
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of the bis(cyclohexenone) core of (−)-lomaiviticin A†
John A.
Rose
a,
Subham
Mahapatra
a,
Xin
Li
a,
Chao
Wang
a,
Lei
Chen
a,
Steven M.
Swick
a and
Seth B.
Herzon
 *ab
*ab
aDepartment of Chemistry, Yale University, New Haven, Connecticut 06520, USA. E-mail: seth.herzon@yale.edu
bDepartment of Pharmacology, Yale School of Medicine, New Haven, Connecticut 06520, USA
First published on 9th July 2020
Abstract
(−)-Lomaiviticin A is a complex C2-symmetric bacterial metabolite comprising two diazotetrahydrobenzo[b]fluorene (diazofluorene) residues and four 2,6-dideoxy glycosides, α-L-oleandrose and N,N-dimethyl-β-L-pyrrolosamine. The two halves of lomaiviticin A are linked by a single carbon–carbon bond oriented syn with respect to the oleandrose residues. While many advances toward the synthesis of lomaiviticin A have been reported, including synthesis of the aglycon, a route to the bis(cyclohexenone) core bearing any of the carbohydrate residues has not been disclosed. Here we describe a short route to a core structure of lomaiviticin A bearing two α-L-oleandrose residues. The synthetic route features a Stille coupling to form the conjoining carbon–carbon bond of the target and a double reductive transposition to establish the correct stereochemistry at this bond. Two synthetic routes were developed to elaborate the reductive transposition product to the bis(cyclohexenone) target. The more efficient pathway features an interrupted Barton vinyl iodide synthesis followed by oxidative elimination of iodide to efficiently establish the enone functionalities in the target. The bis(cyclohexenone) product may find use in a synthesis of lomaiviticin A itself.
Introduction
(−)-Lomaiviticin A (1) is a complex C2-symmetric bacterial metabolite first disclosed in 2001 (Fig. 1).1,2 (−)-Lomaiviticin A (1) possesses four carbohydrate residues, α-L-oleandrose and N,N-dimethyl-β-L-pyrrolosamine, and two diazotetrahydrobenzo[b]fluorene (diazofluorene) residues.3–9 The two halves of (−)-lomaiviticin A (1) are linked by a single carbon–carbon bond. Construction of this bond with the correct stereochemical configuration (syn to the tertiary carbon–oxygen bonds, see blue bonds in 1) constitutes a significant barrier to chemical synthesis.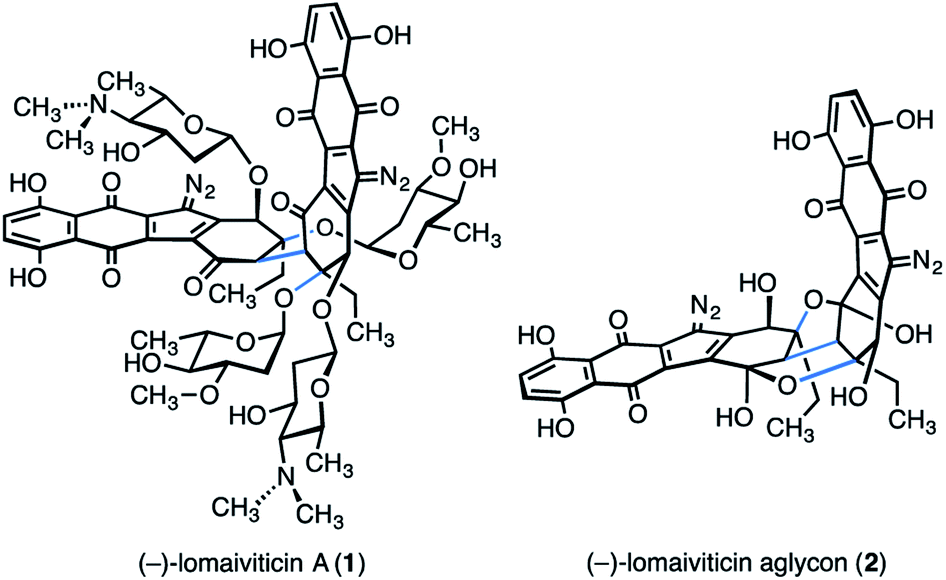 | ||
| Fig. 1 Structures of (−)-lomaiviticin A (1) and (−)-lomaiviticin aglycon (2). | ||
(−)-Lomaiviticin A (1) is cytotoxic with half-maximal inhibitory potencies in the nanomolar–picomolar range against a variety of cultured human cancer cell lines. The cytotoxicity of (−)-lomaiviticin A (1) arises from the induction of double-strand breaks in DNA. Mechanistic studies established that strand cleavage proceeds by the sequential generation of carbon-centered radical intermediates at each diazo carbon, followed by hydrogen atom abstraction from the deoxyribose chain and degradation.9–12
We previously reported a synthesis of (−)-lomaiviticin aglycon (2) by the late stage oxidative coupling of two monomeric diazofluorenes.13 While this strategy was amenable to construction of the aglycon form of the natural product, it has not yet provided a viable means to synthesize (−)-lomaiviticin A (1) itself. Accordingly, we have evaluated strategies wherein the bridging carbon–carbon bond is formed earlier in the synthetic route. Several other laboratories have pursued a similar line of inquiry, and notable advances have been recorded.14–28 However, a synthesis of the bis(cyclohexenone) core of (−)-lomaiviticin A (1) with any of the carbohydrate residues in place has not been disclosed, to our knowledge.
Results and discussion
We initially targeted the bis(oleandrose) derivative 3 (Scheme 1). It was envisioned that 3 could be elaborated to the target by introduction of the aminosugar residues, followed by annulation and global deprotection. We considered installing the aminosugar residues early in the route. However, protected derivatives bearing a carbamate or amide exist as mixtures of E/Z isomers, resulting in mixtures of four possible diastereomers in dimeric structures. In exploratory studies we were unable to identify NMR conditions (solvent, temperature) that provided intelligible spectra of these intermediates.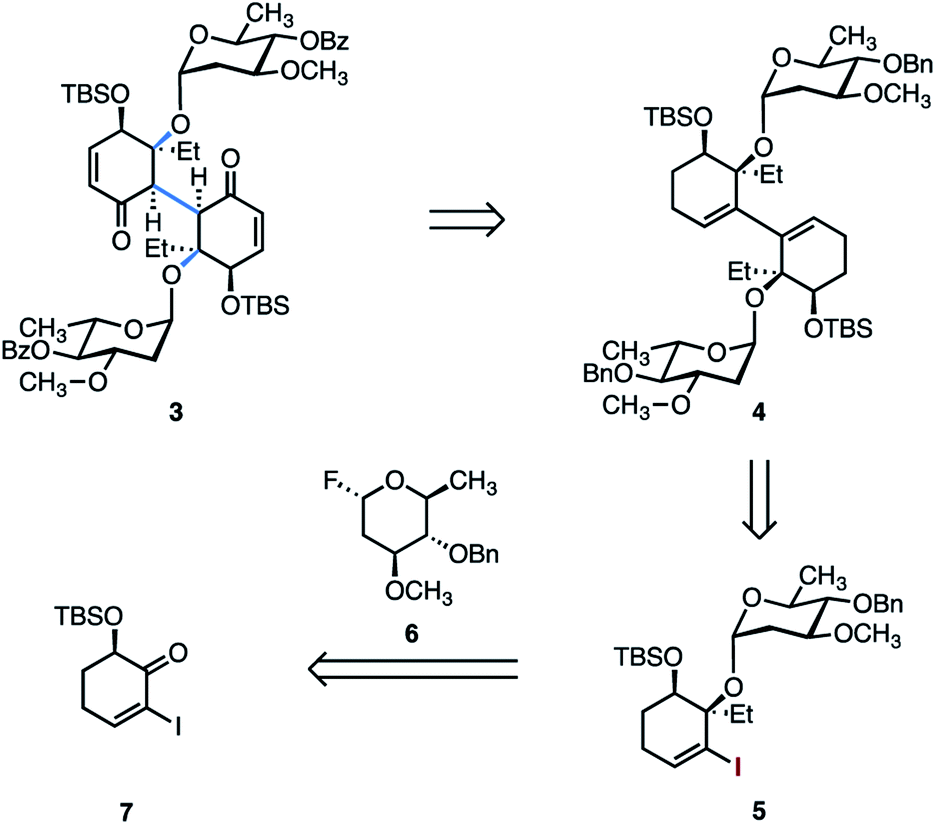 | ||
| Scheme 1 Retrosynthetic analysis of the bis(oleandrose) derivative 3. | ||
Several strategies to access 3 from the 1,3-diene 4 were envisioned. The retrosynthetic conversion of 3 to 4 allows the bridging carbon–carbon bond to be formed by metal-catalyzed formal dimerization of the vinyl iodide 5. The latter was prepared from the oleandrose donor 6 and the known α-silyloxyketone 7.29
Several syntheses of oleandrose have been reported.18,30–38 We developed a new and practical chiral pool approach. Beginning with the commercial reagent 3,4-di-O-acetyl-6-deoxy-L-glucal (8; Scheme 2), site-selective C4 deacetylation and benzylation was achieved under biphasic conditions (benzyl bromide, aqueous sodium hydroxide, dichloromethane).39 Removal of the remaining acetate (potassium carbonate, methanol), provided the differentiated glycal 9 (74%, two steps). Methylation of the C3 alcohol (sodium hydride, methyl iodide) followed by hydroacetoxylation of the enol ether (triphenylphosphine hydrogen bromide, acetic acid)40 formed the glycosyl acetate 10 as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of α- and β-diastereomers (81% over two steps). Finally, fluorodeacetoxylation (hydrogen fluoride–pyridine complex) generated the glycosyl fluoride 6 (>10:1 α:β). The electron-rich glycosyl fluoride 6 was unstable toward purification. Consequently, the unpurified product was used directly in the subsequent glycosylation step.
:1 mixture of α- and β-diastereomers (81% over two steps). Finally, fluorodeacetoxylation (hydrogen fluoride–pyridine complex) generated the glycosyl fluoride 6 (>10:1 α:β). The electron-rich glycosyl fluoride 6 was unstable toward purification. Consequently, the unpurified product was used directly in the subsequent glycosylation step.
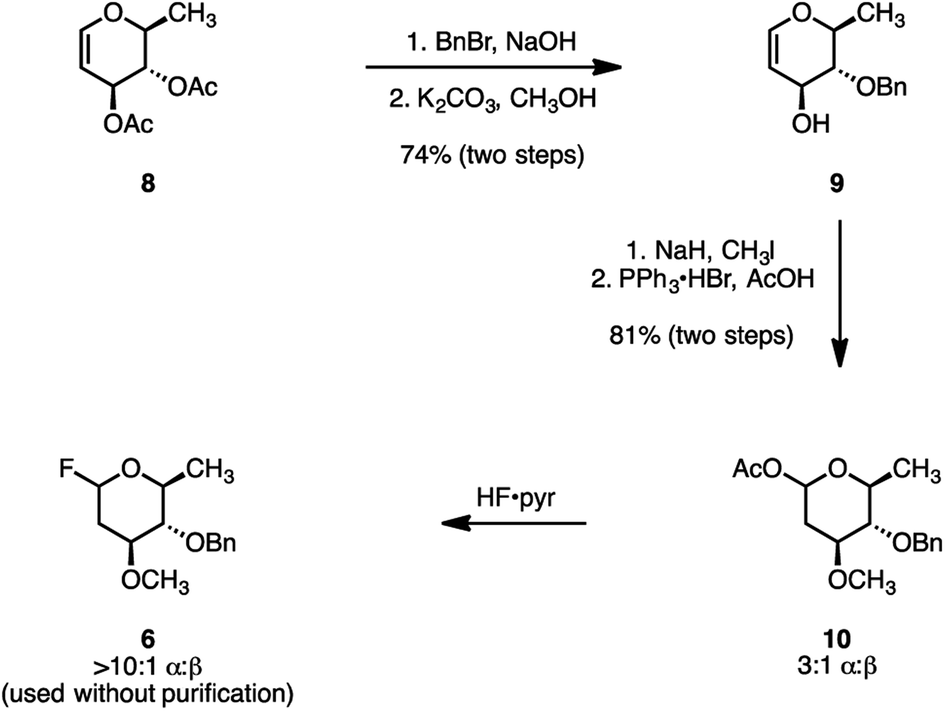 | ||
| Scheme 2 Synthesis of the fluoride donor 6. | ||
The ketone 7 was obtained in four steps, 31% yield, and 99% ee from cyclohex-2-ene-1-one according to a published procedure (Scheme 3).29 We envisioned 1,2-addition of a two-carbon nucleophile to the ketone within 7 to construct the tertiary alcohol of the target. However, consistent with the studies by McIntosh and co-workers,41 significant optimization was required to obtain the cis-1,2-addition product in high yield and stereoselectivity. After much empirical experimentation, we found that the addition of ethylmagnesium bromide to solutions of the ketone 7 in toluene at −78 °C provided the tertiary alcohol 11 in 70% yield and with 4:1 diastereoselectivity in favor of the desired product 11. The relative stereochemistry of 11 was determined by NOE and X-ray crystallographic analysis (vide infra).
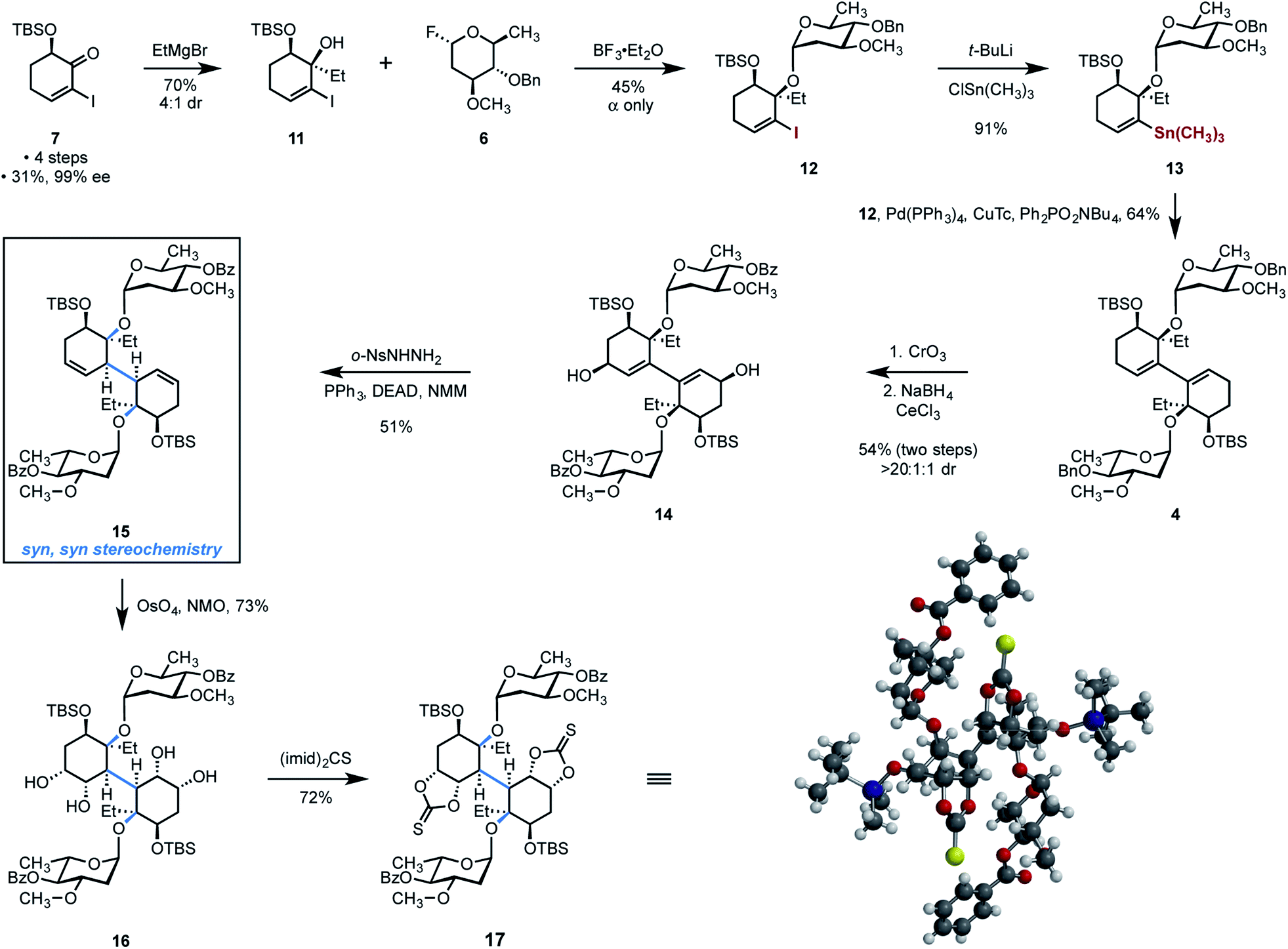 | ||
| Scheme 3 Synthesis of the bis(cyclohexene) 15 and the thiocarbonate 17. | ||
The introduction of the oleandrose residue to the tertiary alcohol in 11 was challenging owing to the steric hindrance created by the silyl ether and vinyl iodide substituents, and the electron-rich nature of the donor 6, which facilitates rapid ionization leading to decomposition. We found that the highest yields of product were obtained upon activation of the donor (nominally 1 equiv.) with boron trifluoride–diethyl etherate complex in the presence of 2.0 equiv. of the acceptor 11 at −25 °C, in tetrahydrofuran as solvent. Under these conditions, the glycoside 12 was obtained in 45% yield as a single detectable α-anomer (1H NMR analysis). We employed an excess of the acceptor 11 to maximize conversion of the donor 6 to the product 12. The majority of unreacted acceptor 11 (>1 equiv.) could be readily-recovered by flash-column chromatography. When the diol 11 was used as limiting reagent, excess donor 6 could not be recovered due to its decomposition under the reaction conditions.
A Stille cross-coupling was uniquely successful among many dimerization conditions evaluated. Accordingly, the vinyl stannane 13 was prepared in 91% yield by metal–halogen exchange (tert-butyllithium) followed by the addition of trimethyltin chloride. Heating a mixture of 12 (1.12 equiv.) and 13 (1 equiv.) in the presence of tetrakis(triphenylphosphine)palladium (0) (25 mol%), copper (I) thiophene-2-carboxylate (1.5 equiv.), and tetra-n-butylammonium diphenyl phosphite42 provided the 1,3-diene 4 in 64% yield on gram scales. Efforts to achieve a more direct dimerization, for example, by the reductive dimerization of 12, were unsuccessful.
With the bridging carbon–carbon bond in place, our attention turned toward diastereoselective reduction or hydrofunctionalization of the diene in order to establish the correct syn,syn stereochemistry at the conjoining bond in the target. Ultimately, a reliable and scalable three-step sequence that achieves the formal stereocontrolled isomerization of 4 to 15 was developed (Scheme 3). First the allylic and benzylic positions in 4 were exhaustively oxidized by treatment with excess chromium trioxide (not shown). Diastereoselective Luche reduction of the oxidation product provided the bis(allylic alcohol) 14 (54%, two steps) as a single detectable diastereomer (1H NMR analysis). The stereochemistry of the reduction was determined by NOE analysis and subsequently confirmed by X-ray crystallography (vide infra). Finally, two-fold reductive transposition43 was achieved by treatment with ortho-nitrobenzenesulfonyl hydrazide (NBSH), triphenylphosphine, and diethylazido dicarboxylate (DEAD) in N-methyl morpholine (NMM) as solvent, to provide the bis(cyclohexene) 15 in 51% yield. The successful synthesis of 15 represents, to our knowledge, the first instance in which the bicyclohexyl core of (−)-lomaiviticin (1) has been constructed with any of the carbohydrate residues in place. Because the Luche reduction generates a single detectable diastereomer, and the reductive transposition step is stereospecific, the product 15 is formed free of any diastereoisomers. The sequence 4 → 15 constitutes a formal stereoselective isomerization of the 1,3-diene within 4. Efforts to achieve the direct hydrofunctionalization of 4 or 14, for example, by hydroboration, hydrogenation, or metal-catalyzed hydrogen atom transfer,44 were unsuccessful. We note parenthetically that we have been unable to identify conditions to introduce the oleandrose residues following dimerization, which highlights the high degree of steric hindrance surrounding the tertiary alcohols in the dimerized products.
Two-fold diastereoselective dihydroxylation of the reductive transposition product 15 provided the tetraol 16 in 73% yield and as a single detectable diastereomer (1H NMR analysis). The bis(thiocarbonate) 17 was obtained in 72% yield by heating solutions of the tetraol 16 and thiocarbonyl bis(imidazole) in toluene. Recrystallization of 17 from dichloromethane–hexanes provided single crystals suitable for X-ray analysis, which rigorously established the relative configuration of the product (see inset, Scheme 3).
With a strategy for the stereocontrolled construction of the bridging carbon–carbon bond of the target in place, our attention shifted toward elaboration of the diene 15 or the tetraol 16 to the bis(cyclohexenone) 3. We initially developed the sequence shown in Scheme 4. Dihydroxylation of 15 (as above) followed by site-selective methanesulfonylation of the less-hindered secondary alcohols formed the bis(mesylate) 18 (59%, two steps). Exposure of 18 to the Dess–Martin periodinane (DMP)45 resulted in oxidation of one of the two hydroxyl substituents. The resulting hydroxy ketone intermediate (not shown) underwent in situ hemiketalization to provide the cyclic hemiketal 19 (90%). Attempts to induce two-fold oxidation of 18 under a variety of conditions were unsuccessful. Accordingly, we evaluated methods to advance the ketal 19. We found that warming solutions of 19, sodium methanesulfonate (50 equiv.), and lithium chloride (25 equiv.) in hexamethyl phosphorotriamide (HMPA) to 85 °C resulted in elimination of one of the methanesulfonate substituents and ring-opening to provide the C1-symmetric diketone 20 in 40% yield (80% based on recovered 19). This reaction may proceed by ionization of the mesylate distal to the hemiketal, 1,2-hydride shift, and ring-opening, as shown. Addition of samarium diiodide to a solution of the ring-opened product 20 in methanol–tetrahydrofuran provided the cyclic enol ether 21 in 80% yield. We hypothesize that this transformation is initiated by site-selective single-electron reduction of the more electron-deficient α-methanesulfonyloxy ketone, elimination of the methanesulfonyloxy substituent, and addition of the resulting enolate to the remaining ketone function. We found that oxidation of the cyclic enol ether 21 under Saegusa–Larock46,47 conditions proceeded smoothly to generate the keto enone 22 (95%). Attempts to further oxidize 22 directly were unsuccessful. However, a second cyclic enol ether, 23, could be obtained in 88% yield by treatment of 22 with trimethylsilyl trifluoromethanesulfonate and 2,6-lutidine. While palladium-mediated oxidation of 23 was unsuccessful, treatment with benzeneselenyl bromide resulted in ring-opening selenylation; oxidation and in situ elimination provided the bis(enone) 3 in 82% yield from 23.
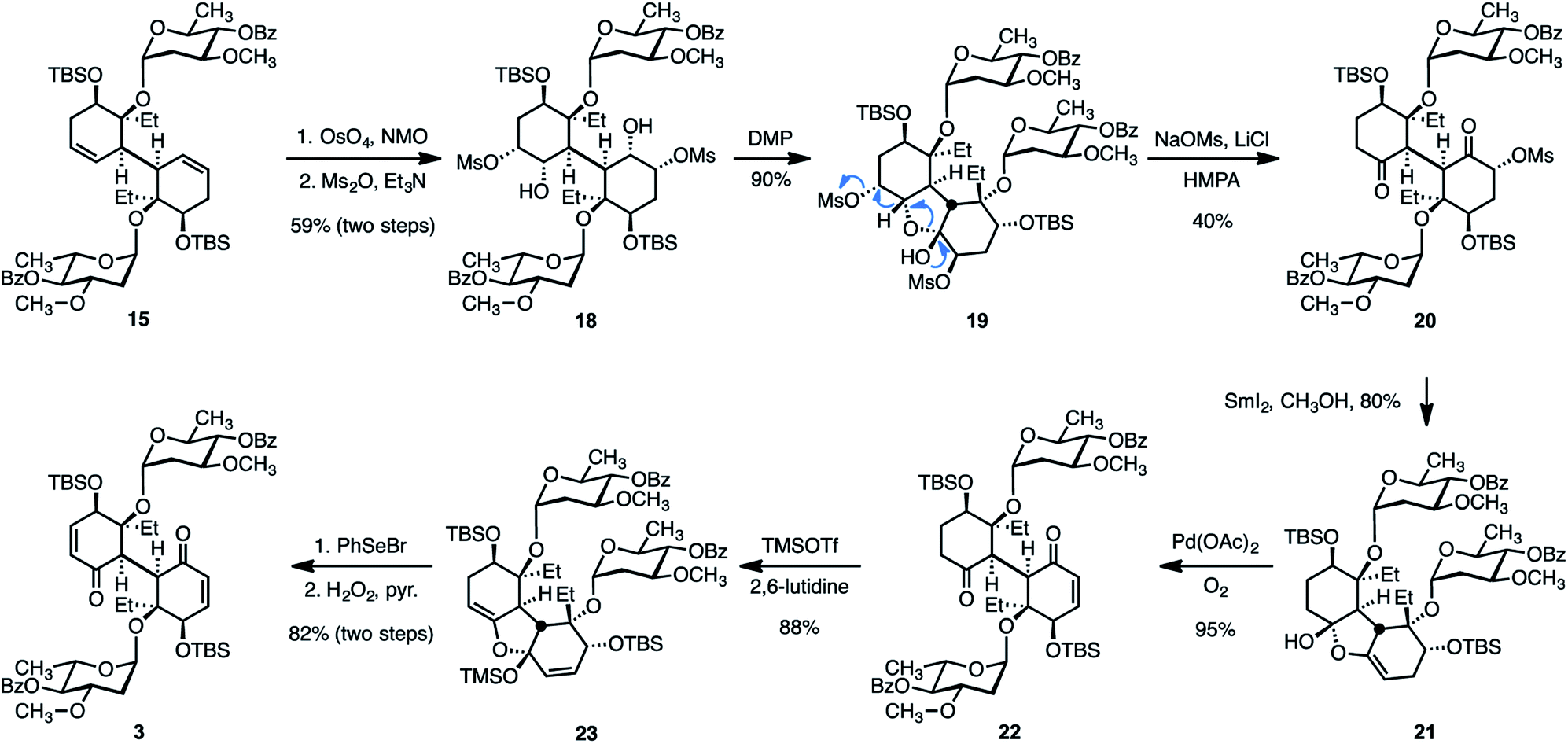 | ||
| Scheme 4 First approach to the synthesis of the bis(cyclohexenone) 3. | ||
The rather lengthy pathway required for the conversion of 15 to the bis(enone) 3 shown in Scheme 4 was a consequence of the cross-reactivity observed between the two cyclohexyl rings, which resulted in differentiation of the pairs of functional groups. In light of these experiences, we pursued more direct pathways that would minimize intramolecular cyclizations and the generation of C1-symmetric intermediates. Toward this end, the bis(α-hydroxyketone) 24 was synthesized by site-selective oxidation of the less-hindered secondary alcohols in 16 (dimethyldioxirane (DMDO), 85%, Scheme 5).48 Treatment of 23 with excess hydrazine provided the bis(hydrazone) 25. The bis(hydrazone) 25 was unstable toward purification and was oxidized directly with molecular iodine. Under these conditions, the bis(α-iodoketone) 30 was obtained in 50% yield and as a single detectable diastereomer (1H NMR analysis) from the diketone 24. While the mechanism of formation of 30 from the bis(hydrazone) 25 has not been rigorously established, we propose that the substrate is first oxidized to the α-iododiazonium salt 26. 1,2-Hydride shift with elimination of dinitrogen, essentially an interrupted Barton vinyl iodide synthesis,49 would generate the α-iodoketone 27. Based on the reactivity of the intermediates shown in Scheme 4, we speculate that 27 undergoes intramolecular cyclization to generate the hemiketal 28. Further oxidation of 28 would provide a second α-iododiazonium salt, 29. 1,2-Hydride shift with elimination of dinitrogen and ring-opening would then generate the observed product 30. Finally, oxidative elimination of the iodide substituents (DMDO)50 provided the target 3.
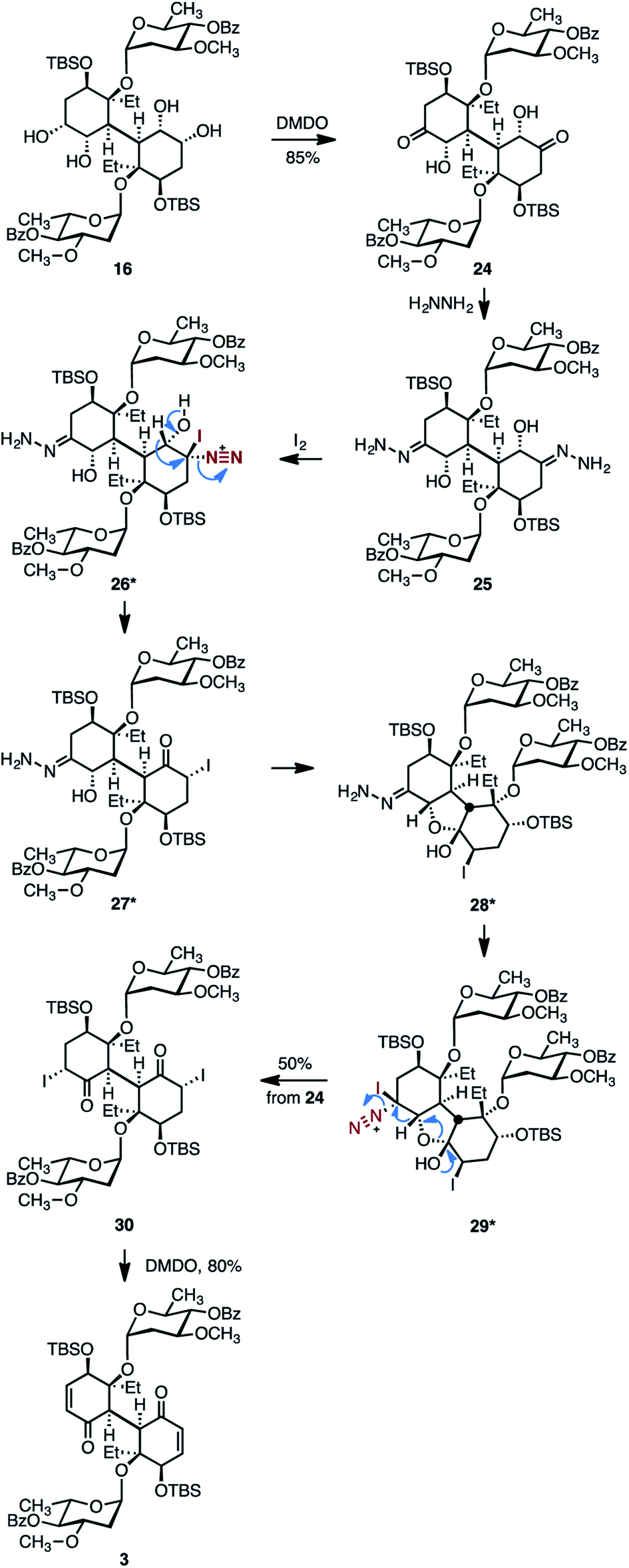 | ||
| Scheme 5 Improved route to the bis(cyclohexenone) 3. Asterisks denote postulated intermediates in the conversion of 25 to 30. | ||
Conclusions
In summary, we have described a short synthetic route to the bis(enone) 3, a potential precursor to (−)-lomaiviticin A (1). The conjoining bond of the target was constructed by a Stille cross-coupling that unites the two cyclohexyl rings, followed by a highly diastereoselective, double reductive transposition sequence to establish the correct relative stereochemistry. The conversion of the reductive transposition product 15 to 3 shown in Scheme 4 underscores the steric congestion of the bicyclic system, which leads to facile transannular reactivity. The improved route to 3 shown in Scheme 5 circumvents many of these reactivity issues and proceeds by iterative increases in oxidation state (15 → 16 → 24 → 30 → 3). It features an unexpected interrupted Barton vinyl iodide synthesis to introduce the ketones of the target. Future studies will focus on elaborating 3 to lomaiviticin itself.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Institutes of Health (R35GM131913) and the National Science Foundation (Predoctoral Fellowship to J. A. R.) is gratefully acknowledged.Notes and references
- H. He, W. D. Ding, V. S. Bernan, A. D. Richardson, C. M. Ireland, M. Greenstein, G. A. Ellestad and G. T. Carter, J. Am. Chem. Soc., 2001, 123, 5362 CrossRef CAS PubMed.
- For determination of the absolute stereochemistry of the aglycon and carbohydrate residues, see: C. M. Woo, N. E. Beizer, J. E. Janso and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 15285 CrossRef CAS PubMed.
- S. J. Gould, Chem. Rev., 1997, 97, 2499 CrossRef CAS PubMed.
- J. Marco-Contelles and M. T. Molina, Curr. Org. Chem., 2003, 7, 1433 CrossRef CAS.
- D. P. Arya, Top. Heterocycl. Chem., 2006, 2, 129 CAS.
- C. C. Nawrat and C. J. Moody, Nat. Prod. Rep., 2011, 28, 1426 RSC.
- S. B. Herzon, in Total Synthesis of Natural Products. At the Frontiers of Organic Chemistry, ed. J. J. Li and E. J. Corey, Springer-Verlag, Berlin Heidelberg, 2012, p. 39 Search PubMed.
- S. B. Herzon and C. M. Woo, Nat. Prod. Rep., 2012, 29, 87 RSC.
- S. B. Herzon, Acc. Chem. Res., 2017, 50, 2577 CrossRef CAS PubMed.
- L. C. Colis, C. M. Woo, D. C. Hegan, Z. Li, P. M. Glazer and S. B. Herzon, Nat. Chem., 2014, 6, 504 CrossRef CAS PubMed.
- C. M. Woo, Z. Li, E. K. Paulson and S. B. Herzon, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2851 CrossRef CAS PubMed.
- M. Xue and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 15559 CrossRef CAS.
- S. B. Herzon, L. Lu, C. M. Woo and S. L. Gholap, J. Am. Chem. Soc., 2011, 133, 7260 CrossRef CAS PubMed.
- K. C. Nicolaou, R. M. Denton, A. Lenzen, D. J. Edmonds, A. Li, R. R. Milburn and S. T. Harrison, Angew. Chem., Int. Ed., 2006, 45, 2076 CrossRef CAS PubMed.
- E. S. Krygowski, K. Murphy-Benenato and M. D. Shair, Angew. Chem., Int. Ed., 2008, 47, 1680 CrossRef CAS PubMed.
- W. Zhang, A. Baranczak and G. A. Sulikowski, Org. Lett., 2008, 10, 1939 CrossRef CAS PubMed.
- W. J. Morris and M. D. Shair, Org. Lett., 2008, 11, 9 CrossRef PubMed.
- S. L. Gholap, C. M. Woo, P. C. Ravikumar and S. B. Herzon, Org. Lett., 2009, 11, 4322 CrossRef CAS PubMed.
- K. C. Nicolaou, A. L. Nold and H. Li, Angew. Chem., Int. Ed., 2009, 48, 5860 CrossRef CAS.
- H. G. Lee, J. Y. Ahn, A. S. Lee and M. D. Shair, Chem.–Eur. J., 2010, 16, 13058 CrossRef CAS.
- W. J. Morris and M. D. Shair, Tetrahedron Lett., 2010, 51, 4310 CrossRef CAS PubMed.
- S. S. Scully and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 9722 CrossRef CAS PubMed.
- A. Baranczak and G. A. Sulikowski, Org. Lett., 2012, 14, 1027 CrossRef CAS.
- S. S. Scully and J. A. Porco, Org. Lett., 2012, 14, 2646 CrossRef CAS PubMed.
- C. M. Woo, S. L. Gholap, L. Lu, M. Kaneko, Z. Li, P. C. Ravikumar and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 17262 CrossRef CAS PubMed.
- K. S. Feldman and B. R. Selfridge, Org. Lett., 2012, 14, 5484 CrossRef CAS PubMed.
- K. S. Feldman and B. R. Selfridge, J. Org. Chem., 2013, 78, 4499 CrossRef CAS PubMed.
- A. S. Lee and M. D. Shair, Org. Lett., 2013, 15, 2390 CrossRef CAS PubMed.
- H. Koizumi, S. Yokoshima and T. Fukuyama, Chem.–Asian J., 2010, 5, 2192 CrossRef CAS PubMed.
- F. Blindenbacher and T. Reichstein, Helv. Chim. Acta, 1948, 31, 2061 CrossRef CAS PubMed.
- G. Berti, G. Catelani, F. Colonna and L. Monti, Tetrahedron, 1982, 38, 3067 CrossRef CAS.
- P. G. M. Wuts and S. S. Bigelow, J. Org. Chem., 1983, 48, 3489 CrossRef CAS.
- D. Ravi, V. R. Kulkarni and H. B. Mereyala, Tetrahedron Lett., 1989, 30, 4287 CrossRef CAS.
- R. L. Tolman and L. H. Peterson, Carbohydr. Res., 1989, 189, 113 CrossRef CAS.
- M. J. Ford and S. V. Ley, Synlett, 1990, 771 CrossRef CAS.
- M. W. Bredenkamp, C. W. Holzapfel and F. Toerien, Synth. Commun., 1992, 22, 2459 CrossRef CAS.
- X. Y. Zhao, M. Ono, H. Akita and Y. M. Chi, Chin. Chem. Lett., 2006, 17, 730 CAS.
- M. Brasholz and H.-U. Reissig, Eur. J. Org. Chem., 2009, 3595 CrossRef CAS.
- J. Wilmot, J. Herrick, D. M. Jones, K. G. Meyer, C. Yao, A. Arnold, J. E. Delorbe, R. L. K. C. Lalonde, J. Renga, J. F. Daeuble Sr, F. Li, K. Bravo-Altamirano, Preparation of macrocyclic picolinamides as fungicide, World Patent WO2016109290A1, 2016.
- V. Bolitt, C. Mioskowski, S. G. Lee and J. R. Falck, J. Org. Chem., 1990, 55, 5812 CrossRef CAS.
- H. A. Lindsay, C. L. Salisbury, W. Cordes and M. C. McIntosh, Org. Lett., 2001, 3, 4007 CrossRef CAS PubMed.
- A. Fürstner, L. C. Bouchez, J.-A. Funel, V. Liepins, F.-H. Porée, R. Gilmour, F. Beaufils, D. Laurich and M. Tamiya, Angew. Chem., Int. Ed. Engl., 2007, 46, 9265 CrossRef PubMed.
- A. G. Myers and B. Zheng, Tetrahedron Lett., 1996, 37, 4841 CrossRef CAS.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS.
- Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011 CrossRef CAS.
- R. C. Larock, T. R. Hightower, G. A. Kraus, P. Hahn and D. Zheng, Tetrahedron Lett., 1995, 36, 2423 CrossRef CAS.
- L. D'Accolti, A. Detomaso, C. Fusco, A. Rosa and R. Curci, J. Org. Chem., 1993, 58, 3600 CrossRef.
- D. H. R. Barton, G. Bashiardes and J.-L. Fourrey, Tetrahedron Lett., 1983, 24, 1605 CrossRef CAS.
- C. A. Horiuchi, S.-J. Ji, M. Matsushita and W. Chai, Synthesis, 2004, 202 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2001251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02770g |
| This journal is © The Royal Society of Chemistry 2020 |