
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in low oxidation state aluminium chemistry
Katie
Hobson
,
Claire J.
Carmalt
 and
Clare
Bakewell
*
and
Clare
Bakewell
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: c.bakewell@ucl.ac.uk
First published on 30th June 2020
Abstract
The synthesis and isolation of novel low oxidation state aluminium (Al) compounds has seen relatively slow progress over the 30 years since such species were first isolated. This is largely due to the significant challenges in isolating these thermodynamically unstable compounds. Despite challenges with isolation, their reactivity has been widely explored and they have been utilized in a wide range of processes including the activation of strong chemicals bonds, as ligands to transition metals and in the formation of heterobimetallic M–M compounds. As such, attempts to isolate novel low oxidation state Al compounds have continued in earnest and in the last few years huge advances have been made. In this review we highlight the remarkable recent developments in the low oxidation state chemistry of aluminium and discuss the variety of new reactions these compounds have made possible.
 Katie Hobson | Katie Hobson received her MSci in Chemistry from University College London in 2018 where her 4th year project focused on the development of n-type transparent conducting oxides under the supervision of Professor Claire Carmalt. Katie remained in the group to undertake a PhD, jointly supervised by Dr Clare Bakewell, investigating the use of main-group complexes as catalysts for synthetically useful reactions. |
 Claire J. Carmalt | Claire Carmalt is a Professor of Inorganic Chemistry at University College London. She obtained her BSc and PhD from Newcastle University and was a postdoctoral fellow at the University of Austin at Texas. She was then awarded a Royal Society Dorothy Hodgkin Fellowship and moved to UCL. She started a lectureship at UCL in 2001 and was promoted to senior lecturer in 2002, reader in 2004 and Professor in 2009. In 2016 she became the 18th Head of Department for Chemistry at UCL and the first woman appointed to the position. She was awarded the RSC 2000 Meldola Medal and the RSC 2019 Applied Inorganic Chemistry Award. |
 Clare Bakewell | Clare Bakewell received her MChem degree from the University of Leeds in 2010, before moving to Imperial College London to conduct a PhD under the supervision of Professor Charlotte Williams, which was followed by an EPSRC Doctoral Prize Fellowship. After spending a year working for start-up company Econic Technologies, she returned to Imperial for a postdoc with Dr Mark Crimmin. In October 2018, she took up a Ramsay Memorial Fellowship at University College London, where her research is focused on using main-group metals in a variety of sustainable catalytic applications. |
Introduction
Recent years have seen a renaissance in main-group chemistry, with novel main-group compounds being shown to adopt unusual electronic configurations, demonstrate application in the activation of strong chemical bonds and facilitate a wide range of catalytic processes.1–3 In particular, the potential for main-group elements to adopt reactivity more commonly associated with transition metals is being widely explored, with redox type processes now becoming less chemical curiosities and more valuable synthetic methodologies.4–7 Aluminium species are one such class of main-group compound whose popularity has soared in recent years. As one of the most abundant elements in the Earth's crust, aluminium's low cost and low toxicity make it an attractive choice for a wide range of applications, from construction and electronics, to a key component in organic synthesis.8,9The use of aluminium in major industrial catalytic processes dates back to the late 19th century, when aluminium(III) chloride was used to mediate the Friedel–Crafts reaction.10 In the 1950s Ziegler then demonstrated the use of aluminium hydrides in the Aufbau (chain growth) reaction of ethylene, a process that revolutionised modern manufacturing.11 In the ensuing years, aluminium's catalytic capabilities has been demonstrated in both hetero- and homogeneous systems for a range of applications spanning polymer chemistry, the synthesis of bulk commodity products and fine chemical production.12–14 Turnover in these homogenous catalytic cycles is typically controlled by the polarity induced between the Lewis acidic metal centre and the reactive metal substituent, Alδ+–Xδ−, thus no change of oxidation at the metal is necessary. It was long believed that stable complexes of aluminium could only exist in the +3 oxidation state, owing to the thermodynamic instability of the +1 and +2 states. However, the late ‘80s’ and early ‘90s’ saw rapid developments towards the isolation of stable, molecular low oxidation state aluminium compounds, with general stabilisation strategies usually involving the use of sterically demanding and/or highly σ- and π-donating ligand systems. This includes work by Uhl and co-workers who reported the first example of an unsupported Al–Al bond [(R2Al-AlR2)] (where R = CH(SiMe3)2) and the low oxidation cluster K2[Al12iBu12].15,16
In 1991, the first molecular Al(I) complex stable at room temperature was reported by Schnöckel and co-workers. The tetrameric complex [(Cp*Al)4] (1, Fig. 1) was synthesised by treatment of a metastable ethereal solution of AlCl with [Mg(Cp*)2], exploiting the steric bulk of the Cp* ligand (Cp* = pentamethylcyclopentadienyl). Further investigation has shown it can also be formed from the reduction of [Cp*AlCl2] and a redox reaction of [Cp*2AlH].17–19 Each Cp* ligand was η5-coordinated to the aluminium metal centre, which adopted a regular tetrahedral geometry. Compound 1 largely maintains its tetrameric structure in solution at room temperature, but can act as a masked source of Al(I) and has been shown to react with some small molecules, such as azides and oxygen, and deliver Cp*Al(I) as a ligand to transition metal centres.20 Its aggregated solution structure does however render it less potent a reducing agent than may be expected. In 2000, Roesky and co-workers made further progress in the advancement of low oxidation state Al chemistry, using the 2,6-diisopropylphenyl (Dipp) β-diketiminate ligand (BDI) to synthesise the first monomeric, solid state Al(I) complex (2, Fig. 1).21 Compound 2 was formed by the reduction of the parent diiodide complex, [BDIAlI2], with potassium metal and is structurally analogous to a carbene. Single crystal X-ray analysis revealed a well-defined monomer, with longer Al–N bond lengths and a more acute N–Al–N bond angle (1.957(2) Å, 89.86°) compared to the related Al(III) dimethylaluminium compound (1.922 Å, 96.18(9)°). These properties are consistent with a change in oxidation state as the Al–N bond becomes less polar due to the reduced positivity of the metal centre. Compound 2 has been extensively investigated as a stoichiometric reducing agent for a range of challenging bond cleavage and small molecule activation reactions. Examples include the oxidative addition of H2 and C–F bonds across the Al(I) centre, as well as the reversible addition of alkenes and facilitation of CO homologation in the presence of a transition metal fragment.22–25 This reactivity has been comprehensively documented in several recent review articles.5,20,26–28 Despite the diverse range of reactivity demonstrated by Al(I), up until recently 1 and 2, and a handful of derivatives thereof, have essentially remained the sole examples of stable sources of this powerful reagent. However, problems with their unreproducible and low yielding syntheses have hampered their use in more scalable synthetic procedures. For instance, 2 is typically formed in ∼20% yield, likely due to over reduction under the harsh reducing conditions. Attempts to make more synthetically attainable and ultimately more powerful sources of low oxidation state aluminium have therefore been pursued. The last few years have seen an explosion in this field, with the isolation and characterisation of a range of complexes that offer exciting new reactivity. In this review, we focus on these monumental advances that have set the stage for the future of main-group chemistry with commentary on the development of (1) aluminium–aluminium multiple bonds; (2) nucleophilic aluminium(I) alumanyl anions; (3) new monomeric aluminium(I) species and (4) the isolation of aluminium(I) hydrides.
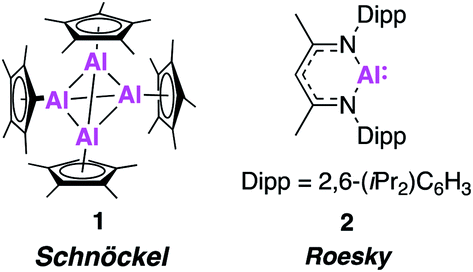 | ||
| Fig. 1 Structures of the first examples of stable aluminium (I) complexes. | ||
Aluminium–aluminium multiple bonds
In addition to the handful of isolated Al(I) species, numerous examples of Al–Al bonded compounds have been reported, some of which have bond orders greater than 1.15,29–33 There have also been examples of homodinuclear multiple bonds of group 13 elements including boron, gallium and indium.34 However until recently there were no known examples of a neutral complex containing a multiple bond between two aluminium centres, an important target if the metal–metal bond is to be partially retained during subsequent reaction.In 2017, Inoue and co-workers reported the first clean and high-yielding synthesis of a stable neutral compound containing an Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al double bond (4, Fig. 2).35 In a similar manner to 1, compound 4 was synthesised by the reduction of an N-heterocyclic carbene (NHC) stabilised dihalide (3) with three equivalents of KC8. As will be shown through forthcoming examples, this use of metal halide precursors and strong reducing agents has become a common strategy to access low oxidation state aluminium centres. Single crystal X-ray diffraction (XRD) revealed a trans-planar geometry with an Al–Al bond length of 2.3943(6) Å. This is significantly shorter than previous examples of structurally characterised dialuminum compounds.30,31,33 Density functional theory (DFT) calculations showed that the highest occupied molecular orbital (HOMO) was the Al–Al π-bond and the HOMO−1 the Al–Al σ-bond. The Wiberg bond index (WBI), a bond order descriptor, was calculated to be 1.70, suggesting significant double bond character. A series of reactivity studies were conducted to prove the formation of an AlAl double bond; firstly 4 was exposed to ethylene gas at room temperature from which a dialuminacyclobutane product was formed via a [2 + 2] cycloaddition process (5, Fig. 2). Compound 4 also reacted with phenylacetylene, this time forming two products, the [2 + 2] cycloaddition product and the terminal C–H insertion product. In all cases, the Al–Al bond was significantly longer than in the parent compound, with WBIs of approximately half, which support the presence of an AlAl double bond in 4. The reactivity of 4 has been further explored, with the stoichiometric activation of carbon dioxide (CO2) shown to be possible under mild conditions (Fig. 2).36 Exposure of 4 to 1 atm of CO2 resulted in the formation of 6 which is the formal [2 + 2] cycloaddition product from CO2 fixation, with single crystal XRD revealing a planar Al–Al–C–O ring. Heating 6 for an additional 60 minutes at 50 °C yielded 7, which was found to contain a completely planar Al–(CO)(O)–Al core, comprised of a bridging carbonyl and oxo unit, representing the first example of a dialuminacarbonyl compound. 7 was calculated to have formed via the isomerisation of 6 in an overall exothermic process. The initial endergonic C–O cleavage step (+24.9 kcal mol−1) was offset by carbon monoxide (CO) recombination and the formation of a 4-membered ring (−42.4 kcal mol−1). Compound 8 was obtained by further heating of 6 under an atmosphere of CO2 and is comprised of a six-membered ring, with a bridging carbonate group and oxygen between the Al centres. The mechanism of formation of 8 was probed computationally and was found to be accessible via two different routes. The first route proceeded via6 through a series of six- and five-membered rings and was found to be exothermic (−77.3 kcal mol−1) and produce 8 as a single isomer. The second route sees 4 react with one equivalent of O2 to form a four-membered planar Al–(O)(O)–Al ring. This was found to be exothermic (−36.5 kcal mol−1) and proceeded via breaking of the 4-membered ring, thus allowing for recombination at either face of the Al-centre and a mixture of products to be obtained. This route could be borne out experimentally, leading to the formation of a 50
Al double bond (4, Fig. 2).35 In a similar manner to 1, compound 4 was synthesised by the reduction of an N-heterocyclic carbene (NHC) stabilised dihalide (3) with three equivalents of KC8. As will be shown through forthcoming examples, this use of metal halide precursors and strong reducing agents has become a common strategy to access low oxidation state aluminium centres. Single crystal X-ray diffraction (XRD) revealed a trans-planar geometry with an Al–Al bond length of 2.3943(6) Å. This is significantly shorter than previous examples of structurally characterised dialuminum compounds.30,31,33 Density functional theory (DFT) calculations showed that the highest occupied molecular orbital (HOMO) was the Al–Al π-bond and the HOMO−1 the Al–Al σ-bond. The Wiberg bond index (WBI), a bond order descriptor, was calculated to be 1.70, suggesting significant double bond character. A series of reactivity studies were conducted to prove the formation of an AlAl double bond; firstly 4 was exposed to ethylene gas at room temperature from which a dialuminacyclobutane product was formed via a [2 + 2] cycloaddition process (5, Fig. 2). Compound 4 also reacted with phenylacetylene, this time forming two products, the [2 + 2] cycloaddition product and the terminal C–H insertion product. In all cases, the Al–Al bond was significantly longer than in the parent compound, with WBIs of approximately half, which support the presence of an AlAl double bond in 4. The reactivity of 4 has been further explored, with the stoichiometric activation of carbon dioxide (CO2) shown to be possible under mild conditions (Fig. 2).36 Exposure of 4 to 1 atm of CO2 resulted in the formation of 6 which is the formal [2 + 2] cycloaddition product from CO2 fixation, with single crystal XRD revealing a planar Al–Al–C–O ring. Heating 6 for an additional 60 minutes at 50 °C yielded 7, which was found to contain a completely planar Al–(CO)(O)–Al core, comprised of a bridging carbonyl and oxo unit, representing the first example of a dialuminacarbonyl compound. 7 was calculated to have formed via the isomerisation of 6 in an overall exothermic process. The initial endergonic C–O cleavage step (+24.9 kcal mol−1) was offset by carbon monoxide (CO) recombination and the formation of a 4-membered ring (−42.4 kcal mol−1). Compound 8 was obtained by further heating of 6 under an atmosphere of CO2 and is comprised of a six-membered ring, with a bridging carbonate group and oxygen between the Al centres. The mechanism of formation of 8 was probed computationally and was found to be accessible via two different routes. The first route proceeded via6 through a series of six- and five-membered rings and was found to be exothermic (−77.3 kcal mol−1) and produce 8 as a single isomer. The second route sees 4 react with one equivalent of O2 to form a four-membered planar Al–(O)(O)–Al ring. This was found to be exothermic (−36.5 kcal mol−1) and proceeded via breaking of the 4-membered ring, thus allowing for recombination at either face of the Al-centre and a mixture of products to be obtained. This route could be borne out experimentally, leading to the formation of a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of both the cis and trans isomers of 8, suggesting the first route to be the most likely mechanism connecting 6 and 8.
