
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new soft-matter material with old chemistry: Passerini multicomponent polymerization-induced assembly of AIE-active double-helical polymers with rapid visible-light degradability†
Jupen
Liu‡
a,
Zhonglong
Luo‡
b,
Le
Yu
a,
Ping
Zhang
a,
Hongqiu
Wei
a and
You
Yu
 *a
*a
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an, China. E-mail: yuyou@nwu.edu.cn
bSchool of Mechanical Engineering, Anhui University of Technology, Ma'anshan, Anhui, China
First published on 15th July 2020
Abstract
Mimicking the superstructures and functions of natural chiral materials is beneficial to understand specific biological activities in living organisms and broaden applications in the fields of chemistry and materials sciences. However, it is still a great challenge to construct water-soluble, double-helical polymers with multiple responsiveness. Herein, we report for the first time a straightforward, general strategy to address this issue by taking advantage of Passerini multicomponent polymerization-induced assembly (PMPIA). The polymerization-induced generation of supramolecular interactions in chiral α-acyloxy amides drives the assembly of polymers and improves their stability in various solvents. This double-helical polymer is sensitive to metal ions, temperature, pH, and solvents, making both the superstructure and the AIE effect reversibly adjustable. Meanwhile, the hydrogen-bonding-assisted cyclization of photolabile α-acyloxy amides accelerates the degradation of helical polymers under visible-light irradiation. It is anticipated that this novel PMPIA strategy opens new horizons to inspire the design of advanced chiral/helical polymers with multiple functions.
Introduction
Helical structures, which are found ubiquitously in nature, play crucial roles in biological activities because such structures guide extraordinary functions of living organisms. Soft-matter materials with water-soluble helices have attracted much interest because most important biological events occur in aqueous media.1,2 For example, hierarchical enzymes can serve as chiral biocatalysts, and they enable achieving the synthesis of functional materials under mild conditions.3 Double-helical DNAs precisely store and replicate genetic information during cell proliferation. Inspired by their unique features, many artificial helical polymers have been designed to mimic the biological superstructures and functions of natural chiral materials.4–8 This has broadened their potential applications in fields such as asymmetric catalysis, chiral recognition, and nonlinear optical materials. Furthermore, the introduction of stimuli-sensitive components, e.g., pH-, concentration-, thermal- and light-sensitive components, into helical architectures, enables the preparation of stimuli-responsive helical polymers, which have important applications in multifunctional smart materials.9–12 In principle, soft materials with multiple helices, e.g., double-helical polymers, can be regarded as assemblies of single strands bound together via noncovalent interactions.13,14 However, compared with polymers that have single-helical conformations, double-helical polymers, especially those with high solubilities in aqueous solutions, are rare because of the limited availability of building blocks, tedious fabrication processes, and insufficient regulation of supramolecular interactions between the two counterparts in water. A one-step general strategy for designing water-soluble, double-helical, multiresponsive polymers is, therefore, a synthetically intriguing target in this field.Multicomponent polymerization is a straightforward and facile method for one-pot construction of complex soft materials that can afford well-defined, structurally diverse polymers.15–17 Polymers with versatile structures, functions, and responsiveness can be easily prepared by inserting specific units into the polymer backbones.18–22 Typically, the metal-free Passerini three-component polymerization of aldehydes, isocyanides, and acids is generally used for the preparation of polymers with predictable topological structures.17 However, to the best of our knowledge, there has been no report on the use of key chiral α-acyloxy amides [–NH–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–CR–O–] from achiral monomers for the construction of chiral/helical polymers. The results of previous studies indicate that the resultant chiral amide can form one hydrogen bond with another electron donor.12,23–25 This drives polymer assembly and improves the stability of chiral/helical structures in various solvents. Also, the generation of supramolecular interactions and self-assembly behavior during polymerization gives more flexibility to regulate polymer conformations for constructing chiral/helical soft-matter materials compared with directly assembling polymers in solutions.
O)–CR–O–] from achiral monomers for the construction of chiral/helical polymers. The results of previous studies indicate that the resultant chiral amide can form one hydrogen bond with another electron donor.12,23–25 This drives polymer assembly and improves the stability of chiral/helical structures in various solvents. Also, the generation of supramolecular interactions and self-assembly behavior during polymerization gives more flexibility to regulate polymer conformations for constructing chiral/helical soft-matter materials compared with directly assembling polymers in solutions.
Inspired by the advantages of Passerini three-component polymerization, we developed a novel one-pot strategy, namely Passerini multicomponent polymerization-induced assembly (PMPIA), for designing a stimuli-responsive, water-soluble, aggregate-induced emission (AIE)-active, double-helical polymer (WADP). This polymer comprises short, soft (–CH2–CH2–O–)3 segments and rigid pyridine units, which are achiral and alternately linked together by chiral α-acyloxy amides. Cooperative hydrogen-bonding and aromatic interactions drive the assembly of the double-helical structures and make them stable in aqueous and polar organic solutions, and endow the polymers with AIE and multiple responsiveness. The hydrogen-bonding-assisted cyclization of photolabile α-acyloxy amides in polymer backbones accelerates the degradation of helical polymers under visible-light irradiation. We believe that the PMPIA strategy will enable the construction of advanced multifunctional, chiral, soft-matter materials.
Results and discussion
As shown in Fig. 1, water-soluble, AIE-active, double-helical polymers were prepared via the PMPIA strategy from three achiral monomers, i.e., two difunctionalized poly(ethylene glycol) (PEG) oligomers and 4-pyridinecarboxaldehyde (PyCHO). These reactants were chosen for the following reasons. (i) PEG monomers are soft, and therefore give highly flexible polymers, enabling regulation of their chiral conformations. (ii) PEG segments, which are soluble in various solvents, endow polymers with excellent water solubility. Different solubilities in typical solvents facilitate precise adjustment of the aggregation of nearby pyridines; therefore helical polymers with controllable optical properties can be obtained.26,27 (iii) The 4-pyridinecarboxaldehyde monomer acts as the electron donor for hydrogen bonding and an H+/OH− acceptor and also provides coordination sites for metal ions. Multi-responsive helical polymers can therefore be prepared.28,29 (iv) Importantly, large, rigid pyridine groups in chiral α-acyloxy amides facilitate the formation of chiral polymers.30 (v) Like other aggregated molecules, the π–π stacked pyridines fluoresce under UV irradiation.27 (vi) Meanwhile, the picolyl ester-containing acyloxy amides in polymer backbones are photolabile, which benefits the synthesis of visible-light-degradable helical polymers.31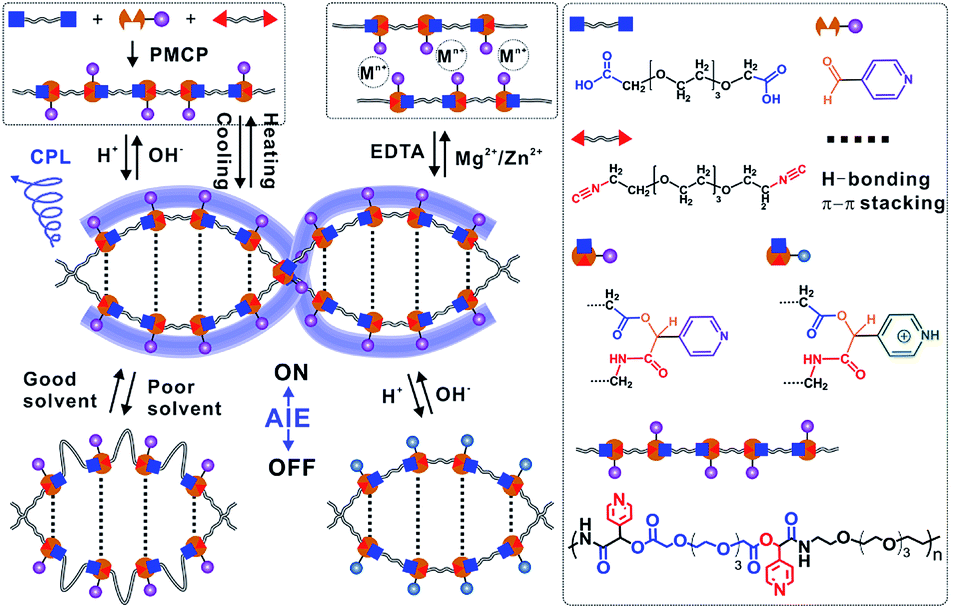 | ||
| Fig. 1 Schematic illustration of synthesizing double-helical, AIE-active polymers with multiple responsiveness via the PMPIA strategy. PMCP: Passerini-multicomponent polymerization. | ||
Carboxylic acid-difunctionalized PEG (HOOC-PEG-COOH, Mn ∼ 250 g mol−1) and PyCHO are commercially available. Isocyanide difunctionalized PEG (CN-PEG-NC) was obtained via amidation of PEG diamine (Mn ∼ 250 g mol−1) and subsequent hydrolysis (Fig. S1†).32 This Passerini three-component polymerization was conducted in tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) because PyCHO is poorly soluble in water (Fig. S2†). However, the as-prepared WADP was hydrophilic and had good solubility in water. Although a higher yield was achieved in DMF (>80 wt%), polymerization in THF gave polymers with higher molecular weight (∼18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and a distribution index of 2.0 (Table S1†). 1H-NMR spectroscopy was used to monitor the synthesis of the WADP. Fig. 2a shows that only three peaks, i.e., from PEG and PyCHO, at 3.5, 7.8, and 8.8 ppm were observed for the initial monomer solution. As the polymerization time increased, the PyCHO peaks gradually split into two sets of four peaks at 7.2–7.8 and 8.2–8.8 ppm, respectively. Two new peaks at 6.2–6.6 ppm appeared. They are ascribed to the hydrogen atoms in the α-acyloxy amides in the WADP (Fig. S3†). This observation indicates that the chemical environment of pyridines has changed, and a new chiral structure has formed during polymerization. This inference is in accord with the circular dichroism (CD) results, which are shown in Fig. 2b and S4.† All the monomers were achiral, but the chirality of the products increased with the increasing polymerization time and concentration in dimethyl sulfoxide (DMSO). This chirality was still observed when the solvent was changed to water but not in Zn2+-containing DMSO (DMSO/Zn2+). These achieved results indicated that the CD signal originated from the PMPIA-induced formation of polymeric superstructures. The relatively low intensity was possibly assigned to the low content of chiral α-acyloxy amides and the long PEG segments between them, which increased the helical pitch of the WADP (shown in Fig. 1).
000 g mol−1) and a distribution index of 2.0 (Table S1†). 1H-NMR spectroscopy was used to monitor the synthesis of the WADP. Fig. 2a shows that only three peaks, i.e., from PEG and PyCHO, at 3.5, 7.8, and 8.8 ppm were observed for the initial monomer solution. As the polymerization time increased, the PyCHO peaks gradually split into two sets of four peaks at 7.2–7.8 and 8.2–8.8 ppm, respectively. Two new peaks at 6.2–6.6 ppm appeared. They are ascribed to the hydrogen atoms in the α-acyloxy amides in the WADP (Fig. S3†). This observation indicates that the chemical environment of pyridines has changed, and a new chiral structure has formed during polymerization. This inference is in accord with the circular dichroism (CD) results, which are shown in Fig. 2b and S4.† All the monomers were achiral, but the chirality of the products increased with the increasing polymerization time and concentration in dimethyl sulfoxide (DMSO). This chirality was still observed when the solvent was changed to water but not in Zn2+-containing DMSO (DMSO/Zn2+). These achieved results indicated that the CD signal originated from the PMPIA-induced formation of polymeric superstructures. The relatively low intensity was possibly assigned to the low content of chiral α-acyloxy amides and the long PEG segments between them, which increased the helical pitch of the WADP (shown in Fig. 1).
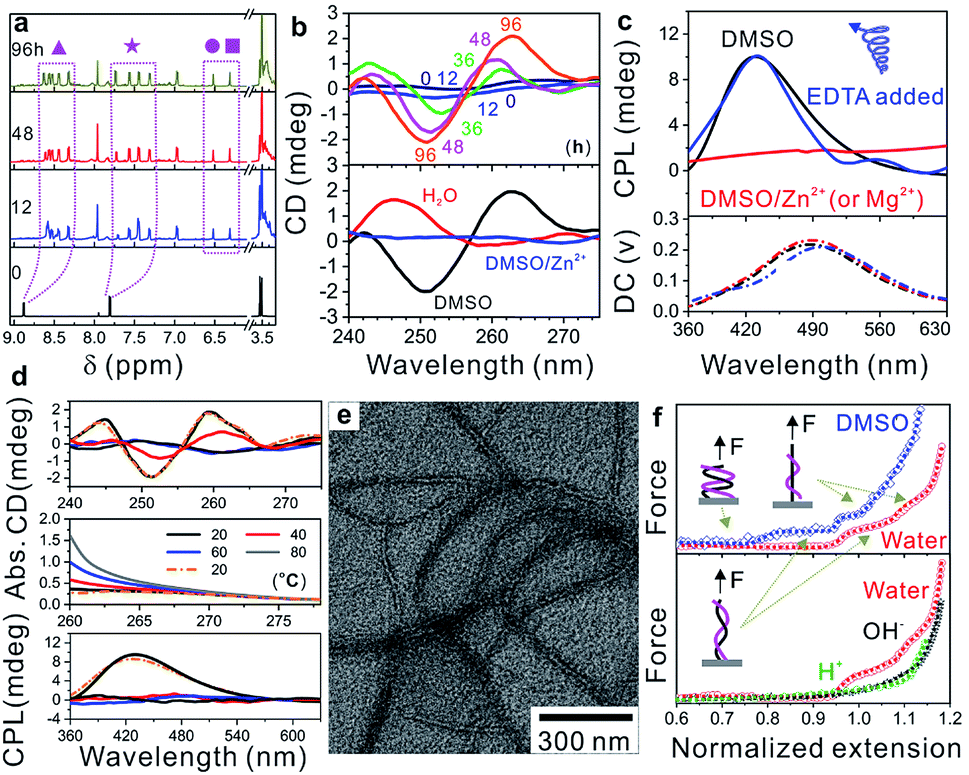 | ||
| Fig. 2 (a) 1H-NMR spectra of the mixed monomers and polymerization products with different reaction times in DMSO-d6. (b) Circular dichroism characterization of the mixed monomers and polymerization products with different reaction times in DMSO, and the WADP in different solvents. The concentration of the samples is 0.25 g L−1. (c) Circularly polarized luminescence characterization of the WADP in DMSO, DMSO/Zn2+ (or Mg2+) and EDTA-added DMSO/Zn2+ (or Mg2+). The concentration of all the samples is 50 g L−1. (d) CD, UV-vis, and CPL spectra of the WADP in DMSO at different temperatures. (e) TEM image of the WADP taken from the THF solution. (f) Single-molecule force spectroscopy characterization of helical polymers in DMSO and water with different pH values. | ||
In contrast, we found that the 1H-NMR spectra of the as-prepared WADP in the above solvents differed (Fig. S5†). Multiple peaks from the pyridine units in the polymer were integrated into two peaks in water, and no characteristic signal was detected in DMSO/Zn2+. This difference implies that the polymers underwent significant conformational changes. The mechanism of the solvent-induced conformational change was investigated by using a circularly polarized luminescence (CPL) technique to detect the signals of the WADP conformations. Fig. 2c shows that the WADP is a left-handed helical polymer in pure DMSO. No signal was detected from the polymer in water because the polymer emitted weak fluorescence under this condition. Considering the better stability of supramolecular interactions in water, we can reasonably speculate that the same helical structure was still present in aqueous solution. However, the chiral signal of CPL disappeared the same as that in the CD and NMR spectra in Fig. 2b and S5† when metal ions, i.e., Zn2+ or Mg2+, were added to the solutions. This is because these metal ions preferentially bind to pyridines, and the formed metal-coordination interactions are stronger than the hydrogen-bonding interactions between amides and pyridines (or between pyridines). As a result, the helical structure in the WADP is destroyed. Because the metal-coordination interactions are reversible, the helical signal can recover to its original state after the introduction of ethylenediaminetetraacetic acid (EDTA) to remove the metal ions. Moreover, considering the instability of supramolecular interactions at high temperature, we monitored the optical changes of the WADP from the CD, UV-vis, and CPL spectra upon gradually increasing the temperatures of solutions from 20 to 80 °C and subsequently decreasing to 20 °C (Fig. 2d). It was found that signal intensities in the CD and CPL spectra decreased, and then mostly recovered to the initial states. But the UV signal increased and then decreased inversely. These results indicated that the WADP was responsive to temperature and underwent reversible disassembly/assembly processes of helical structures. In brief, at higher temperatures, the supramolecular interactions in polymers were destroyed. The paired chiral acyloxy amides were liberated from the assemblies and were in a fully free state whereafter, these interactions regenerated at lower temperatures and drove the reassembly of the WADP.
More importantly, studying the fine superstructure of the helical polymers can clarify the assembly mechanism and enable their applications to be broadened. Fig. 2e shows that the WADP was assembled into soft fibres in THF with diameters from 10–20 nm. However, unlike the case for other chiral polymers, direct observation of the fine helical structure of the WADP in this study was difficult because of its soft nature and weak fluorescence in water.12 Single-molecule force spectroscopy was therefore performed to confirm the helical structure of the WADP in different solvents. This is a powerful tool for in situ monitoring of the conformational transitions of soft-matter materials (e.g., DNA, RNA, and polysaccharides) and involves stretching them at the single-molecule level (Fig. S6a†).33 These transitions in solutions can be identified by plotting the stretching force versus normalized extension (L × L0−1). L0 and L are the stretched and original lengths of the polymer, respectively. In principle, the number of plateaus in the plotted curves indicates the number of conformational transitions in the polymers (Fig. S6b†).34,35Fig. 2f shows that two plateaus were generated when the helical polymer was stretched in both water and DMSO, but no plateau was observed for PEG stretching (Fig. S7†). This shows there are two conformational transitions in the WADP during stretching. The first plateau indicates that the helical polymers changed from a condensed state to a stretched state, and the second plateau reveals the full destruction of the helical structures with applying an external force. It is worth noting that one α-acyloxy amide (containing one amide bond and one pyridine unit) can form one pair of hydrogen bonds with the same structure nearby (Fig. 1 and S8†). These results indicate that the chiral WADP has a double-helical structure.
On the other hand, we tried to use benzaldehyde, which has a molecular structure similar to that of PyCHO, to prepare similar polymers via the same PMPIA strategy (Fig. S9a†). Unfortunately, no signal was observed in the circularly polarized luminescence spectrum, and no characteristic plateau was observed by single-molecular force spectroscopy (Fig. S9b and c†). This control experiment strongly indicates that the noncovalent hydrogen-bonding and aromatic interactions in pyridines promote the formation of the double-helical WADP (Fig. S8†). Furthermore, it was found that this superstructure was sensitive to pH. The two plateaus disappeared under acidic and alkaline conditions but reappeared when the pH was adjusted to 7.0. Meanwhile, the absorbance of the WADP in DMSO (@ <300 nm) gradually increased with the decreasing pH values (<7.0) (Fig. S10†). Such a trend of change in the UV-vis spectra was the same as that achieved at different temperatures in Fig. 2d, indicating that the WADP had conformational transitions from double-strand to single-strand structures at high temperatures or low pH values. On the basis of these results, we speculate that the cooperation between hydrogen-bonding and aromatic interactions in chiral α-acyloxy amides plays a crucial role in the self-assembly of the WADP with a double-helical structure.
In addition to providing high flexibility for assembling the WADP superstructure, PEG segments are sensitive to solvents because of their different solubilities in common solvents, as shown by algorithmic calculations (THF > DMSO > MeOH > water).26 In the helical WADP in this study, two pyridines were linked to the ends of one PEG segment; therefore, the different solubilities of PEG can be used to regulate their conformational states (Fig. 1). Fig. 3a shows that changing the solvent clearly affects the AIE of the WADP (DMSO > DMF > THF > MeOH > water). This order differs slightly from that of the solvency power for PEG because of the generation of new pyridine and α-acyloxy amide structures in the WADP. As shown in Fig. 1, the as-prepared WADP was extendable and flexible in good solvents. It was therefore easy for the pyridines to get close to each other, to form new AIEgens (i.e., stacked pyridines), which give fluorescence emission under UV irradiation. However, when poorer solvents were used, the PEG segment collapsed to a folded conformation, and pyridine π–π stacking was hindered, which decreased the fluorescence intensity.
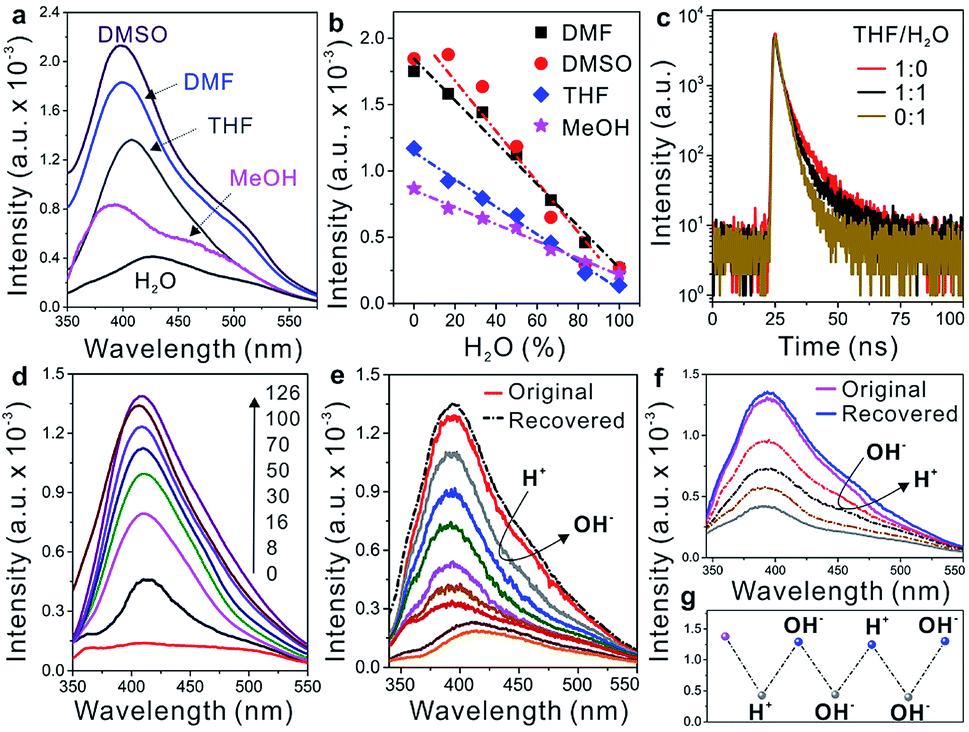 | ||
| Fig. 3 (a) Fluorescence spectra of the WADP in different solvents. (b) Fluorescence intensity of the WADP in DMF, DMSO, THF, and MeOH with varying ratios of the weight of water. (c) Fluorescence lifetime characterization of the WADP in THF, THF/H2O, and H2O. (d) The fluorescence spectra of the polymerization products with the reaction times from 0 to 126 hours. (e and f) Fluorescence spectra of the WADP in THF with different pH values. (g) The change of fluorescence intensity at 400 nm with varying pH values. The concentration of all the samples is 0.25 g L−1. | ||
Unlike other AIEgens that aggregate and fluoresce strongly in water, the fluorescence intensity, lifetime, and UV absorption of the WADP decreased with the increasing water content of these solvents (Fig. 3b and c, and S11†). To further understand this AIE mechanism, real-time fluorescence spectroscopy was used to trace the entire fabrication process of the helical polymer. Fig. 3d shows that the fluorescence intensity of the starting solution was weak but gradually increased with increasing polymerization time. This indicates that new supramolecular interactions were generated, which induced the formation of AIEgens, i.e., stacked pyridines, in the polymer. We also found that the pH, but not metal ions (Zn2+ and Mg2+), affected the fluorescence intensity of this AIE-active polymer (Fig. 3e–g and S12†). When an acid was added, the pyridines in the α-acyloxy amides were salified, and the supramolecular interactions were destroyed simultaneously. This led to a decrease in fluorescence intensity. Thereafter, the intensity recovered to its original state on the addition of an alkaline solution. Interestingly, the fluorescence intensity decreased again when further adding an alkaline solution. The reason can be assigned to the neutralization of hydrogen atoms in amide bonds that leads to the rupture of hydrogen-bonding interactions of the WADP. Moreover, the same fluorescence intensity trend was observed when alkaline and acidic solutions were sequentially added (Fig. 3f). This process can be repeated several times by tuning the pH of the polymer solution because of the reversible destruction–reconstruction of the hydrogen-bonding interactions in the WADP (Fig. 3g). These results indicate that the reasons for the generation of a tunable AIE effect in this polymer are as follows. (i) The generation of new hydrogen-bonding and aromatic interactions leads to the formation of AIEgens, i.e., π–π stacked pyridines, which emit fluorescence under excitation (Fig. S8†). (ii) The WADP solubility in various solvents differs.
Similar to UV-photolabile ortho-nitrobenzyl molecules, picolyl esters and picoliniums are sensitive to visible-light irradiation and can be cleaved via a typical photo-induced electron-transfer process under mild conditions (Fig. S13†).31,36,37 As shown in Fig. 4a, new picolyl groups, which were derived from PyCHO, were generated in the α-acyloxy amides of the WADP. The as-prepared polymer was therefore degraded into small segments under blue-light irradiation with a Ru(II) complex catalyst and ascorbic acid, as previously reported for picolyl compounds. Gel-permeation chromatography (GPC) showed that the elution time increased with the increasing irradiation time, which indicates polymer degradation. Meanwhile, the molecular weight of the WADP decreased from ∼18000 to 800 g mol−1, with a narrow distribution index of 1.13, within 120 min of irradiation (Fig. 4b). The fluorescence intensity decreased ten-fold compared to that of the as-prepared WADP (Fig. S14†). These trends are in accord with the 1H-NMR spectra in Fig. 4c, in which the multiple peaks of pyridines in the chiral α-acyloxy amides gradually merge into a single peak. The double peaks from picolyl groups at 6.2–6.6 ppm disappear, and the integral area at around ∼3.5 ppm increases because of the generation of picolyl amides in the degradation products. These results indicate that the WADP chirality disappeared after photodegradation. We also found that the degradation of this chiral polymer is more rapid than the degradation of polymers previously reported by our group.37,38
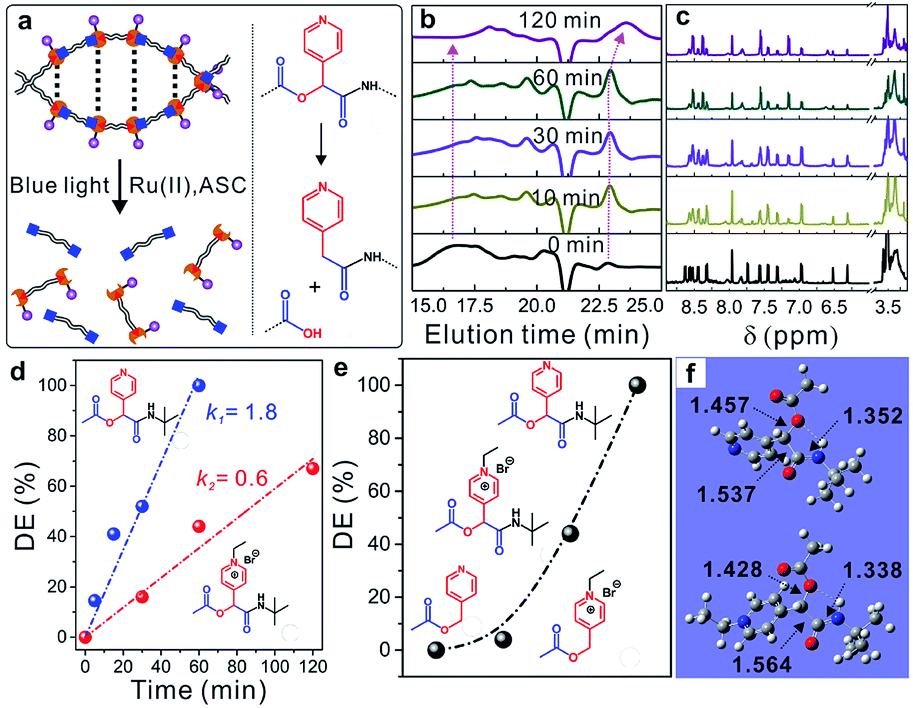 | ||
| Fig. 4 (a) Schematic illustration of degradation of the helical WADP with the catalysis of tris(bipyridine)ruthenium(II) chloride (Ru(II)) and ascorbic acid (ASC) under blue light irradiation (452 nm). (b) Gel permeation chromatography (GPC) characterization and (c) the corresponding 1H-NMR spectra of degradation products with different reaction times. (d) The degradation efficiency of the small model molecules of the amide and ethylated amide with varying times of irradiation. (e) Degradation efficiencies of the small model molecules with a predetermined reaction time of 60 min. (f) The optimized structures of the model amide and ethylated amide from the DFT calculation of B3LYP-D3 and the 6-31G(d) basis set. Normal vibrational mode analysis at the same level of theory confirmed that the optimized structures are minima (zero imaginary frequency). | ||
The WADP degradation mechanism was clarified by synthesizing and degrading four molecules in the same manner (Fig. S15–S17†). Fig. 4d and S18† indicate that, unlike the reported picolyl ester and its ethylation product, although the ethylated amide had a higher reduction potential, the degradation rate of the α-acyloxy amide was three times faster than that of the ethylated sample.39,40 Further study indicated that this amide was degraded completely, whereas the esters in previously reported studies had a degradation efficiency of 0–5% within 60 min (Fig. 4e and S19–S21†). The order of the degradation efficiencies of the model molecules was picolyl amide > ethylated amide > ethylated ester > picolyl ester. Density functional theory (DFT) calculations (Fig. 4f) indicate the presence of hydrogen-bonding-induced five-membered-cyclization structures in the amides and ethylated samples, but not in the reported picolyl esters. This specific cyclized structure promotes degradation of the C–O bonds of esters in amides compared with the degradation of the other two esters. Furthermore, the ethylation of pyridines had an obvious effect on the cyclized amide. After ethylation, the C–O bond lengths decreased from 1.457 to 1.428 Å. Degradation of the ethylated amide was, therefore, more difficult than that of the amide, which resulted in a lower degradation efficiency.
Conclusions
In summary, we report a PMPIA strategy for designing a double-helical, AIE-active polymer with visible-light degradability. The as-prepared WADP is water-soluble and sensitive to metal ions, pH, and solvents. Under these stimuli, the helical structure and AIE effect change reversibly. The WADP therefore could potentially be used in applications such as chemical sensors, information-storage devices, and ion-channel switches. Compared with other reported approaches, this novel method has several advantages. First, this is a metal-free strategy that proceeds spontaneously in water and organic solvents under mild conditions. Biocompatible polymers can be easily prepared without the need for removal of metal-containing catalysts. Secondly, the polymerization process involves three common achiral compounds, namely aldehydes, isocyanides, and acids, which in most cases can be easily synthesized or are commercially available. The classic PMPIA reaction readily enables one-step preparation of well-defined polymers with predicated structures. Thirdly, the generation of chiral α-acyloxy amides and supramolecular interactions greatly promotes the assembly of double helices in polymers, and provides a general method for preparing advanced chiral materials. Furthermore, functional molecules can be used as monomers to construct multi-responsive smart polymers that mimic the functions of materials in living systems, such as DNA, enzymes, and antibodies. We believe that this novel PMPIA strategy opens up new horizons and will inspire the design of advanced multifunctional polymers with chiral/helical structures, and has potential for many applications in biological and materials sciences.Methods
Chemicals
Carboxylic acid and amine difunctionalized PEGs (∼250 g mol−1), 4-pyridinecarboxaldehyde, 4-pyridinemethanol, and tris(bipyridine)ruthenium(II) chloride were purchased from Sigma-Aldrich. Ethyl formate, phosphorus(V) oxychloride, tert-butylamine, ascorbic acid, and the other reagents were of analytical grade and used without further purification. Isocyanide tert-butane was synthesized according to the literature.32Characterization
Fluorescence and circularly polarized luminescence spectra were recorded on F-4500 fluorescence and JASCO CPL-200 spectrophotometers, respectively. Transmission electron microscopy (TEM) characterization was conducted on Talos F200x equipment. Circular dichroism characterization was performed on a J-1500 spectropolarimeter with a 10 mm quartz cuvette. The 1H-NMR measurements were conducted on a 400 MHz spectrometer (Bruker AM-400). Gel permeation chromatography measurements were performed on an Agilent GPC-1260 liquid chromatograph. DMF was used as an eluent at 35 °C, and the flow rate was 1.0 mL min−1. It is worthy of note that, before testing, all the solutions should be dissolved in the corresponding solvents with gentle stirring for 12 h in a dark place.For the single-molecule force spectroscopy (SMFS), the polymers were first dissolved in water at a concentration of 20 g L−1, respectively. Then, ∼100 μL of the WADP solution was allowed to adsorb onto hydroxylated substrates for two hours. After this, the sample was thoroughly rinsed with abundant deionized water to remove the loosely adsorbed polymers, followed by drying with air. The SMFS experiments were performed on a commercial AFM (SHIMADZU, SPM9700-HT), which has a higher sensibility and minimal noise signal. Before the measurements, a certain amount of the corresponding solvent was injected between the sample and the AFM cantilever (MLCT-BIO, Bruker Corp., CA). During the AFM measurement, the data were collected and converted to force–extension curves later. The stretching velocity was 1.0 μm s−1 if not mentioned otherwise. The spring constant of the AFM cantilever was ∼30 pN nm−1.
Synthesis of isocyanide difunctional PEG (CN-PEG-NC) and tert-butane
Isocyanide PEG was synthesized according to the literature with some modifications.32 Briefly, amine difunctionalized PEG (3.0 g, 12 mmol) was slowly dissolved in ethyl formate (50 mL, 0.62 mol) at 0 °C with vigorous stirring and heated to 80 °C for 48 hours. Excessive ethyl formate was removed by rotary evaporation, and the amide product was obtained. Next, this intermediate product and triethylamine (20 mL) were dissolved in dichloromethane and cooled to −20 °C. After this, phosphoryl chloride (3 mL, 32 mmol) in 30 mL of dichloromethane was added dropwise at such a rate that the temperature remained below −10 °C. The mixture was stirred for four hours, then poured into 500 mL of saturated sodium carbonate solution, keeping the temperature below 25 °C, and was continuously stirred for one hour. The organic layer was separated, and the aqueous layer was extracted three times with 50 mL of dichloromethane again. The combined organic layer was dried with Na2SO4, and purified by column chromatography (dichloromethane/methanol, 10/1). Finally, the isocyanide product was obtained. The synthesis of isocyanide tert-butane was the same as that of CN-PEG-NC, but tert-butylamine was used as the amine source.Synthesis of helical polymers
To prepare the helical polymers, the difunctional PEGs of HOOC-PEG-COOH and CN-PEG-NC and 4-pyridinecarboxaldehyde with different ratios were added into THF (or DMF) and reacted at predetermined concentrations and reaction times at room temperature (listed in Table S1†). After completing the polymerization, the mixture was added dropwise to diethyl ether and washed three times to remove the unreacted monomers. Then, the products were dried in a vacuum oven for 24 h. The ethylated polymers were simply synthesized by mixing the polymer and excessive ethyl bromide in acetonitrile and heating to 80 °C for 24 hours. After rotary evaporation, the crude product was purified by dropping into diethyl ether, and a solid product was obtained.Synthesis of small model molecules
The picolyl ester was first synthesized by esterifying 4-pyridinemethanol with acetic acid via the EDC/NHS chemistry.37 As for the model amide, the reactants, including isocyanide tert-butane, 4-pyridinecarboxaldehyde, and acetic acid, were added into THF in a molar ratio of 2:1:1 for 24 h under ambient conditions in a dark place. After vaporizing THF, the mixture was dissolved in dichloromethane and washed with an aqueous solution of NaHCO3. Then, the aqueous phase was further extracted with dichloromethane three times. The organic phases were collected and dried using anhydrous MgSO4. Finally, the product was purified by silica gel column chromatography using an ethyl acetate/hexane mixture (2/1) as the eluting solvent, yielding a white solid amide.
The ethylated molecules were simply synthesized by mixing the model amide (or the picolyl ester) and excessive ethyl bromide in acetonitrile and heating to 80 °C for 24 hours. After rotary evaporation, the crude product was purified by dropping into diethyl ether, and a solid product was obtained.
Photodegradation of polymers and small model molecules
The helical polymers (or model molecules) (1.0 eq.), [Ru(2,2′-bipy)3]Cl2 (0.001 eq.), and ascorbic acid (20 eq.) were dissolved in water and bubbled with argon gas for 10 min to remove the oxygen in a dark place. The mixture was exposed to visible-light irradiation (452 nm) for predetermined times. Then, sodium bicarbonate was added to neutralize the resultant solution. The hydrophobic component was obtained by simply extracting the degradation products with dichloromethane. As for the ethylated model molecules, bis(trifluoromethane)sulfonimide lithium salt (Tf2NLi) was added into the solution in advance. After this, red hydrophobic picoliniums were obtained by extracting with dichloromethane.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the NSFC (21803003, 21604069) and the Nature Science Foundation of Shaanxi Province (2019JM-094, 2020JQ-598) for the financial support of this work.Notes and references
- H. Sun, Q. Luo, C. Hou and J. Liu, Nano Today, 2017, 14, 16–41 CrossRef CAS.
- J. Bella, Biochem. J., 2016, 473, 1001–1025 CrossRef CAS PubMed.
- A. J. Metrano and S. J. Miller, Acc. Chem. Res., 2019, 52, 199–215 CrossRef CAS.
- A. M. Kushner and Z. Guan, Angew. Chem., Int. Ed., 2011, 50, 9026–9057 CrossRef CAS PubMed.
- J. Li, L. Mo, C. H. Lu, T. Fu, H. H. Yang and W. Tan, Chem. Rev., 2016, 45, 1410–1431 CAS.
- S. P. W. Wijnands, W. Engelen, R. P. M. Lafleur, E. W. Meijer and M. Merkx, Nat. Commun., 2018, 9, 65 CrossRef.
- C.-Z. Liu, S. Koppireddi, H. Wang, D. W. Zhang and Z. T. Li, Angew. Chem., Int. Ed., 2019, 58, 226–230 CrossRef CAS PubMed.
- T. Leigh and P. Fernandez-Trillo, Nat. Rev. Chem., 2020, 4, 291–310 CrossRef CAS.
- D. Zhao, T. van Leeuwen, J. Cheng and B. L. Feringa, Nat. Chem., 2017, 9, 250–256 CrossRef CAS PubMed.
- M. Hu, H. T. Feng, Y. X. Yuan, Y. S. Zheng and B. Z. Tang, Coord. Chem. Rev., 2020, 416, 213329 CrossRef CAS.
- X. Lan, T. Liu, Z. Wang, A. O. Govorov, H. Yan and Y. Liu, J. Am. Chem. Soc., 2018, 140, 11763–11770 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- Y. Furusho and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5195–5207 CrossRef CAS.
- J. Tanabe, D. Taura, N. Ousaka and E. Yashima, J. Am. Chem. Soc., 2017, 139, 7388–7398 CrossRef CAS PubMed.
- Z. Zhang, Z.-B. Tan, C. Y. Hong, D. C. Wu and Y. Z. You, Polym. Chem., 2016, 7, 1468–1474 RSC.
- J. F. Lutz, J. M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- P. Theato, Multi-Component and Sequential Reactions in Polymer Synthesis, Springer, 2015 Search PubMed.
- Y. Cui, M. Zhang, F. S. Du and Z. C. Li, ACS Macro Lett., 2017, 6, 11–15 CrossRef CAS.
- H. Xue, Y. Zhao, H. Wu, Z. Wang, B. Yang, Y. Wei, Z. Wang and L. Tao, J. Am. Chem. Soc., 2016, 138, 8690–8693 CrossRef CAS PubMed.
- L. V. Kayser, M. Vollmer, M. Welnhofer, H. Krikcziokat, K. Meerholz and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 10516–10521 CrossRef CAS PubMed.
- B. T. Tuten, L. De Keer, S. Wiedbrauk, P. H. M. Van Steenberge, D. R. Dhooge and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 5672–5676 CrossRef CAS PubMed.
- Q. Gao, L. H. Xiong, T. Han, Z. Qiu, X. He, H. H. Y. Sung, R. T. K. Kwok, I. D. Williams, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 14712–14719 CrossRef CAS PubMed.
- M.-Q. Zhao, Q. Zhang, G. L. Tian and F. Wei, Nanoscale, 2014, 6, 9339–9354 RSC.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- Q. Wang, D. X. Wang, M. X. Wang and J. Zhu, Acc. Chem. Res., 2018, 51, 1290–1300 CrossRef CAS PubMed.
- C. O. D
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) nc, G. Kbarer and A. Güner, J. Appl. Polym. Sci., 2010, 117, 1100–1119 CrossRef.
nc, G. Kbarer and A. Güner, J. Appl. Polym. Sci., 2010, 117, 1100–1119 CrossRef. - J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Rev., 2020, 49, 1144–1172 CAS.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J. M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed.
- M. Horeau, G. Lautrette, B. Wicher, V. Blot, J. Lebreton, M. Pipelier, D. Dubreuil, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2017, 56, 6823–6827 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Acc. Chem. Res., 2013, 46, 927–936 CrossRef CAS PubMed.
- C. Sundararajan and D. E. Falvey, J. Org. Chem., 2004, 69, 5547–5554 CrossRef CAS.
- J. Zhang, M. Zhang, F. S. Du and Z. C. Li, Macromolecules, 2016, 49, 2592–2600 CrossRef CAS.
- Y. Bao, Z. Luo and S. Cui, Chem. Rev., 2020, 49, 2799–2827 CAS.
- S. Cui, J. Yu, F. Kühner, K. Schulten and H. E. Gaub, J. Am. Chem. Soc., 2007, 129, 14710–14716 CrossRef CAS PubMed.
- H. Zhang, X. Li, Y. Lin, F. Gao, Z. Tang, P. Su, W. Zhang, Y. Xu, W. Weng and R. Boulatov, Nat. Commun., 2017, 8, 1147 CrossRef PubMed.
- L. Li, A. Lv, X. X. Deng, F. S. Du and Z. C. Li, Chem. Commun., 2013, 49, 8549–8551 RSC.
- X. Li, H. Zhang, P. Zhang and Y. Yu, Chem. Mater., 2018, 30, 3752–3758 CrossRef CAS.
- T. Zhou, T. Liu, Y. Bao, P. Zhang, C. Yan, F. Yao, S. Cui, Y. Chen, X. Chen and Y. Yu, Polym. Chem., 2018, 9, 138–144 RSC.
- J. B. Borak and D. E. Falvey, J. Org. Chem., 2009, 74, 3894–3899 CrossRef CAS PubMed.
- Y. Yu, X. Kang, X. Yang, L. Yuan, W. Feng and S. Cui, Chem. Commun., 2013, 49, 3431–3433 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting figures and tables. See DOI: 10.1039/d0sc02729d |
| ‡ J. Liu and Z. Luo equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |