
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of crotophorbolone†‡
Tianzi
Yu
,
Ying
Sun
,
Canhui
Tu
,
Ting
Chen
,
Shaomin
Fu
 and
Bo
Liu
*
and
Bo
Liu
*
Key Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Rd., Chengdu, Sichuan 610064, China. E-mail: chembliu@scu.edu.cn
First published on 19th June 2020
Abstract
As a natural diterpenoid, crotophorbolone possesses a challenging trans,trans-5/7/6 framework decorated with six contiguous stereogenic centers and is structurally and biogenetically related to tigliane-type diterpenoids with intriguing bioactivities such as phorbol and prostratin. Based on the convergent strategy, we completed an eighteen-step total synthesis of crotophorbolone starting from (−)-carvone and (+)-dimethyl-2,3-O-isopropylidene-L-tartrate. The key elements of the synthesis involve expedient installation of the six-membered ring and the five-membered ring with multiple functional groups at an early stage, cyclization of the seven-membered ring through alkenylation of the ketone between the five-membered ring and the six-membered ring, functional group-sensitive ring-closing metathesis and final selective introduction of hydroxyls at C20 and C4.
Introduction
Diterpenoid natural products are structurally versatile and can be classified into different families, many of which are biogenetically correlated. Hecker proposed that among them, tigliane might be regarded as the biosynthetic precursor of ingenane, daphnane and rhamnofolane (Scheme 1),1 and this hypothesis was partially supported by co-occurrence of these diterpenoids in the plant families Euphorbiaceae and Thymelaeaceae.2 Tigliane could be biosynthetically achieved initially from abundant geranylgeranyl pyrophosphate (GGPP) via formation of casbene and lathyrane.3 Thus it is intriguing to manifest chemical interconversion among tigliane, ingenane, daphnane and rhamnofolane to probe insightful information on the biosynthetic mechanism. In fact, the Wender group was able to transform crotophorbolone (1) to prostratin (2),4 a tigliane-type diterpenoid used as a potential adjuvant in highly active antiretroviral therapy (HAART) for HIV.5 Interestingly, acid treatment of phorbol (3),6 a typical tigliane-type diterpenoid, led to formation of crotophorbolone as a reaction product as early as in 1934,7 although the first isolation of 1 from natural sources had not been reported until 2010.8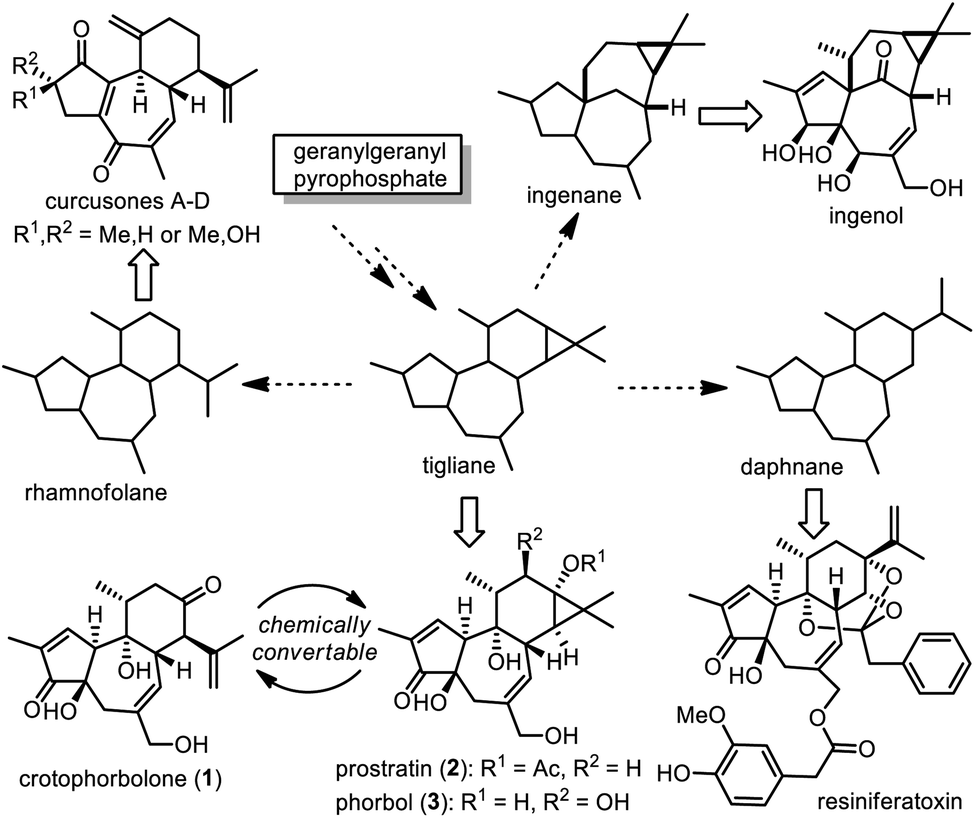 | ||
| Scheme 1 Biogenetic correlation between tigliane, ingenane, daphnane and rhamnofolane. | ||
Allured by their impressive structures and bioactivities, chemists have made numerous synthetic endeavors toward natural ingenane-type, tigliane-type and daphnane-type diterpenoids,9 leading to chemical syntheses of ingenol and its natural derivatives,10 phorbol,11 prostratin,5,12 and resiniferatoxin.13 All of these natural diterpenoids contain a similar tricyclic scaffold embedded with multiple stereogenic centers. Compared to tigliane-type and daphnane-type diterpenoids, rhamnofolane-type diterpenoids belong to a small family with about thirty members.2a,14 As illustrated, crotophorbolone (1) possesses a trans,trans-5/7/6 tricyclic ring system decorated with six contiguous stereogenic centers: two quaternary centers and four tertiary centers. Crotophorbolone (1) is often regarded as a tigliane-type diterpenoid due to its similar oxidation style to phorbol (3) and its biogenesis from phorbol, although it shares a similar carbon skeleton with rhamnofolane-type diterpenoids.
Accompanied by successful chemical syntheses of tigliane-type diterpenoids including prostratin and phorbol,5,11,12 the first total synthesis of crotophorbolone was achieved by the Inoue group.15 In their pioneering work, an impressive strategy was accomplished in thirty four linear steps, featuring smart construction of a unique oxabicyclo[2.2.2]octane intermediate, followed by diastereoselective radical Michael addition to close the middle cycloheptene, which was triggered by cleavage of a bridgehead C–Se bond. Recently, we became interested in developing a convergent synthetic strategy toward natural daphnane-type and rhamnofolane-type diterpenoids,16 based on our persistent research on total synthesis of terpenoids.17 Herein, we would like to present our efforts on convergent total synthesis of crotophorbolone.
Crotophorbolone could be retrosynthetically derived from compound 4 after oxidation of alcohols and alkene isomerization (Scheme 2). Cleavage of the allylic alcohol at C20 and the C6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C7 double bond in 4 would lead to its precursor 5, which could be obtained by coupling 6 and 7 through nucleophilic addition. Fragments 6 and 7 could be synthesized from commercially available (−)-carvone and (+)-dimethyl-2,3-O-isopropylidene-L-tartrate [(+)-8] respectively.
C7 double bond in 4 would lead to its precursor 5, which could be obtained by coupling 6 and 7 through nucleophilic addition. Fragments 6 and 7 could be synthesized from commercially available (−)-carvone and (+)-dimethyl-2,3-O-isopropylidene-L-tartrate [(+)-8] respectively.
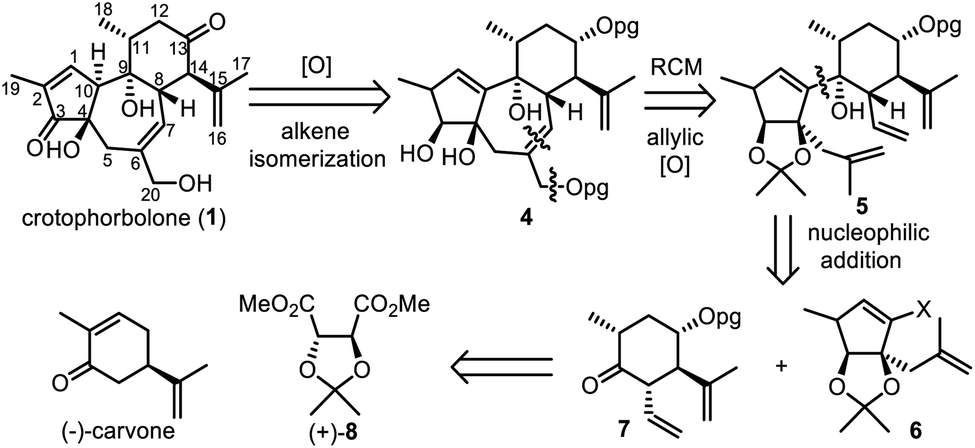 | ||
| Scheme 2 Retrosynthetic analysis. | ||
Results & discussion
Accordingly, we started synthesis of the fully functionalized six-membered ring 12 from compound 9 (Scheme 3), feasibly produced after treating (−)-carvone with a copper–aluminium mixed oxide.18 After silyl protection of the secondary alcohol, the intermediate underwent reduction with sodium dithionite to give a mixture of compounds 10 and 10′,19 which was treated with IBX to afford the pure 10. Coupling compound 10 and 2-(phenylselenyl)-acetaldehyde (11)20 resulted in an aldol intermediate, and mesylation of the resultant secondary alcohol led to elimination to produce compound 12,21 as the equivalent of the fully functionalized six-membered fragment 7.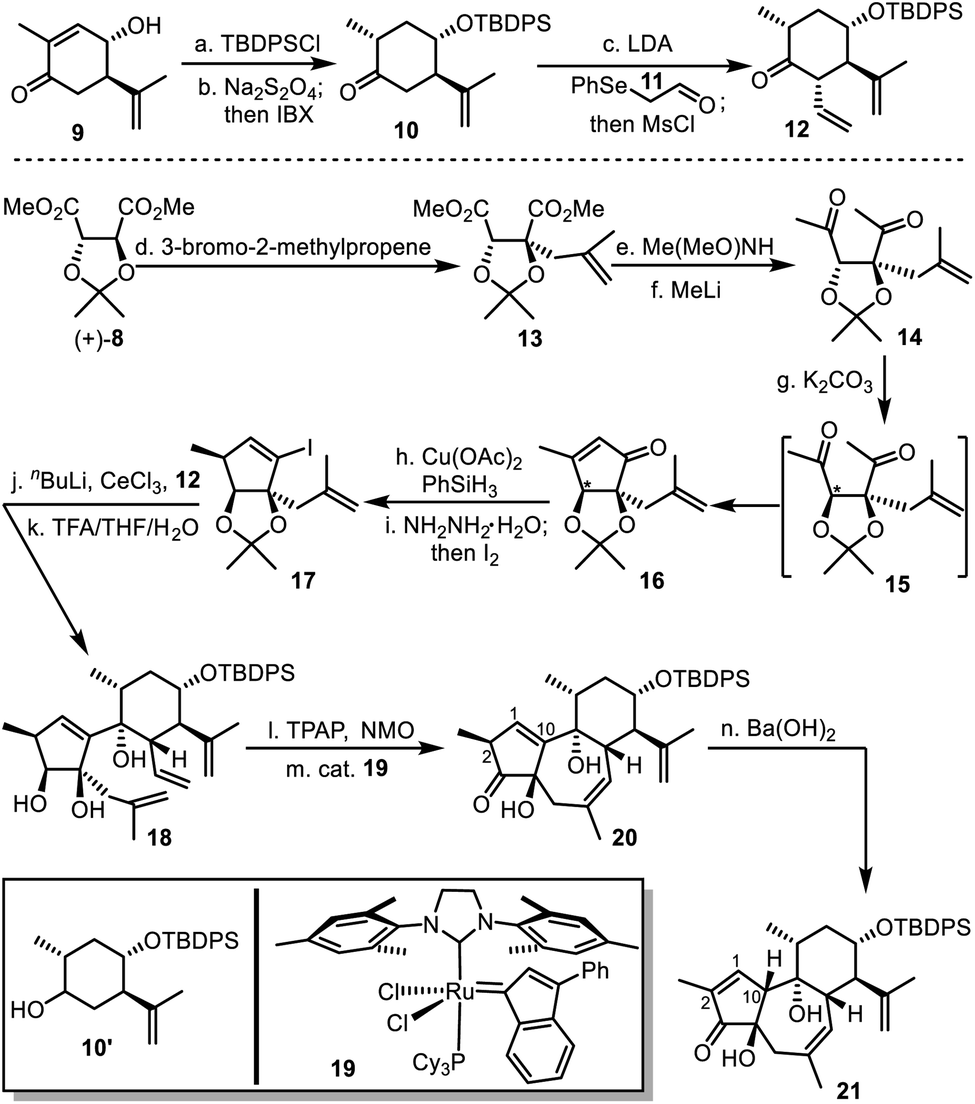 | ||
