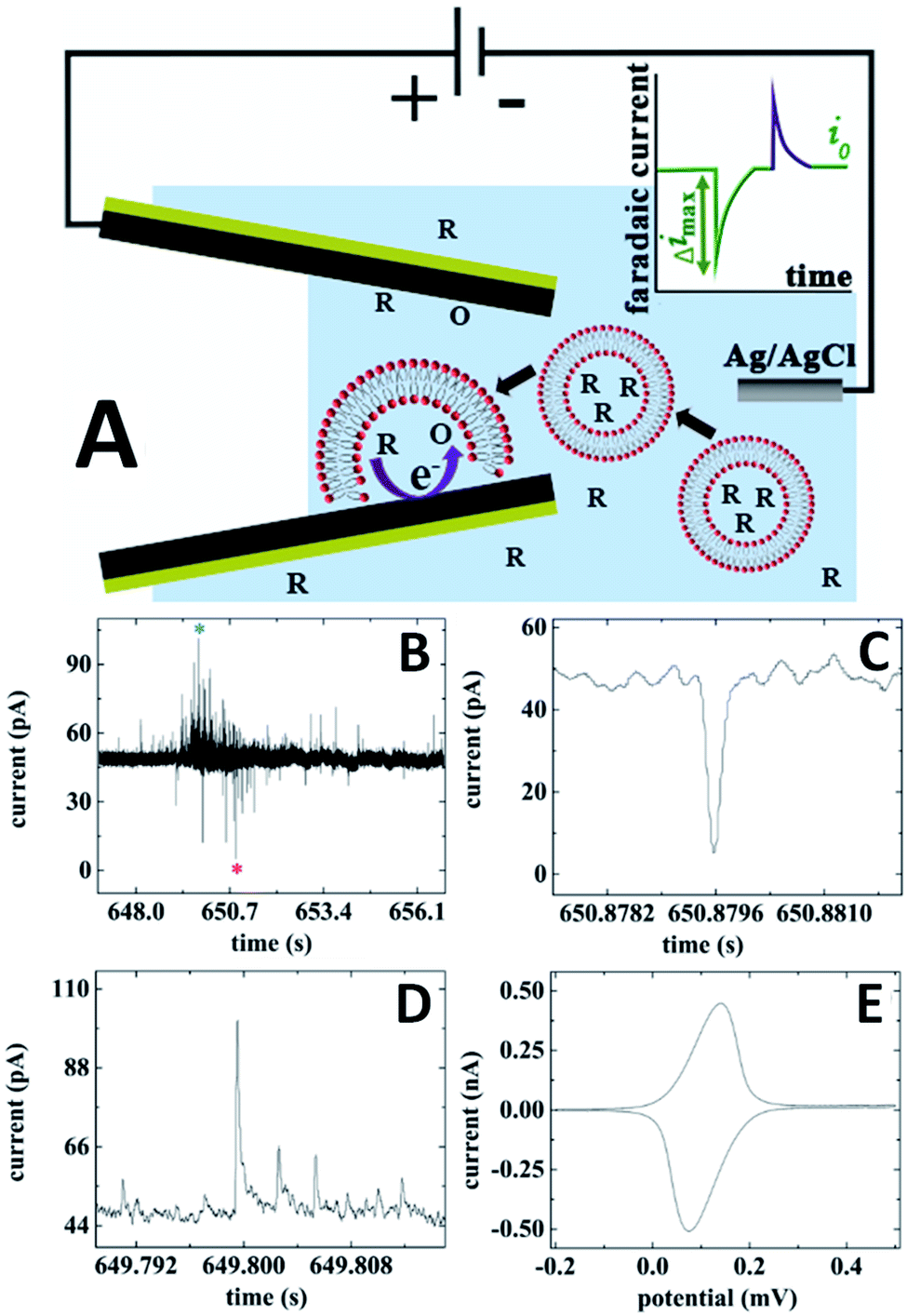 | ||
| Fig. 10 Electrochemical resistive-pulse sensing combined with the dopamine analysis in single liposomes. (A) Schematic representation of the electrochemical resistive-pulse experiment involving translocation of liposomes. The inset shows faradaic current blockage produced by the liposome translocation. (B) Current–time recording obtained with dopamine-loaded liposomes using a 350 nm carbon CNP. (C) Blowup of the individual resistive pulse labeled with a red asterisk in (B). (D) Liposome collision transient labeled with a blue asterisk in (B). CNP potential was 0.5 V vs. Ag/AgCl. (E) Cyclic voltammogram of dopamine. Potential sweep rate, ν = 50 mV s−1. Adapted with permission from ref. 49; copyright 2019, American Chemical Society. | ||
Both upward and downward spikes can be seen in current–time recordings obtained with dopamine-loaded liposomes translocating a CNP (Fig. 10B).49 Typical resistive-pulse and collision transients are shown in Fig. 10C and D, respectively. The integration of the current under the peak in Fig. 10D yields the charge corresponding to the number dopamine molecules oxidized during a specific collision. A cyclic voltammogram obtained with the same pipette immediately after an electrochemical resistive-pulse experiment (Fig. 10E), comprises a pair of symmetrical peaks of exhaustive oxidation/reduction of DA accumulated inside the CNP.
For intracellular resistive-pulse measurements, a CNP has to be positioned within the cell cytoplasm. It is important to make sure that the pipette has penetrated the membrane but has not come too close to the bottom of the cell that would block the diffusion of vesicles to its orifice. A convenient way to monitor the CNP insertion into a cell is to use it as an SECM tip (Fig. 11A). When the nanopipette approaches the cell surface, the steady-state faradaic current decreases with decreasing separation distance (d; in Fig. 11A, it is normalized by the orifice radius, a) because the membrane blocks the diffusion of the redox mediator to the CNP orifice either completely (if the mediator species is hydrophilic) or partially – if it is hydrophobic.31 Although the same hydrophobic redox species (e.g., ferrocenemethanol) could be the source of the tip current outside the cell and the base current for intracellular resistive-pulse measurements, the membrane penetration is harder to detect when the same mediator is present inside and outside the cell.31,36 Thus, in Fig. 11A, two redox mediators—1 mM ferrocenemethanol and 10 mM K4[Fe(CN)6]—were simultaneously present in solution. The experimental approach curve initially fits the theory for negative SECM feedback (solid line in Fig. 11A) and deviates from it when the pipette tip begins to push the cell membrane (positive d correspond to the tip approaching the membrane; negative distances correspond to the tip pushing the membrane and then penetrating the cell). When the tip punctures a hole in the cell membrane, the current drops to the value corresponding to the oxidation of ferrocenemethanol inside the cell.
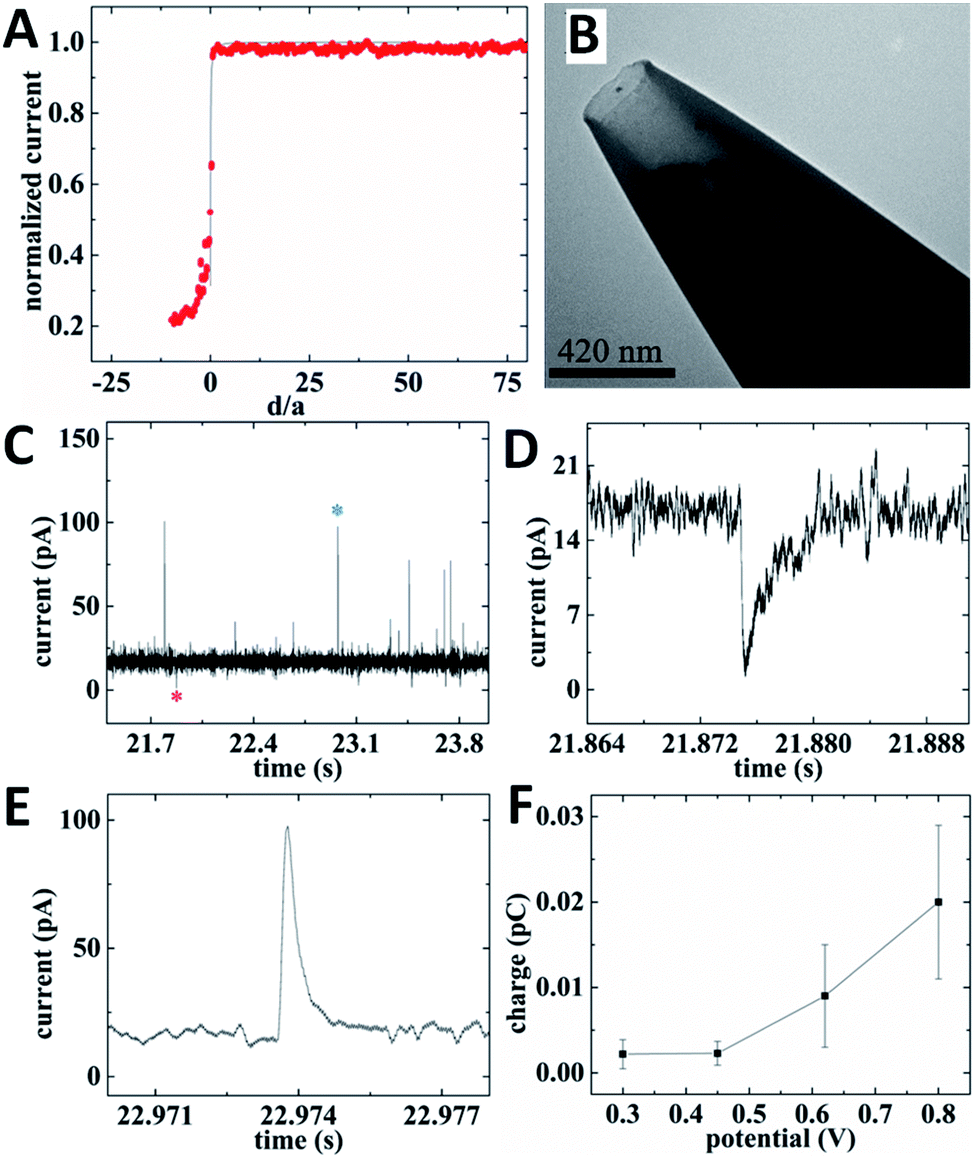 | ||
| Fig. 11 Electrochemical resistive-pulse sensing and amperometric analysis of ROS/RNS in cellular vesicles within a living macrophage cell. (A) SECM current–distance curve obtained with a carbon CNP approaching and penetrating an immobilized macrophage (symbols) fitted to the theory for the negative feedback (solid line). (B) TEM image of a 210 nm-diameter platinized carbon CNP. (C) Current–time recording obtained with the same platinized carbon CNP. (D) Individual resistive-pulse spike labeled with a red asterisk in (C). (E) Representative vesicle collision transient labeled with a blue asterisk in (C). CNP potential was 0.8 V vs. Ag/AgCl. (F) Dependence of the mean vesicle collision charge on CNP potential. Adapted with permission from ref. 28; copyright 2020, American Chemical Society. | ||
While some important analytes (e.g., dopamine or serotonin) can be measured with unmodified carbon CNPs, the carbon surface has to be platinized for determination of ROS/RNS.35,36 To enable simultaneous electrochemical resistive-pulse sensing and amperometric analysis, Pt NPs were deposited on the inner carbon wall, leaving an open path in the middle (Fig. 11B). Both downward and upward spikes can be seen in the current–time recording obtained with a platinized CNP inside a macrophage cell (Fig. 11C). The shape of a representative resistive pulse (Fig. 11D) is similar to those obtained with quartz nanopipettes.28 The current upsurges (Fig. 11C and E) are caused by oxidation of ROS/RNS contained in the vesicles during their collisions with the carbon surface. All four primary ROS/RNS produced in macrophages (i.e. H2O2, ONOO−, NO˙ and NO2−) get oxidized at platinized carbon CNPs biased at 0.8 V vs. Ag/AgCl.36 The integration of the current under a spike (e.g., the one shown in Fig. 11E) yields the charge corresponding to the total amount of ROS/RNS oxidized during a specific collision. To estimate contributions of individual ROS/RNS to the measured charge, the current–time recordings were obtained with the same platinized CNP biased at different potentials. The potential program developed by the Amatore group consisted of four potential steps roughly corresponding to the oxidation of H2O2 (300 mV vs. Ag/AgCl), H2O2 and ONOO− (450 mV), H2O2, ONOO− and NO˙ (620 mV), and all four species (800 mV).61 The average values of charge passed during a single vesicle collision (Fig. 11F) were calculated from current–time recordings obtained with the same platinized CNP at these potential values and used to calculate the corresponding amounts of different ROS/RNS in a vesicle. For a heterogeneous population of vesicles, the vesicle collision frequencies measured at different potentials represent the fractions of vesicles containing measurable amounts of specific ROS/RNS.
Carbon nanofibers33,62 and platinized nanowire electrodes37,63 have also been used for single vesicle amperometry in living cells.7,33,37 The mechanism of the response (i.e. a vesicle colliding with the electrode surface opens and releases its contents) was similar to that shown in Fig. 10A, except for the absence of current blockages. The micrometer-scale length of nanofiber and platinized wire electrodes limits the spatial resolution and prevents such probes from being completely inserted into subcellular compartments.
Similar to conventional nanopore experiments,17,64 electrochemical resistive-pulse sensing can be used to evaluate the size of translocating particles. No signal was obtained when a CNP diameter was smaller than that of a liposome49 or a cellular vesicle.28 When a vesicle can translocate the CNP, the closer its diameter to that of the pipette orifice the larger the magnitude of the resistive pulse. The Ewing group recently combined resistive pulse measurements with vesicle impact electrochemical cytometry (VIEC) to deliver vesicles from a glass nanopipette to the microelectrode surface.65 This approach allows one to simultaneously measure the physical size and count catecholamine molecules in individual nanometer-sized transmitter vesicles. A CNP can be used to make both types of measurements without the need for an additional collector electrode, which is essential for carrying out such experiments inside a living cell or near its surface. Very recent experiments showed that careful deposition of the conductive film that has to cover the inner pipette wall all the way to its orifice is essential for one-to-one matching of a resistive pulse and a faradaic current spike produced by the same vesicle.
5. Conclusions and outlook
In this Perspective we highlighted two major differences between CNP-based techniques and classical nanopore sensing, i.e. CNP capacities for electroanalysis of single entities and localized measurements in small spaces. For decades, resistive-pulse and ion current rectification experiments were only carried out in the bulk solution. Localized resistive-pulse sensing inside biological cells and near their surfaces is a powerful tool for the detection and analysis of nanoscale objects, such as vesicles and nanoparticles, in biological systems. The orifice diameter of a CNP can be varied to match the size of a specific entity for selective and sensitive detection. CNPs also offer significant advantages for qualitative and quantitative analysis of electroactive material sampled within the conductive nanocavity.Recently developed electrochemical resistive-pulse methodology allowed a conductive pipette to end its double life and function simultaneously as a nanopore sensor and an amperometric nanoelectrode. This hybrid technique offers important advantages for single entity detection and analysis. A typical nagging question in single entity electrochemical studies is about the source of the measured signal. For instance, are two successive millisecond-long current transients recorded in a nanoimpact experiment produced by the same NP or by different particles?66,67 Are current spikes measured inside a living cell due to vacuole collisions with a nanoelectrode or a different biological phenomenon?35 Being able to record a resistive pulse produced by a specific single entity, evaluate its size, and then analyze its contents electrochemically should provide answers to such questions and significantly improve our understanding of processes involving various nanoobjects. This approach should be useful for sampling and analyzing physiologically important species in subcellular compartments such as lysosomes40 and synaptic vesicles.7
Major advances in CNP-based single entity sensing can be expected in the near future, including monitoring and analysis of biological vesicles. A potentially important application is to detecting and analyzing of extracellular vesicles released from biological cells that have shown potential for cancer diagnostics and studies of cell signaling.29,68 Extracellular vesicles containing important electroactive analytes (catecholamines, ROS/RNS, etc.) can report on the nature and current state of a specific cell, its response to a therapeutic agent, and provide other valuable biomedical information. The CNP-based techniques are potentially useful for studies of a wide range of other nanoobjects, such as colloid NPs, nanodroplets, and nanobubbles.69
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The support of our CNP research by the National Science Foundation (CHE-1763337) is gratefully acknowledged.References
- Y. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS PubMed.
- A. Wieckowski, E. R. Savinova and C. G. Vayenas, Catalysis and electrocatalysis at nanoparticle surfaces, CRC Press, 2003 Search PubMed.
- O. G. De Jong, S. A. A. Kooijmans, D. E. Murphy, L. Jiang, M. J. W. Evers, J. P. G. Sluijter, P. Vader and R. M. Schiffelers, Acc. Chem. Res., 2019, 52, 1761–1770 CrossRef CAS PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- N. T. N. Phan, X. Li and A. G. Ewing, Nat. Rev. Chem., 2017, 1, 1–18 CrossRef.
- C. F. Stevens, Neuron, 2003, 40, 381–388 CrossRef CAS PubMed.
- S. Gordon, Immunity, 2016, 44, 463–475 CrossRef CAS PubMed.
- H. Bayley and C. R. Martin, Chem. Rev., 2000, 100, 2575–2594 CrossRef CAS PubMed.
- L. Luo, S. R. German, W.-J. Lan, D. A. Holden, T. L. Mega and H. S. White, Annu. Rev. Anal. Chem., 2014, 7, 513–535 CrossRef CAS PubMed.
- W. Shi, A. K. Friedman and L. A. Baker, Anal. Chem., 2017, 89, 157–188 CrossRef CAS PubMed.
- C. Wei, A. J. Bard and S. W. Feldberg, Anal. Chem., 1997, 69, 4627–4633 CrossRef CAS.
- E. A. Heins, Z. S. Siwy, L. A. Baker and C. R. Martin, Nano Lett., 2005, 5, 1824–1829 CrossRef CAS PubMed.
- B. N. Miles, A. P. Ivanov, K. A. Wilson, F. Doğan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15–28 RSC.
- L. Chen, H. He and Y. Jin, Anal. Chem., 2015, 87, 522–529 CrossRef CAS PubMed.
- P. Terejánszky, I. Makra, P. t. Fürjes and R. b. E. Gyurcsányi, Anal. Chem., 2014, 86, 4688–4697 CrossRef PubMed.
- Y. Wang, K. Kececi, M. V. Mirkin, V. Mani, N. Sardesai and J. F. Rusling, Chem. Sci., 2013, 4, 655–663 RSC.
- X. Lin, A. P. Ivanov and J. B. Edel, Chem. Sci., 2017, 8, 3905–3912 RSC.
- C. A. Morris, A. K. Friedman and L. A. Baker, Analyst, 2010, 135, 2190–2202 RSC.
- S. Zhang, M. Li, B. Su and Y. Shao, Annu. Rev. Anal. Chem., 2018, 11, 265–286 CrossRef PubMed.
- M. Chang, G. Morgan, F. Bedier, A. Chieng, P. Gomez, S. Raminani and Y. Wang, J. Electrochem. Soc., 2020, 167, 037533 CrossRef CAS.
- Y. Liu, C. Xu, X. Chen, J. Wang, P. Yu and L. Mao, Electrochem. Commun., 2018, 89, 38–42 CrossRef CAS.
- R. J. Yu, Y. L. Ying, R. Gao and Y. T. Long, Angew. Chem., Int. Ed., 2019, 58, 3706–3714 CrossRef CAS PubMed.
- P. Novak, C. Li, A. I. Shevchuk, R. Stepanyan, M. Caldwell, S. Hughes, T. G. Smart, J. Gorelik, V. P. Ostanin, M. J. Lab, G. W. J. Moss, G. I. Frolenkov, D. Klenerman and Y. E. Korchev, Nat. Methods, 2009, 6, 279–281 CrossRef CAS PubMed.
- M. Shen, Z. Qu, J. DesLaurier, T. M. Welle, J. V. Sweedler and R. Chen, J. Am. Chem. Soc., 2018, 140, 7764–7768 CrossRef CAS PubMed.
- J. Song, C. H. Xu, S. Z. Huang, W. Lei, Y. F. Ruan, H. J. Lu, W. Zhao, J. J. Xu and H. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 13226–13230 CrossRef CAS PubMed.
- R. Pan, K. Hu, R. Jia, S. A. Rotenberg, D. Jiang and M. V. Mirkin, J. Am. Chem. Soc., 2020, 142, 5778–5784 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2016, 34, 359–360 CrossRef CAS PubMed.
- C. C. Harrell, Y. Choi, L. P. Horne, L. A. Baker, Z. S. Siwy and C. R. Martin, Langmuir, 2006, 22, 10837–10843 CrossRef CAS PubMed.
- P. Sun, F. O. Laforge, T. P. Abeyweera, S. A. Rotenberg, J. Carpino and M. V. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 443–448 CrossRef CAS PubMed.
- Y. T. Li, S. H. Zhang, X. Y. Wang, X. W. Zhang, A. I. Oleinick, I. Svir, C. Amatore and W. H. Huang, Angew. Chem., Int. Ed., 2015, 54, 9313–9318 CrossRef CAS PubMed.
- X. Li, S. Majdi, J. Dunevall, H. Fathali and A. G. Ewing, Angew. Chem., Int. Ed., 2015, 54, 11978–11982 CrossRef CAS PubMed.
- J. G. Roberts, E. C. Mitchell, L. E. Dunaway, G. S. McCarty and L. A. Sombers, ACS Nano, 2020, 14, 2917–2926 CrossRef CAS PubMed.
- Y. Li, K. Hu, Y. Yu, S. A. Rotenberg, C. Amatore and M. V. Mirkin, J. Am. Chem. Soc., 2017, 139, 13055–13062 CrossRef CAS PubMed.
- K. Hu, Y. Li, S. A. Rotenberg, C. Amatore and M. V. Mirkin, J. Am. Chem. Soc., 2019, 141, 4564–4568 CrossRef CAS PubMed.
- X. W. Zhang, A. Oleinick, H. Jiang, Q. L. Liao, Q. F. Qiu, I. Svir, Y. L. Liu, C. Amatore and W. H. Huang, Angew. Chem., Int. Ed., 2019, 58, 7753–7756 CrossRef CAS PubMed.
- R. Gao, Y. L. Ying, Y. J. Li, Y. X. Hu, R. J. Yu, Y. Lin and Y. T. Long, Angew. Chem., Int. Ed., 2018, 57, 1011–1015 CrossRef CAS PubMed.
- C. Yang, P. Hinkle, J. Menestrina, I. V. Vlassiouk and Z. S. Siwy, J. Phys. Chem. Lett., 2016, 7, 4152–4158 CrossRef CAS PubMed.
- R. Pan, M. Xu, J. D. Burgess, D. Jiang and H.-Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4087–4092 CrossRef CAS PubMed.
- R. Gao, Y.-L. Ying, Y.-X. Hu, Y.-J. Li and Y.-T. Long, Anal. Chem., 2017, 89, 7382–7387 CrossRef CAS PubMed.
- G. Pérez-Mitta, W. A. Marmisollé, C. Trautmann, M. E. Toimil-Molares and O. Azzaroni, J. Am. Chem. Soc., 2015, 137, 15382–15385 CrossRef PubMed.
- B. M. Kim, T. Murray and H. H. Bau, Nanotechnology, 2005, 16, 1317–1320 CrossRef CAS.
- R. Singhal, S. Bhattacharyya, Z. Orynbayeva, E. Vitol, G. Friedman and Y. Gogotsi, Nanotechnology, 2010, 21, 015304 CrossRef PubMed.
- K. Hu, Y. Wang, H. Cai, M. V. Mirkin, Y. Gao, G. Friedman and Y. Gogotsi, Anal. Chem., 2014, 86, 8897–8901 CrossRef CAS PubMed.
- Y. Yu, J.-M. Noël, M. V. Mirkin, Y. Gao, O. Mashtalir, G. Friedman and Y. Gogotsi, Anal. Chem., 2014, 86, 3365–3372 CrossRef CAS PubMed.
- K. J. Freedman, L. M. Otto, A. P. Ivanov, A. Barik, S.-H. Oh and J. B. Edel, Nat. Commun., 2016, 7, 10217 CrossRef CAS PubMed.
- R. Pan, M. Xu, D. Jiang, J. D. Burgess and H.-Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11436–11440 CrossRef CAS PubMed.
- R. Pan, K. Hu, D. Jiang, U. Samuni and M. V. Mirkin, J. Am. Chem. Soc., 2019, 141, 19555–19559 CrossRef CAS PubMed.
- M. Wanunu and A. Meller, Nano Lett., 2007, 7, 1580–1585 CrossRef CAS PubMed.
- I. Vlassiouk, T. R. Kozel and Z. S. Siwy, J. Am. Chem. Soc., 2009, 131, 8211–8220 CrossRef CAS PubMed.
- S. Liu, Y. Dong, W. Zhao, X. Xie, T. Ji, X. Yin, Y. Liu, Z. Liang, D. Momotenko and D. Liang, Anal. Chem., 2012, 84, 5565–5573 CrossRef CAS PubMed.
- A. Rutkowska, K. Freedman, J. Skalkowska, M. J. Kim, J. B. Edel and T. Albrecht, Anal. Chem., 2015, 87, 2337–2344 CrossRef CAS PubMed.
- D. Wang and M. V. Mirkin, J. Am. Chem. Soc., 2017, 139, 11654–11657 CrossRef CAS PubMed.
- Z. Siwy, L. Trofin, P. Kohli, L. A. Baker, C. Trautmann and C. R. Martin, J. Am. Chem. Soc., 2005, 127, 5000–5001 CrossRef CAS PubMed.
- J. H. Bae, D. Wang, K. Hu and M. V. Mirkin, Anal. Chem., 2019, 91, 5530–5536 CrossRef CAS PubMed.
- K. Hu, D. Wang, M. Zhou, J. H. Bae, Y. Yu, H. Xin and M. V. Mirkin, Anal. Chem., 2019, 91, 12935–12941 CrossRef CAS PubMed.
- C. Yang, K. Hu, D. Wang, Y. Zubi, S. T. Lee, P. Puthongkham, M. V. Mirkin and B. J. Venton, Anal. Chem., 2019, 91, 4618–4624 CrossRef CAS PubMed.
- Y.-L. Ying, Y.-X. Hu, R. Gao, R.-J. Yu, Z. Gu, L. P. Lee and Y.-T. Long, J. Am. Chem. Soc., 2018, 140, 5385–5392 CrossRef CAS PubMed.
- R. Gao, Y. Lin, Y. L. Ying, Y. X. Hu, S. W. Xu, L. Q. Ruan, R. J. Yu, Y. J. Li, H. W. Li, L. F. Cui and Y.-T. Long, Nat. Protoc., 2019, 14, 2015–2035 CrossRef CAS PubMed.
- C. Amatore, S. p. Arbault and A. C. W. Koh, Anal. Chem., 2010, 82, 1411–1419 CrossRef CAS PubMed.
- J. Dunevall, H. Fathali, N. Najafinobar, J. Lovric, J. Wigstrom, A. Cans and A. G. Ewing, J. Am. Chem. Soc., 2015, 137, 4344–4346 CrossRef CAS PubMed.
- X. W. Zhang, Q. F. Qiu, H. Jiang, F. L. Zhang, Y. L. Liu, C. Amatore and W. H. Huang, Angew. Chem., Int. Ed., 2017, 56, 12997–13000 CrossRef CAS PubMed.
- S. R. German, T. S. Hurd, H. S. White and T. L. Mega, ACS Nano, 2015, 9, 7186–7194 CrossRef CAS PubMed.
- X.-W. Zhang, A. Hatamie and A. G. Ewing, J. Am. Chem. Soc., 2020, 142, 4093–4097 CrossRef CAS PubMed.
- S. M. Oja, D. A. Robinson, N. J. Vitti, M. A. Edwards, Y. Liu, H. S. White and B. Zhang, J. Am. Chem. Soc., 2017, 139, 708–718 CrossRef CAS PubMed.
- M. Zhou, Y. Yu, K. Hu, H. L. Xin and M. V. Mirkin, Anal. Chem., 2017, 89, 2880–2885 CrossRef CAS PubMed.
- M. Colombo, G. Raposo and C. Théry, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS PubMed.
- S.-M. Lu, Y.-Y. Peng, Y.-L. Ying and Y.-T. Long, Anal. Chem., 2020, 92, 5621–5644 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |