
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct catalytic asymmetric and anti-selective vinylogous addition of butenolides to chromones†
Jin
Cui
,
Naoya
Kumagai
 ,
Takumi
Watanabe
* and
Masakatsu
Shibasaki
*
,
Takumi
Watanabe
* and
Masakatsu
Shibasaki
*
Institute of Microbial Chemistry (BIKAKEN), Tokyo 141-0021, Japan. E-mail: twatanabe@bikaken.or.jp; mshibasa@bikaken.or.jp
First published on 22nd June 2020
Abstract
An anti-selective catalytic asymmetric Michael-type vinylogous addition of β,γ-butenolides to chromones was developed. The catalyst system developed herein is characterized by tuning of the steric and electronic effects using a proper Biphep-type chiral ligand to invert the diastereoselection, and improvement of the catalyst turnover by a coordinative phenolic additive. The catalytic protocol renders potentially biologically active natural product analogs accessible in good yield with moderate diastereoselectivity and high enantiomeric purity, mostly greater than 99% ee.
Introduction
Well-designed asymmetric transformations under reagent-controlled conditions have revolutionized C–C bond formation with the desired absolute configuration.1 Moreover, catalytic asymmetric reactions in the modern era are in demand because of their sustainability; direct asymmetric catalytic reactions that do not require activation of the substrate(s) by derivatization are ideal processes in terms of atom-economy.2 Our research group has devoted tremendous effort toward developing these valuable transformations, including aldol- and Michael-type catalytic asymmetric reactions. In fact, we reported the first catalytic asymmetric aldol reaction in 1992,3 a catalytic asymmetric nitroaldol reaction promoted by the LaLi3tris(binaphthoxide) (LLB) catalyst comprising BINOL, La, and Li (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 complex). Subsequently, we disclosed the first example of a direct catalytic asymmetric aldol reaction of simple ketones in 1997.4 For conjugate additions, a La–Na–BINOL-catalyzed5 asymmetric Michael reaction was published in 1995, followed by the development of Al–Li–BINOL6 and La-linked-BINOL7 catalyzed versions. Since then, direct catalytic asymmetric aldol and Michael strategies have been applied to various substrates to attain universal applicability; recent endeavors indicate that even notoriously tough pronucleophiles, such as amides, can be tamed by selective activation toward deprotonation through the coordination of chiral copper species to a tactically designed amide-substructure.8
:1:3 complex). Subsequently, we disclosed the first example of a direct catalytic asymmetric aldol reaction of simple ketones in 1997.4 For conjugate additions, a La–Na–BINOL-catalyzed5 asymmetric Michael reaction was published in 1995, followed by the development of Al–Li–BINOL6 and La-linked-BINOL7 catalyzed versions. Since then, direct catalytic asymmetric aldol and Michael strategies have been applied to various substrates to attain universal applicability; recent endeavors indicate that even notoriously tough pronucleophiles, such as amides, can be tamed by selective activation toward deprotonation through the coordination of chiral copper species to a tactically designed amide-substructure.8
Chiral γ-butenolide units that feature a five-membered γ-lactone with unsaturation at α,β-carbon atoms are ubiquitous in natural products and biologically active compounds.9 The direct utilization of the nonactivated α,β- and β,γ-unsaturated butyrolactones as pronucleophiles instead of 2-siloxyfurans avoids the generation of silyl by-products and the preactivation of butenolides, paving the way to optically active butenolide-containing products in an atom-economical manner.10,11 The direct use of γ-butenolide in catalytic asymmetric vinylogous conjugate addition to chromones is a straightforward approach to highly appealing biologically privileged architectures which was pioneered by Trost et al.10 The reported dinuclear zinc-ProPhenol-catalyzed transformation proceeds with high diastereo- and enantioselectivity with α,β- and β,γ-butenolides and a wide variety of chromones to afford natural product-like scaffold, in which the syn-configuration predominated as the newly formed connectivity (vide infra).
The structural features of the products share characteristics of the core components of the chromanone lactone natural products depicted in Fig. 1, including blennolides D and E, microdiplodiasone, gonytolides C and G, and lachnone C.9 Two of the natural products mentioned above have a syn-stereochemistry between the chromanone core and the butanolide-derived 5-membered lactone, but natural products with an anti-configuration are more abundant. Thus, an anti-selective version of the addition reaction of butenolides to chromanone is in high demand. In fact, in the above-mentioned paper, Trost stated that “we believe that the diastereoselectivity is a feature inherent to this type of transformation” in terms of the stereochemical outcome. Fig. 2 illustrates the predominant formation of syn-products attributed to repulsion of the lone pairs embedded in the chromone and butenolide cores. As Trost points out, the same tendency is observed in the cycloaddition reaction of siloxyfurans and benzopyryliums reported by Porco and co-workers.12 In the present report, we disclose a direct catalytic asymmetric vinylogous Michael-type reaction of chromones and β,γ-butenolides to afford anti-adducts with almost complete enantioselectivity using a newly developed catalyst system.
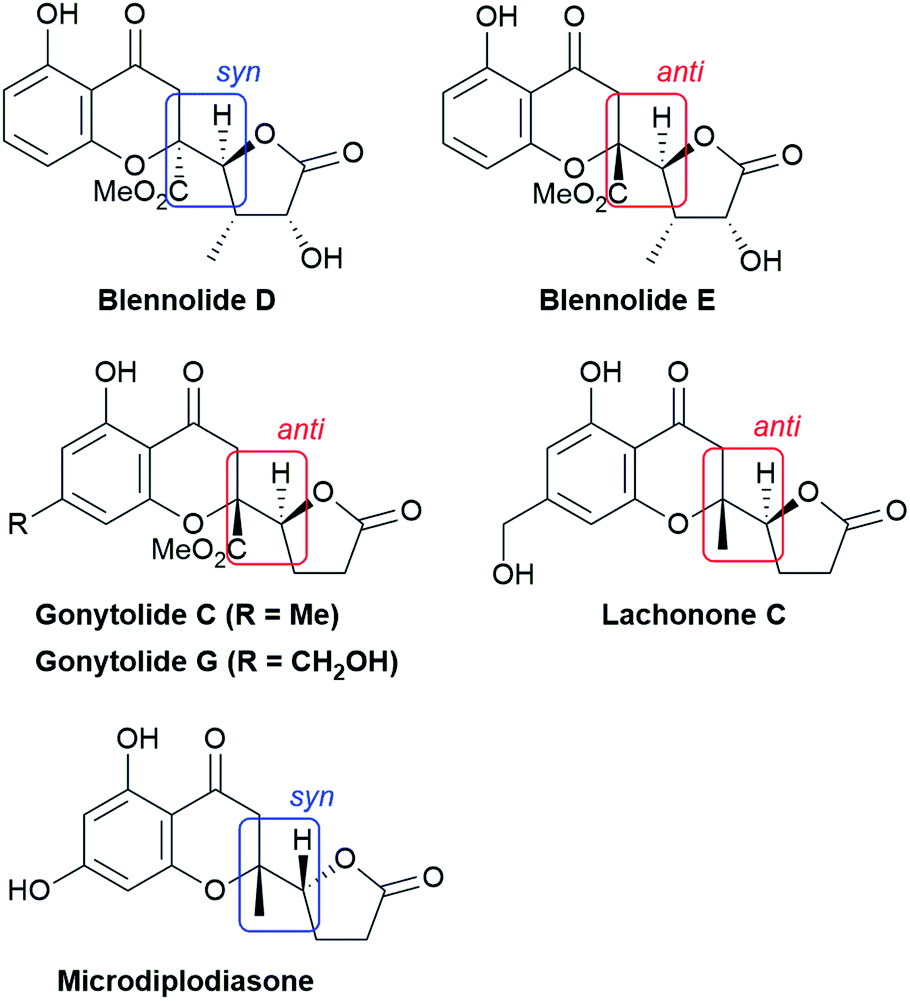 | ||
| Fig. 1 Structure of chromanone lactone natural products. | ||
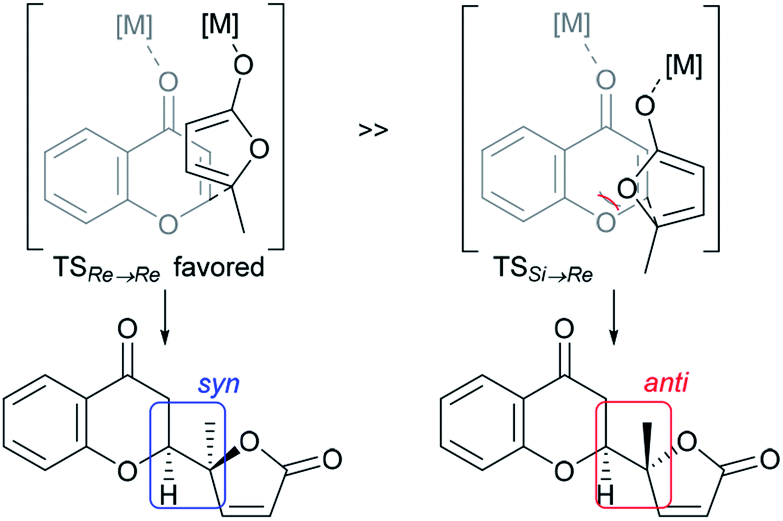 | ||
| Fig. 2 Rationale for intrinsic syn-selectivity of vinylogous addition of butenolide to chromone (ref. 10). | ||
Results and discussion
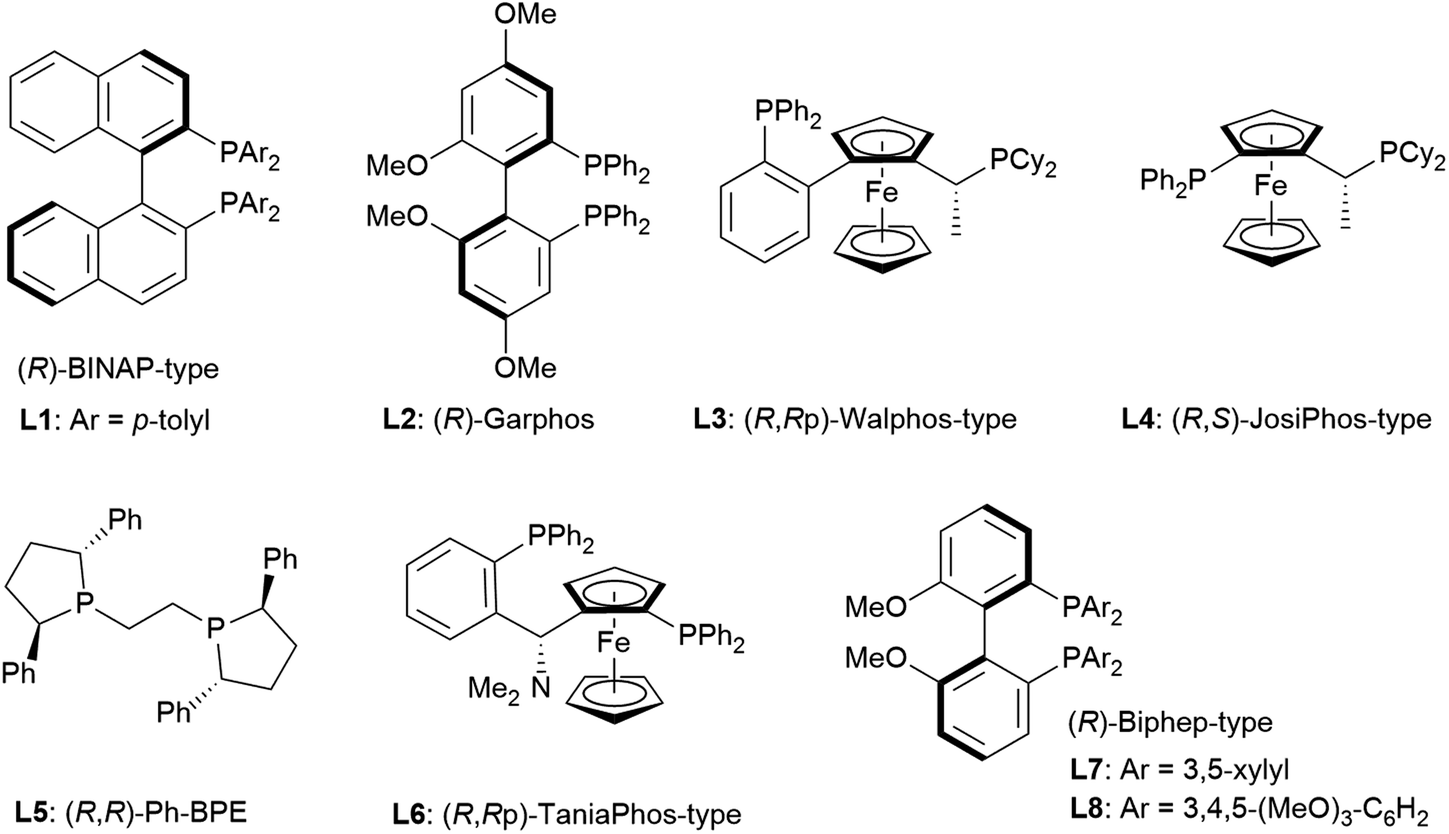
The reaction conditions were first screened using a simple chromone 1a and α-angelica lactone 2a treated with [Cu(CH3CN)]PF6, a cationic copper(I) source, chiral ligand, and Brønsted base (10 mol% each) at a fixed temperature (0 °C) in THF (Table 1). When (R)-tolyl-BINAP (L1) was utilized as a bidentate phosphine ligand with DBU as a base, the inherent syn-selectivity was confirmed (entry 1). Changing the ligand to (R)-Garphos (L2, entry 2) afforded more or less the same result as shown in entry 1 (anti:syn = 1:5.7 in entry 1, and 1:6.7 in entry 2). The enantiomeric excess of the major product (syn-isomer) was at best around 30% (entry 2), and conversion was equally unsatisfactory (only 2 turnovers). The chiral ligands, including (R,Rp)-Walphos- (L3) and (R,S)-JosiPhos-type (L4) ligands, and (R,R)-Ph-BPE (L5) gave the syn-products, exclusively, with a meager chemical yield (entries 3–5). The catalyst system involving a (R,Rp)-Taniaphos-type ligand (L6) resulted in almost no reaction (entry 6). Subsequently, we incorporated Biphep-type biphenyl-based chiral ligands. Our first selection was unfortunately fruitless; the L7 ligand with a 3,5-xylyl group at the phosphorus centers resulted in a similar yield to earlier (entrie 7). Surprisingly, changing the aryl substituents on the phosphorus to 3,4,5-(MeO)3-phenyl (L8) reverted the diastereoselectivity to anti:syn = 2.6:1 with excellent enantioselectivity (>99% ee) of the anti-product13 (89% combined NMR yield of anti- and syn-isomers, entry 8). Decreasing the temperature to −40 °C improved the diastereoselectivity up to 3.5:1 without any loss of enantiopurity (>99% ee), but the chemical yield was reduced to 65% (entry 9). The choice of the Brønsted base was also critical to the reaction outcome. In fact, the use of triethylamine (Et3N) instead of DBU exhibited a detrimental effect on conversion, even at 0 °C (29%, entry 10). Tetramethylguanidine (TMG) slightly increased the yield compared to DBU (72%, entry 11) with the same diastereomeric ratio (3.5:1). Barton's base dramatically improved the conversion to 97%, but the diastereoselectivity was compromised (2.0:1, entry 12). Continuing with Barton's base, the solvent effect was thoroughly investigated. CH2Cl2, toluene, and dimethylformamide (DMF) showed no beneficial effect on anti-selectivity (2.1:1 at best, entries 13–15), whereas 2-Me-THF improved the diastereoselectivity (anti:syn = 3.5:1; entry 16). In each case, the enantiomeric ratio remained at an almost perfect level (>99% ee). The use of DBU in 2-Me-THF improved the diastereoselectivity to 4.1:1, but the conversion was unsatisfactory (60%, entry 17). Increasing the amount of DBU (20 mol%) negatively affected the conversion (40%) and diastereomeric ratio (2.0:1, entry 18).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Ligand | Brønsted base | Solvent | Temperature (°C) | Yielda (%) | dr (anti:syn) |
ee [%] (major isomer) | ee [%] (minor isomer) | |
| a Combined NMR yield of both isomers. b 20 mol% of DBU. c ee of anti-isomer. | |||||||||
| 1 | L1 | DBU | THF | 0 | 22 | 1:5.7 |
93 | −23 | |
| 2 | L2 | DBU | THF | 0 | 17 | 1:6.7 |
90 | −30 | |
| 3 | L3 | DBU | THF | 0 | 7 | 1:>19 |
ND | 5 | |
| 4 | L4 | DBU | THF | 0 | 5 | 1:>19 |
ND | 23 | |
| 5 | L5 | DBU | THF | 0 | 4 | 1:>19 |
ND | −12 | |
| 6 | L6 | DBU | THF | 0 | trace | ND | ND | ND | |
| 7 | L7 | DBU | THF | 0 | 24 | 1:6.7 |
92 | −24 | |
| 8 | L8 | DBU | THF | 0 | 89 | 2.6:1 |
>99 | 33 | |
| 9 | L8 | DBU | THF | −40 | 65 | 3.5:1 |
>99 | 48 | |
| 10 | L8 | Et3N | THF | 0 | 29 | 2.9:1 |
>99 | 33 | |
| 11 | L8 | TMG | THF | −40 | 72 | 3.5:1 |
>99 | 50 | |
| 12 | L8 | Barton's base | THF | −40 | 97 | 2.0:1 |
>99 | 44 | |
| 13 | L8 | Barton's base | CH2Cl2 | −40 | 95 | 1.3:1 |
>99 | 69 | |
| 14 | L8 | Barton's base | Toluene | −40 | 94 | 2.1:1 |
>99 | 80 | |
| 15 | L8 | Barton's base | DMF | −40 | 81 | 1:1 |
>99c | 10 | |
| 16 | L8 | Barton's base | 2-Me-THF | −40 | 95 | 3.5:1 |
>99 | 44 | |
| 17 | L8 | DBU | 2-Me-THF | −40 | 60 | 4.1:1 |
>99 | 48 | |
| 18 | L8 | DBUb | 2-Me-THF | −40 | 40 | 2.0:1 |
>99 | 48 | |
|
|||||||||
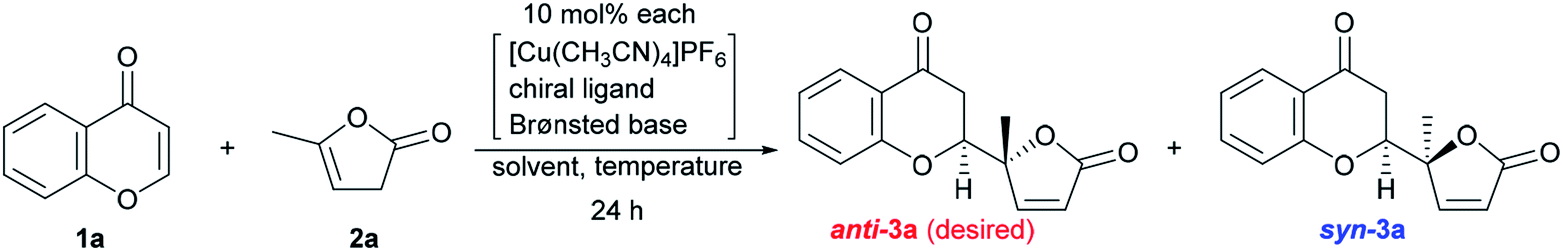
We next undertook a study on substrate generality using the initially optimized reaction conditions shown in Table 1, entry 16, but only disappointing results were obtained. For example, the NMR yield of 6-MeO- (3b), 7-Me- (3h), and 7-MeO-products (3i) was low, around 30−40%, whereas 6-Br- (3f) and 7-AcO-adducts (3j) were afforded in a moderate yield with lower diastereoselectivity (2.3:1, and 2.1:1, respectively). At this stage, we began to fine-tune the reaction conditions using 7-MeO-chromone (1i) as the benchmark substrate with fixed parameters: temperature at −20 °C, reaction duration of 24 h, and 2-Me-THF as the solvent (43% yield, dr of 2.0:1, and >99% ee for major anti-products). Initially, the lithium alkoxide of 2,2,5,7,8-pentamethylchroman-6-ol (4j) was utilized, resulting in 60% NMR yield, 2.4:1 dr, and >99% ee for major anti-isomers (Fig. 3, bottom), which led us to investigate phenolic additives.14
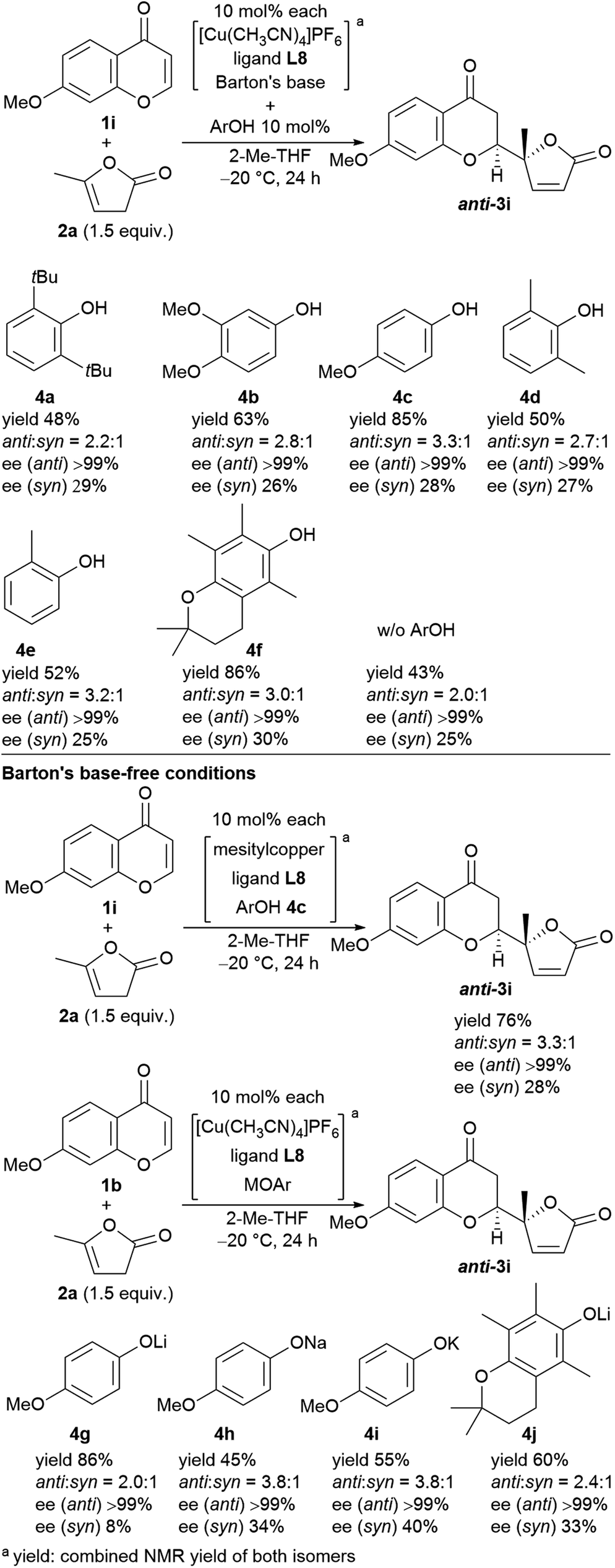 | ||
| Fig. 3 Screening of phenol additives for the direct vinylogous addition of butenolides to chromones and the reactions with Barton's base-free conditions. | ||
Performing the reaction with five phenolic derivatives with varied substitution patterns (4a–4e) in the presence of Barton's base, as shown in Fig. 3, clearly indicate that additive 4c was preferable for achieving high conversion up to 85% with satisfactory diastereoselectivity (3.3:1) and excellent enantioselectivity (>99%). The sterically hindered phenols (4a, 4d, 4e) bearing noncoordinative alkyl groups at the ortho-position yielded slight improvement of the conversion. Moreover, when using 4b as the additive, a moderate conversion was observed. The noticeable effect of a phenolic additive that participates in the catalytic cycle to affect the reaction outcome was also documented in our previous aldol reaction using α-vinyl-appended thioamide and 7-azaindoline amide as substrates.15 In the present reaction, perturbation of the catalyst system by coordination of 4c to the CuI center should be essential for the catalyst turnover from the CuI–alkoxide (Fig. 4a). The deprotonation of butenolide by catalyst L8/CuI-OAr 4c, which was generated from L8/CuI/Barton's base/4c, initiated the catalysis; the CuI dienolate formed a cyclic transition state with the incoming chromone en route to the anti-isomer via the coordination of 4c to the CuI cation and subsequent protonation, followed by regeneration of the catalyst. On the other hand, the catalyst prepared with phenol 4f provided comparable conversion but lower dr as compared with the reaction with 4c. The electron-rich nature of 4f corresponding to the high basicity of the conjugate base (CuI–OAr) is crucial to promote the reaction. This electronic feature compensates for its reduced coordination, caused by the ortho-substituted methyl groups, with CuI cation from the CuI–alkoxide intermediate.
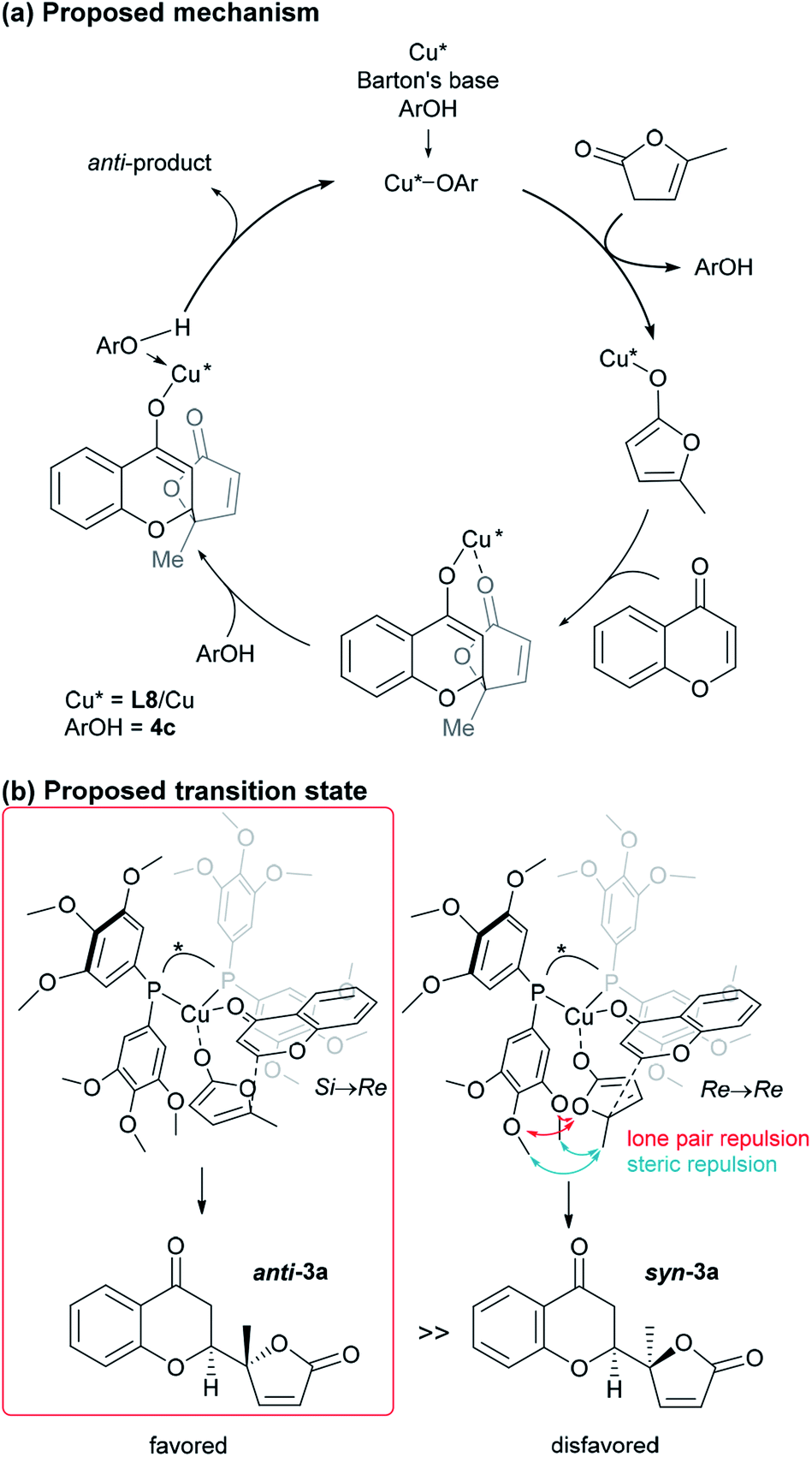 | ||
| Fig. 4 Proposed catalytic cycle and TS of the present vinylogous addition of butenolide to chromone. | ||
Next, we revisited Barton's base-free conditions, but did not obtain superior results (Fig. 3, bottom). A basic copper source, MesCu, decreased the conversion to 76%. Both sodium- (4h) and potassium 4-methoxyphenoxide (4i) exhibited a good diastereoselectivity of 3.8:1, but the turnover was limited. The lithium congener (4g) drove the catalytic cycle efficiently (86% yield), but with only moderate diastereoselectivity (2.0:1). Throughout, the enantioselectivity remained at >99%.
At this point, the factors affecting the reversal of the diastereoselectivity was investigated. As shown in Fig. 2, the topology between chromone and butenolide upon their impending C–C bond formation disfavors the severe repulsion of two ring oxygens in the Si → Re transition state. In principle, this repulsive nonbonding interaction cannot be avoided simply by changing the structure of the substrates, which led Trost to express that this tendency is “inherent”. Intrigued by the change of preferred diastereoselection by L7 and L8, we focused our attention on the substituent effect of the ancillary aromatic rings attached to the two phosphorous atoms of Biphep-type ligands (Table 2). The ligands L9, L10 and L7 exhibited a strong propensity to form syn-isomers (entry 1–3). The proportion of anti-isomers increased dramatically by introducing more sterically demanding substituents; two tert-butyl groups at meta-positions (L11) afforded a 1:1 mixture of anti- and syn-isomers (entry 4). This trend was maintained when another functional group (MeO) was introduced (L12); the anti/syn ratio increased to 1.4:1 (entry 5). We then examined the less sterically demanding ligand L13 having MeO groups at meta-positions which resulted in a similar degree of anti-selectivity (1.7:1, entry 6) to that of entry 5. These results clearly indicate that steric bulkiness cannot be the only factor which govern diastereoselectivity, but electronic profiles are also critical. The trimethoxy substructure in L8 provided the best anti-selectivity (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Yielda [%], dr (anti:syn) |
ee [%] | |
| anti | syn | |||
| a Combined NMR yield of both isomers. | ||||
| 1 | L9: | 57 | 98 | −37 |
| Ar = Ph | 1:9.7 |
|||
| 2 | L10: | 43 | 97 | −21 |
| Ar = p-tol | 1:12 |
|||
| 3 | L7: | 47 | 98 | −29 |
| Ar = 3,5-xylyl | 1:7.5 |
|||
| 4 | L11: | 79 | >99 | 53 |
| Ar = 3,5-tBu2-Ph | 1:1 |
|||
| 5 | L12: | 88 | >99 | 80 |
| Ar = 3,5-tBu2-4-MeO-Ph | 1.4:1 |
|||
| 6 | L13: | 91 | >99 | 35 |
| Ar = 3,5-(MeO)2-Ph | 1.7:1 |
|||
| 7 | L8: | >95 | >99 | 46 |
| Ar = 3,4,5-(MeO)3-Ph | 4.0:1 |
|||
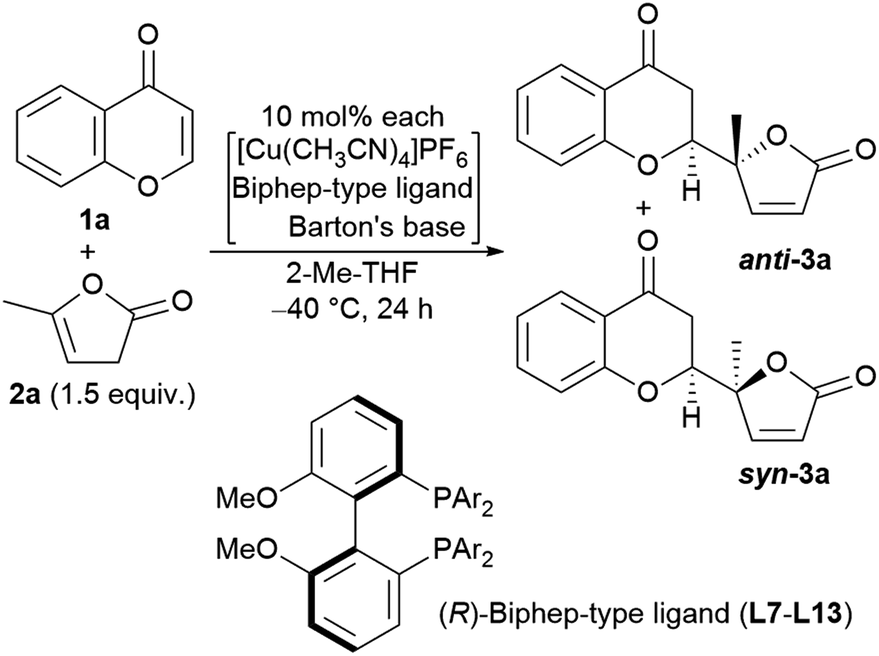
Based on the results described above, we speculate that the present system takes advantage of the steric and electronic repulsion between the substituents at the ancillary benzene rings and the butenolide substrate to tame the “disfavored” transition state (Fig. 4b). Lower anti-selectivity of tert-butly-bearing ligands (L11 and L12) compared to the less bulky ones (L8 and L13) indicated that the steric effect is not determining factor for anti-selectivity. Introduction of the methoxy groups exerted electrostatic repulsion between the oxygens of the butenolide ring and ligand recognized in Re → Re, compared with those from the two ring oxygens in the Si → Re transition state. Thereby, the relative stability of the two transition states was flipped to fulfil the surprising stereochemical switch for the reaction course.16
Encouraged by the results described above, we investigated the scope of the reaction in terms of diversity in both the substrates as shown in Fig. 5. First, all the reactions performed gave >99% ee with one exception: 98% ee (3g). Unsubstituted chromone, 1a, afforded 74% isolated yield of adduct 3a. The 6-substituted products (R1 = MeO (3b), Me (3c), F (3d), Cl (3e), Br (3f), NO2 (3g)) were obtained in fair to good diastereoselectivity (up to 5.2:1), and a reasonable isolated yield of anti-products, from 60% to 73%. The 7-functionalized chromones gave equally good results with a diastereoselectivity up to 3.5:1, and an isolated yield of the desired isomer ranging from 61% to 73% (R1 = Me (3h), MeO (3i), AcO (3j), F (3k), Cl (3l), and Br (3m)). Substitution at the 8-position showed preferential effects for diastereoselectivity: 4.0:1 for 3n (R1 = Me), 6.7:1 for 3o (R1 = Cl) in overall highest isolated yield (82%). The highest diastereoselectivity (7.0:1) was achieved with 8-bromo-substitution (3p). Introduction of an ethyl group instead of the methyl group of the pronucleophile (2b) was also tolerable; with chromone (1a), the diastereoselectivity was 2.8:1 in 70% isolated yield (5a), and with 8-bromochomone, the diastereoselectivity was 3.7:1 in 74% yield (5b, isolated yield for anti-adduct).
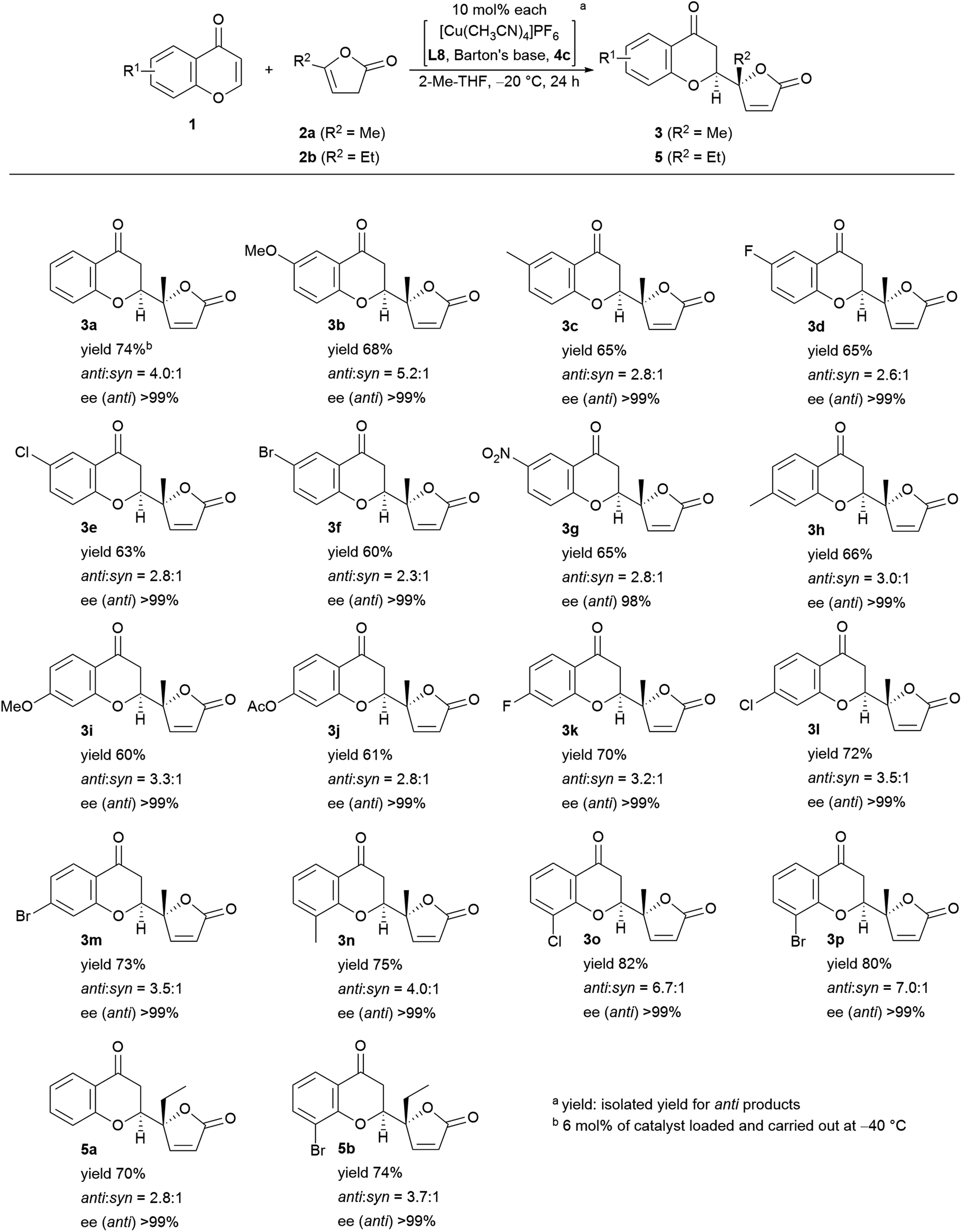 | ||
| Fig. 5 Substrate scope of the vinylogous addition of butenolide to chromone. | ||
This reaction is characterized not only by its broad substrate scope, but also by its scalability. A gram–scale reaction using 1.0 g of chromone 1a and 1.5 equivalent of α-angelica lactone 2a was successfully carried out with even less catalyst (as low as 3 mol%) resulting in a diastereoselectivity of 4.0:1 and an enantioselectivity > 99% at −40 °C (Scheme 1). The only difference in the results from that of the smaller scale reaction with 6 mol% catalyst was an isolated yield of 72%, which was within the experimental fluctuation range.
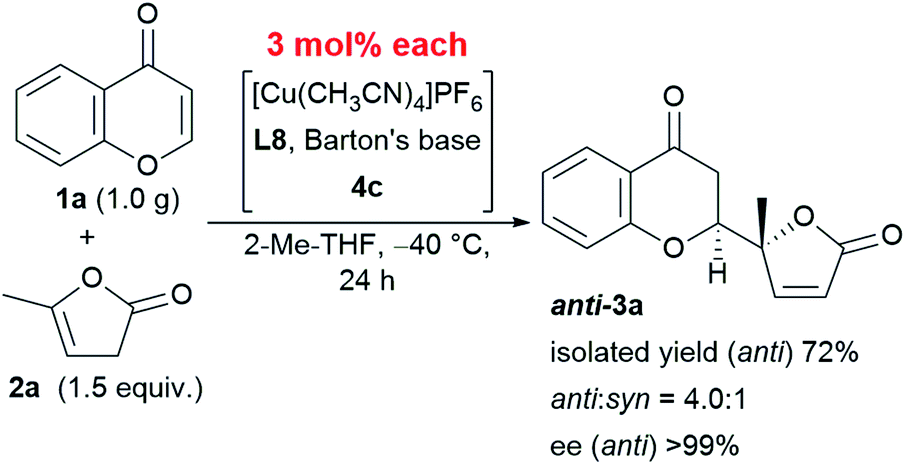 | ||
| Scheme 1 Gram-scale protocol with reduced catalyst loading. | ||
Conclusions
We report an anti-selective catalytic asymmetric vinylogous addition of β,γ-butenolides to chromones. Considering the extremely high intrinsic tendency to produce syn-adducts under a substrate-controlled reaction mechanism, the unexpected selectivity reversal was highly remarkable. The catalyst system developed herein is characterized by (1) tuning of the steric and electronic environment within the stereocontrolling transition state using (R)-3,4,5-(MeO)3-MeOBIPHEP as a chiral ligand to invert diastereoselection, and (2) improvement of the catalyst turnover by a coordinative phenoxide additive to increase the chemical yield. This method will pave the way to the syntheses of natural product-like libraries with high levels of stereoselectivity. Further synthetic studies in this vein will be reported in due course.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J. C. is grateful to JSPS KAKENHI (20K15966). The authors thank Dr Ryuichi Sawa, Ms. Yumiko Kubota, Dr Kiyoko Iijima, and Ms. Yuko Takahashi (BIKAKEN) for the spectroscopic analysis, and Dr Tomoyuki Kimura for X-ray crystallographic analysis.Notes and references
- M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117 CrossRef CAS PubMed.
- For review direct catalytic asymmetric aldol- and Michael-type reaction: Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2017, 47, 4388 RSC; B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS PubMed.
- H. Sasai, T. Suzuki, S. Arai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1992, 114, 4418 CrossRef CAS ; see also H. Sasai, T. Suzuki, N. Itoh, K. Tanaka, T. Date, K. Okamura and M. Shibasaki, J. Am. Chem. Soc., 1993, 115, 10372 CrossRef; H. Sasai, T. Tokunaga, S. Watanabe, T. Suzuki, N. Itoh and M. Shibasaki, J. Org. Chem., 1995, 60, 7388 CrossRef.
- Y. M. A. Yamada, N. Yoshikawa, H. Sasai and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1871 CrossRef CAS ; see also N. Yoshikawa, Y. M. A. Yamada, H. J. Das, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 4168 CrossRef; N. Yoshikawa, N. Kumagai, S. Matsunaga, G. Moll, T. Ohshima, T. Suzuki and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 2466 CrossRef PubMed ; for another pioneering work of direct catalytic asymmetric aldol reaction, see B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003 CrossRef; B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef.
- H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, J. Am. Chem. Soc., 1995, 117, 6194 CrossRef CAS ; see also H. Sasai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1994, 116, 1571 CrossRef.
- T. Arai, H. Sasai, K. Aoe, K. Okamura, T. Date and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1996, 35, 104 CrossRef CAS.
- Y. S. Kim, S. Matsunaga, J. Das, A. Sekine, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 6506 CrossRef CAS.
- N. Kumagai and M. Shibasaki, Synthesis, 2019, 51, 185 CrossRef CAS; K. Weidner, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2014, 53, 6150 CrossRef PubMed; Z. Liu, T. Takeuchi, R. Pluta, F. A. Arteaga, N. Kumagai and M. Shibasaki, Org. Lett., 2017, 19, 710 CrossRef PubMed; H. Noda, F. Amemiya, K. Weidner, N. Kumagai and M. Shibasaki, Chem. Sci., 2017, 8, 3260 RSC; A. Matsuzawa, H. Noda, N. Kumagai and M. Shibasaki, J. Org. Chem., 2018, 82, 8304 CrossRef PubMed; Z. Li, H. Noda, N. Kumagai and M. Shibasaki, Tetrahedron, 2018, 74, 3301 CrossRef.
- W. Zhang, K. Krohn, Zia-Ullal, U. Flörke, G. Pescitelli, L. DiBari, S. Antus, T. Kurtán, J. Rheinheimer, S. Draeger and B. Schulz, Chem. –Eur. J., 2008, 14, 4913 CrossRef CAS PubMed; I. N. Siddiqui, A. Zahoor, H. Hussain, I. Ahmed, V. U. Ahmad, D. Padula, S. Draeger, B. Schulz, K. Meier, M. Steinert, T. Kurtán, U. Flörke, G. Pescitelli and K. Krohn, J. Nat. Prod., 2011, 74, 365 CrossRef PubMed; H. Kikuchi, M. Isobe, M. Sekiya, Y. Abe, T. Hoshikawa, K. Ueda, S. Kurata, Y. Katou and Y. Oshima, Org. Lett., 2011, 13, 4624 CrossRef PubMed.
- B. M. Trost, E. Gnanamani, C. A. Kalnmals, C.-I. J. Hung and J. S. Tracy, J. Am. Chem. Soc., 2019, 141, 1489 CrossRef CAS PubMed.
- Other examples of butenolides as substates for catalytic enantioselective addition reactions by Trost and co-workers, see: B. M. Trost, E. Gnanamani, J. S. Tracy and C. A. Kalnmals, J. Am. Chem. Soc., 2017, 139, 18198 CrossRef CAS PubMed; B. M. Trost, C.-I. Hung and M. J. Scharf, Angew. Chem., Int. Ed. Engl., 2018, 57, 11408 CrossRef PubMed for a recent review, see: B. Mao, M. Fañańas-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef PubMed.
- T. Qin, R. P. Johnson and J. A. Porco Jr, J. Am. Chem. Soc., 2011, 133, 1714 CrossRef CAS PubMed.
- Absolute configuration of anti-3a, and 3g was determined by X-ray crystallographic analysis (see Fig. S1 and 2 in ESI†).
- M. Iwata, R. Yazaki, I.-H. Chen, D. Sureshkumar, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2011, 133, 5554 CrossRef CAS PubMed; M. Iwata, R. Yazaki, Y. Suzuki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 18244 CrossRef PubMed.
- J. Cui, A. Ohtsuki, T. Watanabe, N. Kumagai and M. Shibasaki, Chem. –Eur, J., 2018, 24, 2598 CrossRef CAS PubMed; T. Takeuchi, N. Kumagai and M. Shibasaki, J. Org. Chem., 2018, 83, 5851 CrossRef PubMed.
- The same anti-preference regarding electrostatic effects is expected upon postulation of C-bound form of Cu-butenolide complex (see Fig. S4†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, determination of stereoselectivity, characterization of new compounds, 1H and 13C NMR spectra. CCDC 1988595 and 1988596. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01914c |
| This journal is © The Royal Society of Chemistry 2020 |