Scheme 3 Synthesis of 5/7/6 tricyclic intermediate 21. Reagents and conditions: (a) TBDPSCl, imidazole, DCM, rt, 3 h, 78%; (b) Na2S2O4, NaHCO3, Adogen® 464, PhMe/H2O (1/1), reflux, 1.5 h; and then IBX, EtOAc, 80 °C, 12 h, 77%, dr 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; (c) LDA, PhSeCH2CHO, THF, −78 °C to −55 °C, 2 h; and then MsCl, Et3N, DCM, 0 °C, 2 h, 59%; (d) LiHMDS, 3-bromo-2-methylpropene, HMPA, THF, −78 °C, 4 h, quant., dr > 20:1; (e) Me(MeO)NH·HCl, nBuLi, THF, −55 °C, 1 h; (f) MeLi, THF, −78 to rt, 1.5 h, 72% over two steps; (g) K2CO3, MeOH/EtOH (2/1), rt, 13 h, 72%; (h) Cu(OAc)2, PPh3, PhSiH3, PhMe, rt, 5 h, 69%; (i) NH2NH2·H2O, Et3N, EtOH, 80 °C, 20 h; and then I2, Et3N, THF, 0 °C, 0.5 h, 85%; (j) nBuLi, CeCl3, 12, THF, −78 °C, 1 h; (k) TFA/THF/H2O (3/4/4), rt, 3 h, 65% over two steps; (l) TPAP, NMO, DCM, 0 °C, 3 h, 72%; (m) cat. 19, C6F6, reflux, 2 h, 88%; and (n) Ba(OH)2, MeOH/toluene (2/1), 55 °C, 10 min, 44%. TBDPSCl = tert-butyldiphenylsilyl chloride, Adogen® 464 = methyltrialkyl(C8–C10)ammonium chloride, IBX = 2-iodoxybenzoic acid, MsCl = methanesulfonyl chloride, LiHMDS = lithium bis(trimethylsilyl)amide, TFA = trifluoroacetic acid, TPAP = tetrapropylammonium perruthenate, and NMO = N-methylmorpholine N-oxide. :1; (c) LDA, PhSeCH2CHO, THF, −78 °C to −55 °C, 2 h; and then MsCl, Et3N, DCM, 0 °C, 2 h, 59%; (d) LiHMDS, 3-bromo-2-methylpropene, HMPA, THF, −78 °C, 4 h, quant., dr > 20:1; (e) Me(MeO)NH·HCl, nBuLi, THF, −55 °C, 1 h; (f) MeLi, THF, −78 to rt, 1.5 h, 72% over two steps; (g) K2CO3, MeOH/EtOH (2/1), rt, 13 h, 72%; (h) Cu(OAc)2, PPh3, PhSiH3, PhMe, rt, 5 h, 69%; (i) NH2NH2·H2O, Et3N, EtOH, 80 °C, 20 h; and then I2, Et3N, THF, 0 °C, 0.5 h, 85%; (j) nBuLi, CeCl3, 12, THF, −78 °C, 1 h; (k) TFA/THF/H2O (3/4/4), rt, 3 h, 65% over two steps; (l) TPAP, NMO, DCM, 0 °C, 3 h, 72%; (m) cat. 19, C6F6, reflux, 2 h, 88%; and (n) Ba(OH)2, MeOH/toluene (2/1), 55 °C, 10 min, 44%. TBDPSCl = tert-butyldiphenylsilyl chloride, Adogen® 464 = methyltrialkyl(C8–C10)ammonium chloride, IBX = 2-iodoxybenzoic acid, MsCl = methanesulfonyl chloride, LiHMDS = lithium bis(trimethylsilyl)amide, TFA = trifluoroacetic acid, TPAP = tetrapropylammonium perruthenate, and NMO = N-methylmorpholine N-oxide. | ||
Then synthesis of the five-membered fragment 17 began with methallylation of compound (+)-8 with high yield and diastereoselectivity.22 Sequential Weinreb amidation and nucleophilic addition with methyl lithium led to compound 14. The following treatment with potassium carbonate resulted in the functionalized cyclopentenone 16.23 This process involved inversion of the tertiary stereogenic center and intramolecular aldol condensation to generate the thermodynamically more stable cis-fused 5/5 bicyclic ring system. Copper-catalyzed 1,4-reduction delivered an intermediate, which was transformed to compound 17 as a surrogate of fragment 6 after formation of hydrazone and subsequent iodination.
Inspired by preceding synthetic studies,24 we attempted connection between the six-membered ring 12 and the five-membered ring 17. In the presence of CeCl3, the alkenyl lithium generated from 17 and butyl lithium were added to the ketone 12 to afford a diastereoselective adduct, the acetonide of which was removed to yield compound 18. It was then converted into compound 20 with the desired 5/7/6 tricyclic skeleton, after oxidation of the secondary alcohol and ring-closing metathesis (RCM) in C6F6 (ref. 25) using Nolan's ruthenium catalyst 19 to achieve high yield.26 With compound 20 in hand, we had expected that the desired trans-5,7-fused ring system with α-H at C10 would be constructed after alkene isomerization from C1C10 to C1C2,15a,27 at least co-existing with the cis-5,7-fused ring system (21). Unfortunately, we failed to gain the desired compound after numerous trials. Treating 20 with Ba(OH)2 in hot methanol only led to compound 21 in 44% yield, while recovery or decomposition of compound 20 was observed in other cases.
To introduce proper stereochemistry at C10, we decided to invert the absolute configuration of the five-membered fragment by starting the synthesis from (−)-8 (Scheme 4). Thus compound ent-17, prepared by the same synthetic procedure as compound 17, was coupled with compound 12. The resultant intermediate was converted into compound 22 after acidic removal of the acetonide. Then TPAP oxidation and RCM cyclization smoothly afforded compound 23. In contrast to low conversion of 20 into 21 by using Ba(OH)2, treating 23 with DBU promoted alkene isomerization to deliver compound 24 in 94% yield, as a cis-5/7 ring system with identical stereochemistry at C10 to 1. At this stage, directly inverting the stereochemistry of the quaternary stereogenic center at C4 was unfeasible. So the α-OH at C4 in 24 was cleaved with samarium(II) iodide to afford compound 25, whose relative stereochemistry was unambiguously established by single crystal X-ray diffraction. Subsequently, we proposed that a primary allylic alcohol at C20 be selectively introduced to generate compound 26 in the presence of the other two alkenes. However, oxidation with stoichiometric selenium dioxide in THF at 50 °C resulted in 26′ instead, while its application in other solvents and the combination of catalytic SeO2 and tBuOOH led to decomposition of 25. Then White's protocol with Pd(OAc)2-sulfoxide catalysis was attempted,28 but no reaction was observed. Finally, 26 was obtained by means of the Schenck ene reaction with singlet oxygen,29 followed by Re2O7-mediated rearrangement.30 Notably, allylic positions adjacent to C1C2 and C15C16 were inert, probably due to the electron-deficient properties of C1C2 and the shielding effect of neighbouring TBDPS ether near C15C16 respectively.
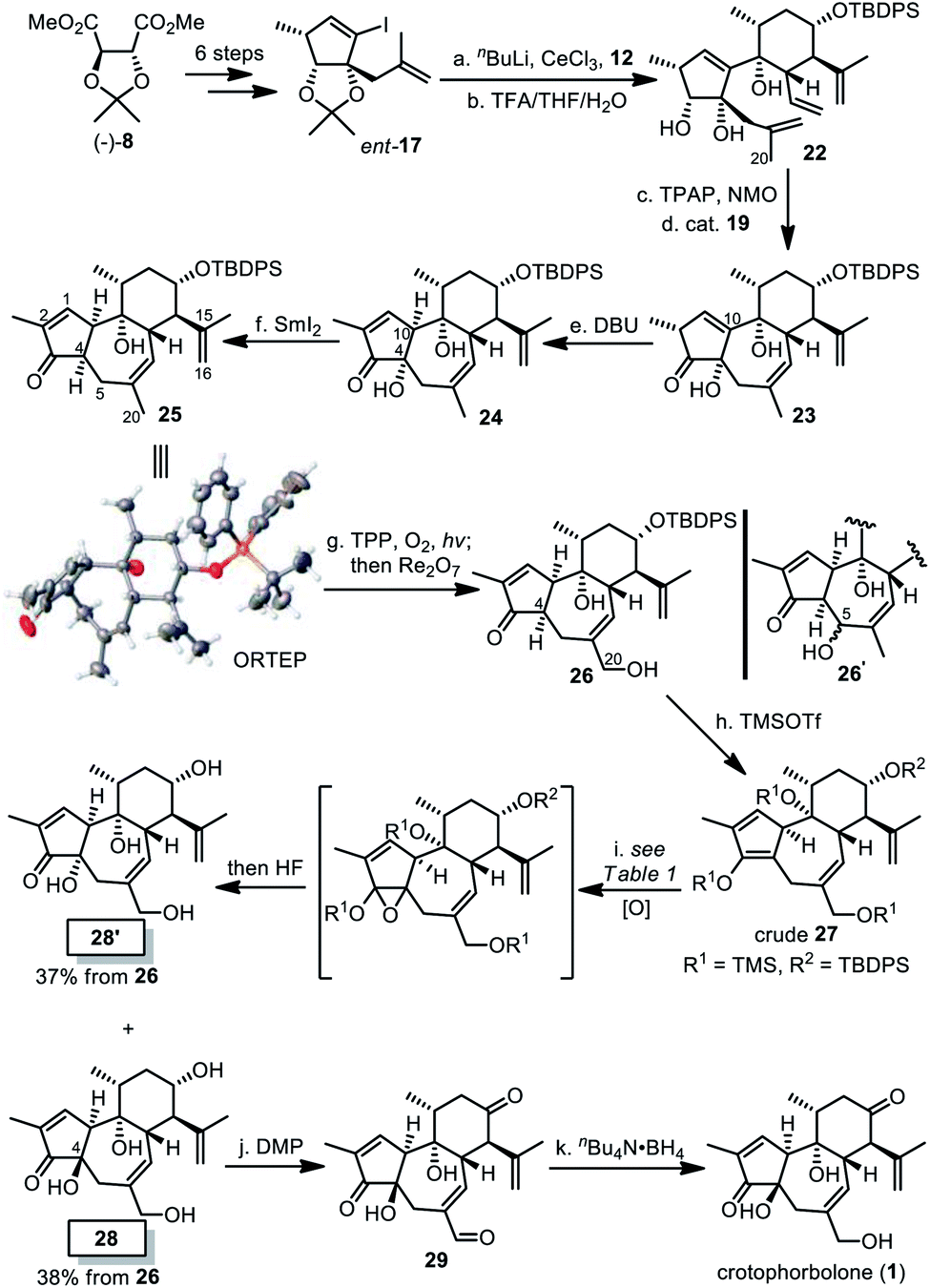 | ||
| Scheme 4 Total synthesis of crotophorbolone. Reagents and conditions: (a) nBuLi, CeCl3, 12, THF, −78 °C, 1 h; (b) TFA/THF/H2O (3/4/4), rt, 4 h, 86% over two steps; (c) TPAP, NMO, DCM, 0 °C, 2.5 h, 78%; (d) cat. 19, C6F6, reflux, 3 h, 96%; (e) DBU, MeOH, 0 °C to rt, 1.5 h, 94%; (f) SmI2, THF, 0 °C, 20 min, 63% (brsm 90%); (g) TPP, O2, hν, DCE, 12 h, PPh3; and then Re2O7, 15 min, 32% (brsm 45%) after two cycles; (h) TMSOTf, Et3N, DCM, 0 °C, 3 h, 72%; (i) see Table 1, then 40% HF/MeCN (1/4), 60 °C, 4 h, 28/28′ = 38%/37%; (j) Dess–Martin periodinane, NaHCO3, DCM, rt, 3 h, quant.; and (k) nBu4N·BH4, MeOH, −40 °C, 10 min, 92%. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TPP = 5,10,15,20-tetraphenylporphyrin, DCE = 1,2-dichloroethane, and brsm = based on the recovery of the starting material. | ||
To introduce a hydroxyl group at C4, we decided to examine the feasibility of a three-step sequence involving silyl enolation, diastereoselective epoxidation and global deprotection to afford the desired compound 28. Crude 27 was first obtained in situ by silylation of compound 26. As summarized in Table 1, in the presence of oxone or MeReO3/H2O2, epoxidation of crude 27 only resulted in the undesired cis-product 28′ after desilylation (entries 1 and 2). Although application of meta-chloroperbenzoic acid (mCPBA) at −78 °C afforded no reaction (entry 3), and epoxidation with it at higher temperature did take place, which was followed by global desilylation to afford a mixture of diastereomers 28 and 28′ in almost a 1:1 ratio (entries 4 and 5). To our delight, we found that direct addition of mCPBA in one batch into the DCM solution of crude 27 at 0 °C provided the optimal result, eventually producing 28 in 38% yield and 28′ in 37% yield from 26 (entry 6). Finally, oxidation with Dess–Martin periodinane gave compound 29, and selective reduction of the aldehyde accomplished the total synthesis of crotophorbolone (1).
| Entry | Conditions in the epoxidation step | Resulta |
|---|---|---|
| a Overall isolated yield from 27. b N. R. = no reaction. c Overall isolated yield from 26. mCPBA = meta-chloroperbenzoic acid. | ||
| 1 | Oxone (1.5 equiv.), NaHCO3 (3.0 equiv.) acetone, 0 °C | 28′ (67%) |
| 2 | MeReO3 (0.25 equiv.), pyridine (2.5 equiv.), H2O2 (2.5 equiv.), MeCN/AcOH (19/1), 0 °C | 28′ (57%) |
| 3 | NaHCO3 (2 equiv.), DCM, dropwise addition of mCPBA (1.05 equiv.) in DCM, −78 °C | N. R.b |
| 4 | NaHCO3 (2 equiv.), DCM, dropwise addition of mCPBA (1.05 equiv.) in DCM, −40 °C | 28/28′ (1/1.4) 67% |
| 5 | NaHCO3 (2 equiv.), DCM, dropwise addition of mCPBA (1.05 equiv.) in DCM, 0 °C | 28/28′ (1.4/1) 59% |
| 6 | NaHCO3 (2 equiv.), DCM, one-batch addition of mCPBA (1.05 equiv.), 0 °C | 28 (38%)c |
| 28′ (37%)c | ||
Surprisingly, similar to that of 24, treatment of 21 with SmI2 in THF led to 25 as the sole isolable product. This indicated the unexpected thermodynamic stability of 25 over 30′ although both compounds contain cis-fused 5/7 ring systems, which was further evidenced by base-mediated conversion of 30 into 25 without 30′ being detected (Scheme 5).
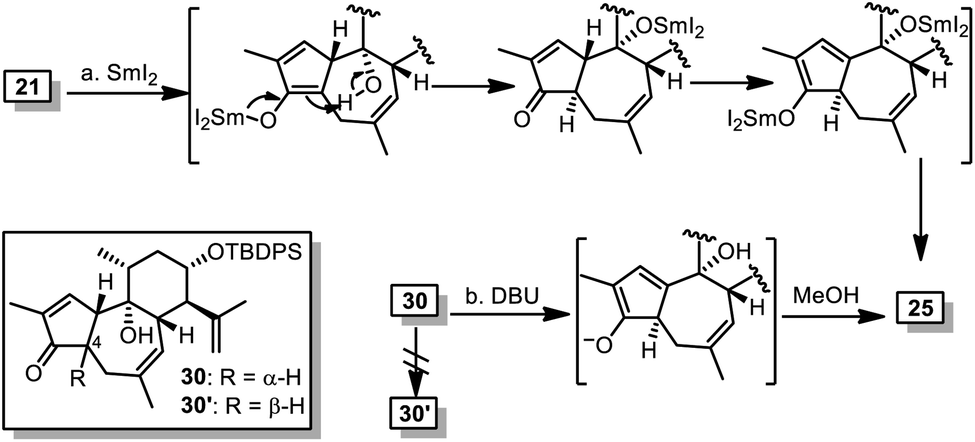 | ||
| Scheme 5 Thermodynamic stability of 25 over 30′. Reagents and conditions: (a) SmI2, THF, 0 °C, 20 min, 74%; and (b) DBU, MeOH, rt-40 °C, 16 h, 63%. | ||
Actually, to properly install the hydroxyl group at C4, we first attempted deprotonation of compound 26 and coupled the resulting enolate with different oxaziridines including the Davis' reagent (+)-31 (Scheme 6A). Unfortunately, under these conditions, either no reaction was observed, or only a trace amount of 32′ with the cis-5/7 ring system was generated instead of the desired 32. By supposing that protection of free alcohols and enhancement of opposite steric hindrance might induce favored diastereoselective hydroxylation, we transformed 26 to 33 to test the practicability (Scheme 6B). In most cases, enolation of 33 with strong bases, followed by treatment with oxaziridine, led to either no reaction or decomposition of the reactant. The best 20% (brsm 31%) yield of the desired compound 34 was achieved when 33 was treated with sodium hexamethyldisilazide (NaHMDS) and (+)-31 in THF at 0 °C. Fortunately, a three-step manipulation was developed to successfully transform 26 to 28 as illustrated in Scheme 4.
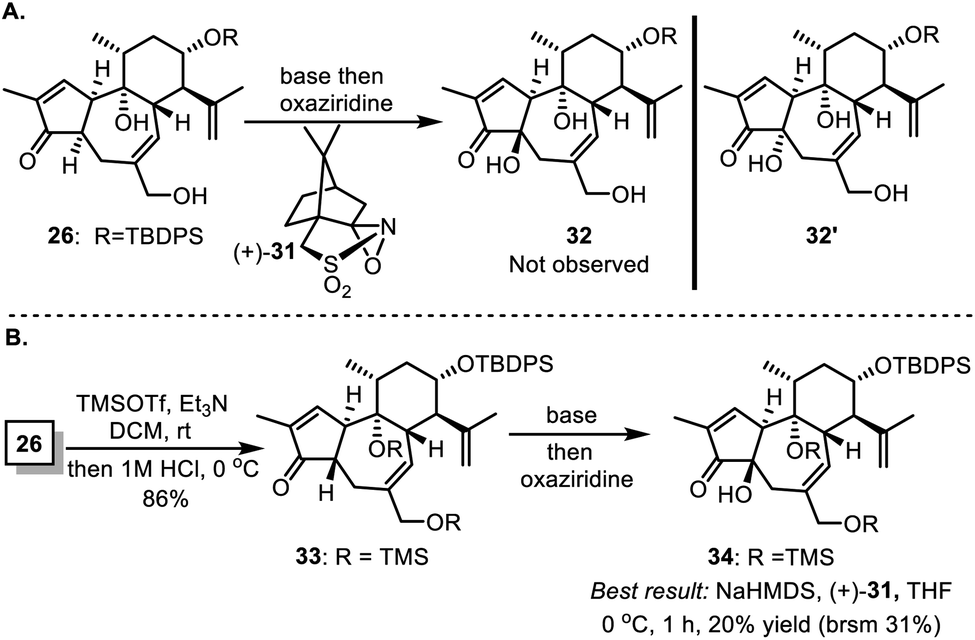 | ||
| Scheme 6 Attempts to install β-OH at C4. | ||
Conclusions
In general, we have completed a convergent total synthesis of crotophorbolone in eighteen longest linear steps. The synthesis features expedient construction of the fully functionalized substructures, i.e. the six-membered fragment 12 through diastereoselective hydroxylation and vinylation, the five-membered fragment 17 through selective methallylation and aldol condensation, and the 5/7/6 tricyclic framework through nucleophilic coupling and RCM cyclization. Selective installation of alcohols at C20 and C4 proved challenging but accessible. Undoubtedly, our discovery on stability of 25 over 30′ would benefit the design of concise routes in the future total synthesis of crotophorbolone and other structurally and biosynthetically related diterpenoids.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We acknowledge financial support from the NSFC (21925106, 21672153, 21921002) and Fundamental Research Funds for the Central Universities. We thank the Analytical & Testing Center of Sichuan University for NMR recording and X-ray diffraction work. We are grateful to Dr Daibing Luo at Sichuan University for his help in single crystal analysis.Notes and references
- (a) E. Hecker, Pure Appl. Chem., 1977, 49, 1423–1431 CAS; (b) W. Adolf and E. Hecker, Isr. J. Chem., 1977, 16, 75–83 CrossRef CAS.
- (a) H. B. Wang, X. Y. Wang, L. P. Liu, G. W. Qin and T. G. Kang, Chem. Rev., 2015, 115, 2975–3011 CrossRef CAS PubMed; (b) G. Appendino, Prog. Chem. Org. Nat. Prod., 2016, 102, 1–90 CAS; (c) Y. X. Jin, L. L. Shi, D. P. Zhang, H. Y. Wei, Y. Si, G. X. Ma and J. Zhang, Molecules, 2019, 24, 1842 CrossRef CAS PubMed; (d) S. G. Liao, H. D. Chen and J. M. Yue, Chem. Rev., 2009, 109, 1092–1140 CrossRef CAS PubMed; (e) H. A. Abdelgadir and J. Van Staden, S. Afr. J. Bot., 2013, 88, 204–218 CrossRef CAS; (f) R. K. Devappa, H. P. S. Makkar and K. Becker, J. Am. Oil Chem. Soc., 2011, 88, 301–322 CrossRef CAS; (g) F. J. Evans and C. J. Soper, Lloydia, 1978, 41, 193–233 CAS.
- R. J. Schmidt, Bot. J. Linn. Soc., 1987, 94, 221–230 CrossRef.
- G. A. Miana, M. Riaz, S. Shahzad-ul-Hussan, R. Z. Paracha and U. Z. Paracha, Mini-Rev. Med. Chem., 2015, 15, 1122–1130 CrossRef CAS PubMed.
- P. A. Wender, J. M. Kee and J. M. Warrington, Science, 2008, 320, 649–652 CrossRef CAS PubMed.
- G. Goel, H. P. S. Makkar, G. Francis and K. Becker, Int. J. Toxicol., 2007, 26, 279–288 CrossRef CAS PubMed.
- (a) B. Flaschentrager and F. F. Falkenhausen, Justus Liebigs Ann. Chem., 1934, 514, 252–260 CrossRef CAS; (b) H. W. Thielmann and E. Hecker, Justus Liebigs Ann. Chem., 1969, 728, 158–183 CrossRef CAS.
- H. B. Wang, W. J. Chu, Y. Wang, P. Ji, Y. B. Wang, Q. Yu and G. W. Qin, J. Asian Nat. Prod. Res., 2010, 12, 1038–1043 CrossRef CAS PubMed.
- (a) R. Liu, J. Feng and B. Liu, Acta Chim. Sin., 2016, 74, 24–43 CrossRef CAS; (b) Y. Li, M. Wei and M. Dai, Tetrahedron, 2017, 73, 4172–4177 CrossRef CAS PubMed; (c) L. V. Nguyen and A. B. Beeler, Org. Lett., 2018, 20, 5177–5180 CrossRef CAS PubMed; (d) Z. G. Brill, Y.-M. Zhao, V. H. Vasilev and T. J. Maimone, Tetrahedron, 2019, 75, 4212–4221 CrossRef CAS; (e) K. Watanabe, Y. Suzuki, K. Aoki, A. Sakakura, K. Suenaga and H. Kigoshi, J. Org. Chem., 2004, 69, 7802–7808 CrossRef CAS PubMed; (f) X. Liu, J. Liu, J. Zhao, S. Li and C. C. Li, Org. Lett., 2017, 19, 2742–2745 CrossRef CAS PubMed; (g) K. Murai, S.-i. Katoh, D. Urabe and M. Inoue, Chem. Sci., 2013, 4, 2364–2368 RSC; (h) J. K. Cha and O. L. Epstein, Tetrahedron, 2006, 62, 1329–1343 CrossRef CAS; (i) S. Chow, T. Krainz, P. V. Bernhardt and C. M. Williams, Org. Lett., 2019, 21, 8761–8764 CrossRef CAS PubMed; (j) A. Pascual-Escudero, M. González-Esguevillas, S. Padilla, J. Adrio and J. C. Carretero, Org. Lett., 2014, 16, 2228–2231 CrossRef CAS PubMed; (k) A. J. Catino, A. Sherlock, P. Shieh, J. S. Wzorek and D. A. Evans, Org. Lett., 2013, 15, 3330–3333 CrossRef CAS PubMed; (l) C. Stewart, R. McDonald and F. G. West, Org. Lett., 2011, 13, 720–723 CrossRef CAS PubMed; (m) G. L. Carroll and R. D. Little, Org. Lett., 2000, 2, 2873–2876 CrossRef CAS PubMed.
- (a) J. D. Winkler, M. B. Rouse, M. F. Greaney, S. J. Harrison and Y. T. Jeon, J. Am. Chem. Soc., 2002, 124, 9726–9728 CrossRef CAS PubMed; (b) K. Tanino, K. Onuki, K. Asano, M. Miyashita, T. Nakamura, Y. Takahashi and I. Kuwajima, J. Am. Chem. Soc., 2003, 125, 1498–1500 CrossRef CAS PubMed; (c) A. Nickel, T. Maruyama, H. Tang, P. D. Murphy, B. Greene, N. Yusuff and J. L. Wood, J. Am. Chem. Soc., 2004, 126, 16300–16301 CrossRef CAS PubMed; (d) I. Kuwajima and K. Tanino, Chem. Rev., 2005, 105, 4661–4670 CrossRef CAS PubMed; (e) T. Ohyoshi, S. Funakubo, Y. Miyazawa, K. Niida, I. Hayakawa and H. Kigoshi, Angew. Chem., Int. Ed., 2012, 51, 4972–4975 CrossRef CAS PubMed; (f) L. Jorgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878–882 CrossRef CAS PubMed; (g) S. J. McKerrall, L. Jorgensen, C. A. Kuttruff, F. Ungeheuer and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5799–5810 CrossRef CAS PubMed; (h) Y. Jin, C. H. Yeh, C. A. Kuttruff, L. Jorgensen, G. Dunstl, J. Felding, S. R. Natarajan and P. S. Baran, Angew. Chem., Int. Ed., 2015, 54, 14044–14048 CrossRef CAS PubMed.
- (a) P. A. Wender, H. Kogen, H. Y. Lee, J. D. Munger, R. S. Wilhelm and P. D. Williams, J. Am. Chem. Soc., 1989, 111, 8957–8958 CrossRef CAS; (b) P. A. Wender and F. E. McDonald, J. Am. Chem. Soc., 1990, 112, 4956–4958 CrossRef CAS; (c) P. A. Wender, K. D. Rice and M. E. Schnute, J. Am. Chem. Soc., 1997, 119, 7897–7898 CrossRef CAS; (d) K. Lee and J. K. Cha, J. Am. Chem. Soc., 2001, 123, 5590–5591 CrossRef CAS PubMed; (e) S. Kawamura, H. Chu, J. Felding and P. S. Baran, Nature, 2016, 532, 90–93 CrossRef CAS PubMed.
- (a) G. Tong, Z. Liu and P. Li, Chem, 2018, 4, 2944–2954 CrossRef CAS; (b) G. Tong, Z. Ding, Z. Liu, Y. Ding, L. Xu, H. Zhang and P. Li, J. Org. Chem., 2020, 85, 4813–4837 CrossRef CAS PubMed.
- (a) P. A. Wender, C. D. Jesudason, H. Nakahira, N. Tamura, A. L. Tebbe and Y. Ueno, J. Am. Chem. Soc., 1997, 119, 12976–12977 CrossRef CAS; (b) P. A. Wender, N. Buschmann, N. B. Cardin, L. R. Jones, C. Kan, J. M. Kee, J. A. Kowalski and K. E. Longcore, Nat. Chem., 2011, 3, 615–619 CrossRef CAS PubMed; (c) S. Hashimoto, S.-i. Katoh, T. Kato, D. Urabe and M. Inoue, J. Am. Chem. Soc., 2017, 139, 16420–16429 CrossRef CAS PubMed.
- A. Vasas and J. Hohmann, Chem. Rev., 2014, 114, 8579–8612 CrossRef CAS PubMed.
- (a) T. Asaba, Y. Katoh, D. Urabe and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 14457–14461 CrossRef CAS PubMed; (b) D. Urabe, T. Asaba and M. Inoue, Bull. Chem. Soc. Jpn., 2016, 89, 1137–1144 CrossRef CAS.
- (a) J. Feng, H. Yin, Y. Man, S. Fu and B. Liu, Chin. J. Chem., 2018, 36, 831–836 CrossRef CAS; (b) J. Feng, T. Yu, Z. Zhang, J. Li, S. Fu, J. Chen and B. Liu, Chem. Commun., 2018, 54, 7665–7668 RSC.
- (a) C. Yuan, B. Du, L. Yang and B. Liu, J. Am. Chem. Soc., 2013, 135, 9291–9294 CrossRef CAS PubMed; (b) L. Song, G. Zhu, Y. Liu, B. Liu and S. Qin, J. Am. Chem. Soc., 2015, 137, 13706–13714 CrossRef CAS PubMed; (c) H. Deng, W. Cao, R. Liu, Y. Zhang and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 5849–5852 CrossRef CAS PubMed; (d) C. Yuan, B. Du, H. Deng, Y. Man and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 637–640 CrossRef CAS PubMed; (e) B. Du, Z. Huang, X. Wang, T. Chen, G. Shen, S. Fu and B. Liu, Nat. Commun., 2019, 10, 1892 CrossRef PubMed.
- A. Leticia Garcia-Cabeza, R. Marin-Barrios, R. Azarken, F. Javier Moreno-Dorado, M. J. Ortega, H. Vidal, J. M. Gatica, G. M. Massanet and F. M. Guerra, Eur. J. Org. Chem., 2013, 2013, 8307–8314 CrossRef.
- F. Camps, J. Coll and J. Guitart, Tetrahedron, 1986, 42, 4603–4609 CrossRef CAS.
- H. J. Reich and F. Chow, J. Chem. Soc., Chem. Commun., 1975, 790–791 RSC.
- C. J. Kowalski and J. S. Dung, J. Am. Chem. Soc., 1980, 102, 7950–7951 CrossRef CAS.
- R. Naef and D. Seebach, Angew. Chem., Int. Ed., 1981, 20, 1030–1031 CrossRef.
- (a) A. I. Meyers and B. A. Lefker, Tetrahedron Lett., 1987, 28, 1745–1748 CrossRef CAS; (b) A. I. Meyers and B. A. Lefker, Tetrahedron, 1987, 43, 5663–5676 CrossRef CAS; (c) A. I. Meyers and C. A. Busacca, Tetrahedron Lett., 1989, 30, 6977–6980 CrossRef CAS.
- (a) L. A. Paquette, F. Gallou, Z. Zhao, D. G. Young, J. Liu, J. Yang and D. Friedrich, J. Am. Chem. Soc., 2000, 122, 9610–9620 CrossRef CAS; (b) T. V. Ovaska, S. E. Reisman and M. A. Flynn, Org. Lett., 2001, 3, 115–117 CrossRef CAS PubMed; (c) O. L. Epstein and J. K. Cha, Angew. Chem., Int. Ed., 2005, 44, 121–123 CrossRef CAS PubMed.
- C. Samojlowicz, M. Bieniek, A. Zarecki, R. Kadyrov and K. Grela, Chem. Commun., 2008, 6282–6284 RSC.
- L. Jafarpour, H. J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5416–5419 CrossRef CAS.
- X. Huang, L. Song, J. Xu, G. Zhu and B. Liu, Angew. Chem., Int. Ed., 2013, 52, 952–955 CrossRef CAS PubMed.
- (a) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346–1347 CrossRef CAS PubMed; (b) M. S. Chen, N. Prabagaran, N. A. Labenz and M. C. White, J. Am. Chem. Soc., 2005, 127, 6970–6971 CrossRef CAS PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- I. Volchkov and D. Lee, Chem. Soc. Rev., 2014, 43, 4381–4394 RSC.
Footnotes |
| † Dedicated to the 70th anniversary of the Shanghai Institute of Organic Chemistry and in memory of Prof. Wei-Shan Zhou. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and details of the electrochemical apparatus employed. CCDC 1989322 and 1989323. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02829k |
| This journal is © The Royal Society of Chemistry 2020 |