:50 mixture of both the cis and trans isomers of 8, suggesting the first route to be the most likely mechanism connecting 6 and 8.
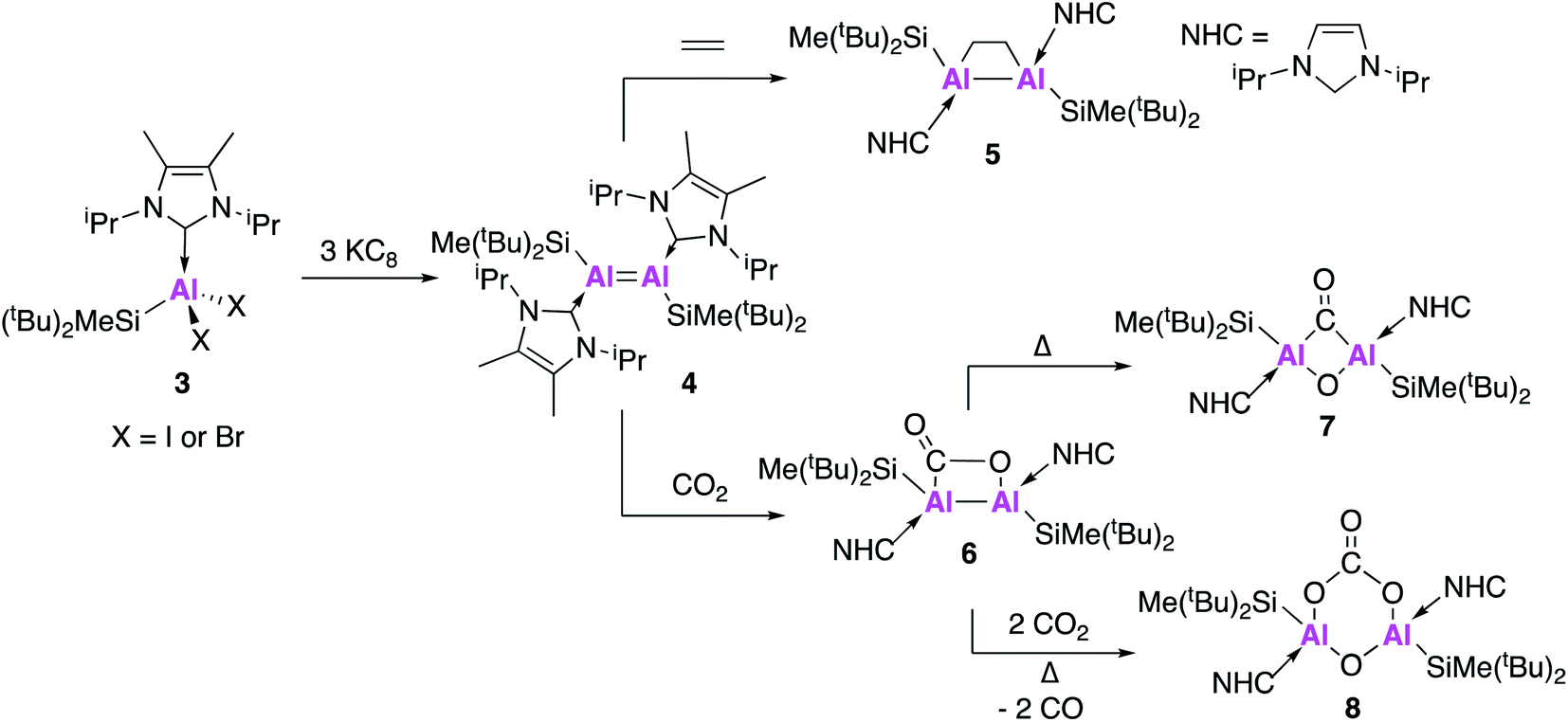 | ||
| Fig. 2 The synthesis of AlAl doubly bonded compound 4 and its reactions with small molecules. | ||
Compound 8, formed in situ from 4, was found to be active towards the catalytic reduction of CO2 using 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (HBpin) as a substrate (Fig. 3). A combined mechanistic and computational study revealed an overall exothermic transformation (−13.4 kcal mol−1), proceeding via initial coordination of HBpin to the exocyclic oxygen of the carbonate fragment. An endergonic hydride transfer, found to be the rate determining step (+22.2 kcal mol−1) was proposed, which was energetically offset by the formation of the reduced carbonate species (−26.6 kcal mol−1). Finally, CO2 coordination to the opposite plane of Al⋯Al allowed for reformation of 8 and released the formic acid derivative as the CO2 reduction product (Fig. 3).
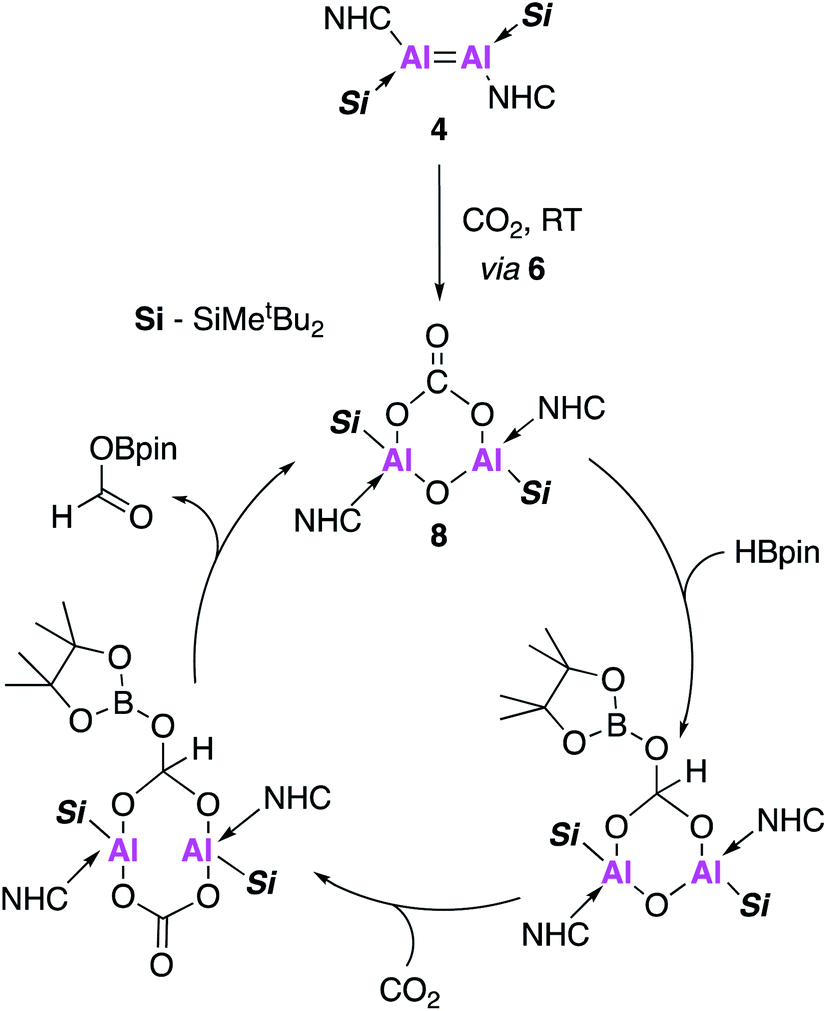 | ||
| Fig. 3 The proposed catalytic cycle for the hydroboration of CO2 using 4 as a pre-catalyst. | ||
Further derivatisation of 4 was targeted in order to investigate the effects of different ligands on the geometry and reactivity of the AlAl bond. Compound 9 (Fig. 4) was synthesised in an analogous fashion to 4, with crystallographic analysis revealing a trans-bent and twisted geometry with nearly perpendicular arrangement of the NHC groups.37 This was notably different to 4, whose NHC groups were orientated parallel to each other. The Al–Al bond length of 2.4039(8) Å was also slightly longer than in 4. DFT analysis saw a reduced HOMO–LUMO gap between 9 (1.86 eV) and 4 (2.24 eV), with differences in the electronic structures attributed to the difference in steric demands of the stabilising ligands (silyl vs. aryl). Compound 9 reacts with ethylene in an analogous fashion to 4, however reaction with phenylacetylene saw formation of only the [2 + 2] cycloaddition product (10). Interestingly, 10 decomposed in solution to form styrene, which is proposed to form via an intramolecular C–H insertion pathway. Enhanced reactivity of 9 was demonstrated by the facile reaction with diphenylacetylene at room temperature to generate the [2 + 2] cycloaddition product 11 (no reaction was seen with 4 at elevated temperature). This enhanced reactivity was attributed to the increased electrophilicity of 9 and flexibility of the Tipp ligand allowing for easier access of sterically demanding reagents to the AlAl core. Similarly, two equivalents of 2,6-dimethylphenylisocyanide reacted with 9 to form the bridging XylC = N 12, whilst the same reaction with 4 yielded an indistinct mixture of products.
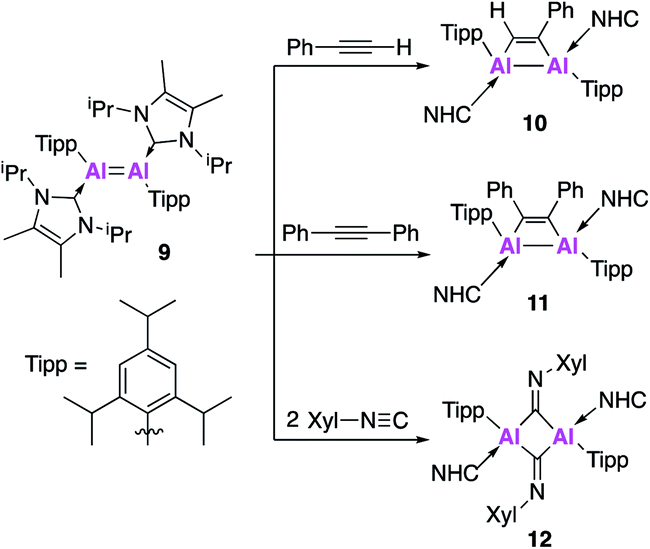 | ||
| Fig. 4 Reactivity of 9 (an analogue of 4) with unsaturated molecules. | ||
Compound 9 underwent a [2 + 2] cycloaddition reaction with CO2 to form 13, via a CO2 fixation product analogous to 6 (Fig. 5). However, the formation of both products could only be confirmed by the 13C NMR spectra due to the formation of numerous other compounds, attributed to the increased reactivity of 9. The dioxo product 14 was obtained upon reaction of 9 with oxygen and, as was observed for 4, further reaction with CO2 yielded carbonate complex 13. Interestingly, compound 14 was not directly formed from the reaction of 9 with nitrous oxide (N2O) but instead formed a bridging aluminium(II) mono-oxide species which, when further exposed to N2O, formed 14. Reaction of 9 with dihydrogen (H2) was also explored; spectroscopic analysis indicated addition of H2 across the AlAl double bond to form 15. Single crystal XRD was not possible, but comparison of the observed IR frequencies with related terminal and bridging structures combined with DFT supported the formation of a structure with terminal hydrides and an Al–Al single bond. The activity of 9 was also tested in catalysis towards the hydroboration of CO2 and amine-borane dehydrogenation. The hydroboration of CO2 led to the facile formation of a number of reduction products, the ratio of which varied with respect to temperature. With 4, only the singly reduced formic acid equivalent product was observed to form and this required long reaction times and high catalyst loading (Fig. 5). Amine borane dehydrogenation also saw an increased rate of conversion to a number of dehydrogenation products. The increased reactivity of 9 was attributed to its increased flexibility allowing greater access to the reactive core and nicely demonstrates how making simple changes to the ligand system can have a dramatic effect on reactivity. Further derivatisation of this class of Al–Al double bonded compounds has the potential to unlock even broader reactivity.
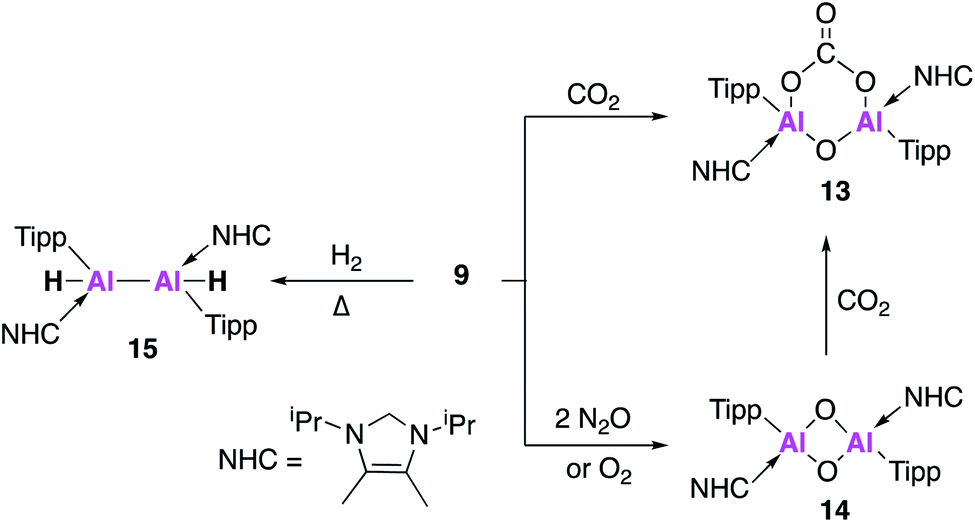 | ||
| Fig. 5 Reactions of 9 with small molecules. | ||
Nucleophilic aluminium(I) alumanyl anions
Examples of charged low oxidation state aluminium complexes have also been the centre of much investigation in recent years. In 2018, the remarkable discovery by Aldridge, Goicoechea and co-workers of the first example of an anionic Al(I) nucleophile, [K{Al(NON)}]2, (17, NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene, Fig. 6) was reported.38 This was synthesised by reduction of the aluminium iodide (16) with excess KC8 to obtain 17 as a centrosymmetric dimer. Compound 17 is comprised of two [Al(NON)]− units and is stabilised by potassium–arene contacts. In an effort to rule out the presence of metal-bound hydrogens, [K{H2Al(NON)}]2 was synthesised, and all analysis supported the formation of two chemically distinct compounds. The Al–N bond distances in 17 (1.956(2) and 1.963(2) Å) were found to be longer than in 16 (1.846(2) Å), consistent with the formation of an Al(I) species. A series of reactions with electrophilic reagents was carried out, including H2 and benzene, resulting in the formation of multiple new aluminium-element bonds (Al–H, 18; Al–C, 19). This is in direct contrast to 2, which only activates the strong C–H bonds of benzene in the presence of a palladium catalyst, highlighting its superior performance as a synthetic reagent.39 The highly donating alumanyl (sometimes referred to as aluminyl) complex was also employed as a ligand to stabilise electron rich transition metal compounds. Compound 17 reacted with one equivalent of [tBu3PAuI] to form the two-coordinate gold complex 20, which contained an Al–Au covalent bond.40 This structure was found to adopt a near linear geometry, with an Al–Au bond length of 2.402(3) Å. The effective atomic charges of the Al and Au centres were calculated to be +2.21 and −0.82, respectively, supporting the formation of a highly polarised Auδ−Alδ+ bond. Complex 20 therefore should react as a gold centred nucleophile, an assumption which was ratified by the reductive insertion of CO2 and diisopropylcarbodiimide into the Al–Au bond, forming products in which the gold centre was bound to the carbon.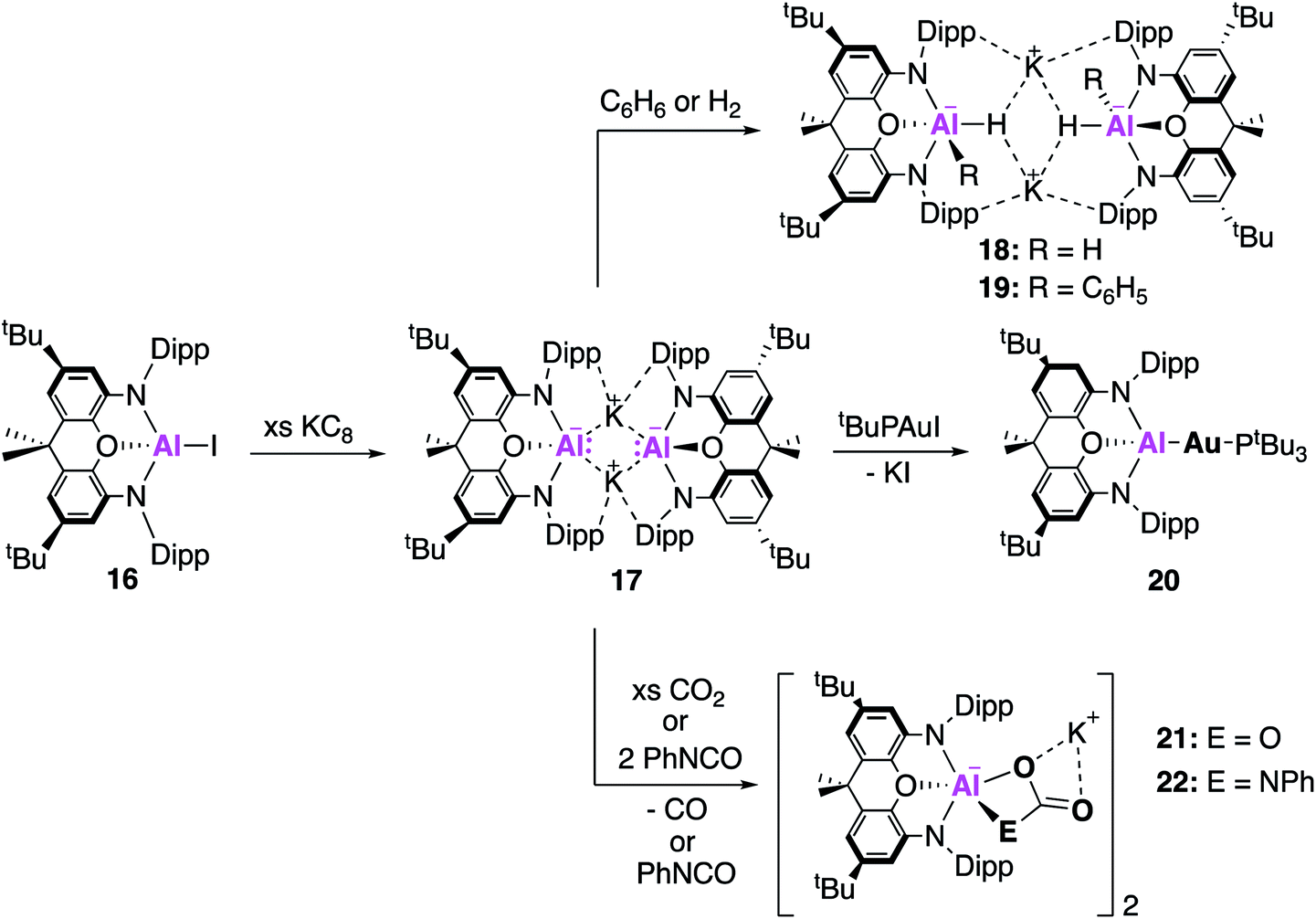 | ||
| Fig. 6 The synthesis and reactivity of the nucleophilic potassium alumanyl 17. | ||
The potassium alumanyl complex, 17, could also be used to access an aluminium oxide ion, a compound that is of immense interest given the industrial use of aluminium oxide compounds in catalysis. The reaction of 17 with CO2 or phenyl isocyanate (PhNCO) gives the aluminium carbonate or carbamate products 21 and 22, respectively (Fig. 6).41 Formation of both products was proposed to proceed via an aluminium oxide intermediate, though this could not be observed experimentally.
The aluminium oxide was therefore targeted directly; reaction of 17 with N2O at low temperature in tetrahydrofuran (THF) gives the THF ligated aluminium oxide 23 (Fig. 7). Single crystal XRD reveals a dimeric structure with two Al–O bond lengths measuring ∼1.68 Å, assigned as Al–O single bonds on the basis of DFT calculations which support the description of a highly polarised terminal bond. Compound 23 reacts with both CO2 and PhNCO at room temperature, to give the aforementioned products 21 and 22, though no mechanistic investigation was conducted. Compound 23 was also able to react with H2 at room temperature and pressure, to give the aluminium hydride hydroxide 24, the first isolated example of dihydrogen activation across an Al–O bond.
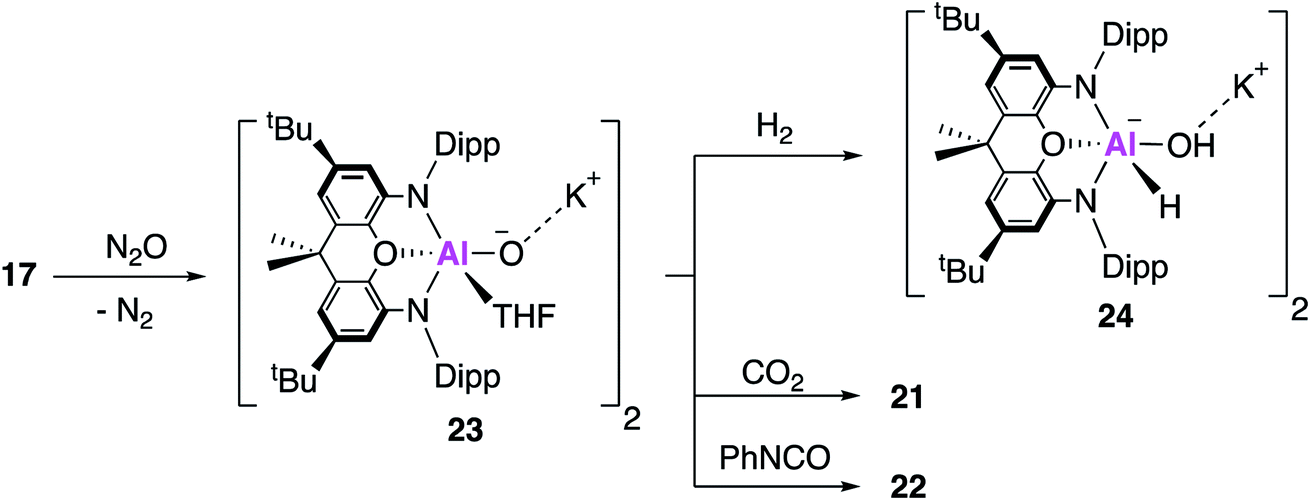 | ||
| Fig. 7 The formation of the aluminium oxide 23 and its subsequent reactivity with small molecules. | ||
In related work, it was proposed that targeting the analogous imide complex, with a more shielded and less polarised Al–E bond, would lead to a greater degree of stability and reaction control than was offered by the aluminium oxide 23. The imide complex, 25, was synthesised from the reaction of 17 with the sterically hindered and less σ-donating 2,6-diisopropylphenylazide (DippN3) (Fig. 8); use of the smaller trimethylsilylazide led to the formation of the tetrazene complex or benzylic C–H activation of toluene.42 X-ray crystallographic analysis revealed 25 to be a centrosymmetric dimer, comprised of two anionic [(NON)Al(NDipp)]− units bridged by K+ counterions, with Al–N bond lengths of 1.723(2) Å. Whilst the authors highlight this short bond length may suggest multiple bond character, they rule it out based on DFT which suggests the Al–N bond was best described as a highly polarised σ-bond. The propensity of 25 to activate small molecules was probed using dihydrogen and carbon monoxide. The reaction with H2 proceeded in an analogous fashion to 23, but only after heating, to form the (amido)aluminium hydride 26. Exposure of 25 to one atmosphere of carbon monoxide gave 27 as the sole product, formally derived from the assimilation of two CO molecules. The X-ray structure showed that the C–O bond lengths were consistent with the formation of a carboxylate bound through oxygen and the presence of a CN double bond at the exocyclic carbon. This was proposed to proceed via carbon–oxygen triple bond cleavage and C–C bond formation; detailed DFT suggest an end-on approach of the first CO molecule to form a cyclic three-membered Al–N–C intermediate. The rate determining step was calculated to be isomerisation of this intermediate, forming a second intermediate where DippNCO was bound via the CO π-bond. CO bond cleavage led to a third intermediate, classed as the DippNC adduct of the analogous oxide [(NON)AlO]−, from which a barrierless assimilation of a further molecule of CO formed 27.
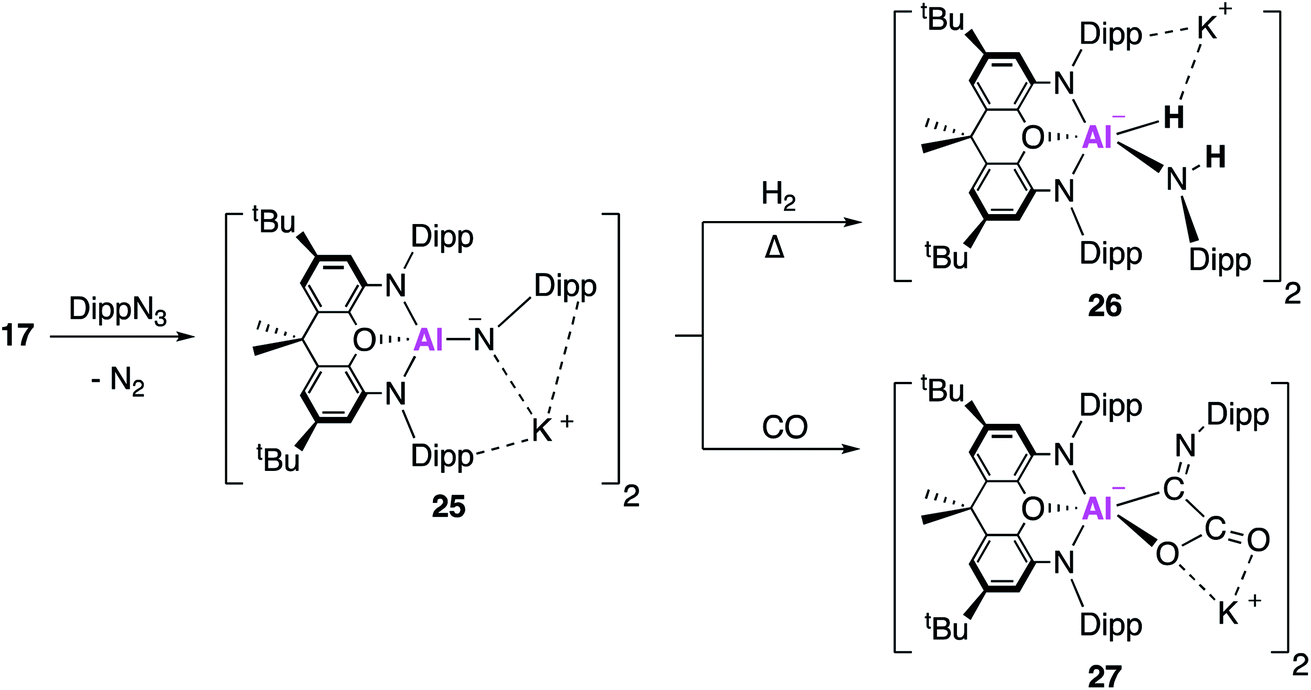 | ||
| Fig. 8 The formation of the aluminium imide 25 and its reactivity with CO2 and H2. | ||
With the reactivity of 17 as a nucleophilic source of aluminium investigated extensively, a structurally analogous mononuclear species was targeted in order to study the effect of complex aggregation. The resultant monomeric species (28, Fig. 9) was synthesised via the addition of the K+ sequestering agent [2.2.2]cryptand to 17.43 X-ray analysis revealed no short Al–Al or K–Al contacts whilst the NMR spectra exhibited characteristic differences compared to 17 and free [2.2.2]cryptand. Despite these differences, compounds 17 and 28 were both found to possess similar coordination geometries at the aluminium centre, consistent with the retention of an Al(I) species. Hence, 28 was best described as solution separated ions of both [K(2.2.2)crypt]+ and [(NON)Al]−. Assessment of the reactivity saw significant divergent reactivity between 17 and 28. Reaction of 28 with benzene was found to proceed at room temperature and resulted in the activation of an aromatic C–C bond, to give 29. This is in direct contrast to the observed reaction with 17, which yielded 19via the formal oxidative cleavage of a benzene C–H bond (vide supra) and mirrors reactivity observed in photolysis reactions between 6-membered aromatic molecules and a dialkylsilylene.44 The resultant seven membered metallocycle, [AlC6H6] (29), had Al–C bond lengths consistent with a vinyl-aluminium species (1.980(2) Å), in conjunction with alternating double and single C–C bond lengths. The reversibility of C–C bond activation was probed by reacting complexes 28 and 29 with naphthalene. Both reactions yielded the 1- and 2- C–H activated naphthyl products in roughly equal ratios, supporting reversible C–C bond activation. Calculations revealed that the C–H activation of benzene was a significantly more exergonic process than C–C bond activation (−34.9 and −4.1 kJ mol−1), thus 29 forms as the kinetic product. Compound 29 was calculated to form from 28via a bicyclic intermediate from an initial [2 + 1] cycloaddition of benzene at the Al centre. The high energy HOMO of 28 is believed to facilitate the initial [2 + 1] cycloaddition step. In contrast, the same orbital in compound 17 was significantly stabilised by the dimeric K+-bridged [K(NON)Al]2 units, thus accounting for the observed pathways of reactivity. Expanding this reactivity further, it was proposed that this C–C bond cleavage could be exploited to form acyclic complexes via the reaction of benzene with electrophiles. The reaction of 29 with tin halides was shown to proceed at room temperature and yielded a di-tin species bridged by an acyclic C6H6 fragment, with retention of the benzene stereochemistry (Fig. 9).
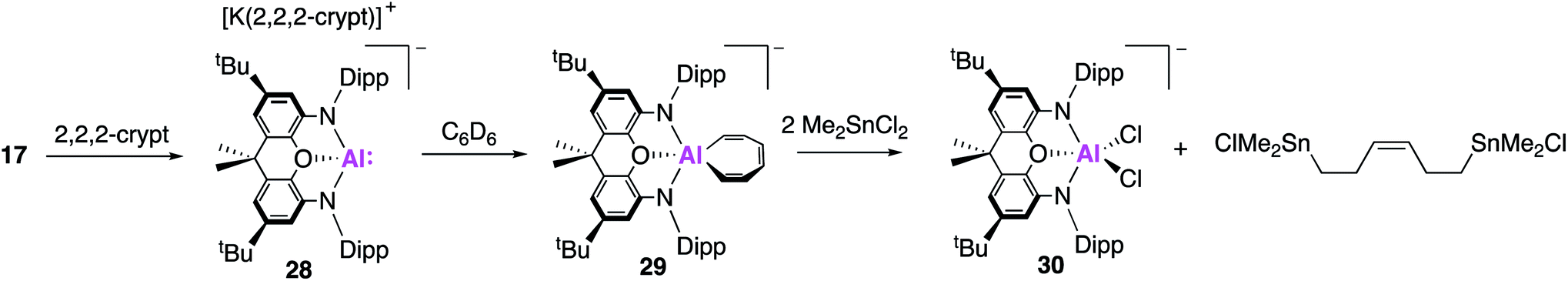 | ||
| Fig. 9 The formation of the monomeric alumanyl compound 28 and the C–C bond activation and functionalisation of benzene (29–30). | ||
Following on from the seminal isolation of NON–Al complexes, Coles and co-workers expanded this work by investigating the reactivity of a novel alumanyl anion with 1,3,5,7-cyclooctatetraene (COT).45 In an analogous fashion, the aluminium iodide (31) was reduced with potassium metal to form the alumanyl anion, 32, which exists as dimer containing two anionic [Al(NONAr)]− units (Fig. 10). Unlike in 17, the siloxane oxygen of the ligand backbone was found to be non-bonding, with Al⋯O distances of 3.356(2) and 3.418(2) Å and 32 can thus be assigned as a N-heterocyclic alumanyl anion. Reaction of 32 with COT led to the polymeric structure [K{Al(NONAr)-(COT)}] (33), the first [Al–COT] complex with significant aromatic character. The Al–C bond lengths from the metal centre to the COT ligand (2.061(3) Å and 2.071(3) Å) were found to be longer than in other known Al(III) compounds, confirming a η2-coordination mode. The crown ether, 18-crown-6, was added to a solution of 33 in an attempt to disrupt the polymer and as such the discrete [K(18-c-6)][Al(NONAr)(COT)] (34) monomer was isolated. Interestingly, the coordination mode of the COT ligand changed to form the addition product 9-aluminabicyclo[4.2.1]nona-2,4,7-triene, which is reminiscent of the reactivity of 2 and related cyclic alkenes.46
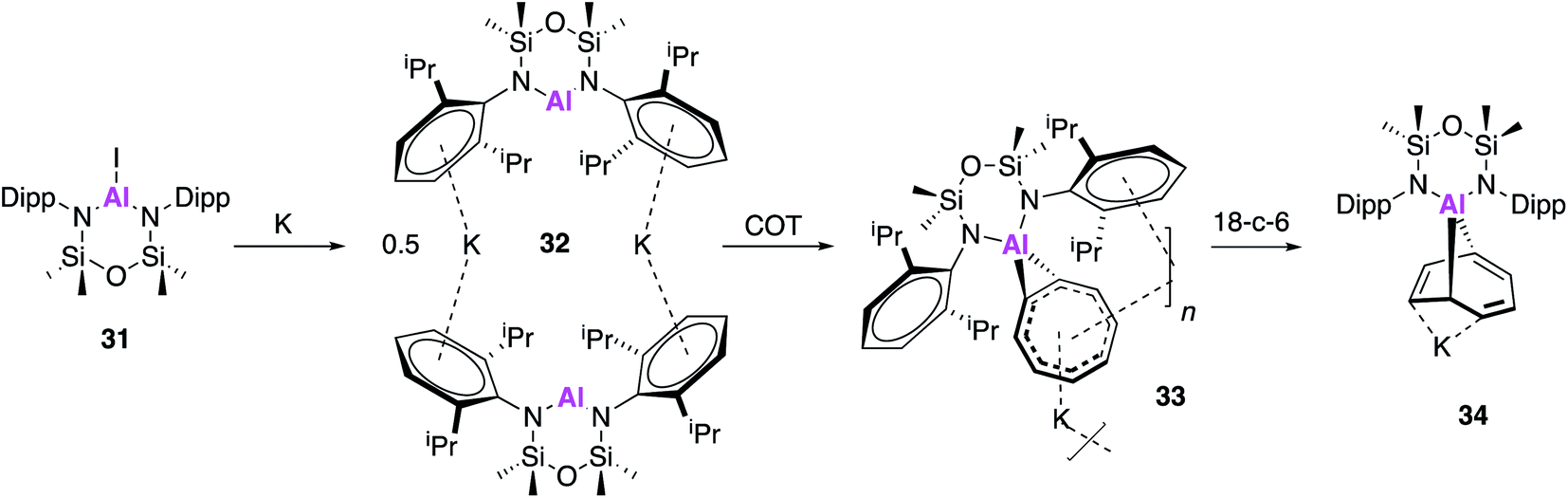 | ||
| Fig. 10 The synthesis and reactivity of the potassium alumanyl 32. | ||
In work published simultaneously to that of Aldridge and Goicoechea (17, Fig. 6–8), Coles and Anker sought to investigate the reactivity of 32 with CO2 and N2O. When 32 was exposed to one atmosphere of 13CO2 an analogous aluminium carbonate product 35 was observed to form via a [2 + 2] cycloaddition (Fig. 11).47 In an attempt to isolate the proposed aluminium oxide intermediate, 32 was also exposed to one atmosphere of N2O which resulted in formation of the dimer 36, which comprised of [Al(NONAr)(O)]− anions and K+ counterions. The terminal Al–O bonds (1.6362(14) Å) were shorter than those of the related aluminium monoalumoxane species,48 and slightly shorter than those of compound 23. The authors suggest this indicates multiple bond character, however both the calculated Wiberg bond index and NPA charges suggest ionic single bond character (note: 23 is assigned as an Al–O single bond). 36 reacts with 13CO2 to form 35, supporting the hypothesis that a terminal Al–O bond is an intermediate in the formation of the carbonate species; this is analogous to reactivity observed with 23 to form 21. Compound 36 was also found to react further with N2O in the presence of [2.2.2]cryptand to yield 37via a cycloaddition across the Al–O bond. These results suggest that the structurally analogous Al–O bonds of 23 and 36 may be described as having both single and multiple bond character.
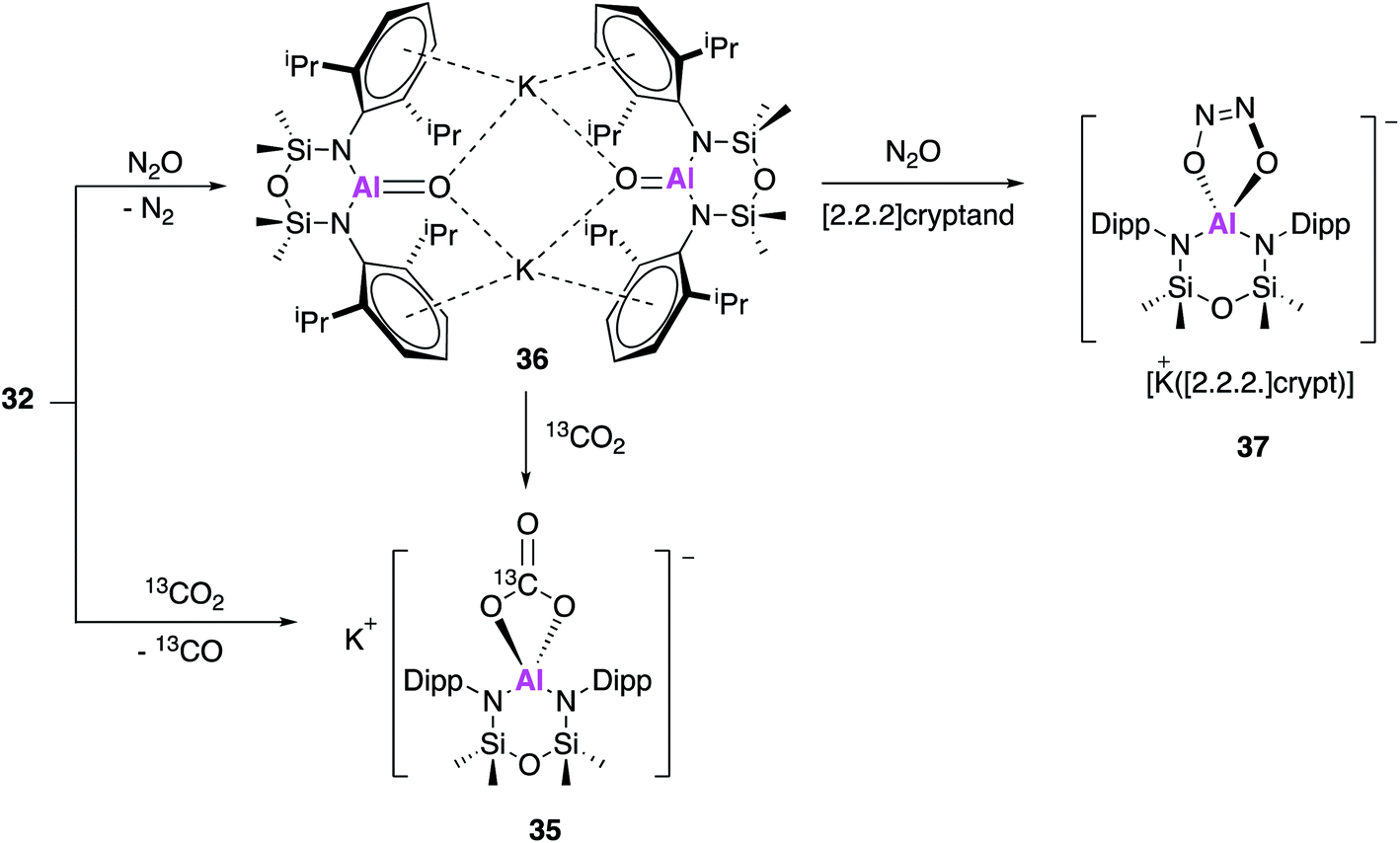 | ||
| Fig. 11 The reactivity of potassium alumanyl 32 with N2O and CO2. | ||
Once again in simultaneously published research to 17, 32 was reacted with mesityl azide, to yield an analogous three-coordinate aluminium complex containing a terminal aluminium-imide (38, Fig. 12).49 Again, single crystal XRD showed that 38 crystallised as a dimer, comprising of the aluminium imide salt K[Al(NONAr)(NMes)] with an Al–N bond length of 1.7251(11) Å. This is comparable to the bond length in 25, which was described as an Al–N single bond. DFT was used to probe the electronic structure of 38 and as with 25, the WBI and NPA suggest a significant ionic component, however Kohn–Sham molecular orbitals indicate π-character. Therefore 38 (and 25) may be better considered in resonance between Al−N and Al–N−. With analogous reactivity to 36, exposure of 38 to an atmosphere of CO2 saw complete conversion to the aluminium carbamate complex 39, which was proposed to form via a [2 + 2] cycloaddition between the aluminium–nitrogen bond of the imide with a CO of carbon dioxide.
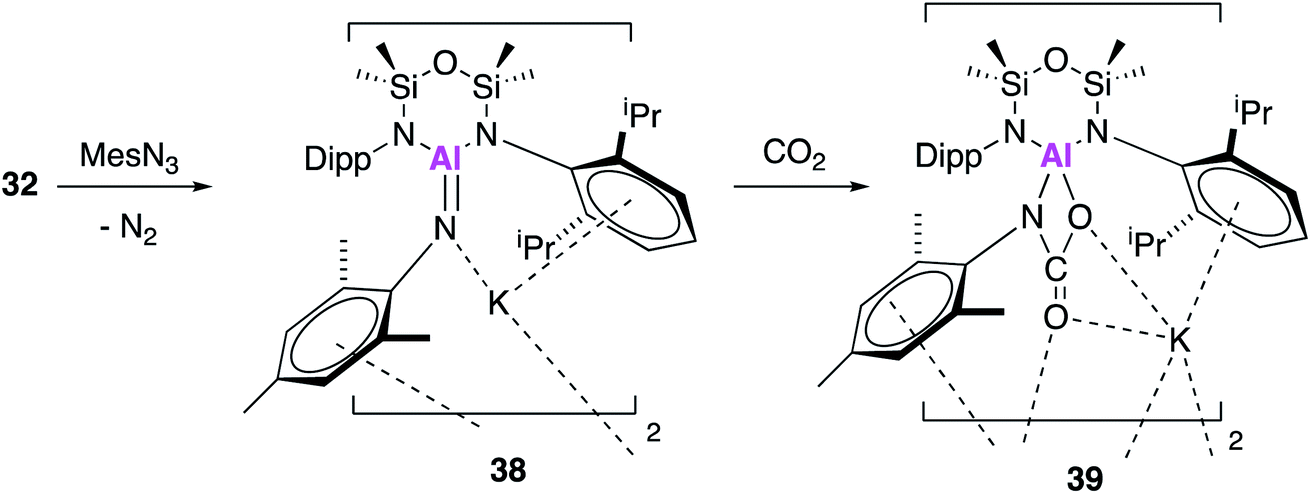 | ||
| Fig. 12 The reaction of potassium alumanyl 32 with mesityl azide. | ||
Compound 32 has also been used to access isoelectronic ‘heavy’ analogues of carbonyl functionalities.50 Reaction with one equivalent of selenium and catalytic amounts of tri-n-butylphosphine, as a selenium transfer agent, yielded the aluminium selenide (40, Fig. 13). Single crystal XRD revealed [Al(NONAr)(Se)]− anions linked to [K(THF)]+ cations through Se–K and π-arene interactions. The AlSe bond length (2.2253(11) Å) was in good agreement with the sum of the molecular covalent double bond radii and shorter than terminal Al–SeH bonds. As with previous reactivity (Fig. 10 and 11), a sequestering agent ([2.2.2]cryptand) could be used break the coordination polymer and isolate AlSe free from through space interactions. Compound 41 adopted a distorted trigonal planar geometry and a terminal selenium atom which exhibited the shortest Al–Se bond length ever reported (2.2032(6) Å). This was then used to target the selenium analogue of a dioxirane, by reaction with a further equivalent of selenium to afford 42. Interestingly, 42 could also be obtained directly from the reaction of 32 with two equivalents of selenium and in the presence of [2.2.2]cryptand. The anionic components of 41 and 42 were probed computationally, and the calculated Al–Se bond lengths were found to be in good agreement with those observed experimentally. The Wiberg bond index of the Al–Se bond in 41 was calculated to be greater than that of an analogous Al–S bond indicating the presence of multiple bond character. This was further corroborated upon analysis of the orbitals whereby the HOMO was found to correspond to a π-symmetry lone pair on selenium, and the HOMO−1 contained lobes orthogonal to the HOMO extended across the Al–Se bond. The ability of this family of aluminium compounds to access such structures undoubtedly comes from the anionic nature of the aluminium fragment, allowing stabilisation of these isoelectronic heavy carbonyl species. Overall, investigations conducted with the alumanyl 32 emphasise the closely related reactivity with that of 17, indicating that the differing ligand structure is having a negligible effect on the aluminium centre.
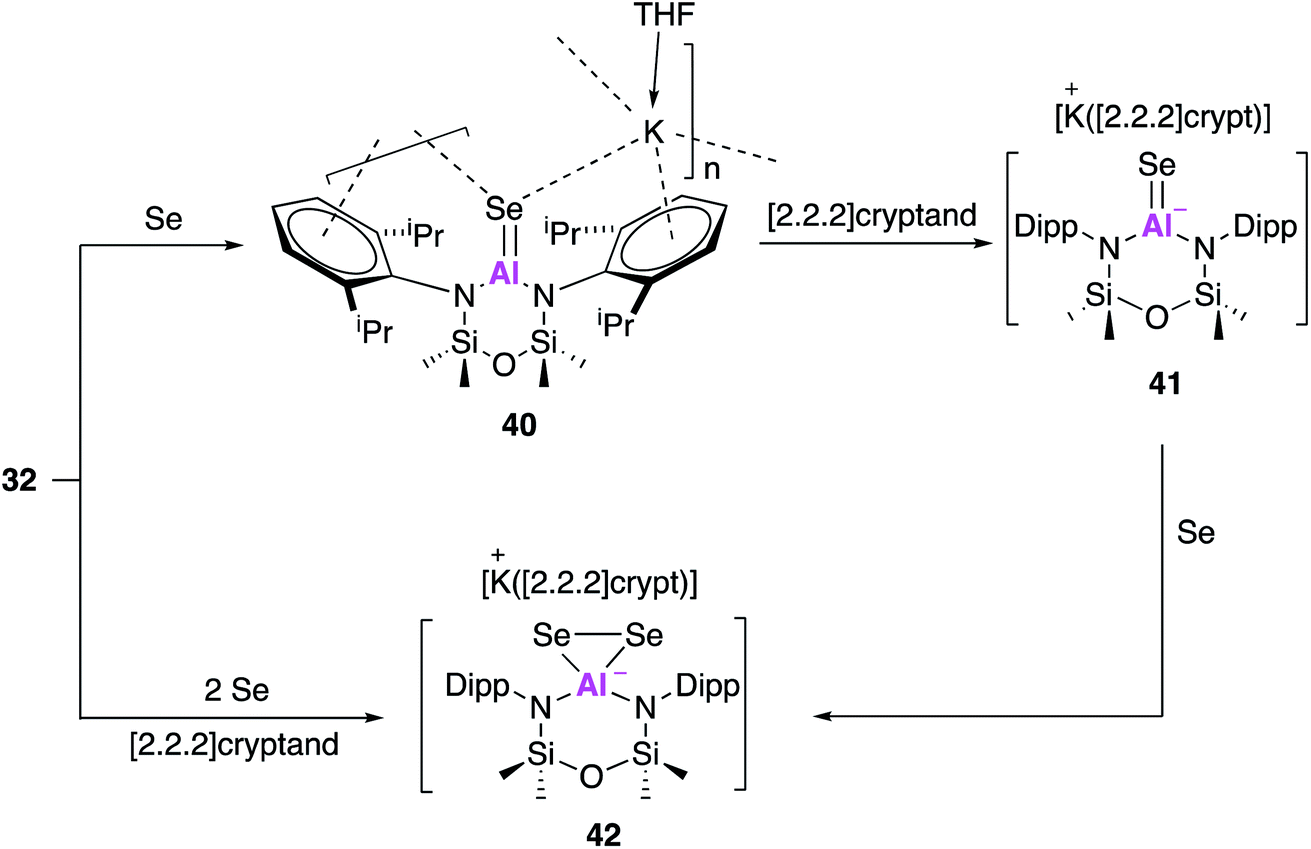 | ||
| Fig. 13 The reaction of potassium alumanyl 32 with selenium and related reactivity. | ||
In related work by Hill and co-workers, this class of nucleophilic aluminium reagents was expanded to include a seven-membered N,N′-heterocyclic potassium alumanyl species (44), which does not contain a bonding moiety in the ligand backbone.51 Compound 44 was synthesised analogously to 32, via reduction of the aluminium iodide 43 over a potassium mirror (Fig. 14). Drawing inspiration from the reactivity of Roesky's [(DippBDI)Al] complex with related β-diketiminate complexes,52–54 the propensity of 44 to form metal–metal bonds with group 2 complexes was investigated. Alkaline earth alumanyls of the form [{SiNDipp}Al–M(DippBDI)] (M = Mg, 45; Ca, 46) were synthesised upon addition of 44 to metal tetraphenylborate derivatives ([(DippBDI)MBPh4]; M = Mg or Ca). Single crystal XRD showed both complexes to comprise of an alkaline earth-aluminium bond, with each metal centre adopting a three-coordinate geometry. The Mg–Al bond length (2.798(6) Å) was longer than that of [(NONAr)AlMg(mesBDI)] (2.696(1) Å), formed by reaction of 17 and (mesBDI)MgI(OEt2), but comparable to the Mg–Al bond in the previous reported compound [(DippBDI)Al(Me)Mg(DippBDI)].52 The Al–Ca bond in 46 (3.1664(4) Å) was longer than the sum of covalent radii for the metal centres, but was found to be in good agreement with a previously calculated structure for [(DippBDI)Al(H)Ca(DippBDI)] (3.12 Å).53 Complex 46 contains an additional η6 interaction between the calcium and the aryl substituent of the aluminium-coordinated diamide ligand which were not observed in 45. DFT calculations were used to probe each structure further, and Pipek–Mezey orbital calculations revealed an intrinsic polarisation to both Mg–Al and Ca–Al σ-bonds. Natural bond orbital (NBO) calculations showed the Al–Ca interaction of 46 was slightly more ionic than the corresponding Mg–Al bond. Initial investigations into the reactivity of complexes 45 and 46 revealed that neither species reacted with aromatic solvents. Compound 45 remained stable whilst suspended in ethereal solvents but addition of THF to 46 in methylcyclohexane yielded a charge separated species comprised of [(DippBDI)Ca(THF)3]+ cations and [(SiNDipp)AlO(CH2)4]− anions. This was formally described as the oxidative addition product of a THF C–O bond to the Al(I) centre. Similarly, reaction of 46 with 1,3,5,7-cyclooctatetraene (COT) resulted in the two-electron aromatisation of the COT species and formed a heterobimetallic inverse sandwich species, whilst 45 was found to be unreactive. As the reactivity of 44 with small molecules such as H2 and CO2 has, to date, not been reported, it is difficult to draw conclusions on the effect of ligand variation compared to 17 and 32. However, similarities in Al–Mg bond formation suggest parallels in reactivity are to be expected.
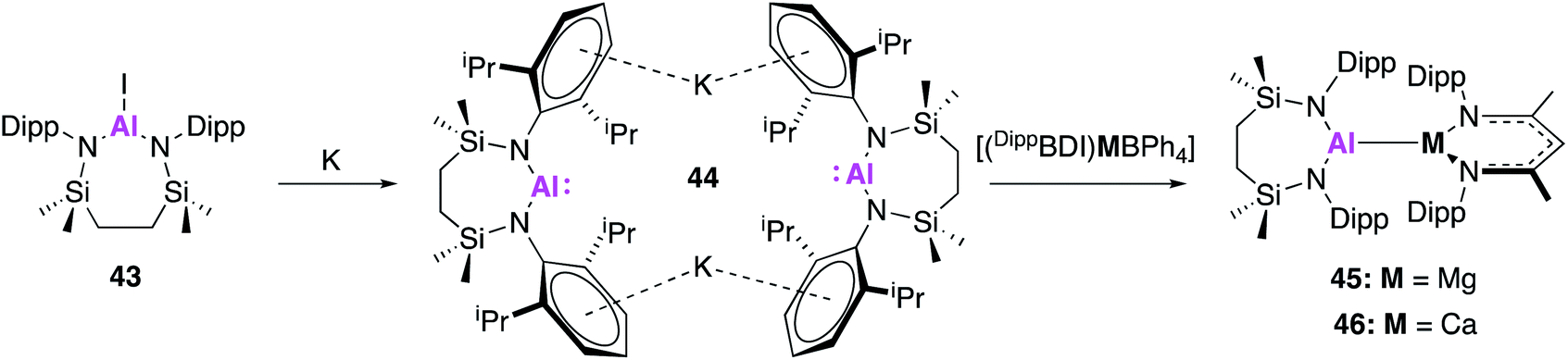 | ||
| Fig. 14 The synthesis of the potassium alumanyl 44 and subsequent formation of Al–M bonds. | ||
All examples of base stabilised nucleophilic aluminium species discussed thus far contain nitrogen ligand systems, that offer additional stabilisation to the metal centre. Yamashita and co-workers proposed to explore the possibility of creating an ‘electronically unbiased’ group 13 anion, rationalising the tetrakis(trimethylsilyl)butylene ligand system to be an appropriate choice given its successful implementation in silylene chemistry.55,56 The corresponding aluminium iodide 47 could be synthesised in a two-step process from 1,1-bis(trimethylsilyl)ethylene, firstly by lithiation to form an etherate of 1,4-dilithio-1,1,4,4-tetrakis(trimethylsilyl)butane, followed by a salt metathesis with aluminium triiodide. Compound 47 was reacted with excess lithium to form the dialumane 48, with subsequent treatment with sodium–potassium alloy (NaK) resulting in the formation of the potassium alumanyl 49 (Fig. 15). Single crystal XRD found 49 to exist as a monomeric Al–K complex, in contrast to the dimeric structures previously reported (17, 32, 44). The Al–K bond of length (3.4549(5) Å) was slightly longer than the sum of the covalent radii, 3.28 Å, and much shorter than observed in 17, 32 and 44.38,45,51 The Al–C bond lengths in 49 were longer than in 48 and the C–Al–C bond angle was wider, indicating a less electropositive Al centre. DFT analysis revealed the HOMO to be the lone pair of electrons centred mainly on the aluminium and the lowest unoccupied molecular orbital (LUMO) to be comprised of the π* orbitals of the potassium coordinated toluene molecules. The vacant 3p orbital of Al was found in the LUMO+8. The HOMO was higher in energy and the HOMO–LUMO gap was found to be significantly smaller than in 17 and 32. This was proposed to be due to the absence of electron withdrawing nitrogen atoms and the associated Al–N pπ–pπ interactions in addition to the presence of the low-lying π* toluene orbitals. Both natural population analysis (NPA) and atoms in molecules (AIM) analysis show the Al–K bond in 49 can be considered as highly polarized. Compound 49 was found to deprotonate benzene under mild conditions (2.5 h, r.t.) to give the C–H oxidative addition product 50, significantly faster than the related reaction between 17 and benzene (60 °C, 4 days). Reaction with methyl triflate lead to compound 51, demonstrating the nucleophilicity of the aluminium centre. Interestingly, 49 was also found to activate C–F bonds in hexafluorobenzene, undergo nucleophilic substitution with the chloride atom in benzyl chloride and to facilitate [1 + 4] and [1 + 2] cycloaddition reactions with unsaturated hydrocarbons (Fig. 16).57 For instance, reaction of 49 and naphthalene gave the 1 + 4 cycloaddition product, 52. Reaction of 49 with (E)- or (Z) stilbene gave the same trans-diphenylaluminacyclopropane [1 + 2] cycloaddition product 53, with the trans-selectivity being confirmed by both single crystal XRD and 1H NMR analysis. DFT shows (E)-stilbene to react directly via a concerted pathway, but reaction of (Z)-stilbene proceeds via the formation of carbanionic intermediate containing a single C–C bond. This facilitates rotation around the C–C bond leading to the more thermodynamically favourable trans-product.
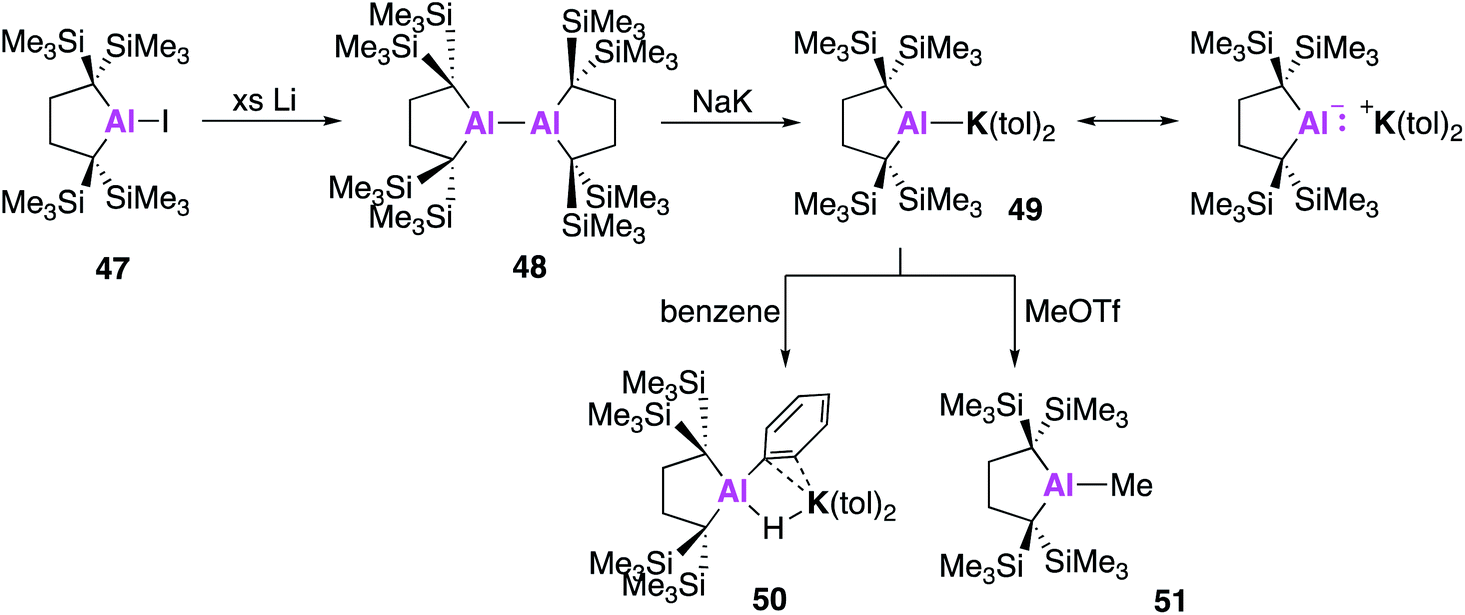 | ||
| Fig. 15 The synthesis of potassium alumanyl 49 and reactivity with benzene and MeOTf. | ||
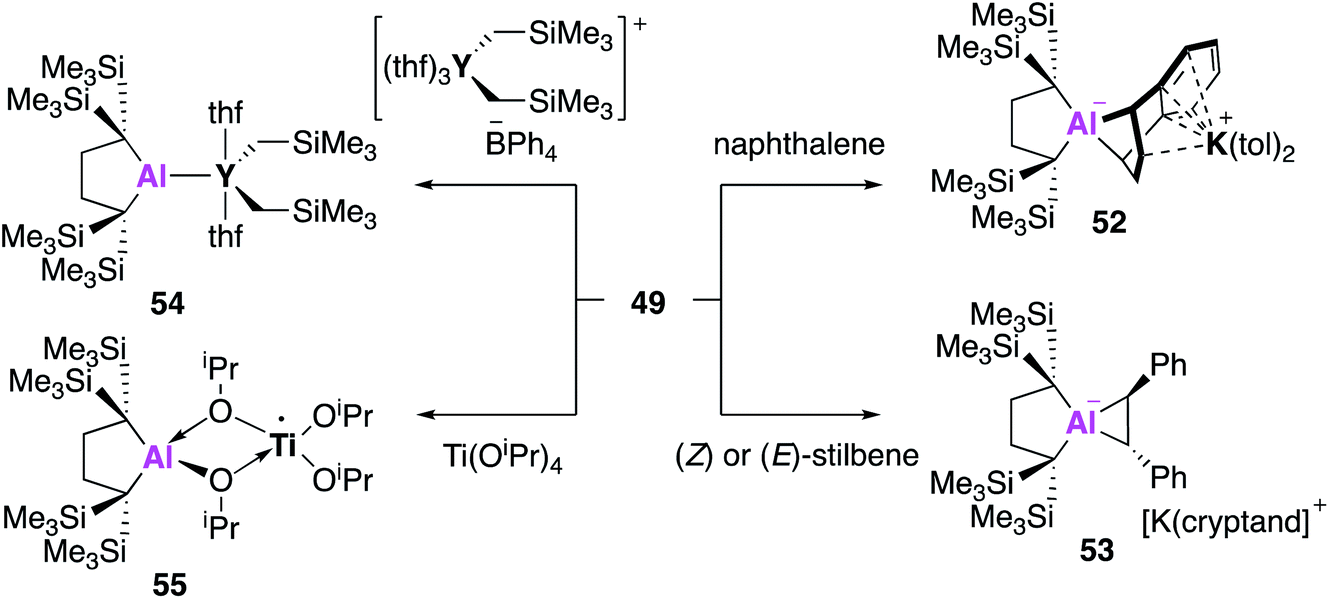 | ||
| Fig. 16 The reactivity of potassium alumanyl 49. | ||
Reaction of 49 could also be extended to the formation of unsupported metal–metal bonds.58 Reaction of 49 with [Y(CH2SiMe3)2(THF)3][BPh4] led to the formation of 54, the first example of a single aluminium–yttrium bond. The aluminium–yttrium bond length was found to be 3.1870(8) Å and the aluminium centre exhibited trigonal planar geometry. DFT was used to determine the electronic structure of 54, with the HOMO representing the 2c–2e Al−–Y+ bond and the LUMO overlapping vacant 3p and 3d-orbitals. AIM calculations reveal a stronger Al–M bond in 54 than in 49. In further reactions with metal precursors, 49 was also recently found to reduce titanium tetraisopropoxide to the trivalent titanium complex 55.59 Overall, 49 has been shown to have enhanced reactivity towards molecules such as benzene than the dimeric alumanyl compound 17 (reactivity of 32 and 44 not reported) and a broad reaction scope. The use of the tetrakis(trimethylsilyl)butylene ligand system has led to a monomeric structure with a reduced HOMO–LUMO gap, leading to this heightened reactivity. However, recent work by Harder and co-workers has shown an anionic potassium alumanyl, formed via deprotonation of the BDI ligand in 2 with KN(SiMe3)2, to be capable of benzene C–H activation.60 Here, the potassium anion was found to stabilise the transition state, thus the role of potassium and possibility of bimetallic mechanism should be explored across this family of compounds.
Even further extension of the structural diversity of Al(I) anions was subsequently reported by Kinjo and Koshino, who targeted an aluminium analogue of a cyclic (alkyl)(amino)carbene (CAAC). CAACs are known to be more σ-donating and π-accepting than conventional NHCs and thus have the potential to have divergent activity from the aforementioned N,N-stabilised alumanyls (17, 32, 44) and the carbene analogue 49. In a similar fashion, the formation of the cyclic (alkyl)(amino)alumanyl anion (CAAAl) 58 was achieved by reduction of the aluminium bromide 56 with potassium graphite in an apolar solvent, which yielded the dialane compound 57 (Fig. 17).61 This was followed by addition of diethyl ether and three further equivalents of potassium graphite, followed by the addition of 12-crown-4 ether to give the alumanyl anion product 58. Compound 58 exists as a separated ion pair, similar to 28, with sp3 hybridised C and N atoms bound to the Al centre. DFT of the aluminium anion revealed the HOMO to be the lone-pair at Al and the empty p-orbital to be the LUMO+1. The HOMO was found to be higher in energy than that of compounds 28 and 49, though the HOMO–LUMO gap was larger. The reactivity of 58 was tested towards BH3·SMe2 and strong σ-bonds, the former leading to the formation of 59, which contains a trigonal AlB2 ring. This type of reactivity has never before been observed in the main-group and 59 is the first isolated example of an aluminium complex of diborane. Reaction of 58 with phenylsilane or ammonia led to oxidative addition of an E–H bond to the Al centre, leading to 60 and 61 respectively. Formation of 60 represents the first example of a hydridic Si–H activation by an anionic aluminium species, though related reactions are known with the neural compound, 2.22 Oxidative reactivity could also be extended to apolar σ-bonds and 58 was found to effect the C–C bond activation of biphenylene to form 62. This diverse reactivity indicates 58 has the potential to access a wide range of aluminium bound molecular fragments that may be used in further synthesis, although the substrate scope investigated to date does not allow for direct comparison with other alumanyl species.
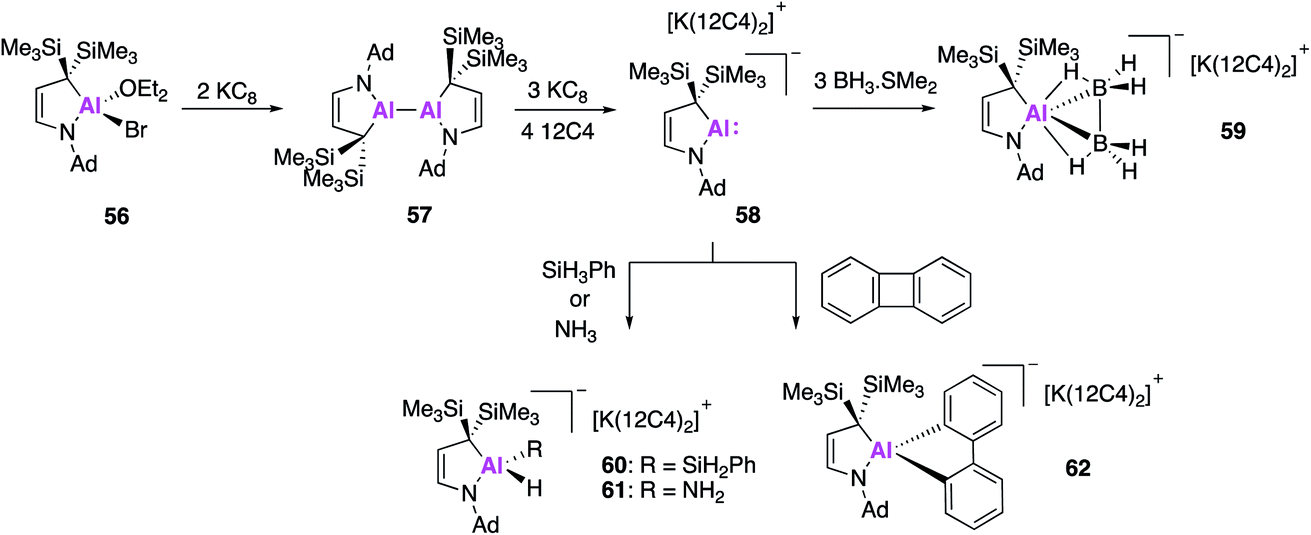 | ||
| Fig. 17 The formation of alumanyl 58 and its reactivity with small molecules. | ||
New monomeric aluminium(I) complexes
Whilst huge advances have been made in the development of alumanyl anions, the isolation of new examples of monomeric aluminium(I) compounds remains an attractive target, especially considering the broad and well-defined reactivity displayed by Roesky's β-diketiminate complex (2). As mentioned previously, Schnöckel's Al(I) complex (1) is known to exist as a tetramer both in the solid state and in solution at room temperature, though an equilibrium with the monomeric form can be achieved by varying reaction temperature. However, differing substituents of the Cp ring allows for perturbation of this equilibrium which is dependent on both sterics and temperature.62,63 The tetrameric structure can hamper the reactivity of 1, and over the years its use in small molecule activation has not been as widely explored as for 2, whose discrete solid and solution state structure and ready solubility in common organic solvents renders it more user friendly.Attempts to isolate monomeric forms of 1 by changing the sterics of the Cp ligand framework had not previously led to an easily isolatable monomeric Al(I) species. Accordingly, Braunschweig and co-workers targeted the formation of a monomeric Al(I) species that could be readily synthesised and isolated.64 The sterically demanding 1,3,5-tri-tert-butylcyclopenta-dienyl (Cp3tBu) ligand was employed, and reaction of (Cp3tBu)2Mg with AlBr3 saw the formation of the aluminium dibromide (Cp3tBu)AlBr2 (63, Fig. 18). Compound 63 was shown to be a monomer in the solid state, where as the Cp* analogue is dimeric. The low coordinate Al(I) species 65 was then isolated first by a conproportionation reaction with (Cp*Al)4 to form 64, followed by addition of a strong Lewis base (CAAC), after traditional reduction routes proved unsuccessful. The 27Al NMR spectrum showed a resonance at −161 ppm, indicative of monomeric Al(I) with no evidence of the tetramer and whilst formation of single crystals was not possible the 1H DOSY spectra also pointed towards a monomeric solution structure.
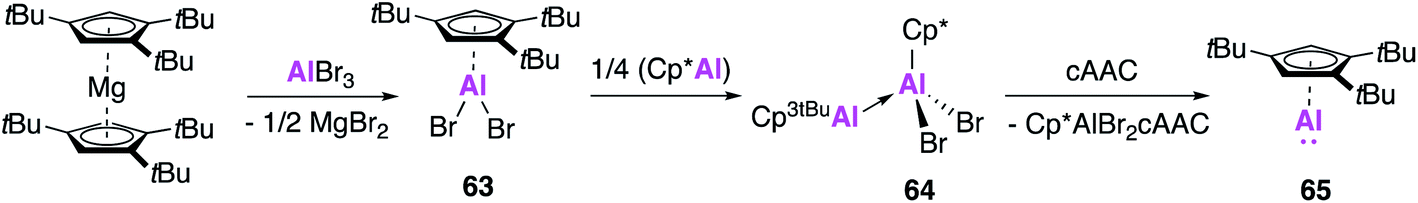 | ||
| Fig. 18 The synthesis of monomeric Al(I) species 65. | ||
The reactivity of 65 was investigated with a number of aluminium precursors and small molecules (Fig. 19). Analysis of the reaction of 65 with 63 indicates the presence of valence tautomerism between the Al(I)–Al(III) product 66 and the Al(II)–Al(II) product 67, which appear to exist in rapid equilibrium but can be selectively isolated at different temperatures. Conversely, reaction with tBuAlCl2 or AlBr3 only led to formation of the Al(I)–Al(III) Lewis acid–base adducts 68 and 69, respectively. In an effort to target a monomeric Al–O containing complex, 65 was oxygenated with N2O, however what instead formed was the 6-membered heterocycle 70, the formal oligomerisation product of Cp3tBuAlO. Further reaction of 65 with phenyl azide led to the formation of the imidoalane 71, which was found to be dimeric in the solid state but showed evidence of monomeric solution behaviour. These reactions all proceed faster and often with more control than analogous reactions with (Cp*Al)4, highlighting the benefits of using the monomeric Cp-based Al(I) in synthesis.
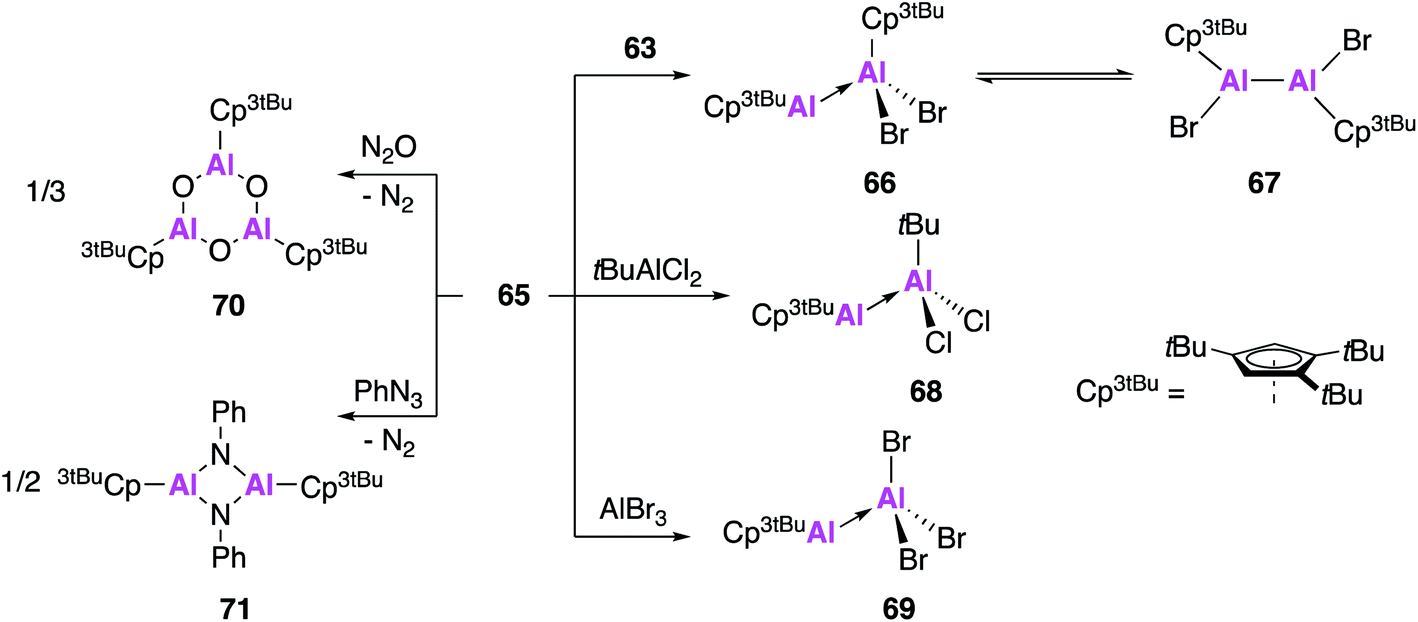 | ||
| Fig. 19 The reactivity of monomeric Al(I) species 65 with aluminium precursors and small molecules. | ||
Braunschweig and co-workers further extended the reactivity of 65, targeting compounds with direct unsupported aluminium–boron bonds (Fig. 20).65 Reaction of 65 with B(C6F5)3 also led to the formation of the acid–base adduct 72, for which the analogous product was observed with both (Cp*Al)4 (1) and 2.66,67 However, reaction with the other boron sources led to significantly divergent behaviour. The B–Cl bond of bis-2,6-(2,4,6-triisopropylphenyl)-phenyl dichloroborane was oxidatively added to 65, leading to the covalent bora–alane complex 73, which is possibly favoured over the adduct due to steric congestion at boron. The utility of 65 as a reducing agent was also demonstrated, with the redox formation of borylene–alane complexes. The reaction of 65 with the NHC stabilised boron species [(IMes)B(Br)NMe2]Br yielded the oxidative addition product 74, which forms with oxidation of aluminium and reduction of boron (Al(III)–B(I)). A redox reaction also occurred in the reaction of 65 with BI3, this time resulting from the rearrangement of substituents, leading to an AlI3 borylene adduct (75). This range of reactivity with boron underlines the highly versatile nature of 65, and the benefits of using a monomeric source of CpAl(I).
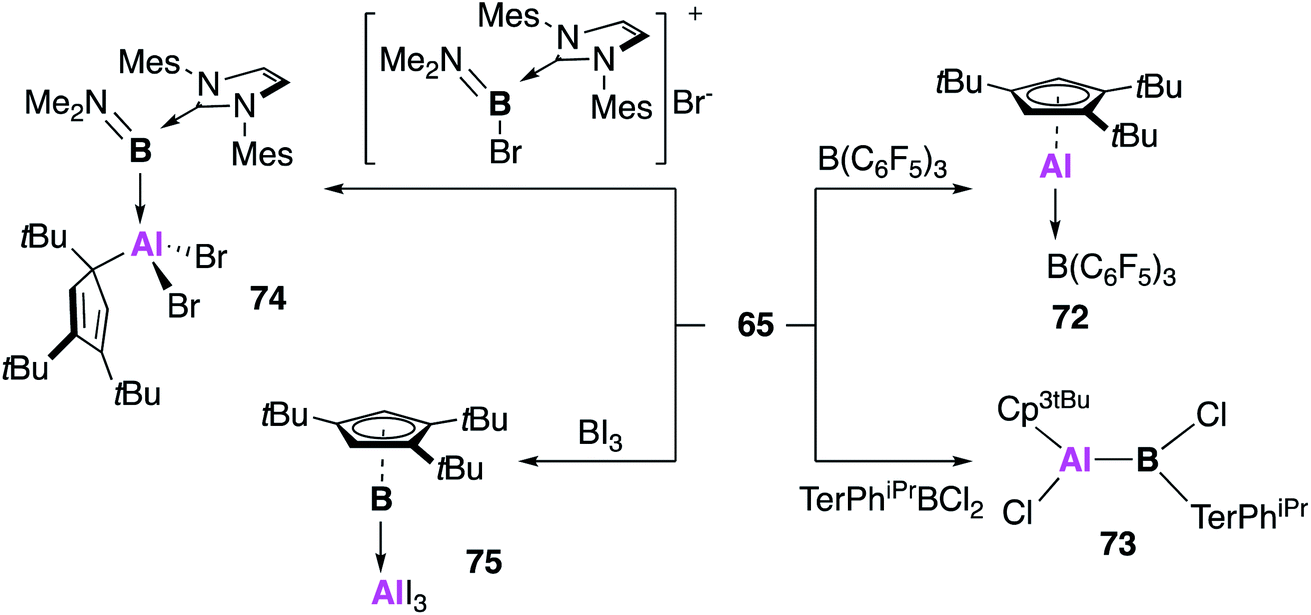 | ||
| Fig. 20 The reactivity of monomeric Al(I) 65 with boron(III) species. | ||
The isolation of aluminium(I) hydrides
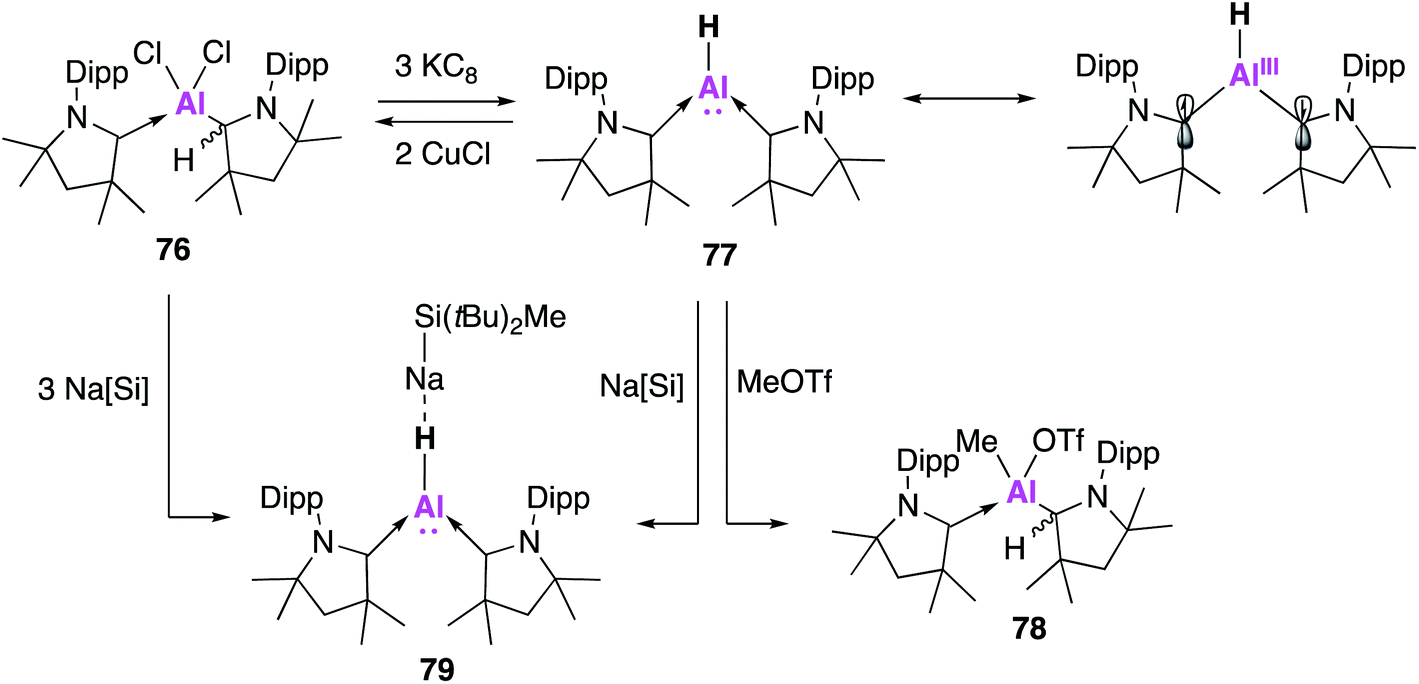
Whilst examples of some classes of Al(I) species are slowly becoming more established, the Al(I) hydride ‘aluminene’ has remained more elusive. Al(I)–H has previously been studied in a matrix, but attempts to isolate it synthetically have proven challenging.68 In 2018, Jones and co-workers reported the isolation of an Al6H6 cluster, formed through the reaction of a magnesium(I) dimer and an aluminium(III) dihydride.69 This was characterised in the solid state as a [Al6H6(HC(NDipp)2)2]2− dianion stabilised by two [BDIMg]+ cations and is the first example of a stable Al(I)–H containing complex. A lack of solubility in common hydrocarbons precluded solution state characterisation and to date further reactivity has not been reported. Driess and co-workers have also invoked the transient formation of an NHC stabilised complex aluminene bound to an iron carbonyl, but isolation of this species was not possible.70 Instead it readily partook in the C–H activation of the THF solvent.Braunschweig and co-workers sought to exploit the exceptional σ-donating and π-accepting properties of the cyclic (alkyl)(amino) carbene (CAAC) ligand system in order to stabilise an aluminene fragment under the harsh reaction conditions required.71 Reduction of the alkyldichloroalane precursor (76) with potassium graphite (KC8) saw the ready formation of an aluminene species stabilised by two CAAC ligands (77, Fig. 21). Both the hydride and deuteride analogues could be synthesised, with the hydride transferred from the protonated CAAC ligand as confirmed by deuteration experiments. This reaction was remarkably high yielding (70%) when compared with other monomeric Al(I) syntheses. Compound 77 was found to be centrosymmetric in the solid-state, with a trigonal planar geometry at the aluminium centre with a highly electron rich hydride bound to aluminium. DFT calculations revealed that 77 was best described as an Al(I) hydride with a non-negligible contribution (approximately 1/3) from an open-shell Al(III) diradical resonance form (Fig. 21). Despite the nucleophilicity of 77, its reaction with a series of small molecules, including H2, CO2 and CO, led to a variety of decomposition products; this is marked contrast to most other examples of low oxidation state aluminium complexes. However, methyl triflate (MeOTf) underwent nucleophilic substitution at aluminium to form 78. Nucleophilic attack at the methyl group is proposed to form an unstable Al(III) cation, thus the Al-bound hydride migrates back to a CAAC centre. The reversible 2-fold oxidation of 77 was observed in the presence of CuCl, whilst reaction with Na[SitBuMe2] formed the 1:1 complex 79 as determined by single crystal XRD. Interestingly, dissolution of single crystals of 79 in C6D6 revealed only resonances of free 77 and Na[SitBuMe2], suggesting that 79 formed via the co-crystallisation of 77 and the sodium silane, which was likely facilitated by electrostatic interactions between the Al–H and the sodium atom.
 | ||
| Fig. 21 The synthesis of CAAC stabilised aluminene 77 and its reactivity. | ||
Conclusions and outlook
Over just the last four years a wealth of new research into the low oxidation state chemistry of aluminium has emerged. This has largely been possible through the rational design of complexes, with judicious choice of the ancillary ligands which are essential to enabling stabilisation of these highly reactive compounds. This research has included the formation of the first aluminium–aluminium double bond (4), isolation of several examples of alumanyl anions (17, 32, 44, 49 and 58) and the first isolatable example of an aluminene species (77). These highly unusual compounds have demonstrated unique reactivity towards the activation of small molecules and have enabled the formation of both bimetallic and heterobimetallic compounds. In particular, Aldridge and Goicoechea's potassium alumanyl species has shown utility in a wide range of bond activation processes and has been used to provide access to unusual chemical structures such as the seven membered metallocycle formed through the C–C bond activation of benzene. Aluminium–aluminium doubly bonded compounds have also proven to be efficient pre-catalysts for the hydroboration of CO2 as well as the dehydrogenative coupling of amine boranes. This remarkable breadth of reactivity is largely driven by the ready accessibility of the frontier molecular orbitals (FMOs), with combinations of high energy HOMOs/low-lying LUMOs leading to narrow HOMO–LUMO gaps. These FMOs are able to be tuned by the employment of ligands with different electronic and steric properties, with lessons taken from other aspects of main-group and transition metal chemistry. Here, a careful balance must be struck between complex stabilisation and facilitating access to the powerful reaction potential of these compounds. Through expansion of the scope of ligands used to stabilise low oxidation state aluminium complexes we can expect to delve further into the chemical capabilities of such molecules.That said this is a challenging pursuit. Nearly 30 years after Schnöckel reported the first stable Al(I) molecule, there remain only two types of mono-anionic ligand capable of stabilising monomeric Al(I) centres. This will continue to be a valuable synthetic target due to their diverse and well-defined reactivity; employing ligands that are not susceptible to further reaction (β-diketiminates are known to undergo deprotonation/bond activation) will be particularly important in this regard. Additionally, whilst the group of low oxidation state aluminium compounds documented herein has found utility in a large range of bond activation processes, the further reactivity and transfer of the molecular fragments formed has yet to be much explored. The development of aluminium chemistry lags much behind boron in this respect, which is widely used to deliver organic building blocks for the formation of more complex molecules across academia and chemical industry. Further challenges within the field include creating synthetic protocols that are cheap, accessible and yield high quantities of the reagent, something which has hindered the more universal use of low oxidation state aluminium as a reagent. This will also assist with establishing the more wide-spread use of such aluminium compounds as stoichiometric reducing agents. Finally, the isolation of stable Al(I) species presents obvious questions about whether aluminium can be used in redox based catalysis. Examples of reversible redox reactivity at aluminium centres provide some hope in this respect, but without the presence of additional empty orbitals or vacant coordination sites it is difficult to envisage how transition metal like redox catalysis could be achieved; thus, further complex development is undoubtably required to reach such heights. Nevertheless, with such landmark discoveries made on a yearly basis, the future of aluminium chemistry looks very bright indeed.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the Ramsay Memorial Trust and UCL Chemistry for their support (CB). KH thanks UCL Chemistry for providing a departmental studentship. The EPSRC is thanked for funding via the Impact Acceleration Account award to UCL 2017–20 (EP/R511638/1) and for grant EP/L017709 (CJC).References
- R. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- G. E. Totten and D. S. MacKenzie, Handbook of Aluminum: Volume 2: Alloy Production and Materials Manufacturing: Alloy Production and Materials Manufacturing, 2003, vol. 2 Search PubMed.
- S. Saito, Main Group Metals in Organic Synthesis, Aluminium in Organic Synthesis, 2004 Search PubMed.
- G. A. Olah, V. P. Reddy and G. K. Prakash, Kirk-Othmer Encyclopedia of Chemical Technology, Friedel-Crafts Reactions, 2000 Search PubMed.
- R. Mulhaupt, Macromol. Chem. Phys., 2003, 204, 289–327 CrossRef CAS.
- G. I. Nikonov, ACS Catal., 2017, 7, 7257–7266 CrossRef CAS.
- A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- J. Wu, T. Yu, C. Chen and C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS.
- W. Uhl, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 1113–1118 CAS.
- W. Hiller, K.-W. Klinkhammer, W. Uhl and J. Wagner, Angew. Chem., Int. Ed., 1991, 30, 179–180 CrossRef.
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed., 1991, 564–565 CrossRef.
- S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed., 1993, 32, 1729–1731 CrossRef.
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Chem. Commun., 2013, 49, 2858–2860 RSC.
- Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS.
- C. Cui, H. W. Roesky, H. G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452–2458 RSC.
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed.
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351–1364 RSC.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed.
- W. Uhl, Angew. Chem., Int. Ed., 1993, 32, 1386–1397 CrossRef.
- R. J. Wehmschulte, K. Ruhlandt-Senge, M. M. Olmstead, H. Hope, B. E. Sturgeon and P. P. Power, Inorg. Chem., 1993, 32, 2983–2984 CrossRef CAS.
- C. Pluta, K.-R. Porschke, C. Kruger and K. Hildenbrand, Angew. Chem., Int. Ed., 1993, 32, 388–390 CrossRef.
- R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS PubMed.
- R. J. Wright, M. Brynda and P. P. Power, Angew. Chem., Int. Ed., 2006, 45, 5953–5956 CrossRef CAS PubMed.
- R. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed.
- C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, Angew. Chem., Int. Ed., 2019, 58, 10961–10965 CrossRef CAS PubMed.
- C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- T. N. Hooper, M. Garcon, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 5435–5440 RSC.
- J. Hicks, A. Mansikkamaki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- J. Hicks, A. Heilmann, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 17265–17268 CrossRef CAS PubMed.
- A. Heilmann, J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 4897–4901 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 41, 11000–11003 CrossRef PubMed.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2002, 124, 3830–3831 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- C. Bakewell, M. Garcon, R. Y. Kong, L. O'Hare, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2020, 59, 4608–4616 CrossRef CAS PubMed.
- M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 18261–18265 CrossRef CAS PubMed.
- D. Neculai, H. W. Roesky, A. M. Neculai, J. Magull, B. Walfort and D. Stalke, Angew. Chem., Int. Ed., 2002, 41, 4294–4296 CrossRef CAS PubMed.
- M. D. Anker, R. J. Schwamm and M. P. Coles, Chem. Commun., 2020, 56, 2288–2291 RSC.
- M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 13452–13455 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- C. Bakewell, B. J. Ward, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 2348–2356 RSC.
- S. Brand, H. Elsen, J. Langer, S. Grams and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 15496–15503 CrossRef CAS PubMed.
- A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 1999, 121, 9722–9723 CrossRef CAS.
- K. Sugita, R. Nakano and M. Yamashita, Chem.–Eur. J., 2020, 26, 2174–2177 CrossRef CAS PubMed.
- K. Sugita and M. Yamashita, Chem.–Eur. J., 2020, 26, 4520–4523 CrossRef CAS PubMed.
- K. Sugita and M. Yamashita, Organometallics, 2020, 39(11), 2125–2129 CrossRef CAS.
- S. Harder, S. Grams, J. Eyselein, J. Langer and C. Färber, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006693.
- K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142(19), 9057–9062 CrossRef CAS PubMed.
- C. Dohmeier, E. Baum, A. Ecker, R. Koppe and H. Schnöckel, Organometallics, 1996, 15, 4702–4706 CrossRef CAS.
- H. Sitzmann, M. F. Lappert, C. Dohmeier, C. Uffing and H. Schnöckel, J. Organomet. Chem., 1998, 561, 203–208 CrossRef CAS.
- A. Hofmann, T. Tröster, T. Kupfer and H. Braunschweig, Chem. Sci., 2019, 10, 3421–3428 RSC.
- A. Hofmann, C. Pranckevicius, T. Tröster and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 3625–3629 CrossRef CAS PubMed.
- J. D. Gorden, A. Voigt, C. L. B. Macdonald, J. S. Silverman and A. H. Cowley, J. Am. Chem. Soc., 2000, 122, 950–951 CrossRef CAS.
- Z. Yang, X. Ma, R. B. Oswald, H. W. Roesky, H. Zhu, C. Schulzke, K. Starke, M. Baldus, H. G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2005, 44, 7072–7074 CrossRef CAS PubMed.
- G. V. Chertihin and L. Andrews, J. Phys. Chem., 1993, 97, 10295–10300 CrossRef CAS.
- S. J. Bonyhady, D. Collis, N. Holzmann, A. J. Edwards, R. O. Piltz, G. Frenking, A. Stasch and C. Jones, Nat. Commun., 2020, 1–6 Search PubMed.
- G. Tan, T. Szilvási, S. Inoue, B. Blom and M. Driess, J. Am. Chem. Soc., 2020, 136, 9732–9742 CrossRef PubMed.
- S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, I. Krummenacher, Q. Ye, V. Engel, B. Engels and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 16954–16960 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |