
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoretention in styrene heterodimerisation promoted by one-electron oxidants†
Xinglong
Zhang‡
 a and
Robert S.
Paton
*ab
a and
Robert S.
Paton
*ab
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK
bDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA. E-mail: robert.paton@colostate.edu
First published on 17th August 2020
Abstract
Radical cations generated from the oxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π-bonds are synthetically useful reactive intermediates for C–C and C–X bond formation. Radical cation formation, induced by sub-stoichiometric amounts of external oxidant, are important intermediates in the Woodward–Hoffmann thermally disallowed [2 + 2] cycloaddition of electron-rich alkenes. Using density functional theory (DFT), we report the detailed mechanisms underlying the intermolecular heterodimerisation of anethole and β-methylstyrene to give unsymmetrical, tetra-substituted cyclobutanes. Reactions between trans-alkenes favour the all-trans adduct, resulting from a kinetic preference for anti-addition reinforced by reversibility at ambient temperatures since this is also the thermodynamic product; on the other hand, reactions between a trans-alkene and a cis-alkene favour syn-addition, while exocyclic rotation in the acyclic radical cation intermediate is also possible since C–C forming barriers are higher. Computations are consistent with the experimental observation that hexafluoroisopropanol (HFIP) is a better solvent than acetonitrile, in part due to its ability to stabilise the reduced form of the hypervalent iodine initiator by hydrogen bonding, but also through the stabilisation of radical cationic intermediates along the reaction coordinate.
C π-bonds are synthetically useful reactive intermediates for C–C and C–X bond formation. Radical cation formation, induced by sub-stoichiometric amounts of external oxidant, are important intermediates in the Woodward–Hoffmann thermally disallowed [2 + 2] cycloaddition of electron-rich alkenes. Using density functional theory (DFT), we report the detailed mechanisms underlying the intermolecular heterodimerisation of anethole and β-methylstyrene to give unsymmetrical, tetra-substituted cyclobutanes. Reactions between trans-alkenes favour the all-trans adduct, resulting from a kinetic preference for anti-addition reinforced by reversibility at ambient temperatures since this is also the thermodynamic product; on the other hand, reactions between a trans-alkene and a cis-alkene favour syn-addition, while exocyclic rotation in the acyclic radical cation intermediate is also possible since C–C forming barriers are higher. Computations are consistent with the experimental observation that hexafluoroisopropanol (HFIP) is a better solvent than acetonitrile, in part due to its ability to stabilise the reduced form of the hypervalent iodine initiator by hydrogen bonding, but also through the stabilisation of radical cationic intermediates along the reaction coordinate.
1. Introduction
Cyclobutanes are ubiquitous structural motifs in natural products and bioactive molecules.1,2 They can be considered useful synthons for 3-membered and 5-membered rings via regio- and/or stereoselective transformations by ring-contraction or ring-expansion reactions to form otherwise challenging products.3,4 The inherent ring strain of cyclobutene of 26 kcal mol−1 is predominantly due to angular (Baeyer) strain5 and makes its selective cleavage to access both acyclic and cyclic systems highly amenable. Although the construction of 4-membered cycloalkanes via thermal [2 + 2] cycloaddition is symmetry-forbidden based on the Woodward–Hoffmann rules, alternative methods to access this highly versatile motif, with precise chemo-, regio- and stereocontrol continue to emerge. Since the first report of photochemical [2 + 2] cycloaddition accessing a 4-membered carbocycle by Liebermann in 1877,6 the construction of 4-membered cyclobutyl rings from photo-cycloaddition of alkenes has become arguably the most employed technique for cyclobutanation.7–12 Cyclobutanation can now also be readily achieved via organocatalysis,13–15 organometallic catalysis,7,16–18 as well as electrocatalysis19–22 (Scheme 1). Until recently when the synthesis of substituted cyclobutanes could be realised via selective C–H functionalisation of unsubstituted cyclobutanes,23–26 formal [2 + 2] cycloaddition remained the main strategy for the synthesis of complex, tetra-substituted cyclobutyl rings.27–30 For the latter strategy, the formation of an unsymmetrical, tetra-substituted cyclobutane can arise from intermolecular heterodimerisation of two substituted alkenes. This, however, remains a challenge as a varied mixture of products can result from the reaction of two alkenes due to a lack of control on homo- and hetero-[2 + 2]-dimerisation and the possibility of regiochemical (head-to-head vs. head-to-tail) and stereochemical (cis vs. trans) variations. Many examples of both head-to-head and head-to-tail coupled cyclobutanes exist in biologically active natural products, such as argenteoside family of compounds.31 Synthetic methods for cyclobutane formation with precise control over selectivity are therefore of intense interest.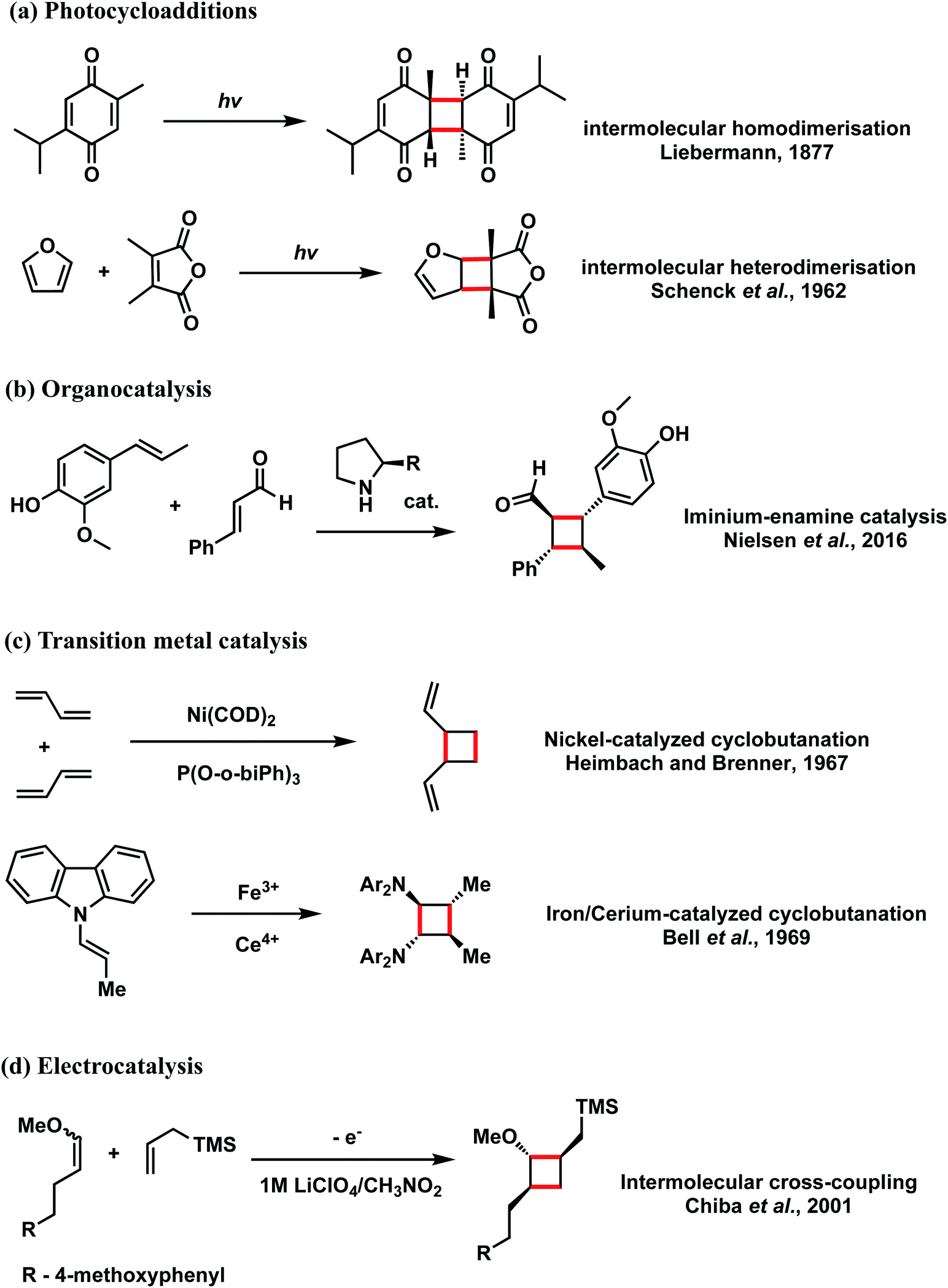 | ||
| Scheme 1 Selected examples for [2 + 2] cyclobutanation. | ||
Radical cations constitute an important class of reactive intermediate in forging C–C and C–X bonds.32–37 Their formation is typically achieved via single-electron transfer (SET) from a neutral substrate's π-bond or lone-pair, creating a “hole” in the system. This process can be mediated using transition metals, either by themselves7,38–40 or coupled with organic41 or organometallic42–45 photoredox catalysts; it can also be mediated by organic oxidants such as aminium radical cationic salts.46 In this last approach, these powerful oxidants can easily decompose or induce undesirable side reactions.47–49 Due to these limitations, the use of oxidants for SET-initiation and catalysis of cyclobutanation is very much underexplored compared to photochemical means. In a recent study, Donohoe and co-workers reported the use of a hypervalent iodine oxidants (phenyliodine diacetate or Dess–Martin periodinane (DMP)) to promote homo and hetero-dimerisations of styrenes to access unsymmetrical, tetra-substituted cyclobutyl rings.13,50 The regio- and stereochemical outcomes of this oxidant-promoted, SET-catalysed heterodimerisation of two unsymmetrical alkenes present an interesting avenue for detailed mechanistic study (Scheme 2(a)). The all-trans stereochemical outcome of the hypervalent-iodine promoted reaction between trans-anethole and trans-β-methylstyrene (Scheme 2(a)(i)) is consistent with trans-anethole homodimerisation promoted by an aminium cation at temperatures of 0 °C and above (Scheme 2(b)(ii)). At lower temperatures evidence for cis-addition is seen (Scheme 2(b)(i)), but complete transfer of alkene stereochemical information occurs at all temperatures. The reaction between cis-anethole and trans-β-methylstyrene (Scheme 2(b)(iii)), however, shows incomplete stereochemical transfer, giving 20% of the all-trans product. Note that in all these products, only the so-called “head-to-head” cyclobutanes are observed.
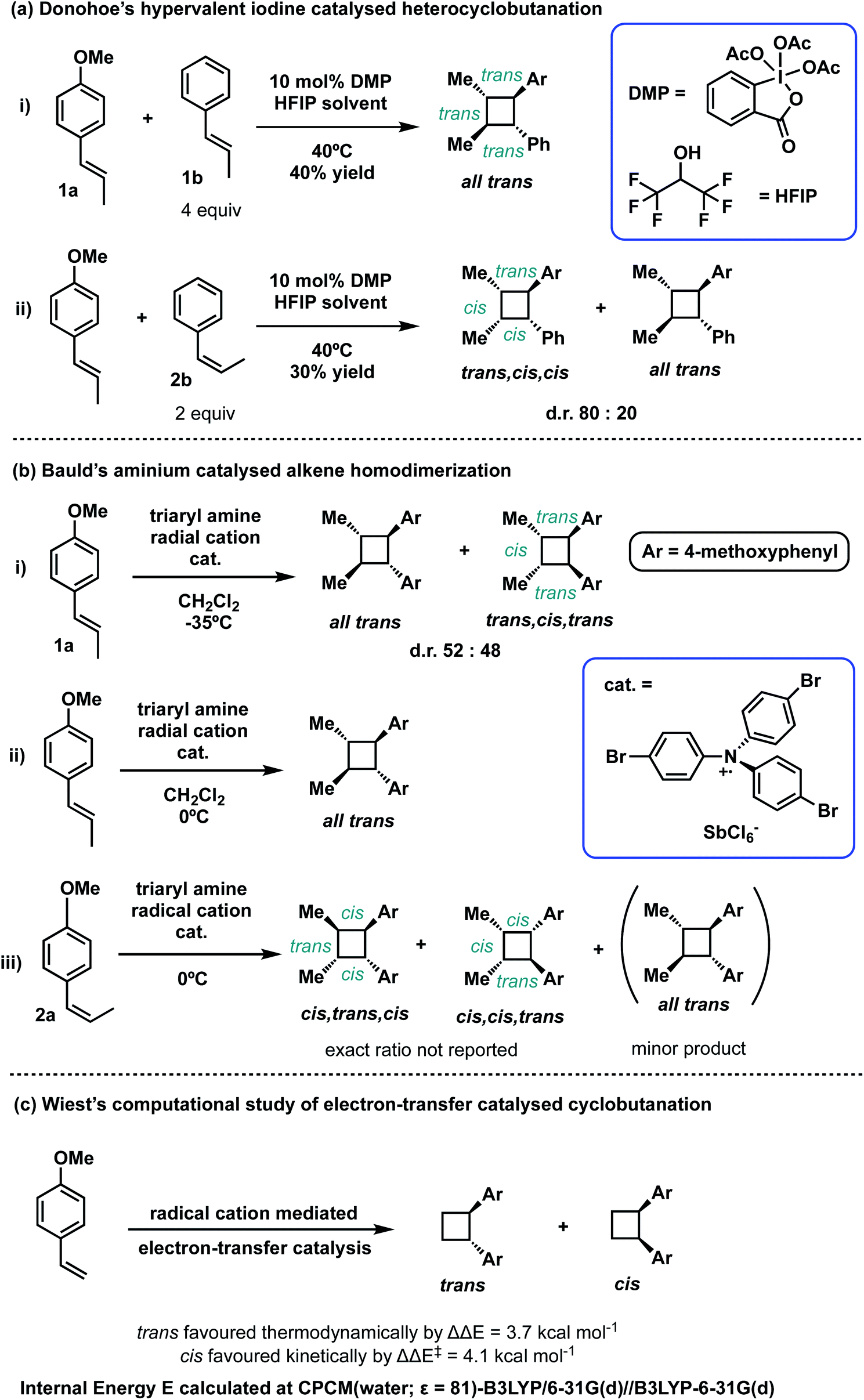 | ||
| Scheme 2 Stereochemical outcomes from (a) hetero- and (b) homo-coupling of electron-rich alkenes under single electron transfer (SET) catalysis and (c) computational studies of prototypical cyclobutanation from methoxystyrene, a terminal alkene. | ||
The detailed mechanism of intermolecular radical cation cycloaddition has been probed experimentally, where both stepwise51–54 and concerted, asynchronous46,55–57 pathways have been proposed. The homo-dimerisation of 4-methoxystyrene to form a disubstituted cyclobutene via a radical cationic pathway has been studied computationally (with density functional theory, DFT) by Wiest and co-workers, where a stepwise mechanism was obtained.58 The study reported that the cis-adduct is favoured kinetically by an activation barrier ΔΔE‡ of 4.1 kcal mol−1 whereas the trans-adduct is favoured thermodynamically by a reaction energy ΔΔE of 3.7 kcal mol−1 (Scheme 2(c)). Following this study, Metzger and co-workers in 2008 detected a distonic (where charge and radical sites are separated) radical cationic intermediate via extractive electrospray ionisation mass spectrometry, lending direct support to the stepwise mechanism.59 Further computational mechanistic investigations looking into intramolecular cyclobutanation of (bis)styrene60 and intermolecular cyclobutanation of unsubstituted styrene61via radical cation catalysis further confirmed the stepwise nature of such mechanism.
In this computational study, we focus on the effects of alkene cis- and trans-configurations on the mechanism of the intermolecular radical cation dimerisation of styrenes, since to the best of our knowledge β-substituents have yet to feature in theoretical studies. In the experimental work by Donohoe and co-workers, one particularly intriguing aspect has been the reaction shown in Scheme 2(a)(ii), wherein 20% of all-trans cyclobutyl ring product is formed, although the alkenes used do not both have the trans geometry. We envisioned that the reaction proceeds via initiation, creating a hole in one of the neutral alkenes, followed by propagation, forming radical cationic cyclobutanation, and finally termination, where the radical cationic cyclobutyl product gets reduced to its neutral form (Scheme 3). We herein report a theoretical investigation into the origins governing the chemical reactivities and selectivities effecting the observed stereochemical outcomes in the heterodimerisation of two unsymmetrical, electron-rich alkenes (Scheme 2(a))
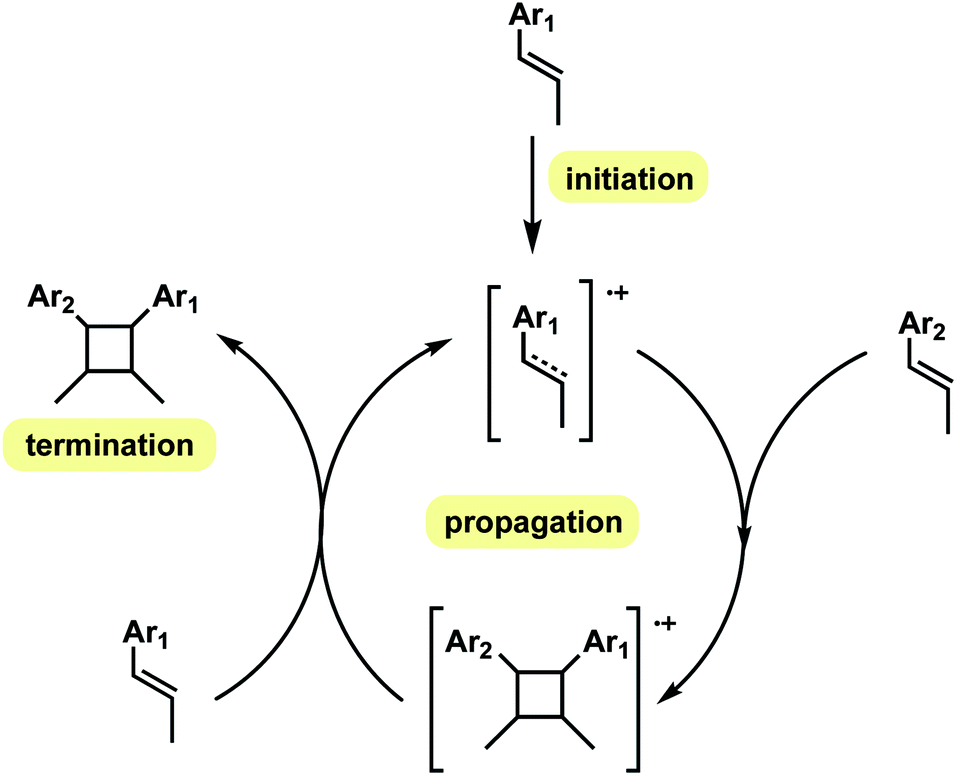 | ||
| Scheme 3 Schematic outline of catalytic cycle. In the initiation step, one of the alkenes gets oxidised to radical cation, which then attacks the other, neutral alkene to form the radical cationic cyclobutene in the propagation step. The termination step occurs when the radical cationic cyclobutene gets reduced to its neutral form. | ||
2. Results and discussion
2.1 Computational electrochemical potentials
The feasibility of radical cation generation in the initiation step and the reduction of the radical cation product in the termination step can be quantified using the electrochemical potentials of these species. For a reaction between two different alkenes, their electrochemical potentials directly determine their role selectivity as caticogen (one that readily forms radical cation under oxidation/SET conditions) or caticophile (species that attacks the radical cation). The calculation of reduction potentials provides a direct measure of the thermodynamic feasibility of these steps.Ab initio computations of reduction potentials of various systems have been widely explored.62–72 We adopt the thermodynamic cycle for redox potential computation73,74 and found that M06-2X functional gives the best agreement (smallest mean unsigned error, MUE) with the experimental redox potential values for substrates trans-anethole 1a and trans-β-methylstyrene 1b in MeCN solvent amongst 8 functionals tested (ESI section 2†). This is in agreement with a study of both experimental and computational electrochemical potentials for over 180 organic substrates where M06-2X functional gives an R2 value of 0.97 for the correlation between the experimental and calculated redox potentials.75 M06-2X was used for all subsequent DFT calculations.
The redox potential of the hypervalent iodine oxidant, Dess–Martin Periodinane (DMP, chemical structure in Scheme 2(a)), was computed at SMD(solvent)-M06-2X/def2-TZVPP//M06-2X/GenECP(LanL2DZ for I atom and 6-31G(d) for other atoms) where each of the MeCN and HFIP solvents was separately calculated. The results, together with the redox potentials for our starting materials and cyclobutyl ring products, are given in Table 1. From the table, we can see that in both solvents, trans-anethole 1a is oxidised more easily than trans-β-methylstyrene 1b to their respective radical cation. For example, in HFIP, the oxidation of 1a to [1a]˙+ by DMP has Ecell of 0.306–1.467 = −1.161 V and the oxidation of 1b to [1b]˙+ has Ecell of 0.306–1.879 = −1.573 V, the latter being much less favourable thermodynamically (by about 10 kcal mol−1). Thus, 1a will act as a caticogen where it is oxidised to its radical cationic form whereas the neutral 1b will act as a caticophile, attacking the radical cationic [1a]˙+ in the subsequent cyclobutanation steps (vide infra).
The effect of solvent has a greater influence on the redox potential of the iodinated oxidant DMP, whose reduction potential becomes less negative (by 205 mV) in HFIP than in MeCN, indicating its greater oxidising strength in the fluorinated solvent, in agreement with an electrochemical study on the enhanced oxidising power of similar hypervalent iodine oxidants in HFIP than in MeCN.50 The molecular origin of that enhanced reactivity can be traced to the formation of a strong hydrogen-bonded oxidant–HFIP complex.50 By including one molecule of HFIP explicitly in our recalculation of redox potentials of DMP (see Fig. 1 for optimised structures), we found that the computed redox potential of DMP becomes even more positive, being now 223 mV more favoured than without an explicit molecule of HFIP (75 mV more favoured in HFIP than in MeCN). This can be attributed to the strengthening of the intermolecular H-bond in the DMP–HFIP complex upon reduction, with a reduction in the O–H distance from 1.79 to 1.51 Å (Fig. 1). The ease of reduction of DMP in MeCN is also augmented by 353 mV by the inclusion of an H-bonded molecule of HFIP, although experimentally using HFIP as an additive in MeCN solvent does not lead to significant cyclobutanation (<5% yield) potentially because the MeCN molecule could be a better H-bond acceptor than the hypervalent iodine oxidant for HFIP molecule.50 The formation of radical cationic [1a]˙+ can be more easily achieved in HFIP (with an overall Ecell of 0.306–1.467 = −1.161 V) than in MeCN (overall Ecell of −0.122 − 1.388 = −1.510 V without HFIP), by ca. 8 kcal mol−1, illustrative of the promoting influence of HFIP on initiation.50,76,77 Additionally, tight complexation of DMP with HFIP may also prohibit ion-pairing of the reduced form with radical cations, leading to enhanced reactivity.78 SET from anethole to DMP is computed as endergonic, as reflected by the overall negative electrochemical cell potential. The weakly oxidising iodinated reagent is employed to ensure that only a small amount of radical cation is produced in situ to avoid dimerisation and further oxidation to dications.50,79,80
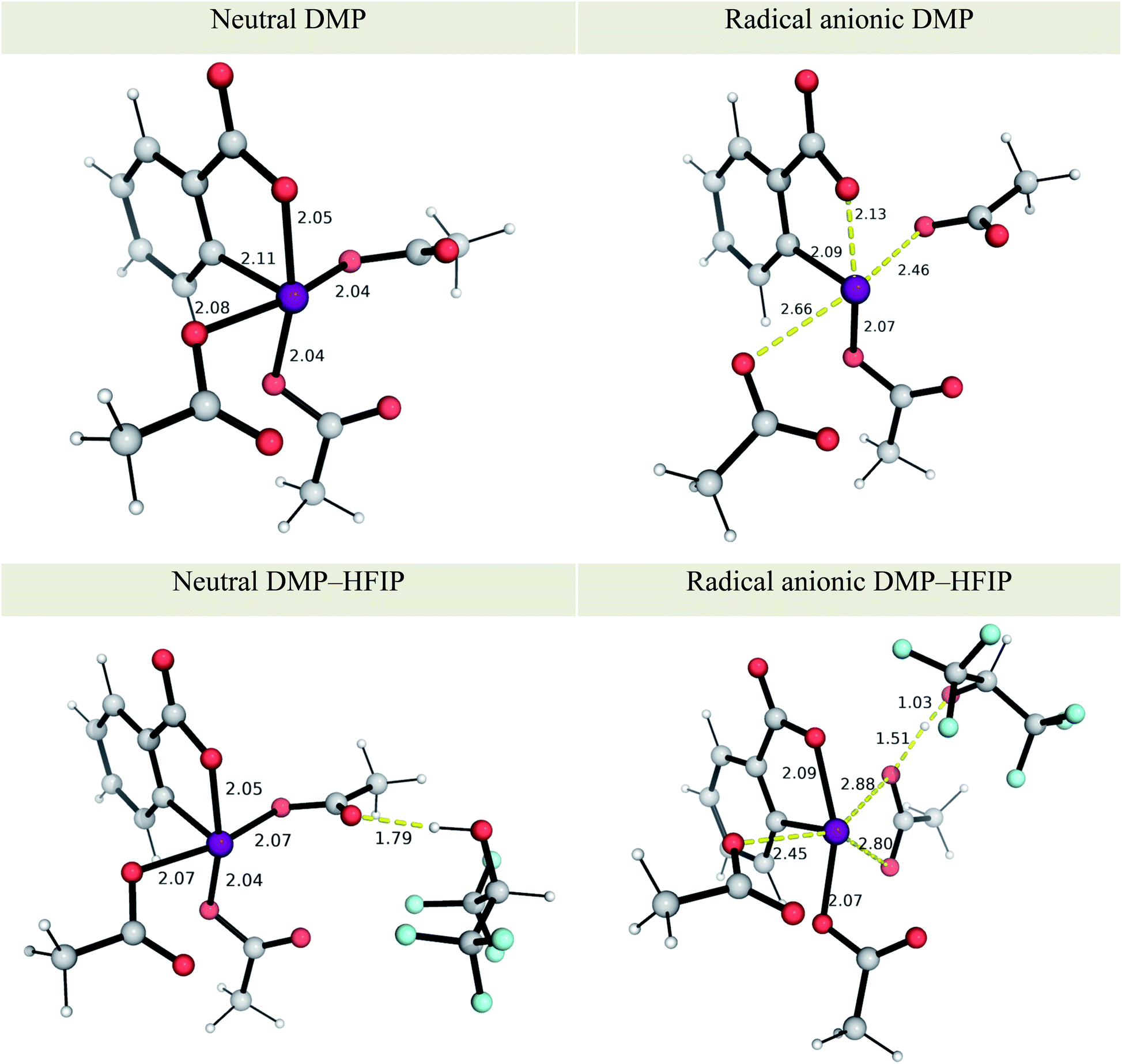 | ||
| Fig. 1 M06-2X optimised structures of neutral and radical anion forms of Dess–Martin periodinane (DMP) and DMP–HFIP. See details of conformational considerations in ESI section 2.1.† Key distances are shown in Å. | ||
2.2 Reactivity and selectivity between trans-anethole and trans-β-methylstyrene (P1)
For the reaction between trans-anethole 1a and trans-β-methylstyrene 1b (reaction P1) as shown in Scheme 2(a)(i), electron-rich alkene 1a is preferentially oxidised by DMPvia SET to give distonic radical cation [1a]˙+, which adds to the neutral alkene partner 1b that is present in excess. Consistent with Wiest's earlier studies with B3LYP, no concerted 4-membered TS could be found on the M06-2X potential energy surface (PES). For the stepwise mechanism, both head-to-head and head-to-tail cyclobutanes are possible. In addition, syn- and anti-adducts can be formed depending on the orientation of the reacting alkenes when they approach each other. The Gibbs energy profile for both reaction pathways is given in Fig. 2. Possible conformations of the TSs were explored systematically (Fig. S2†) and the lowest energy conformers are presented here. The turnover-frequency determining transition state (TDTS) along the entire PES occurs during the first C–C bond forming step. The head-to-head TSs (ts1 and ts1′) are kinetically more favourable, with a lower activation barrier than the head-to-tail TSs (ts1-g1 and ts1-g2), by at least 4.7 kcal mol−1 (a selectivity of 410![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, using transition state theory), although the neutral, closed-shell cyclobutyl ring products lie closer in energy (within 1.1 kcal mol−1 of the observed product int3n, Fig. 2). For the head-to-head products, the anti-adduct int3n is favoured over the syn-adduct int3′n both kinetically (by 1.1 kcal mol−1) and thermodynamically (by 2.1 kcal mol−1). Although oxidation of the hypervalent iodine catalyst (DMP˙−) is easier than neutral trans-anethole, the reduction of radical cationic intermediates to the neutral, closed-shell cyclobutanes is more likely to involve anethole, present in larger amounts than the initiator. Additionally, cyclic voltammetry studies show irreversible reduction of the hypervalent iodine species taking place.50 With this in mind, our computed energy profile uses trans-anethole as the reductant to complete the catalytic cycle.
:1, using transition state theory), although the neutral, closed-shell cyclobutyl ring products lie closer in energy (within 1.1 kcal mol−1 of the observed product int3n, Fig. 2). For the head-to-head products, the anti-adduct int3n is favoured over the syn-adduct int3′n both kinetically (by 1.1 kcal mol−1) and thermodynamically (by 2.1 kcal mol−1). Although oxidation of the hypervalent iodine catalyst (DMP˙−) is easier than neutral trans-anethole, the reduction of radical cationic intermediates to the neutral, closed-shell cyclobutanes is more likely to involve anethole, present in larger amounts than the initiator. Additionally, cyclic voltammetry studies show irreversible reduction of the hypervalent iodine species taking place.50 With this in mind, our computed energy profile uses trans-anethole as the reductant to complete the catalytic cycle.
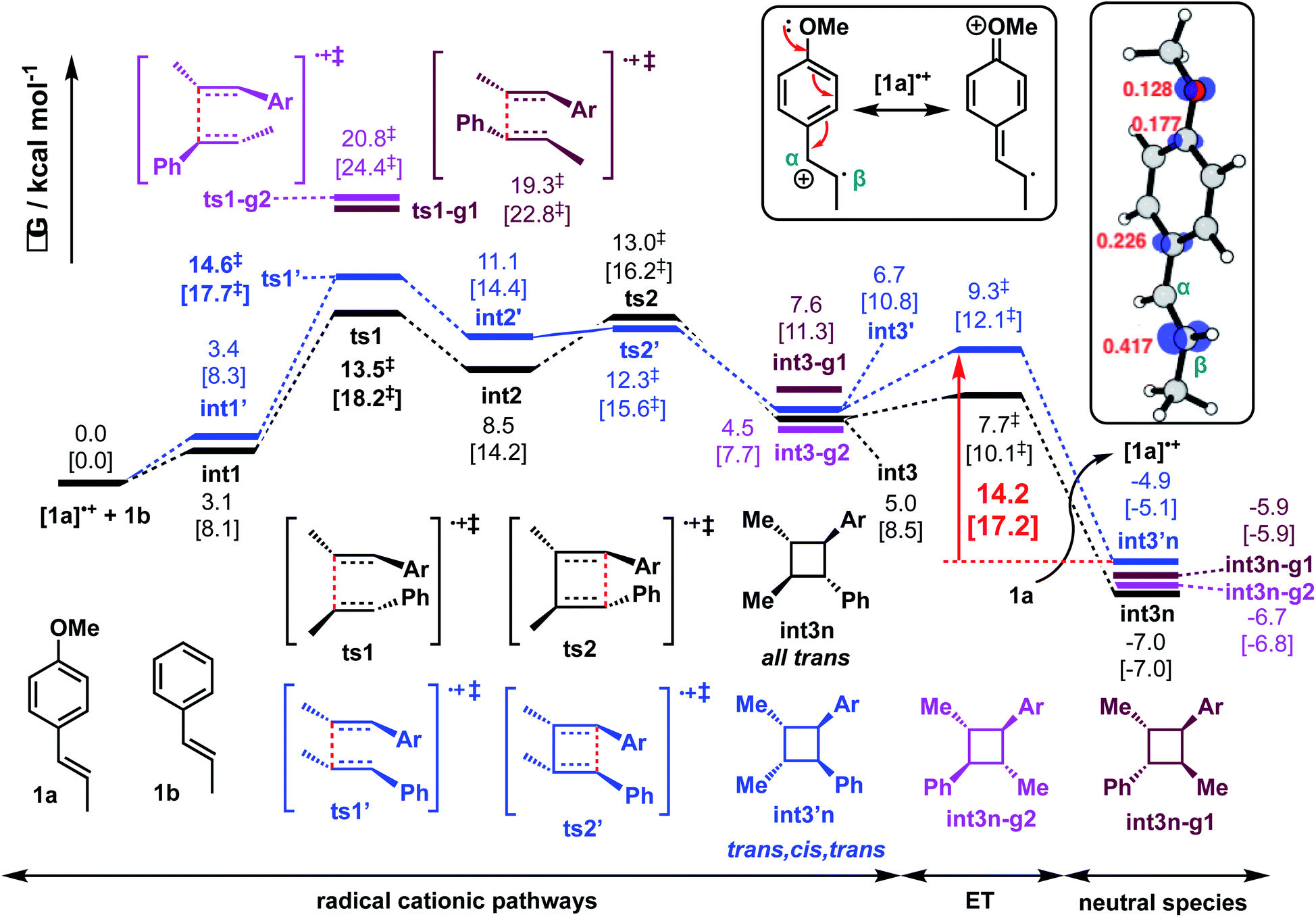 | ||
| Fig. 2 Gibbs energy profile for reaction between trans-anethole and trans-β-methylstyrene computed at SMD(solvent)-M06-2X/def2-TZVPP//M06-2X/6-31G(d) level of theory. The values in HFIP are given together with values in MeCN in square brackets. ET = electron transfer barrier heights estimated using Marcus-Hush theory at an intermolecular separation of 3 Å (see ESI† for details). Spin density plots (at an isovalue of 0.02 a.u.) and the Mulliken spin density values of radical cationic trans-anethole is also shown. | ||
The spin density in radical cation [1a]˙+ is predominantly localised on the β-carbon (Fig. 2 box). The benzylic carbocation is additionally stabilised by the p-methoxy lone pair. C–C bond formation occurs first at the β-carbon of both styrene partners (ts1 and ts1′, Fig. S3†). This head-to-head combination forms an intermediate in which the positive charge and unpaired electron are stabilised at the two benzylic positions of int2. There is one benzylic position available for head-to-tail attack (ts1-g1 and ts1-g2, Fig. S3†). Thus, the head-to-head TSs have much lower activation barriers (by more than 4.7 kcal mol−1). The regioselectivity (head-to-head vs. head-to-tail) is kinetically controlled, as this first step is irreversible. Additionally, the ring-closed head-to-tail products int3n-g1 and int3n-g2 are close in energy to int3n but they are not observed, further supporting the absence of any interconversion of these products due to any reversibility of C–C formation. The electron transfer (ET) barrier for the reduction of the radical cationic products to their neutral form can be estimated using Marcus-Hush (MH) theory.81–84 It was found that these barriers (on the order of 1–3 kcal mol−1) are much smaller than any bond-forming barriers in the PES (ESI section 5†). Care should be noted, however, that the use of static, single-particle, DFT calculation in ET barrier estimation could possibly neglect effects of other electrons on the transfer.85 The assumption of linear solvent response and the same curvature of reactant and product PESs could further introduce errors in our MH estimate.86,87 Nevertheless, ET barriers on the order of 1–3 kcal mol−1 have been observed experimentally.88–90 This suggests that this product-forming SET step is not overall rate-limiting. The reverse of this elementary step, oxidation of the cyclic products by the anethole radical cation, has an estimated (MH) barrier of about 14 kcal mol−1. This is comparable to the barrier of the TDTS (Fig. 2) and suggests that this electron transfer can occur reversibly. This has stereochemical implications, since the overall barrier for cycloreversion (viats1′ from int3′n) of the minor diastereomer is 19.5 kcal mol−1 in HFIP (22.8 kcal mol−1 in MeCN). This permits equilibration to form the thermodynamically more favourable all-trans diastereomer at 40 °C. At lower temperatures (e.g. −35 °C) the diastereoselectivity is predicted to be lower, consistent with Bauld's experimental results (Scheme 2(b)).
To further understand the steric and electronic factors influencing the head-to-head vs. head-to-tail regioselectivity, we applied the distortion-interaction91,92/activation strain (DI–AS) model92–96 to the TSs for the formation of first C–C bond for the present reaction (Fig. 3). As we can see, although the head-to-tail TSs have later transition states, evidenced by their shorter C–C bond in the TSs, the energetic penalty to distort the reacting molecules as they come together in the TSs, i.e., the activation strain or the distortion energy (in blue), for all 4 TSs are similar; the head-to-tail TSs are disfavoured due to their less favourable interaction energies (in green) than the head-to-head TSs. Comparing the selectivity of ts1 over ts1′, the distortion energies are identical whereas ts1 has slightly more favourable interactions. The non-covalent interaction (NCI) plots in these TSs (Fig. 5) suggests that ts1 benefits from favourable π–π stacking interaction (slightly staggered aryl rings) thus making it more favourable than ts1′ having no such stacking interactions. This π–π stacking interaction is also observed in ts6′ (but not ts6) (Fig. 5) such that the syn-addition ts6′ has a lower activation barrier than the anti-addition ts6 (activation strain model, Fig. S15†) (vide infra).
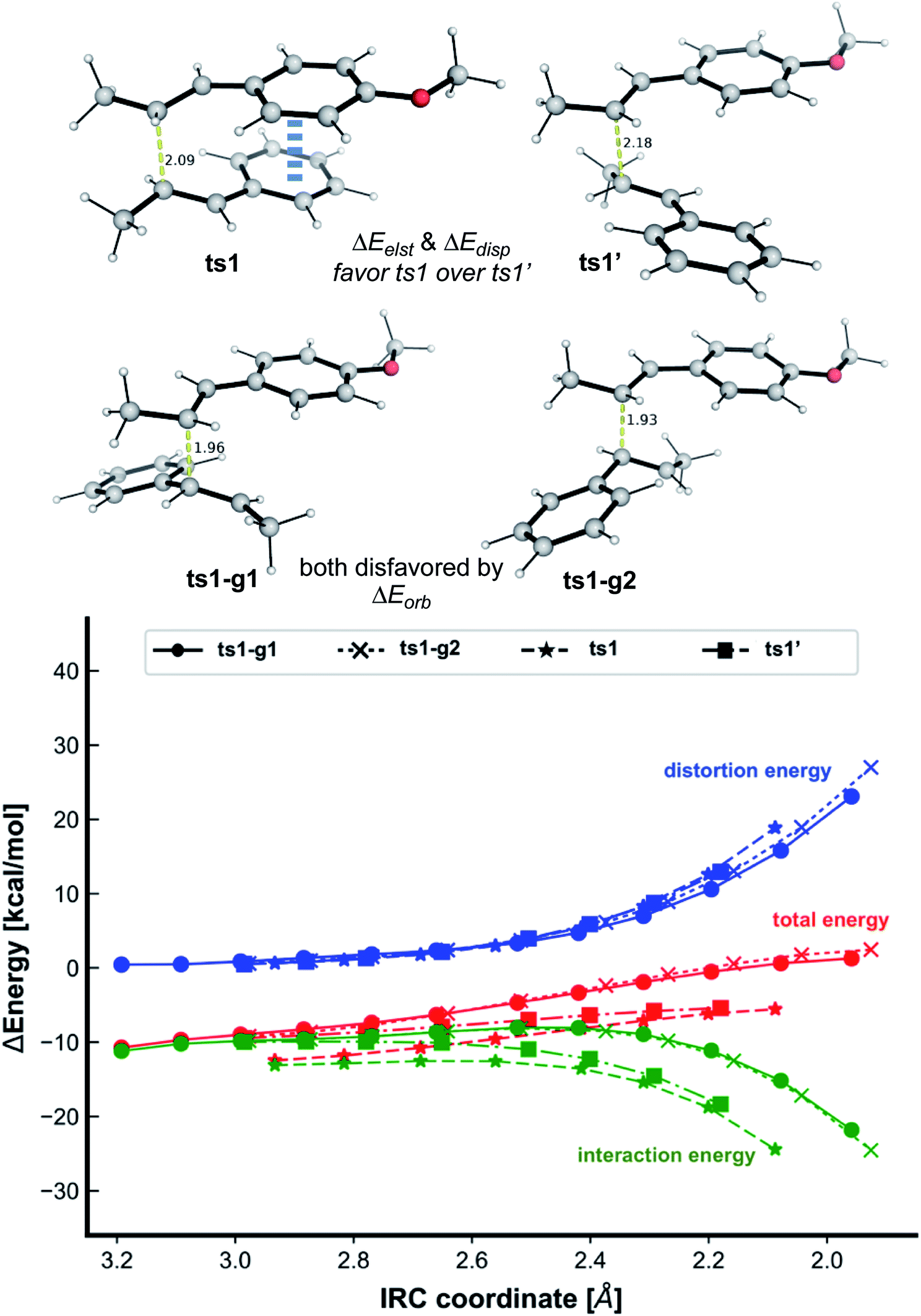 | ||
| Fig. 3 The activation strain or distortion-interaction analyses applied to transition structures for both head-to-head (ts1 and ts1′) and head-to-tail (ts1-g1 and ts1-g2) first C–C bond formation TSs (optimised structures given). All energies are calculated at UM062X/def2TZVPP//UM062X/6-31G(d) and used without any further corrections. | ||
We further applied the method of energy decomposition analysis (with the ALMO-EDA method),97–99 as applied in other similar systems,94,96,100–102 to break down the contributions to the interaction energy between the reacting fragments into the repulsive exchange energy due to Pauli's principle, EPauli, the (semi-)classical electrostatic interaction energy between the charge densities of the fragments, Eelec, the orbital interaction energies between the fragments as the TS occurs, Eorb, and the dispersion energy between the fragments Edisp. Comparing the EDA between the major, anti-adduct viats1 to other TSs (Fig. S9–S11†) in pathway P1, we see that despite having larger Pauli repulsion, the much lower interaction energy in ts1 is due to its much favourable dispersion (in agreement with NCI results) and electrostatic energies (not picked up by NCI analysis), although the orbital interaction energy is similar. In the comparison between ts1 and ts1′ for example, favourable non-covalent interactions stabilising ts1 gains further support from the ALMO-EDA analysis (Fig. 4), which shows that the intermolecular dispersion energy is more stabilising for ts1 than for ts1′ along the reaction coordinate (green). There is in addition to a preferential electrostatic stabilisation (blue) in ts1. However, little difference in the orbital contribution (red) to the interaction energy is seen in both ts1 and ts1′, since they have the same regiochemistry and bond formation occurs between the same pair of atoms. In the comparison of ts1 with other regioisomeric transition structures (e.g., ts1vs.ts1-g1, Fig. S10†), we see the additional contribution of the intermolecular orbital interaction energy (red) in favouring ts1 along the reaction coordinate. The orbital preference for the major regioisomer can be understood from FMO-based arguments. The neutral alkene preferentially reacts with a cationic reagent at the β-position since this site has the larger HOMO coefficient. The radical cationic alkene reacts preferentially at the β-position since this is the site with higher spin density and greater SOMO coefficient.
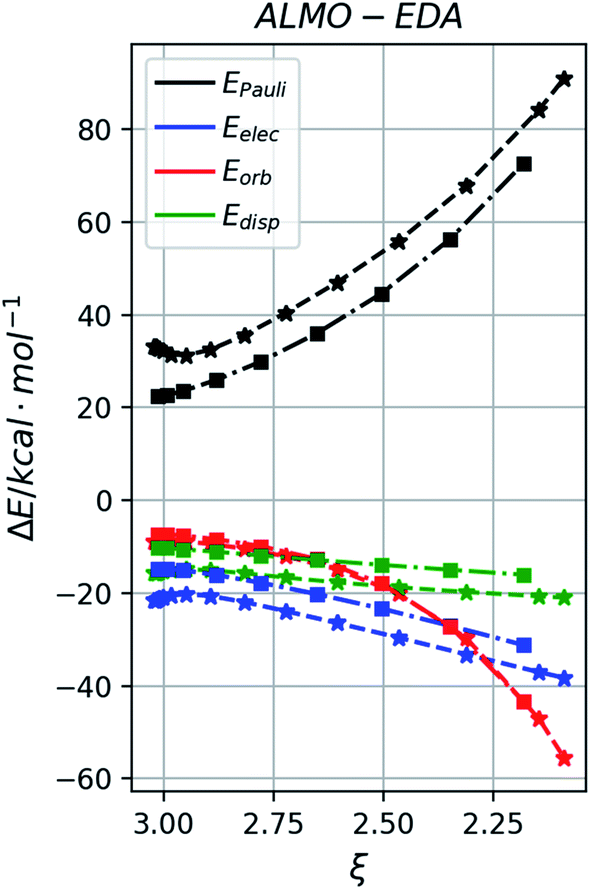 | ||
| Fig. 4 Comparison of the absolutely-localized molecular orbitals energy decomposition analysis (ALMO-EDA) for ts1 (star marker) and ts1′ (square marker) in reaction P1. | ||
Gibbs energies for all radical cationic intermediates in the reaction are preferentially stabilised in HFIP solvent than in MeCN solvent (by 3.2 to 5.7 kcal mol−1), indicating the former solvent's ability in stabilising the charged intermediates;50,76,77 the energies for the neutral species (int3n, int3′n, int3n-g1 and int3n-g2), on the other hand, do not significantly differ in either solvent system (within 0.2 kcal mol−1). This is not the result of bulk electrostatics, since it is MeCN that has a higher dielectric constant than HFIP (ε = 38.8 vs. 16.7). Rather, differences captured by parameters used by the SMD solvation model103 relate to a greater Abraham's hydrogen bond acidity of HFIP than MeCN (1.96 vs. 0.07) and electronegative halogenicity (0.6 vs. 0.0), which reflect the difference in Gibbs energy change associated with cavitation, dispersion and solvent structure for these two solvents.104–106
2.3 Reactivity and selectivity between trans-anethole and cis-β-methylstyrene (P2)
We similarly investigated the SET hole-catalysed reaction between trans-anethole 1a and cis-β-methylstyrene 2b (reaction P2) as shown in Scheme 2(a)(ii). From the calculated redox potentials for these substrates in Table 1, trans-anethole 1a will be oxidised preferentially to cis-β-methylstyrene 2b by the hypervalent iodine reagent in the fluorinated solvent. The Gibbs energy profile for both head-to-head and head-to-tail cyclobutanation is shown in Fig. 5 (See Fig. S12† for all possible conformations). The syn-addition (blue pathway) has the lowest activation barrier, at 15.1 kcal mol−1, with ts7′ (the second C–C bond formation) as the overall TDTS. The anti-addition (black pathway) has ts6 (first C–C bond formation) as the overall TDTS, at 17.1 kcal mol−1. Comparing the first C–C bond formation TSs ts6 and ts6′, the syn-addition ts6′ is lower than the anti-addition ts6, by 3.3 kcal mol−1. The TS ts6′ benefits from π–π stacking interaction that is absent in ts6 as evidenced by NCI plots in Fig. 6. This is similar to pathway P1, where the anti-addition ts1 is lower in activation barrier than syn-addition ts1′, where π–π stacking interaction is also observed for ts1, as discussed previously.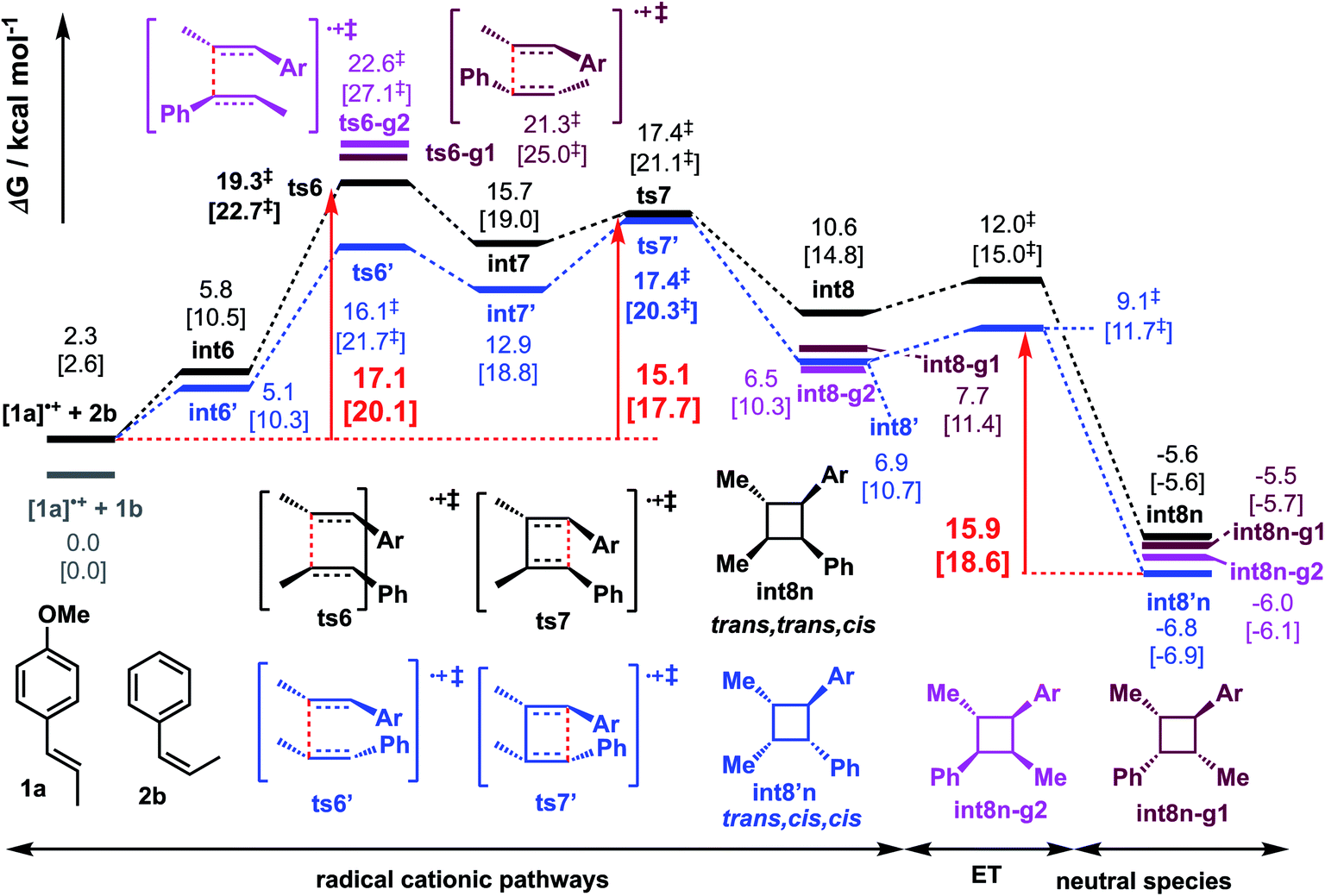 | ||
| Fig. 5 Gibbs energy profile for reaction between trans-anethole and cis-β-methylstyrene computed at SMD(solvent)-M06-2X/def2-TZVPP//M06-2X/6-31G(d) level of theory. Same energy zero is used as previously. ET = electron transfer barrier heights estimated using Marcus-Hush theory at an intermolecular separation of 3 Å (see ESI† for details). The values in HFIP are given together with values in MeCN in square brackets. | ||
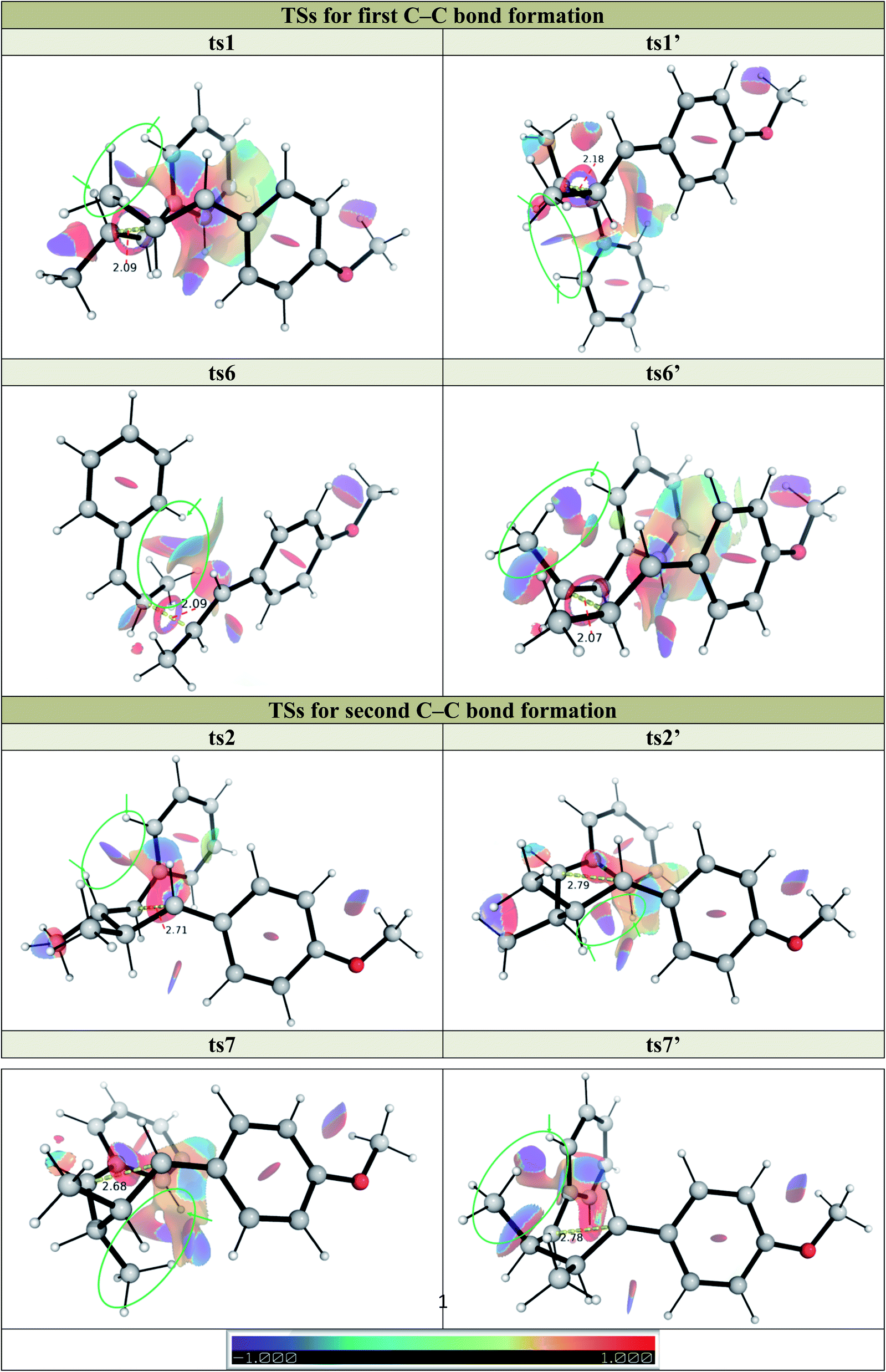 | ||
| Fig. 6 Non-covalent interaction (NCI) plots for key TSs in pathways P1 and P2 at gradient isosurface value of s = 0.5 a.u. | ||
Activation barriers for head-to-head additions are again lower due to the preferential location of the radical cation at the two benzylic α-positions (spin density plots, Fig. S13†). DI–AS analysis of the first C–C bond formation TSs shows again that the interaction energy is more favourable in head-to-head isomers than in head-to-tail isomers whereas the distortion energies are similar in all cases, as in pathway P1. ALMO-EDA comparison between the major syn-addition pathway (viats6′) and other pathways (Fig. S20–S22†) suggests that dispersion and electrostatic interactions contributes favourably to ts6′ selectivity despite its higher Pauli repulsion, as observed for ts1 in reaction P1. We note that no trans,trans,cis product, int8n, is observed experimentally. Product int8′n is kinetically favoured by 2.0 kcal mol−1 and thermodynamically favoured by 1.2 kcal mol−1 than product int8n.
The Gibbs energy profile in Fig. 5 explains the selectivity in favour of the formation of trans,cis,cis cyclobutane int8′n arising from syn-addition. Experimentally, in addition to this product, the all-trans cyclobutane int3n is also formed. The diastereomeric ratio of int8′n:int3n is 4:1 (Scheme 2(a)(ii)). The cycloadduct int3n cannot be formed directly from the starting materials in the present transformation; two possibilities could occur to yield int3n: either cis-β-methylstyrene undergoes isomerisation to give trans-β-methylstyrene before formal [2 + 2] cycloaddition, or the intermediates after first C–C bond formation undergo rotation before the second C–C bond formation/cyclisation to yield product int3n. The isomerisation of neutral cis-β-methyl-styrene to its trans-form cannot be achieved thermally at the reaction temperature, requiring ca. 60 kcal mol−1.107,108 Alternatively, cis-to-trans isomerisation of β-methylstyrene via hole-catalysed rotation can be achieved more readily than in the neutral counterpart,109 however, the higher redox potential of cis-β-methylstyrene 2b than trans-anethole 1a (521 mV difference in HFIP, Table 1) implies the latter would preferentially form a radical cation first (a preference of ca. 12 kcal mol−1), which subsequently attacks the neutral cis-β-methylstyrene, present in excess. The 1:2 stoichiometric ratio of the two alkenes 1a:2b (even though this increases slightly during the reaction as reactants are consumed) is not enough to outweigh the intrinsic difference in oxidation potentials such that we consider the possibility in the isomerisation of radical cationic cis-β-methylstyrene unlikely.
We therefore focused our attention on the rotation of the acyclic radical cation intermediates formed after the first step. Free rotation about C–C single bonds in radical cationic systems are known to be relatively facile;57,86 these rotations can be more easily achieved in radical cationic species than in their neutral counterparts.109Scheme 4 shows the reactions between trans-anethole 1a and either trans-β-methylstyrene 1b (P1) or cis-β-methylstyrene 2b (P2). These two reactions are interconvertible via a rotation about C1–C2 bond in the radical cationic intermediate following first C–C bond formation. In the reaction between [1a]˙+ and 1b for example (P1), at the intermediate int2 after the first C–C bond formation, direct second C–C bond formation (ts2, 13.0 kcal mol−1) has a much lower activation barrier than the rotational barrier to access pathways P2 (ts-rot-12, 20.3 kcal mol−1), such that only int3n is formed as the sole product.
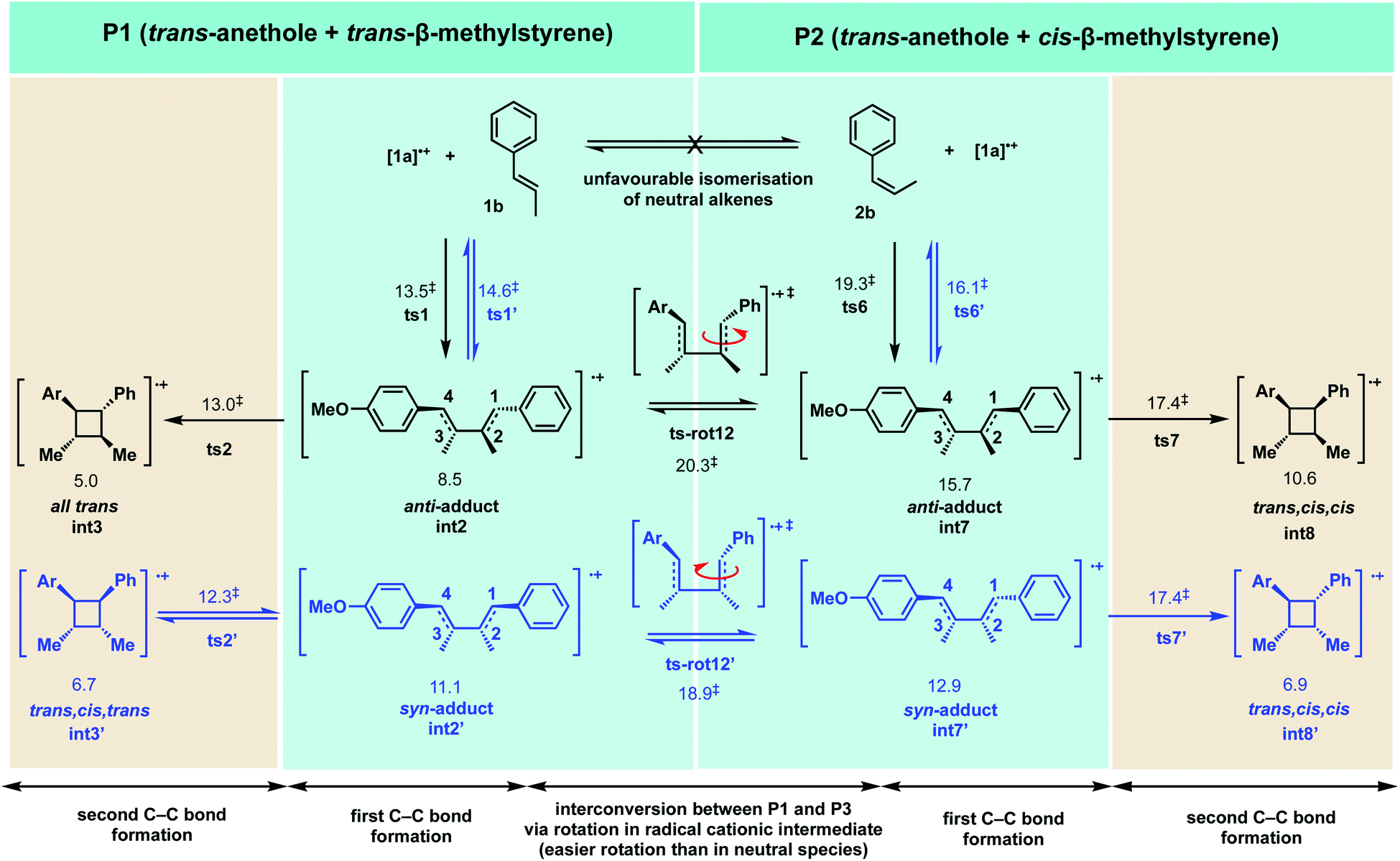 | ||
| Scheme 4 Interconversion between reaction pathways involving trans-anethole and trans-β-methylstyrene (P1) and trans-anethole and cis-β-methylstyrene (P2) via rotations in the radical cationic intermediates. Gibbs energies in HFIP solvents are given in kcal mol−1. | ||
In the reaction between [1a]˙+ and 2b (P2), after the first reversible C–C bond formation, the intermediate int7′ can undergo a direct second C–C bond formation ts7′, with an activation barrier of 17.4 kcal mol−1. In this scenario, however, the rotational barrier is much more competitive (ts-rot-12′, 18.9 kcal mol−1) to give int2′, allowing the reaction to access pathway P1. This intermediate int2′ could form neutral cyclobutyl ring int3′n directly due to the low barriers for the second C–C bond formation and the electron transfer to reduce the radical cationic cyclobutyl int3′. This neutral cyclobutene int3′n could also be reduced back to its radical cationic int3′ with a barrier of 14.2 kcal mol−1, which is (slightly) lower than the overall TDTS ts1′ at 14.6 kcal mol−1. The cycloreversion110,111 of int3′ would give [1a]˙+ and 1b, which subsequently forms int3n as the thermodynamic product. In a way, we can think of the radical cationic species int7′ as an intermediate for the conversion of cis-β-methylstyrene to trans-β-methylstyrene before further reaction to give the all-trans cyclobutane. Note that structures int3n and int8′n are almost isoenergetic and are favoured both kinetically (by 1.6–2.0 kcal mol−1) and thermodynamically (by 1.2–2.1 kcal mol−1) over int3′n and int8n. The activation barrier difference ΔΔG‡ of 1.5 kcal mol−1 between second C–C bond formation/cyclisation (ts7′, 17.4 kcal mol−1) and rotation (ts-rot-12′, 18.9 kcal mol−1) gives a selectivity ratio of 6.8:1 under kinetic control; this is close to the experimentally observed ratio of 4:1. The exact ratio is likely influenced by dynamical effects in such radical cationic intermediates, shown in a quantum classical dynamics study of radical cationic Diels–Alder cycloaddition.112
Comparing reactions P1 and P2, we note that for both the first and second C–C bond formations, the TSs for P1, where both alkenes involved have trans-geometry, are lower in activation barrier than the TSs for P2, where one of the alkenes has cis-geometry whereas the other trans-geometry. From Fig. 6 (and Fig. S2 and S12†), the bond lengths in TSs for first (ts1, ts1′, ts6 and ts6′) and second (ts2, ts2′, ts7 and ts7′) C–C bond formations are similar for both pathways P1 and P2 (to within 0.1 Å). The TSs in P1 (ts1 and ts1′ for first C–C bond formation and ts2 and ts2′ for second C–C bond formation), however, all have lower activation barriers than the corresponding TSs in P2 (ts6 and ts6′ for first C–C bond formation and ts7 and ts7′ for second C–C bond formation), since there is less steric clashes between the O–H atom on the phenyl ring and the H atom on β-carbon of trans-β-methylstyrene in P1 than between the O–H atom on the phenyl ring and the methyl group on the β-carbon cis-β-methylstyrene in P2, giving greater reactivity in reaction P1 than reaction P2.
3. Conclusions
We have studied the heterodimerisation of styrenes promoted by hypervalent iodine oxidants with DFT calculations. The homodimerisation of anethole is computed to occur around 56 times faster than heterodimerisation (ΔΔG‡ = 2.5 kcal mol−1). Experimentally, low concentrations of anethole are necessary to avoid homodimerisation. Anethole is added (dropwise) to a solution of an otherwise unreactive alkene in excess (2 equiv.) to ensure heterodimerisation occurs. Computed reduction potentials with implicit and explicit models of solvation indicate that HFIP solvation creates a more powerful oxidant by stabilising the radical anionic form of the hypervalent iodine species, forming strong intermolecular hydrogen bonds. The regioselectivity for head-to-head dimerisation results from a kinetic preference in the first C–C bond forming step. In the heterodimerisation of two trans-styrenes (reaction P1) complete stereoretention is observed. The all-trans diastereomer is favoured kinetically over the trans–cis–trans form in the first step, and this preference is further reinforced by the greater thermodynamic stability of the all-trans product and the potential for overall reversibility at ambient temperatures and above. TSs that result in the loss of alkene stereochemistry by an exocyclic rotation in the acylic radical cation intermediates lie more than 4 kcal mol−1 above all C–C bond forming TSs and do not play a role for these substrates. In contrast, C–C forming TSs in the reaction between a trans- and a cis-alkene (reaction P2) lie higher in energy, such that rotation is now competitive with ring-closure. This results in the formation of products that are normally formed from the reaction of two alkenes of trans-geometry. In addition, anti-addition (giving trans-geometry across the first C–C bond formation) is favoured when the reacting alkenes are both of trans-geometry whereas syn-addition (cis-geometry across the first C–C bond formation) is favoured when one of the reacting alkenes is of cis-geometry. These calculations agree with the stereochemical results of recent studies with hypervalent iodine oxidants, and with previous studies using amininum oxidants.We have shown that the radical cation mediated hole-catalysed cycloaddition is predominantly influenced by electronic factors and is under both kinetic and thermodynamic controls. The regioselectivity for head-to-head over head-to-tail isomer formations is under kinetic control, as both these neutral 4-membered rings have rather close energies. For the head-to-head isomers (syn- vs. anti-adducts), although the major isomer is favoured both kinetically and thermodynamically, the product selectivity is controlled by thermodynamics of the neutral products since the experimentally observed product distribution agrees better with the thermodynamic energetic differences rather than the kinetic activation barrier differences.
Alkene heterodimerisation with “hole catalysis” is intrinsically challenging since the more electron-rich reagent is both easier to oxidize and more reactive towards the radical cation once it has been formed. We predict that the homodimerisation pathway for trans-anethole is kinetically favoured by 2.5 kcal mol−1 over the heterodimerisation with β-methylstyrene where the concentrations of the two alkenes are identical. Practical solutions to this challenge have thus far focused on increasing the amount of the less electron-rich alkene and adding the more electron-rich component dropwise. Based on our analysis of the competing transition structures, and in particular those features that favour heterodimerisation ts1, we propose enhancing attractive dispersive and electrostatic interactions as an alternative way to increase the likelihood of heterodimerisation. For example, we expect that incorporation of larger π-systems or aromatic substituents known to provide greater dispersive interactions into the less easily oxidised alkene will stabilise heterodimerisation ts1. Additionally, through-space electrostatic interactions could also be optimised for the less electron-rich component by the introduction of polar functional groups that are not in direct conjugation with the alkene. Such groups could then stabilise the heterodimerisation pathway without a large change in oxidation potential to this component. Since dispersive and electrostatic contributions also stabilise ts1 over diasteromeric pathways, tuning these interactions could also be used to ensure high levels of diastereoselectivity. The understanding of this mechanism opens up possibilities of experimentally controlling the chemoselectivity, regioselectivity and stereoselectivity in oxidant-promoted SET hole-catalysed heterodimerisation of electron-rich alkenes in accessing complex, tetra-substituted cyclobutyl rings. Using computational calculations of redox potentials of different alkenes and understanding their inherent steric and electronic properties, we envision that intermolecular heterodimerisation of different alkenes can be efficiently controlled, with precise chemical selectivity, to efficiently access tetra-substituted cyclobutanes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Agency for Science, Technology and Research (A*STAR), Singapore (X. Z.) is gratefully acknowledged. X. Z. and R. S. P. acknowledge the EPSRC Centre for Doctoral Training in Theory and Modelling in Chemical Sciences (EP/L015722/1) and the use of Dirac cluster. X. Z. thanks Dr Maria Koiyoni for her help with HFIP solvent parametrisation. R. S. P. acknowledges computational resources from the RMACC Summit supercomputer supported by the National Science Foundation (ACI-1532235 and ACI-1532236), the University of Colorado Boulder and Colorado State University, and the Extreme Science and Engineering Discovery Environment (XSEDE) through allocation TG-CHE180056. XSEDE is supported by the National Science Foundation (ACI-1548562). We thank Prof. Aqeel Hussain for sharing information with us.113References
- V. M. Dembitsky, Bioactive Cyclobutane-Containing Alkaloids, J. Nat. Med., 2008, 62(1), 1–33 CrossRef CAS PubMed.
- A. Sergeiko, V. V. Poroikov, L. O. Hanus and V. M. Dembitsky, Cyclobutane-Containing Alkaloids: Origin, Synthesis, and Biological Activities, Open Med. Chem. J., 2008, 2(1), 26–37 CrossRef CAS PubMed.
- J. C. Namyslo and D. E. Kaufmann, The Application of Cyclobutane Derivatives in Organic Synthesis, Chem. Rev., 2003, 103(4), 1485–1538 CrossRef CAS PubMed.
- T. Seiser, T. Saget, D. N. Tran and N. Cramer, Cyclobutanes in Catalysis, Angew. Chem., Int. Ed., 2011, 50(34), 7740–7752 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Chapter 2: Strain and Stability, in Modern Physical Organic Chemistry, University Science, Sausalito, CA, 2006, pp. 100–109 Search PubMed.
- C. Liebermann, Ueber Polythymochinon, Ber. Dtsch. Chem. Ges., 1877, 10(2), 2177–2179 CrossRef.
- S. Farid and S. E. Shealer, Radical Cations: Photochemical and Ferric Ion-Induced Formation and Reactions of Indene Radical Cation, J. Chem. Soc., Chem. Commun., 1973, 0(18), 677–678 RSC.
- S. Kuwata, Y. Shigemitsu and Y. Odaira, Photosensitized Cyclodimerization of Phenyl Vinyl Ether, J. Chem. Soc., Chem. Commun., 1972, 0(1), 2 RSC.
- R. A. Caldwell, K. Mizuno, P. E. Hansen, L. P. Vo, M. Frentrup and C. D. Ho, Photochemistry of the Phenanthrene-Stilbene System. Cycloaddition and Singlet-Sensitized Isomerization, J. Am. Chem. Soc., 1981, 103(24), 7263–7269 CrossRef CAS.
- T. Bach and J. P. Hehn, Photochemical Reactions as Key Steps in Natural Product Synthesis, Angew. Chem., Int. Ed., 2011, 50(5), 1000–1045 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, How Cyclobutanes Are Assembled in Nature-Insights from Quantum Chemistry, Chem. Soc. Rev., 2014, 43(14), 5042–5050 RSC.
- S. Poplata, A. Tröster, Y. Q. Zou and T. Bach, Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 +2] Photocycloaddition Reactions, Chem. Rev., 2016, 116(17), 9748–9815 CrossRef CAS PubMed.
- I. Colomer, R. Coura Barcelos and T. J. Donohoe, Catalytic Hypervalent Iodine Promoters Lead to Styrene Dimerization and the Formation of Tri- and Tetrasubstituted Cyclobutanes, Angew. Chem., Int. Ed., 2016, 55(15), 4748–4752 CrossRef CAS PubMed.
- Ł. Albrecht, G. Dickmeiss, F. C. Acosta, C. Rodríguez-Escrich, R. L. Davis and K. A. Jørgensen, Asymmetric Organocatalytic Formal [2 + 2]-Cycloadditions via Bifunctional H-Bond Directing Dienamine Catalysis, J. Am. Chem. Soc., 2012, 134(5), 2543–2546 CrossRef PubMed.
- A. J. Nielsen, H. A. Jenkins and J. McNulty, Asymmetric Organocatalytic Stepwise [2 + 2] Entry to Tetra-Substituted Heterodimeric and Homochiral Cyclobutanes, Chem. - Eur. J., 2016, 22(27), 9111–9115 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113(7), 5322–5363 CrossRef CAS PubMed.
- M. Lautens, W. Klute and W. Tam, Transition Metal-Mediated Cycloaddition Reactions, Chem. Rev., 1996, 96(1), 49–92 CrossRef CAS PubMed.
- V. A. Schmidt, J. M. Hoyt, G. W. Margulieux and P. J. Chirik, Cobalt-Catalyzed [2π + 2π] Cycloadditions of Alkenes: Scope, Mechanism, and Elucidation of Electronic Structure of Catalytic Intermediates, J. Am. Chem. Soc., 2015, 137(24), 7903–7914 CrossRef CAS PubMed.
- K. Chiba, T. Miura, S. Kim, Y. Kitano and M. Tada, Electrocatalytic Intermolecular Olefin Cross-Coupling by Anodically Induced Formal [2 + 2] Cycloaddition between Enol Ethers and Alkenes, J. Am. Chem. Soc., 2001, 123(45), 11314–11315 CrossRef CAS PubMed.
- M. Arata, T. Miura and K. Chiba, Electrocatalytic Formal [2 + 2] Cycloaddition Reactions between Anodically Activated Enyloxy Benzene and Alkenes, Org. Lett., 2007, 9(21), 4347–4350 CrossRef CAS PubMed.
- T. Miura, S. Kim, Y. Kitano, M. Tada and K. Chiba, Electrochemical Enol Ether/Olefin Cross-Metathesis in a Lithium Perchlorate/Nitromethane Electrolyte Solution, Angew. Chem., Int. Ed., 2006, 45(9), 1461–1463 CrossRef CAS PubMed.
- K. Chiba and M. Tada, Diels–Alder Reaction of Quinones Generated in situ by Electrochemical Oxidation in Lithium Perchlorate-Nitromethane, J. Chem. Soc., Chem. Commun., 1994, 2485–2486 RSC.
- W. R. Gutekunst and P. S. Baran, Applications of C–H Functionalization Logic to Cyclobutane Synthesis, J. Org. Chem., 2014, 79(6), 2430–2452 CrossRef CAS PubMed.
- Q. F. Wu, X. B. Wang, P. X. Shen and J. Q. Yu, Enantioselective C–H Arylation and Vinylation of Cyclobutyl Carboxylic Amides, ACS Catal., 2018, 8(3), 2577–2581 CrossRef CAS PubMed.
- J.-L. Hu, L.-W. Feng, L. Wang, Z. Xie, Y. Tang and X. Li, Enantioselective Construction of Cyclobutanes: A New and Concise Approach to the Total Synthesis of (+)-Piperarborenine B, J. Am. Chem. Soc., 2016, 138(40), 13151–13154 CrossRef CAS PubMed.
- X. Yang, G. Shan, Z. Yang, G. Huang, G. Dong, C. Sheng and Y. Rao, One-Pot Synthesis of Quaternary Carbon Centered Cyclobutanes via Pd(II)-Catalyzed Cascade C(sp3)–H Activations, Chem. Commun., 2017, 53(9), 1534–1537 RSC.
- V. Mascitti and E. J. Corey, Total Synthesis of (±)-Pentacycloanammoxic Acid, J. Am. Chem. Soc., 2004, 126(48), 15664–15665 CrossRef CAS PubMed.
- P. Lu and T. Bach, Total Synthesis of (+)-Lactiflorin by an Intramolecular [2 + 2] Photocycloaddition, Angew. Chem., Int. Ed., 2012, 51(5), 1261–1264 CrossRef CAS PubMed.
- P. Zhang, Y. Wang, R. Bao, T. Luo, Z. Yang and Y. Tang, Enantioselective Biomimetic Total Syntheses of Katsumadain and Katsumadain C, Org. Lett., 2012, 14(1), 162–165 CrossRef CAS PubMed.
- S. E. Reisman, J. M. Ready, A. Hasuoka, C. J. Smith and J. L. Wood, Total Synthesis of (±)-Welwitindolinone A Isonitrile, J. Am. Chem. Soc., 2006, 128(5), 1448–1449 CrossRef CAS PubMed.
- F. D. Piaz, A. Vassallo, A. Temraz, R. Cotugno, M. A. Belisario, G. Bifulco, M. G. Chini, C. Pisano, N. De Tommasi and A. Braca, A Chemical-Biological Study Reveals C9-Type Iridoids as Novel Heat Shock Protein 90 (Hsp90) Inhibitors, J. Med. Chem., 2013, 56(4), 1583–1595 CrossRef CAS PubMed.
- L. Eberson, Catalysis by Electron Transfer in Organic Chemistry, J. Mol. Catal., 1983, 20(1), 27–52 CrossRef CAS.
- M. A. Ischay and T. P. Yoon, Accessing the Synthetic Chemistry of Radical Ions, Eur. J. Org. Chem., 2012, 2012(18), 3359–3372 CrossRef CAS.
- S. Lin, S. D. Lies, C. S. Gravatt and T. P. Yoon, Radical Cation Cycloadditions Using Cleavable Redox Auxiliaries, Org. Lett., 2017, 19(2), 368–371 CrossRef CAS PubMed.
- Y. Yamaguchi, Y. Okada and K. Chiba, Understanding the Reactivity of Enol Ether Radical Cations: Investigation of Anodic Four-Membered Carbon Ring Formation, J. Org. Chem., 2013, 78(6), 2626–2638 CrossRef CAS PubMed.
- O. Cortezano-Arellano, L. Quintero and F. Sartillo-Piscil, Total Synthesis of Cephalosporolide E via a Tandem Radical/Polar Crossover Reaction. The Use of the Radical Cations under Nonoxidative Conditions in Total Synthesis, J. Org. Chem., 2015, 80(5), 2601–2608 CrossRef CAS PubMed.
- M. S. Alehashem, C. G. Lim and N. F. Thomas, The Radical Cation Mediated Cleavage of Catharanthine Leading to the Vinblastine Type Alkaloids: Implications for Total Synthesis and Drug Design, RSC Adv., 2016, 6(22), 18002–18025 RSC.
- F. A. Bell, R. A. Crellin, H. Fujii and A. Ledwith, Cation-Radicals: Metal-Catalysed Cyclodimerisation of Aromatic Enamines, J. Chem. Soc. D, 1969, 6, 251–252 RSC.
- L.-Q. Q. Liu, D.-Q. Q. Sun and J.-K. K. Yang, Efficient Additives to Improve the Catalytic Effects of Ferric(III) Chloride on the Reaction of N-Vinylcarbazole: Hydroquinone for Methanolysis, Hydrogen Peroxide for Cyclobutanation Respectively, J. Phys. Org. Chem., 2012, 25(4), 272–277 CrossRef CAS.
- Y. Yu, Y. Fu and F. Zhong, Benign Catalysis with Iron: Facile Assembly of Cyclobutanes and Cyclohexenes: Via Intermolecular Radical Cation Cycloadditions, Green Chem., 2018, 20(8), 1743–1747 RSC.
- M. Riener and D. A. Nicewicz, Synthesis of Cyclobutane Lignans via an Organic Single Electron Oxidant-Electron Relay System, Chem. Sci., 2013, 4(6), 2625–2629 RSC.
- Z. Lu and T. P. Yoon, Visible Light Photocatalysis of [2 + 2] Styrene Cycloadditions by Energy Transfer, Angew. Chem., Int. Ed., 2012, 51(41), 10329–10332 CrossRef CAS PubMed.
- J. Du and T. P. Yoon, Crossed Intermolecular [2 + 2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis, J. Am. Chem. Soc., 2009, 131(41), 14604–14605 CrossRef CAS PubMed.
- M. A. Ischay, M. S. Ament and T. P. Yoon, Crossed Intermolecular [2 + 2] Cycloaddition of Styrenes by Visible Light Photocatalysis, Chem. Sci., 2012, 3(9), 2807–2811 RSC.
- A. E. Hurtley, Z. Lu and T. P. Yoon, Cycloaddition of 1,3-Dienes by Visible Light Photocatalysis, Angew. Chem., Int. Ed., 2014, 53(34), 8991–8994 CrossRef CAS PubMed.
- N. L. Bauld and R. Pabon, Cation Radical Catalyzed Olefin Cyclodimerization, J. Am. Chem. Soc., 1983, 105(3), 633–634 CrossRef CAS.
- P. G. Gassman and D. A. Singleton, Distinction between Aminium Cation Radical and Protic Acid Catalyzed Diels-Alder Reactions, J. Am. Chem. Soc., 1984, 106(25), 7993–7994 CrossRef CAS.
- D. W. Reynolds, K. T. Lorenz, H. S. Chiou, D. J. Bellville, R. A. Pabon and N. L. Bauld, Mechanistic Diagnosis of Aminium Salt Initiated Diels-Alder Cycloadditions in the Diene/Diene Format, J. Am. Chem. Soc., 1987, 109(16), 4960–4968 CrossRef CAS.
- N. L. Bauld, J. Yang and D. Gao, Diels–Alder Cycloadditions of the N-Vinylcarbazole Radical Cation, J. Chem. Soc., Perkin Trans. 2, 2000,(2), 207–210 RSC.
- I. Colomer, C. Batchelor-Mcauley, B. Odell, T. J. Donohoe and R. G. Compton, Hydrogen Bonding to Hexafluoroisopropanol Controls the Oxidative Strength of Hypervalent Iodine Reagents, J. Am. Chem. Soc., 2016, 138(28), 8855–8861 CrossRef CAS PubMed.
- A. Ledwith, Cation Radicals in Electron Transfer Reactions, Acc. Chem. Res., 1972, 5(4), 133–139 CrossRef CAS.
- S. L. Mattes and S. Farid, Photooxygenation via Electron Transfer. 1,1-Dimethylindene, J. Am. Chem. Soc., 1982, 104(5), 1454–1456 CrossRef CAS.
- S. L. Mattes and S. Farid, Photosensitized Electron-Transfer Reactions. Interception of the Geminate Radical Ion Pair, J. Am. Chem. Soc., 1983, 105(5), 1386–1387 CrossRef CAS.
- S. L. Mattes and S. Farid, Photochemical Electron-Transfer Reactions of 1,1-Diarylethylenes, J. Am. Chem. Soc., 1986, 108(23), 7356–7361 CrossRef CAS.
- D. W. Reynolds, B. Harirchian, H.-S. Chiou, B. K. Marsh and N. L. Bauld, The Cation Radical Vinylcyclobutane Rearrangement, J. Phys. Org. Chem., 1989, 2(1), 57–88 CrossRef CAS.
- N. P. Schepp and L. J. Johnston, Nanosecond and Picosecond Dynamics of the Radical Cation Mediated Dimerization of 4-Methoxystyrene, J. Am. Chem. Soc., 1994, 116(15), 6895–6903 CrossRef CAS.
- L. J. Johnston and N. P. Schepp, in Advances in Electron Transfer, ed. P. S. Mariano, JAI Press, New York, 1996, vol. 6, pp. 41–102 Search PubMed.
- L. L. O'Neil and O. Wiest, Acyclic or Long-Bond Intermediate in the Electron-Transfer-Catalyzed Dimerization of 4-Methoxystyrene, J. Org. Chem., 2006, 71(23), 8926–8933 CrossRef PubMed.
- C. A. Marquez, H. Wang, F. Fabbretti and J. O. Metzger, Electron-Transfer-Catalyzed Dimerization of Trans-Anethole: Detection of the Distonic Tetramethylene Radical Cation Intermediate by Extractive Electrospray Ionization Mass Spectrometry, J. Am. Chem. Soc., 2008, 130(51), 17208–17209 CrossRef CAS PubMed.
- C. Guo, L. Cui, B. Chen, J. Yuan and Z. Tian, A Theoretical Study on the Stereoconvergency of the Intramolecular Radical Cation [2 + 2] Cycloadditions of Bis(Styrenes), RSC Adv., 2012, 2(26), 9932–9937 RSC.
- Y. Li, C. Guo and B.-Z. Chen, A Theoretical Study on Intermolecular [2 + 2] Radical Cation Cycloaddition Reactions and the Competition between Concerted and Stepwise Mechanisms, Comput. Theor. Chem., 2016, 1078, 163–172 CrossRef CAS.
- M. H. Baik and R. A. Friesner, Computing Redox Potentials in Solution: Density Functional Theory as a Tool for Rational Design of Redox Agents, J. Phys. Chem. A, 2002, 106(32), 7407–7412 CrossRef CAS.
- P. Winget, C. J. Cramer and D. G. Truhlar, Computation of Equilibrium Oxidation and Reduction Potentials for Reversible and Dissociative Electron-Transfer Reactions in Solution, Theor. Chem. Acc., 2004, 112(4), 217–227 Search PubMed.
- Y. Fu, L. Liu, H.-Z. Yu, Y.-M. Wang and Q.-X. Guo, Quantum-Chemical Predictions of Absolute Standard Redox Potentials of Diverse Organic Molecules and Free Radicals in Acetonitrile, J. Am. Chem. Soc., 2005, 127(19), 7227–7234 CrossRef CAS PubMed.
- M. Schmidt Am Busch and E. W. Knapp, One-Electron Reduction Potential for Oxygen- and Sulfur-Centered Organic Radicals in Protic and Aprotic Solvents, J. Am. Chem. Soc., 2005, 127(45), 15730–15737 CrossRef CAS PubMed.
- P. Jaque, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Computational Electrochemistry: The Aqueous Ru3+|Ru2+ Reduction Potential, J. Phys. Chem. C, 2007, 111(15), 5783–5799 CrossRef CAS.
- A. S. Dutton, J. M. Fukuto and K. N. Houk, Theoretical Reduction Potentials for Nitrogen Oxides from CBS-QB3 Energetics and (C)PCM Solvation Calculations, Inorg. Chem., 2005, 44(11), 4024–4028 CrossRef CAS PubMed.
- J. L. Hodgson, M. Namazian, S. E. Bottle and M. L. Coote, One-Electron Oxidation and Reduction Potentials of Nitroxide Antioxidants: A Theoretical Study, J. Phys. Chem. A, 2007, 111(51), 13595–13605 CrossRef CAS PubMed.
- L. E. Roy, E. Jakubikova, M. Graham Guthrie and E. R. Batista, Calculation of One-Electron Redox Potentials Revisited. Is It Possible to Calculate Accurate Potentials with Density Functional Methods?, J. Phys. Chem. A, 2009, 113(24), 6745–6750 CrossRef CAS PubMed.
- G. Gryn'ova, J. M. Barakat, J. P. Blinco, S. E. Bottle and M. L. Coote, Computational Design of Cyclic Nitroxides as Efficient Redox Mediators for Dye-Sensitized Solar Cells, Chem.–Eur. J., 2012, 18(24), 7582–7593 CrossRef PubMed.
- A. V. Marenich, A. Majumdar, M. Lenz, C. J. Cramer and D. G. Truhlar, Construction of Pourbaix Diagrams for Ruthenium-Based Water-Oxidation Catalysts by Density Functional Theory, Angew. Chem., Int. Ed., 2012, 51(51), 12810–12814 CrossRef CAS PubMed.
- B. T. Psciuk and H. B. Schlegel, Computational Prediction of One-Electron Reduction Potentials and Acid Dissociation Constants for Guanine Oxidation Intermediates and Products, J. Phys. Chem. B, 2013, 117(32), 9518–9531 CrossRef CAS PubMed.
- J. Ho, Are Thermodynamic Cycles Necessary for Continuum Solvent Calculation of PK a s and Reduction Potentials?, Phys. Chem. Chem. Phys., 2015, 17(4), 2859–2868 RSC.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Computational Electrochemistry: Prediction of Liquid-Phase Reduction Potentials, Phys. Chem. Chem. Phys., 2014, 16(29), 15068–15106 RSC.
- H. G. Roth, N. A. Romero, D. A. Nicewicz, H. G. Roth, N. A. Romero, D. A. Nicewicz, H. G. Roth, N. A. Romero and D. A. Nicewicz, Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry, Synlett, 2016, 27(05), 714–723 CAS.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Hexafluoroisopropanol as a Highly Versatile Solvent, Nat. Rev. Chem., 2017, 1(11), 0088 CrossRef CAS.
- L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Making Radical Cations Live Longer, Chem. Commun., 1996, 18, 2105–2112 RSC.
- N. Shida, Y. Imada, S. Nagahara, Y. Okada and K. Chiba, Interplay of Arene Radical Cations with Anions and Fluorinated Alcohols in Hole Catalysis, Commun. Chem., 2019, 2(1), 1–8 CrossRef.
- D. Chong, M. Stewart and W. E. Geiger, Cycloaddition Reactions of Unactivated Olefins Catalyzed by an Organorhenium Electron-Transfer Mediator, J. Am. Chem. Soc., 2009, 131(23), 7968–7969 CrossRef CAS PubMed.
- M. P. Stewart, K. Lam, D. Chong and W. E. Geiger, CElectron-Transfer Catalyzed Cycloaddition Reactions of Unactivated Cyclic Olefins in Weakly Coordinating Anion Electrolyte, J. Electroanal. Chem., 2015, 743, 68–77 CrossRef CAS.
- R. A. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I, J. Chem. Phys., 1956, 24(5), 966–978 CrossRef CAS.
- R. A. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. III. Applications to Data on the Rates of Organic Redox Reactions, J. Chem. Phys., 1957, 26(4), 872–877 CrossRef CAS.
- R. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Election Transfer V. Comparison and Properties of Electrochemical and Chemical Rate Constants - Correction, J. Phys. Chem., 2007, 67(12), 2889 CrossRef.
- N. S. Hush, Homogeneous and Heterogeneous Optical and Thermal Electron Transfer, Electrochim. Acta, 1968, 13(5), 1005–1023 CrossRef CAS.
- R. Reslan, K. Lopata, C. Arntsen, N. Govind and D. Neuhauser, Electron Transfer beyond the Static Picture: A TDDFT/TD-ZINDO Study of a Pentacene Dimer, J. Chem. Phys., 2012, 137, 22–502 CrossRef PubMed.
- H. X. Zhou and A. Szabo, Microscopic Formulation of Marcus' Theory of Electron Transfer, J. Chem. Phys., 1995, 103(9), 3481–3494 CrossRef CAS.
- J. Zwickl, N. Shenvi, J. R. Schmidt and J. C. Tully, Transition State Barriers in Multidimensional Marcus Theory, J. Phys. Chem. A, 2008, 112(42), 10570–10579 CrossRef CAS PubMed.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, Contemporary Issues in Electron Transfer Research, J. Phys. Chem., 1996, 100(31), 13148–13168 CrossRef CAS.
- S. F. Nelsen, R. F. Ismagilov, K. E. Gentile and D. R. Powell, Temperature Effects on Electron Transfer within Intervalence Bis(Hydrazine) Radical Cations, J. Am. Chem. Soc., 1999, 121(30), 7108–7114 CrossRef CAS.
- M. E. Walther and O. S. Wenger, Tuning the Rates of Long-Range Charge Transfer across Phenylene Wires, ChemPhysChem, 2009, 10(8), 1203–1206 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity, J. Am. Chem. Soc., 2007, 129(35), 10646–10647 CrossRef CAS PubMed.
- F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56(34), 10070–10086 CrossRef CAS PubMed.
- F. M. Bickelhaupt, Understanding Reactivity with Kohn-Sham Molecular Orbital Theory: E2-SN2 Mechanistic Spectrum and Other Concepts, J. Comput. Chem., 1999, 20(1), 114–128 CrossRef CAS.
- I. Fernández and F. M. Bickelhaupt, The Activation Strain Model and Molecular Orbital Theory: Understanding and Designing Chemical Reactions, Chem. Soc. Rev., 2014, 43(14), 4953–4967 RSC.
- L. P. Wolters and F. M. Bickelhaupt, The Activation Strain Model and Molecular Orbital Theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5(4), 324–343 CAS.
- P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Understanding Chemical Reactivity Using the Activation Strain Model, Nat. Protoc., 2020, 15(2), 649–667 CrossRef CAS PubMed.
- K. Morokuma, Molecular Orbital Studies of Hydrogen Bonds. III. CO⋯H–O Hydrogen Bond
in H2CO⋯H2O and H2CO⋯2H2O, J. Chem. Phys., 1971, 55(3), 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, On the Calculation of Bonding Energies by the Hartree Fock Slater Method - I. The Transition State Method, Theor. Chim. Acta, 1977, 46(1), 1–10 CrossRef CAS.
- M. von Hopffgarten and G. Frenking, Energy Decomposition Analysis, Wiley Interdisciplinary Reviews: Computational Molecular Science, John Wiley & Sons, Ltd, January 1, 2012, pp. 43–62 Search PubMed.
- I. Fernández, Combined Activation Strain Model and Energy Decomposition Analysis Methods: A New Way to Understand Pericyclic Reactions, Phys. Chem. Chem. Phys., 2014, 7662–7671 RSC.
- I. Fernández and F. M. Bickelhaupt, Origin of the “Endo Rule” in Diels-Alder Reactions, J. Comput. Chem., 2014, 35(5), 371–376 CrossRef PubMed.
- T. A. Hamlin, B. J. Levandowski, A. K. Narsaria, K. N. Houk and F. M. Bickelhaupt, Structural Distortion of Cycloalkynes Influences Cycloaddition Rates Both by Strain and Interaction Energies, Chem.–Eur. J., 2019, 25(25), 6342–6348 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- J. Zhang, H. Zhang, T. Wu, Q. Wang and D. Van Der Spoel, Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents, J. Chem. Theory Comput., 2017, 13(3), 1034–1043 CrossRef CAS PubMed.
- H. Zhang, T. Tan and D. Van Der Spoel, Generalized Born and Explicit Solvent Models for Free Energy Calculations in Organic Solvents: Cyclodextrin Dimerization, J. Chem. Theory Comput., 2015, 11(11), 5103–5113 CrossRef CAS PubMed.
- X. Chen, W. Qiao, W. Miao, Y. Zhang, X. Mu and J. Wang, The Dependence of Implicit Solvent Model Parameters and Electronic Absorption Spectra and Photoinduced Charge Transfer, Sci. Rep., 2020, 10(1), 1–8 CrossRef PubMed.
- T. L. Cottrell, The Strengths of Chemical Bonds, Butterworths, London, 2nd edn, 1958 Search PubMed.
- S. W. Benson III, Bond Energies, J. Chem. Educ., 1965, 42(9), 502 CrossRef.
- J. S. J. Tan and R. S. Paton, Frontier Molecular Orbital Effects Control the Hole-Catalyzed Racemization of Atropisomeric Biaryls, Chem. Sci., 2019, 10(8), 2285–2289 RSC.
- O. Wiest, Structure and [2 + 2] Cycloreversion of the Cyclobutane Radical Cation, J. Phys. Chem. A, 1999, 103(39), 7907–7911 CrossRef CAS.
- N. J. Saettel, J. Oxgaard and O. Wiest, Pericyclic Reactions of Radical Cations, Eur. J. Org. Chem., 2001, 2001(8), 1429–1439 CrossRef.
- J. S. J. Tan, V. Hirvonen and R. S. Paton, Dynamic Intermediates in the Radical Cation Diels–Alder Cycloaddition: Lifetime and Suprafacial Stereoselectivity, Org. Lett., 2018, 20(10), 2821–2825 CrossRef CAS PubMed.
- During the preparation of this manuscript we became aware of related studies and we are very grateful to Prof. Hussein for sharing information with us, A. A. Hussein, A. Al-Yasari and Y. Ma, Thermodynamics Control Reactivity of Hypervalent Iodine-Mediated Styrene Hetero- and Homodimerization: Mechanistic Insights, 2020, DOI:10.26434/chemrxiv.12350879.v4.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03059g |
| ‡ Present address: Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. |
| This journal is © The Royal Society of Chemistry 2020 |