
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe key role of R–NHC coupling (R = C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species
Victor M.
Chernyshev
a,
Ekaterina A.
Denisova
b,
Dmitry B.
Eremin
 bc and
Valentine P.
Ananikov
*ab
bc and
Valentine P.
Ananikov
*ab
aPlatov South-Russian State Polytechnic University (NPI), Prosveschenya 132, Novocherkassk, 346428, Russia
bN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, 119991 Moscow, Russian Federation
cThe Bridge@USC, University of Southern California, 1002 Childs Way, Los Angeles, California 90089-3502, USA
First published on 19th June 2020
Abstract
Complexes of metals with N-heterocyclic carbene ligands (M/NHC) are typically considered the systems of choice in homogeneous catalysis due to their stable metal–ligand framework. However, it becomes obvious that even metal species with a strong M–NHC bond can undergo evolution in catalytic systems, and processes of M–NHC bond cleavage are common for different metals and NHC ligands. This review is focused on the main types of the M–NHC bond cleavage reactions and their impact on activity and stability of M/NHC catalytic systems. For the first time, we consider these processes in terms of NHC-connected and NHC-disconnected active species derived from M/NHC precatalysts and classify them as fundamentally different types of catalysts. Problems of rational catalyst design and sustainability issues are discussed in the context of the two different types of M/NHC catalysis mechanisms.
1. Introduction
Homogeneous metal catalysis is a highly valuable tool of modern organic synthesis. It is used for preparation of fine chemicals, pharmaceuticals and agrochemicals, transformations of natural compounds, syntheses of monomers, polymers and advanced materials.1 Metal catalysts facilitate hundreds of unique C–C and C-heteroatom bond-forming reactions under mild conditions and with high selectivities.2Tremendous progress in fine organic synthesis has been achieved owing to the development of well-defined, stable and easy to use precatalysts based on the complexes of transition metals with organic ligands.3 Important roles of these ligands are numerous: they stabilize active metal species, ensure their solubility, secure favorable electronic states of the metal center, provide sterically defined binding pockets for reagents and substrates, and control chemo-, regio-, stereo- and enantioselectivity during the reaction.4,5 In addition, multifunctional ligands can participate in metal–ligand cooperative catalysis,6 or purposefully change properties of a catalyst under the action of an external stimulus (pH, light, oxidation-reduction, etc.).4,5
Over the last three decades, N-heterocyclic carbenes (NHCs) have been increasingly appreciated as excellent ligands for metal catalysis.5,7–18 Global impact of N-heterocyclic carbene (NHC) ligands is comparable with the impact of phosphines in the 1970s through the 1990s.5 The main benefits of NHCs over phosphines and some other ligands are their relatively easy preparation, lower toxicity, high tunability of electronic and steric parameters, ability to incorporate many additional functions, and, undoubtedly, the enhanced stability of M/NHC complexes arising from strong metal–NHC bonding with a variety of transition metals.11,12,19 In addition, M/NHC complexes are typically less prone to reversible dissociation than complexes with phosphine ligands and are less sensitive to oxidation in solution.11,12
Nevertheless, M/NHC complexes may undergo decomposition during catalysis, with the cleavage of the metal–NHC bond despite its high strength.20–25 Reductive elimination of NHC ligands was described by Cavell and co-workers.26 It was noted that the processes of M–NHC bond cleavage may cause deactivation of M/NHC catalytic systems.20–23,25
However, it has been recently demonstrated that M–NHC bond cleavage may also produce “ligandless” active metal species and thus can be considered as M/NHC precatalyst activation.27 Further studies revealed a diverse range of metal species without NHC ligands in a variety of M/NHC-catalyzed reactions and these literature will be considered in the present review.
Indeed, as one may expect, the reactions of metal–NHC bond cleavage have a great impact on the activity and stability of M/NHC catalytic systems. The processes of catalyst evolution via reductive elimination of NHC ligands (known as R–NHC couplings) drastically change the nature of catalytically active species. Other types of M–NHC bond cleavage were also described and may affect ligand environment of metal centers. In-depth analysis of literature including recent publications shows a number of R–NHC bond formations and several types of M–NHC bond cleavage reactions, which can have remarkable importance for catalysis. We believe the time is ripe now to analyze the impact of these processes on catalytic reactions. In this review we provide systematization of representative M–NHC bond cleavage reactions as well as discuss their potential influence on catalysis. Distinction between the NHC-connected and NHC-disconnected active species is of great importance to design highly efficient M/NHC catalytic systems.
2. State-of-the-art catalysis by metal complexes with NHC ligands
The general concept of M/NHC catalysis has long been based on the assumption of high stability of the M–CNHC framework during catalysis. NHCs were considered as supporting ligands that stabilize and at the same time activate the metal centers. The active centers were commonly imagined as activated molecular (NHC)nM(L)x complexes (Fig. 1) by analogy with phosphine complexes.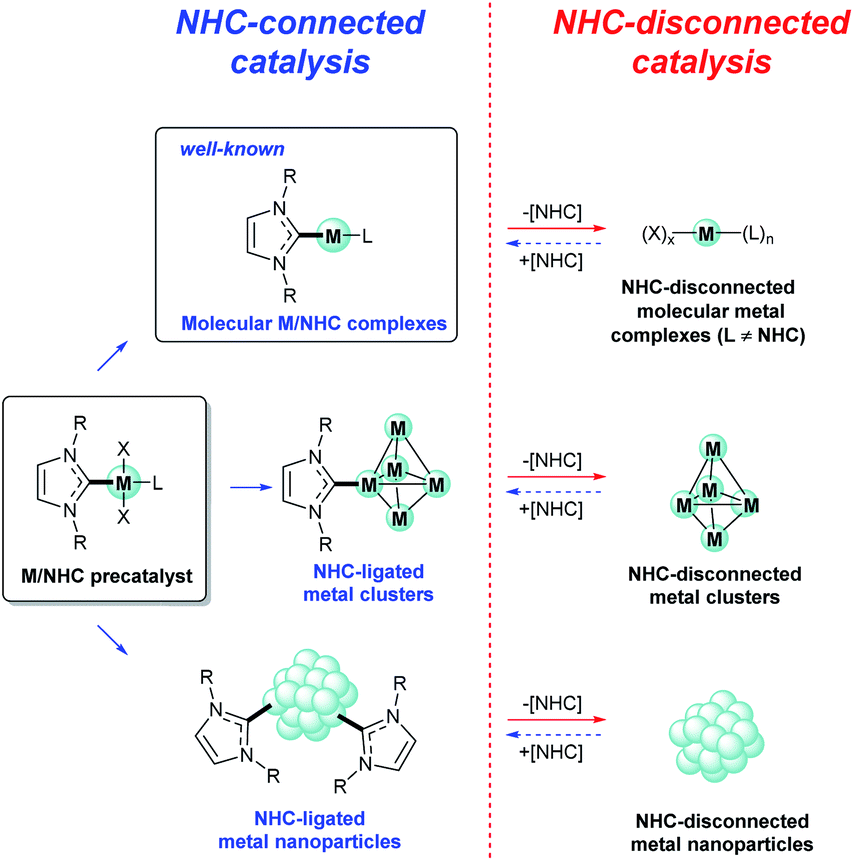 | ||
| Fig. 1 Two types of active species in NHC-connected and NHC-disconnected modes of M/NHC catalysis. | ||
Here we discuss that M/NHC complexes show variable behavior, and the paths of their activation are diverse. The mechanisms of M/NHC catalysis can be divided into two main types depending on the structure of the active centers (Fig. 1):
(i) The NHC-connected mechanisms with active centers containing a typical metal–CNHC sigma bond;
(ii) The NHC-disconnected mechanisms with active centers containing no metal–CNHC sigma bonds.
It should be noted, that similar discussions related to well-defined homogeneous catalysts vs. cocktail-type behavior are ongoing in recent years for other types of catalysts.24,28–44 However, M/NHC catalysis is a least studied topic in the area of dynamic catalysis, since until recently only molecular mode was predominantly considered.
First, let us briefly consider the main features of the above mentioned two types of M/NHC catalysis.
2.1. NHC-connected mechanisms
Undoubtedly, NHC-connected mechanisms, in which active metal species contain NHC ligands connected with metal atoms via the M–CNHC bond, play an important role in M/NHC catalysis. In these mechanisms, active centers are usually represented by molecular M/NHC complexes (molecular M/NHC catalysis, Fig. 1). NHC–ligated metal clusters and nanoparticles may form in the course of catalytic process as a result of partial M/NHC complexes decomposition.45 The NHC–ligated metal clusters and nanoparticles can also act as NHC-connected active centers (Fig. 1).46–48 For example, recent study demonstrated significant impact of the NHC ligand structure on the catalytic activity of Pd/Al2O3 heterogeneous catalysts composed of NHC–ligated Pd nanoparticles in bromobenzene hydrogenolysis and Buchwald–Hartwig amination of aryl halides.46 Comparative DFT calculations for neat and NHC–ligated Pd13 clusters revealed that coordinated NHCs convey electron density to nanoclusters thus lowering the energy barriers of aryl halide oxidative addition.46 Similar effects were observed in electrochemical reduction of CO2 on NHC–ligated Pd electrodes; the role of coordinated NHCs was supported by DFT calculations of the reaction pathways on surface models of Pd(111) and Pd(111)–NHC.47The main feature of the NHC-connected mechanisms is that, after pre-catalyst activation (Fig. 2A), the M–CNHC framework directly participates in the catalytic cycle, or, more specifically, in transition states of the catalyzed reaction (Fig. 2B). Under this condition, electronic and steric parameters of the NHC ligand directly influence the metal center and significantly affect the activation energy.
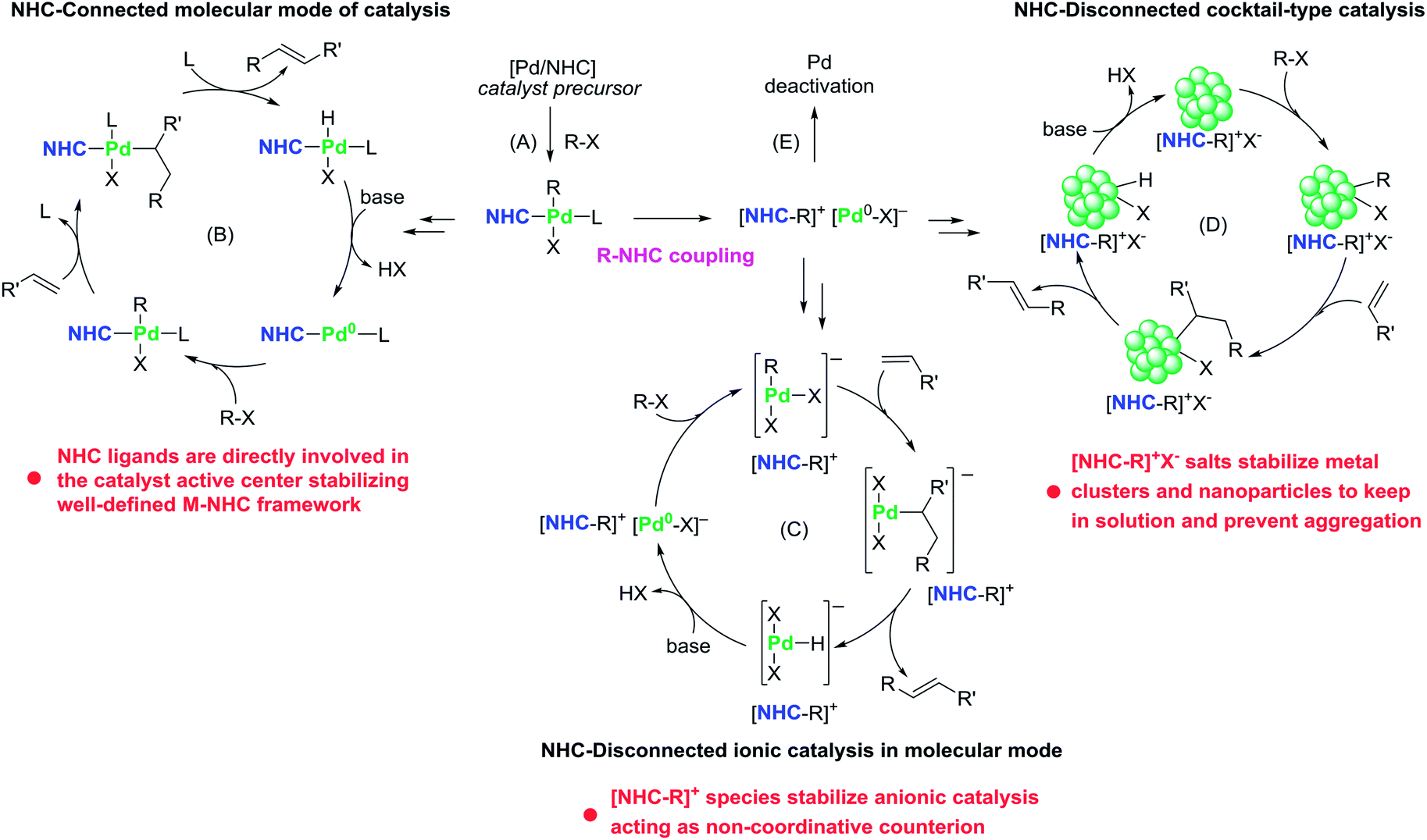 | ||
| Fig. 2 Different modes of operation of Pd/NHC complexes are shown using the Mizoroki–Heck reaction as an example: (A) – initial stage of precatalyst activation; (B) – NHC-connected molecular catalysis; (C) – NHC-disconnected molecular catalysis; (D) – NHC-disconnected cocktail-type catalysis involving nanoparticles; (E) – catalyst degradation after R–NHC coupling (other pathways of catalyst degradation also exist in each of the catalytic cycles; not shown here to avoid picture overload). | ||
Typical examples of catalytic systems operating by the NHC-connected mechanisms are Pd/NHC- and Ni/NHC-catalyzed cross-coupling and CH-functionalization reactions of non-activated aryl chlorides e.g. the Buchwald–Hartwig amination,10,18,49–52 C–S cross-coupling of thiols,53–55 CH–arylation of ketones,56,57 among several other examples.
Catalytic efficacy of M/NHC complexes in the reactions operating by the NHC-connected mechanisms is highly dependent on the electronic effect and steric bulkiness of the NHC ligands.
2.2. NHC-disconnected mechanisms
The alternative, NHC-disconnected mode, is often mentioned as “NHC-free” catalysis or “ligandless” M/NHC catalysis.24 The name indicates that the active metal species (molecular metal complexes, metal clusters, or metal nanoparticles) are formed by decomposition of M/NHC precatalysts and contain no metal–CNHC sigma bonds (Fig. 1).27,54,58–64 In this type of mechanisms, M/NHC complexes serve as precursors or reservoirs of the active metal species. The metal–CNHC framework breaks at the activation stage and it does not participate in the catalytic cycle. A typical example of NHC-disconnected metal catalysis is Pd/NHC catalyzed Mizoroki–Heck reaction (Fig. 2).27,58,59,62At the activation stage, Pd/NHC complexes suffer reductive elimination of NHC ligands via C–NHC coupling or H–NHC coupling to give azolium salts [NHC–R]+X− (R = H, aryl, etc.),27,59,62 or via O–NHC coupling under the action of strong oxygen bases to give azolones.58 The NHC-disconnected Pd(0) active species, that form after the Pd–CNHC bond cleavage, effectively catalyze the Mizoroki–Heck reaction. Under certain reaction conditions and Pd/NHC loadings, molecular M/NHC catalysis cannot be neglected (Fig. 2B);62 however, in such cases, the homogeneous NHC-disconnected catalysis by ionic Pd complexes (Fig. 2C)62 or the cocktail-type NHC-disconnected catalysis by nanoparticles (Fig. 2D) come into effect and make the main contribution to the product formation.27,59
The efficacy of M/NHC complexes in the catalytic systems operating by NHC-disconnected mechanisms depend on the rate of the metal–CNHC bond cleavage and the stability of the forming NHC-disconnected active metal species (considered further in Section 4).27,59,62
Overall, it is evident that the metal–CNHC bond cleavage reactions may have paramount impact on the catalytic systems operating by NHC-connected and NHC-disconnected catalytic mechanisms. Understanding the M–NHC bond cleavage reactions is pivotal for the efficient tuning of activity and stability of the M/NHC catalytic systems.
3. Organometallic chemistry behind the H–NHC, C–NHC and X–NHC couplings
M/NHC complexes can undergo metal–CNHC bond cleavage to give different products depending on the structure of the complex and reaction conditions (Table 1). Here we attempt to classify these reactions considering the type of forming R–NHC bond (C–NHC, H–NHC, and X–NHC), the changes in oxidation state of the metal at the stage of M–CNHC bond cleavage, and some other features of the plausible reaction mechanism. It should be emphasized that the number of detailed studies of the M–NHC bond cleavage reactions is still limited (either by R–NHC coupling or direct M–NHC bond dissociation). Some of the reports elucidate the major NHC conversion products without accounting for the metal-containing products, which complicates conclusions on the stoichiometry and mechanism of reaction.| Metal | M–NHC → C–NHC | M–NHC → H–NHC | M–NHC → X–NHC |
|---|---|---|---|
| Li | Csp2–NHC104 | Ref. 105–109 | P–NHC110 |
| Csp3–NHC105,108 | As–NHC110 | ||
| Na | Csp3–NHC111 | — | — |
| Mg | — | Ref. 112 | — |
| Al | Csp2–NHC113,114 | Ref. 115–119 | — |
| Csp3–NHC120 | |||
| Ga | — | Ref. 105 and 116 | — |
| Ge | — | Ref. 121 and 122 | B–NHC122 |
| In | — | Ref. 116 and 119 | — |
| Tl | — | — | Cl–NHC123 |
| Br–NHC123 | |||
| Sc | Csp2–NHC124 | — | — |
| Y | — | Ref. 125 | — |
| Ce | — | Ref. 125 | — |
| Eu | Csp2–NHC126 | — | — |
| Cr | — | Ref. 127 | I–NHC127 |
| Mo | — | Ref. 127–130 | I–NHC127 |
| W | — | Ref. 127 | — |
| Mn | — | Ref. 131 | — |
| Fe | Csp–NHC132 | Ref. 133–136 | B–NHC137 |
| Csp2–NHC80,135,138 | |||
| Ru | Csp2–NHC73,74,139 | Ref. 140–143 | — |
| Co | — | Ref. 144 | O–NHC145 |
| Rh | Csp2–NHC75–79,146 | Ref. 147–149 | — |
| Csp3–NHC147 | |||
| Ir | — | — | B–NHC150 |
| Ni | Csp2–NHC72,151 | Ref. 152 and 153 | B–NHC154 |
| Csp3–NHC67,82,155,156 | P–NHC157 | ||
| S–NHC99,100 | |||
| Pd | Csp2–NHC27,58,61,62,82,155,156,158–165 | Ref. 59,165–171 | O–NHC97,172 |
| Csp3–NHC26,82,155,156,160,166,173–175 | Cl–NHC83 | ||
| Si–NHC175 | |||
| Pt | — | Ref. 59 and 176 | — |
| Cu | Csp2–NHC71,81,177 | Ref. 71,178–182 | O–NHC71,181–183 |
| Br–NHC81,101,184 | |||
| Cl–NHC101,184 | |||
| I–NHC101 | |||
| N–NHC103 | |||
| P–NHC185 | |||
| S–NHC186–188 | |||
| Ag | — | Ref. 189–205 | B–NHC205 |
| N–NHC206 | |||
| S–NHC207,208 | |||
| Au | — | Ref. 209 | — |
| Zn | Csp2–NHC210–212 | Ref. 212 and 213 | — |
3.1. Metal–NHC bond cleavage reactions with the metal reduction
Reductive M–NHC bond cleavage reactions are typical for complexes of metals in higher oxidation states with a higher redox potential. Such reactions are very important for M/NHC catalysis as they lead to formation of ligandless M0 species which can serve as alternative active centers.24 On the other hand, these reactions often lead to formation of metal precipitates and cause deactivation of M/NHC catalytic systems.22,23,25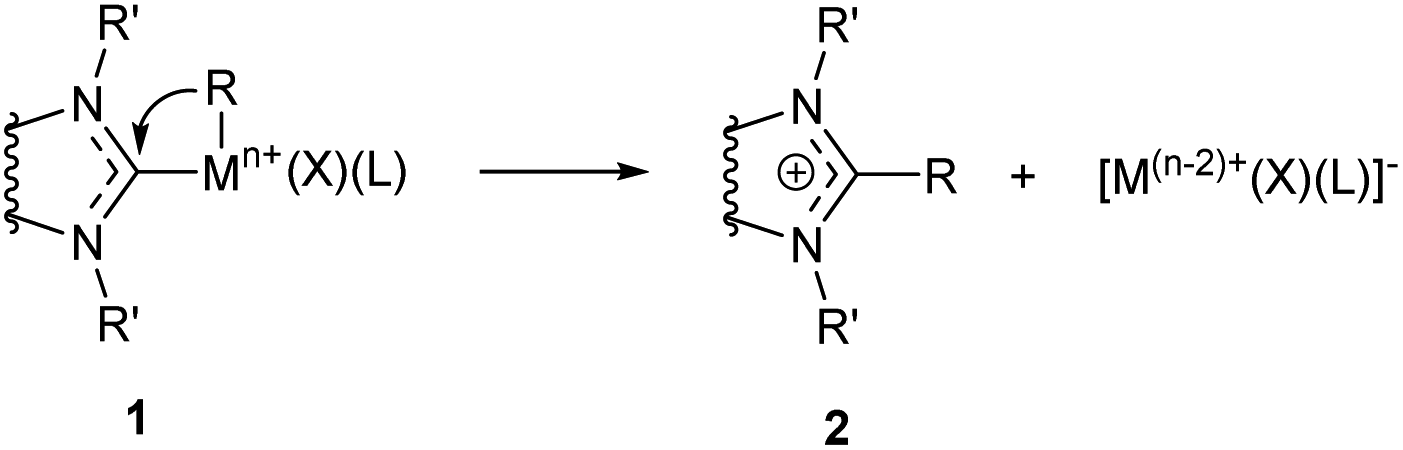 | ||
| Scheme 1 General scheme of the NHC reductive elimination reactions. | ||
The C–NHC coupling reactions were most extensively studied for PdII/NHC complexes (Table 1 column 2). These reactions proceed via cis arrangement of NHC and R groups; kinetic studies and DFT calculations are consistent with the concerted reductive elimination mechanism.82 Susceptibility of (NHC)Pd(R)(Br)Py complexes to R–NHC coupling decreases as R = vinyl > ethynyl > Me ∼ Ph.58 The calculated energy barriers of Ph–NHC coupling is in the range of 17.9–25.1 kcal mol−1 for different NHCs.64 In many catalytic reactions conducted at 50–100 °C such barriers are readily overpassed and the stability of complexes is determined by thermodynamic factors. Reversibility of the C–NHC coupling was confirmed experimentally on an example of the CH3–NHC bond activation catalyzed by palladium nanoparticles.61 Bulky N-substituents in NHC ligands usually increase the activation barriers;58 however, the effects of bulkiness can be more complex, as a significant increase in steric bulkiness can induce dissociation of the stabilizing co-ligands. For example, DFT calculations of Ph–NHC coupling in (NHC)Pd(Ph)(I)DMF complexes predicted lower ΔE≠ for the bulky IPr ligand (19.2 kcal mol−1) than for the non-bulky IMe ligand (20.9 kcal mol−1) owing to the splitting of DMF molecule from the complex with IPr ligand.64
The effect of metal and its oxidation state on the C–NHC coupling efficiency has been evaluated by DFT calculations for MII/NHC and MIV/NHC complexes of Ni, Pd and Pt.60 The results indicate that thermodynamic and kinetic stabilities of both MII and MIV complexes 1 against C–NHC coupling decrease as Pt > Pd > Ni. Besides, complexes 1 with a higher oxidation state of the metal are thermodynamically and kinetically less stable than corresponding complexes with metals in lower oxidation states. Thus, in (NHC)2MIV(Ph)(Br)3 complexes (NHC = 1,3-dimethylimidazol-2-ylidene), Ph–NHC coupling is facilitated dramatically from Pt (ΔG≠ = 37.5 kcal mol−1, ΔG = −36.9 kcal mol−1) to Pd (ΔG≠ = 18.3 kcal mol−1, ΔG = −61.5 kcal mol−1) and Ni (ΔG≠ = 4.7 kcal mol−1, ΔG = −80.2 kcal mol−1). In similar complexes (NHC)2MII(Ph)(Br), corresponding values change in a smaller extent from Pt (ΔG≠ = 50.1 kcal mol−1, ΔG = 34.0 kcal mol−1) to Pd (ΔG≠ = 30.8 kcal mol−1, ΔG = 15.8 kcal mol−1) and Ni (ΔG≠ = 30.1 kcal mol−1, ΔG = 16.6 kcal mol−1). The poor thermodynamic and kinetic stabilities of the regular PdIV and NiIV complexes 1 against R–NHC coupling indicate high probability of the MIV–NHC bond cleavage and implementation of the NHC-disconnected catalytic scenario in the reactions comprising MIV intermediates.60
A similar decrease in the stability against R–NHC coupling was observed for the bis-C–NHC couplings in Pd, Fe and Cu complexes (Scheme 2). Treatment of PdII/NHC complex 3 with chlorine results in formation of cyclic 4,4-biimidazolium salt 5 and release of PdII species; the reaction apparently proceeds via reductive elimination of both NHC ligands from the PdIV/NHC intermediate 4.83 Reductive elimination of both NHC ligands in the stable bis-NHC FeII complexes 6 upon one-electron oxidation with Th+ gives dicationic 2,2′-biimidazolium salts 7, presumably along with unstable FeI species that has not been isolated.80 A similar C–C coupling leading to compound 10 occurs between two NHC ligands in the bis-NHC CuIII complex 9 that forms in situ from the silver complex 8.81
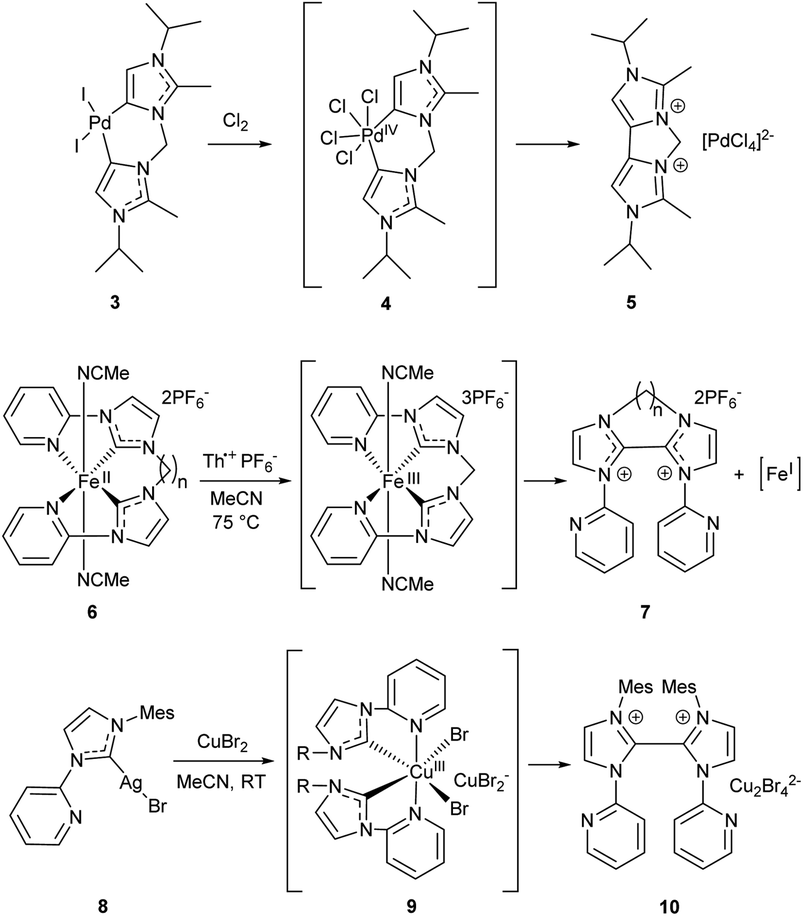 | ||
| Scheme 2 The CNHC–CNHC coupling reactions in bis-NHC complexes of metals in higher oxidation states.80,81,83 | ||
It should be noted that C–NHC coupling reactions have great importance for accessing various functionalized heterocycles.84–89 Numerous metal-catalyzed CH-functionalizations of nitrogen heterocycles like alkylation, arylation and etc. in fact proceed via R–NHC coupling of in situ formed M–NHC complexes in which heterocyclic substrate acts as a protic NHC–ligand.86–89
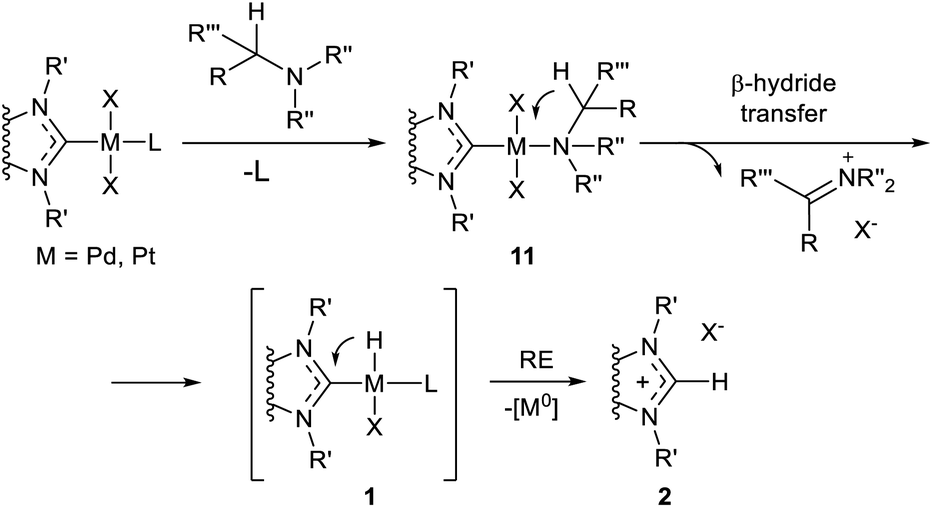 | ||
| Scheme 3 The aliphatic amine-induced H–NHC coupling of MII/NHC complexes.54,59 | ||
Theoretical modeling of the H–NHC coupling and reverse reaction for Ni, Pd and Pt complexes by DFT methods have been reported.62,90,91 Predicted activation and reaction energies vary depending on the structure of complexes 1 (R = H); however, all of the studies emphasize the endergonic character of the H–NHC coupling reactions. For example, a low activation barrier of H–NHC coupling (ΔG≠ = 6.3 kcal mol−1), with corresponding reaction energy of ΔG = 4.4 kcal mol−1, were calculated for (NHC)Pd(H)I (NHC = 1,3-dimethylbenzimidazol-2-ylidene); the reaction leads to ionic aggregate [NHC–H]+[PdI]−. The reversibility of the H–NHC coupling has been confirmed experimentally.21,92,93 Bulky N-substituents in the NHC ligands usually stabilize the Pd/NHC hydride complexes 1.92,94–96 This finding indirectly supports the reductive elimination mechanism (H–NHC coupling) as the predominant pathway of the observed decomposition of hydride complexes 1 into [NHC–H]+ cations and Pd0 species in solutions at moderate temperatures. The alternative mechanism (dissociation of PdII/NHC or Pd0/NHC complexes) would mean a positive correlation between decomposition and steric bulkiness, which contradicts the experimental findings. It should be noted, however, that the alternative pathway to azolium salts 2via the M–NHC bond dissociation and NHC ligand protonation must not be excluded in many cases.
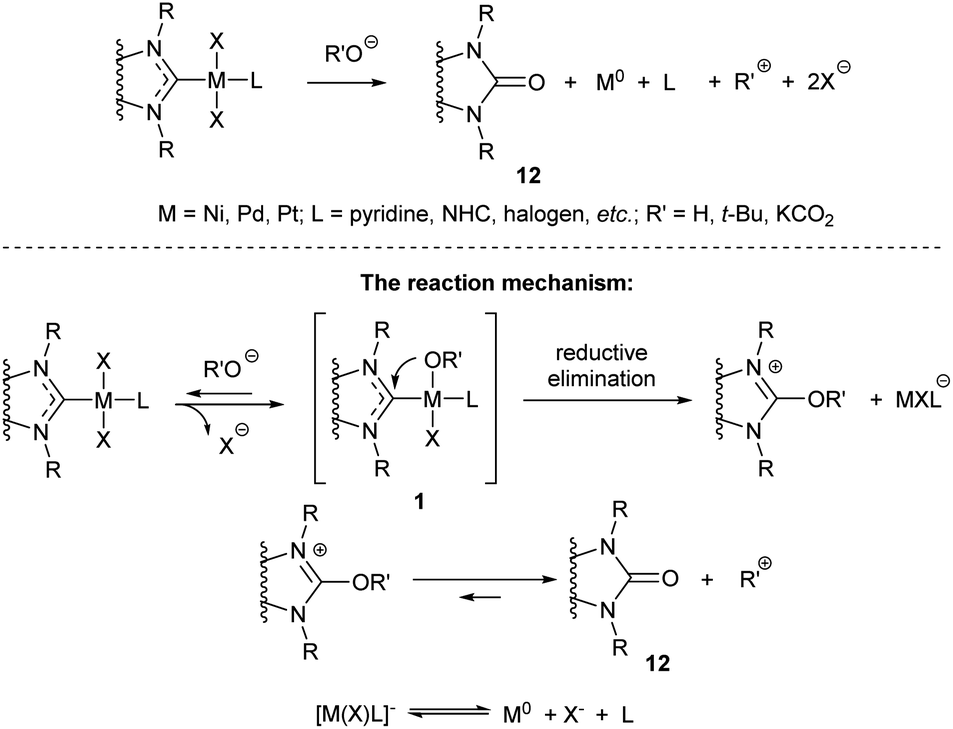 | ||
| Scheme 4 The oxygen-base-mediated O–NHC coupling reactions of M/NHC complexes.97 | ||
The MII/NHC complexes (M = Ni, Pd, Pt) react by reductive elimination of RO and NHC ligands to give M0 species that are typically transformed into metal precipitates and [NHC–OR]+ cations; the latter afford azolones (the oxo-substituted azoles) via dissociation or solvolysis of the R–O bond (Scheme 4).58 In this reaction, NHC ligands play a role of two-electron intramolecular reductants of the coordinated metal dications. The reaction mechanism was confirmed by experiments with 18O labelled potassium hydroxide and observation of key intermediates 1 (R = OH, M = Pd) by ESI-MS with their direct transformations into azolones 12 in MS/MS experiments (Scheme 4).58 Among the studied metal complexes, Pd complexes were found to be the most reactive, and thus more prone to the base-induced O–NHC coupling. Mono-NHC complexes and the halogen-bridged Pd complexes containing non-bulky N-substituents were decomposed by KOH or tBuOK within 10–20 min at 40–100 °C, while bis-NHC complexes (L = NHC) and the complexes with bulky substituents in NHC ligands suffered appreciable conversions only within several hours. S–NHC coupling was observed in the reactions of arylthiols with PdII/NHC and NiII/NHC, as well as in the reactions of S,S′-dimethyl disulfide with (IMes)2Ni0 complex (Scheme 5).55,98,99 For example, S-aryl-imidazolium salt 14 was afforded by heating dithiolate complex 13 in C6D6 (Scheme 5).98 Compound 14 very likely forms via reductive elimination of NHC and thiolate ligands.98 The reaction of Ni0(Mes)2 complex with MeSSMe affords imidazoline-2-thione 15 along with the trinuclear S-bridged complex 16.99 Imidazoline-2-thione 15 is reportedly formed by Ni/NHC-catalyzed hydrothiolation of alkynes.100 Remarkably, trinuclear Pd complexes similar to 16 were observed in the reactions of Pd–PEPPSI complexes with arylthiols, along with 2-arylthioimidazolium salts analogous to 14.55,98 The undesired S–NHC coupling can significantly affect the stability of M/NHC complexes during catalysis of C–S bond formation and activation reactions. However, the detailed evaluation is difficult, as the mechanisms of these reactions are still poorly studied. In the first instance, formation of azoline-2-thiones implies the cleavage of S-aryl and S-alkyl bonds in the starting thiols or disulfides;99,100 the mechanism of this cleavage is unclear. Secondly, reductive elimination of SR and NHC ligands from MII/NHC complexes should produce NHC-disconnected M0 species, no isolation or detection of which have ever been reported. Complexes 16, the only metal-containing products of determined structure reported for this process (Scheme 5),55,98,99 apparently are not the final products of S–NHC coupling. It is quite probable, that the forming NHC-disconnected M0 species are unstable under the reaction conditions and further react with sulfur compounds to give metal polysulfides.100 Overall, the S–NHC coupling reactions require detailed mechanistic investigation.
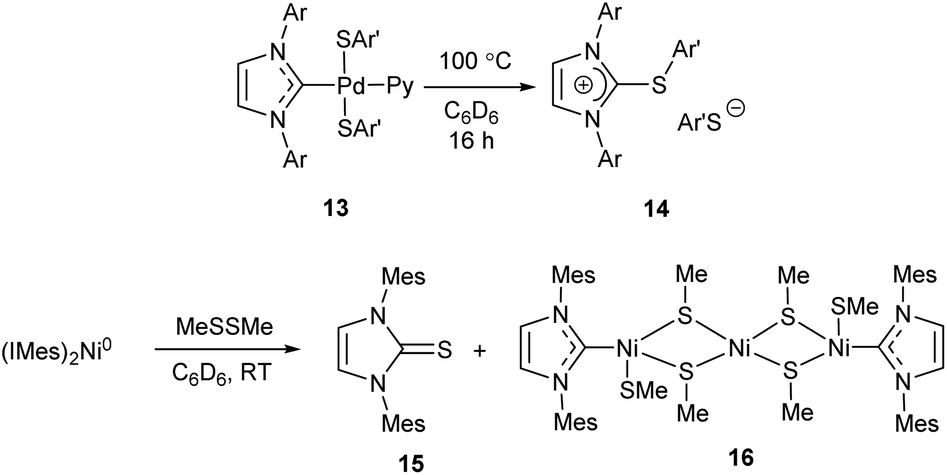 | ||
| Scheme 5 Major products of S–NHC coupling reactions.98,99 | ||
Halogen–NHC coupling is also of great interest (Scheme 6).23,25,81,83 For example, treatment of Pd/NHC complex 17 with chlorine affords salt 18, very likely by reductive elimination of Cl and NHC from the PdIV intermediate similar to 4.83 The authors emphasize the critical influence of a slight change in the NHC bulkiness on the pathway of PdIV/NHC intermediates decomposition (Schemes 2 and 6). Oxidation of (NHC)CuIX complexes 19 with various oxidizers afforded [NHC–X]+Y− salts 20.101 DFT calculations supported the high probability of the reductive elimination mechanism.101 Br–NHC coupling was also observed in reaction of Ag/NHC complex 21 with excess of CuBr2 (Scheme 6).81 The proposed mechanism of this reaction includes formation of CuIII/NHC complex 22 which suffers reductive elimination of Br and NHC ligands to give imidazolium salt 23. This mechanism was supported by DFT calculations.81
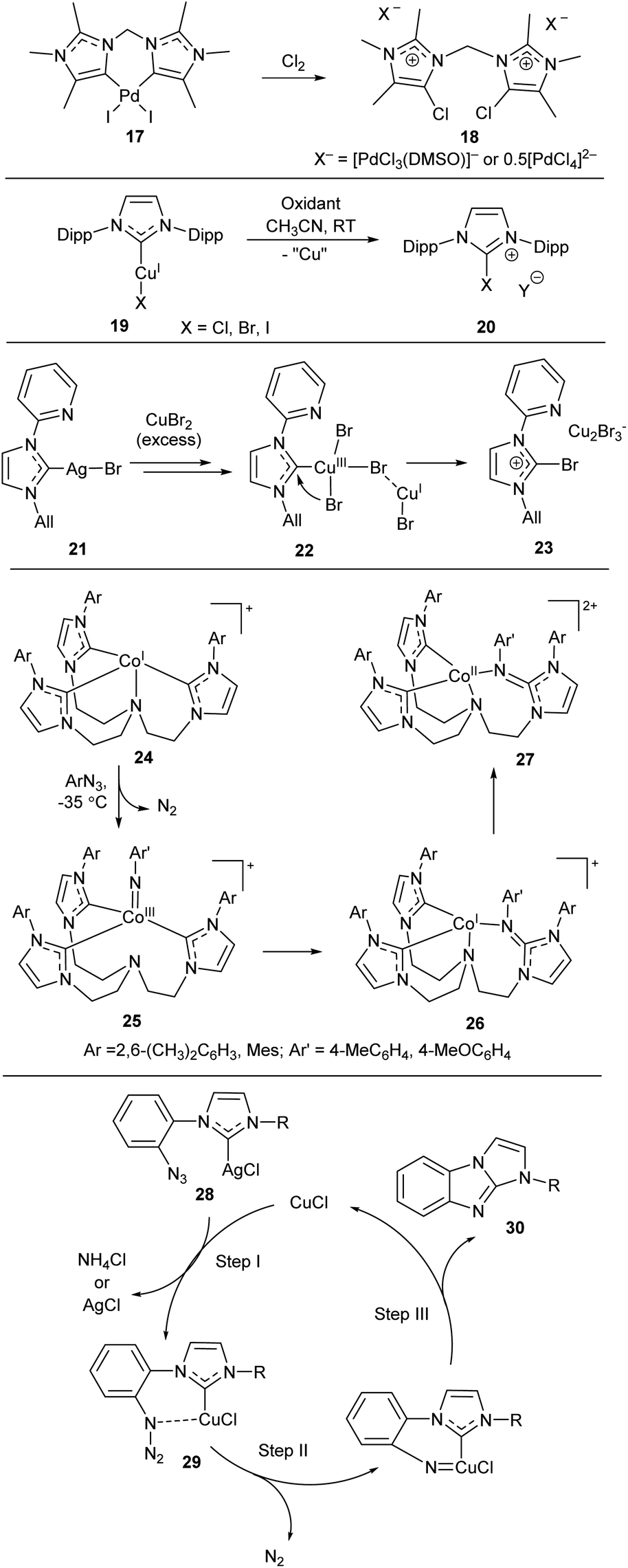 | ||
| Scheme 6 Representative examples of X–NHC coupling reactions.81,83,101–103 | ||
N–NHC bond-forming reactions may proceed by nitrene insertion into M–NHC bond.102,103 For example, interaction of arylazides with CoI/NHC complexes 24 results in formation of corresponding CoIII/NHC intermediates 25 successfully isolated and characterized at low temperatures (Scheme 6).102 Intermediates 25 undergo nitrene insertion into the Co–NHC bond (considered formally as reductive elimination of nitrene and NHC ligands) to give CoI/NHC complexes 26 (detected and characterized in situ). Complexes 26 are unstable and undergo disproportionation to give CoII complexes 27 and other products.102 A similar intramolecular reaction yields CuI/NHC complexes 29 from Ag/NHC complexes 28in situ (Scheme 6).103 Subsequent transformations lead to compound 30; a plausible mechanism for this sequence was supported by DFT calculations.103
3.2. Cleavage of the metal–NHC bond with oxidation of the metal
Reports on metal oxidation at the M–NHC bond cleavage stage are rare. Certain oxidizer-induced reactions may very well proceed with metal oxidation at the M–NHC bond cleavage stage; however, no substantial mechanistic evidence on this point is available.The interaction of [bis(NHC)](silylene)Ni0 complex 31 with catechol borane producing NiII complex 32 is an intriguing example of oxidative cleavage reaction (Scheme 7).154 DFT calculations show that the concerted transfer of Cl from Si to Ni and NHC from Ni to B is a key event in this multistep reaction.154
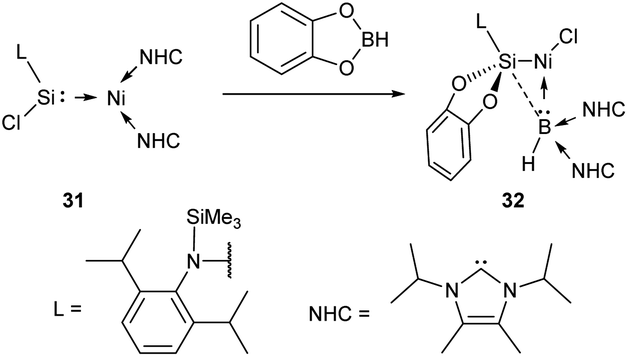 | ||
| Scheme 7 The oxidative Ni–NHC bond cleavage reaction.154 | ||
3.3. Cleavage of the metal–NHC bond without alteration in the oxidation state of the metal
The majority of such reactions can be conventionally classified as (i) M–NHC bond dissociation or (ii) insertion into M–NHC bond. Alternative mechanisms (notably substitution of the metal species through an attack at the carbene carbon) can also be encountered in the literature;23,25,183 it should be noted that mechanistic details are frequently missing.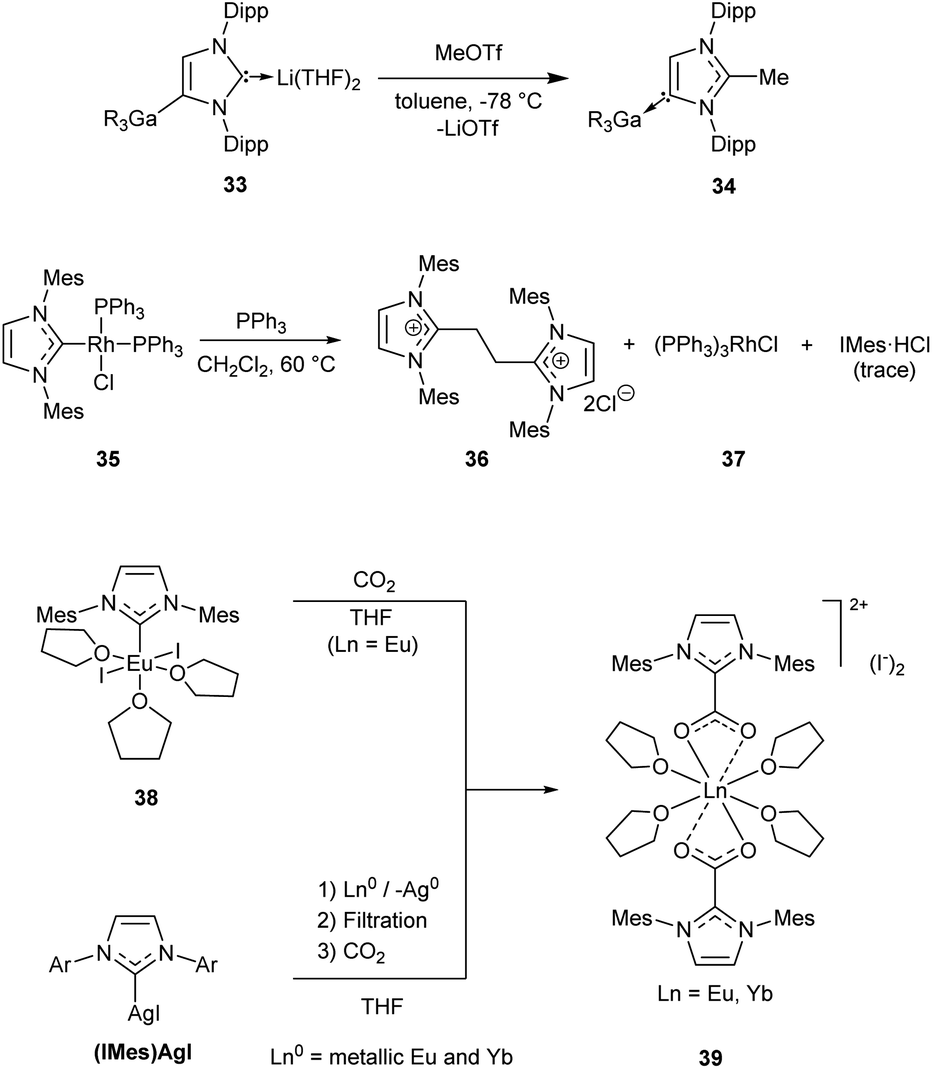 | ||
| Scheme 8 Representative examples of C–NHC bond forming reactions without alteration of the metal oxidation state.105,126,147 | ||
Treatment of a EuII/NHC complex 38, or YbII/NHC complex prepared in situ from (IMes)AgI, with CO2 affords insertion products 39 (Scheme 8).126 Similar reactions were described for ScIII/NHC complexes.124 Reductions of CO2 with hydride ZnII/NHC complexes are accompanied by formation of zwitterionic NHC–COO adducts.210,211
Facile insertions of aldehydes, isocyanates, and carbodiimides into AlIII–NHC bond can also be found in the literature.113,114,120
Protonolysis reactions proceed very easily for Li/NHC105,108 and Mg/NHC112 complexes. Other complexes, including AgI/NHC,191,193,197–199,201 AlIII/NHC,119 InIII/NHC,119 YIII/NHC125 and CeIII/NHC,125 MnI/NHC, ZnII/NHC,213 MoVI/NHC130 and NiII/NHC,214 are also susceptible to protonolysis; the efficiency depends on the structure of NHC ligands, co-ligands and reaction conditions (Table 1 column 3). For example, (NHC)2NiX2 complexes (X = Cl, Br, I) suffer facile hydrolysis in aqueous MeCN or THF at 70 °C to give azolium salts and Ni(OH)2.214 Half-life of the complex decomposition reactions varies from several minutes for the complexes with non-bulky NHCs to about 2 days for (IMes)2NiCl2.214
Even quite stable PdII/NHC168,170 and RuII/NHC141,215 complexes suffer protonolysis under highly acidic conditions to give azolium salts and the corresponding NHC-disconnected MII species. Remarkably, protolytic cleavage of the PdII–CNHC bond induced by traces of DCl can be occasionally observed in CDCl3 solution at 40 °C.215
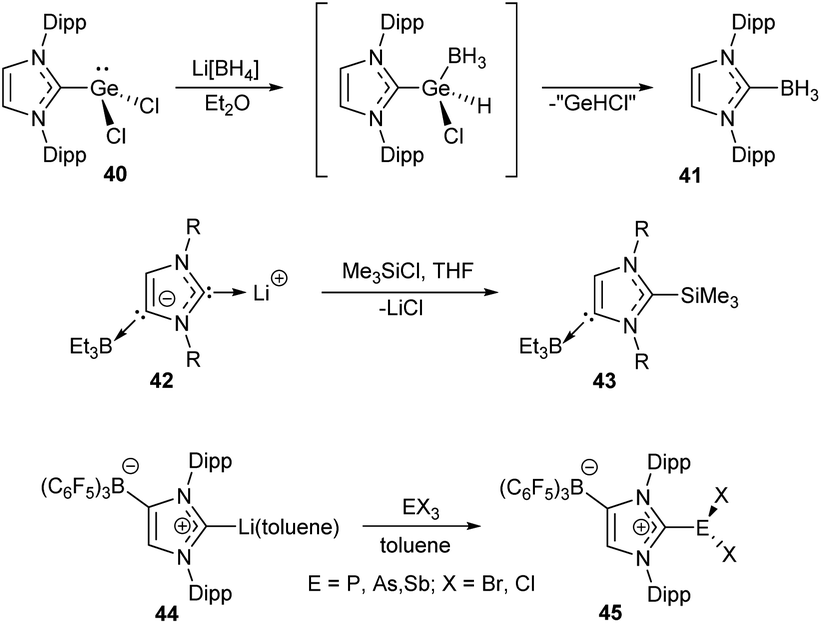 | ||
| Scheme 9 Representative examples of the X–NHC bond forming reactions preserving oxidation state of the metal.108,110,122 | ||
Pd–NHC bond cleavage in PdII/NHC complexes under the action of molecular iodine can be also mentioned in this context.216 In this reaction, NHC is oxidized with iodine to give the NHC·I2 adduct, while Pd atom retains the 2+ oxidation state. Two feasible mechanisms for this reaction were proposed on the basis of DFT calculations.216 One of them involves direct electrophilic attack of I2 at the Pd–NHC bond followed by formation of the NHC·I2 adduct. The second mechanism involves dissociation of the Pd–NHC bond and subsequent electrophilic attack of I2 at the free NHC.
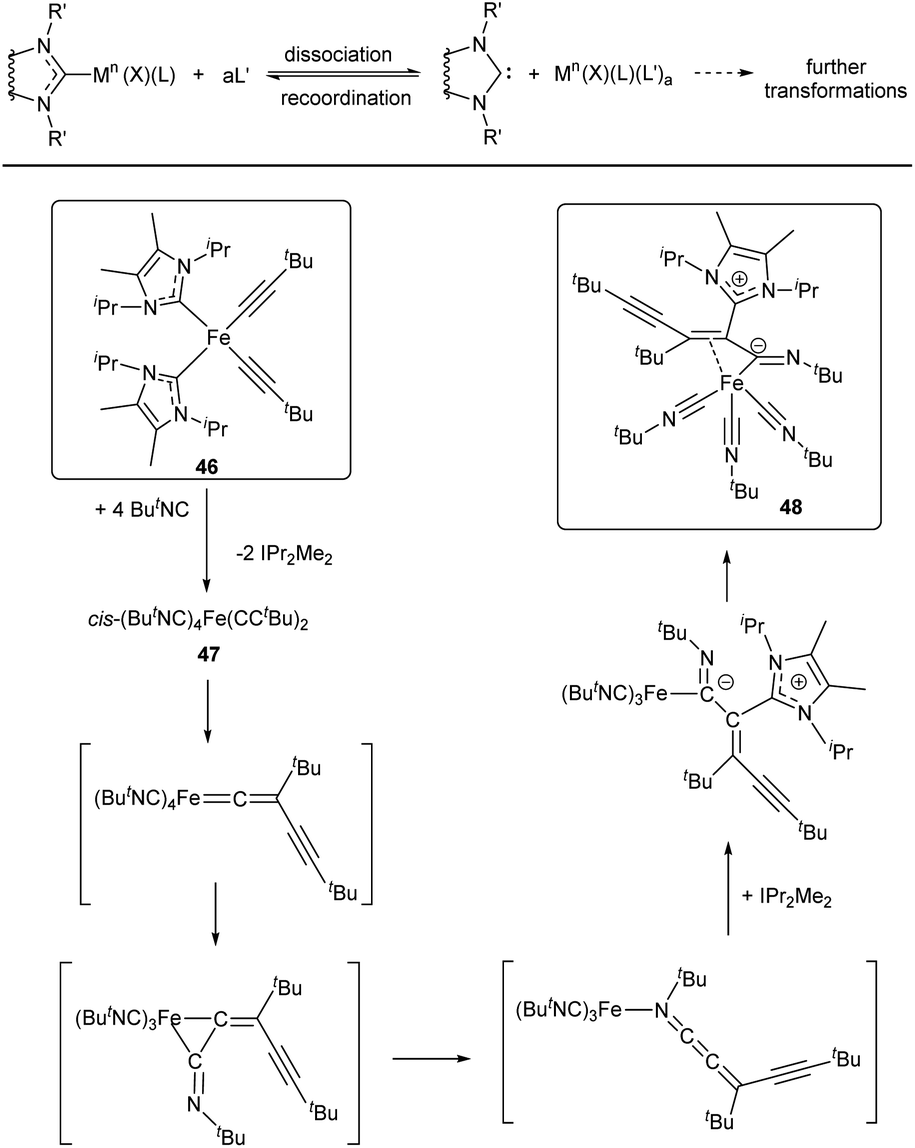 | ||
| Scheme 10 General scheme of the M–NHC bond cleavage reactions induced by dissociation of NHC ligands and reaction of complex 46 with tBuNC.138 | ||
It should be noted that, although M–NHC bond dissociation does not affect the metal oxidation state, the formed NHC-disconnected metal species may undergo subsequent redox transformations (Scheme 10). To illustrate this point, an interesting case of FeII/NHC complex 46 reacting with ButNC to give compound 48 can be mentioned.138 This reaction may be considered as a simple ligand displacement with subsequent transformations of the released species. At the first stage of the reaction, NHC is displaced by ButNC ligand. Dissociation of the FeII–NHC bond thus results in formation of free NHCs and complex 47, which undergoes a cascade of migratory insertion and migration reactions accompanied by reduction of FeII and nucleophilic addition of NHC at the final step (Scheme 10).
3.4. Other cases of the metal–NHC bond cleavage
Some M–NHC bond cleavage reactions are not attributable to any of the considered types but nevertheless highly relevant.These include various transformations leading to the NHC ring opening products;23,25 the mechanisms are multistep and often puzzling. For example, transformation of complex 49 under the action of alkynes leads to compound 50 and is accompanied by release of propylene (Scheme 11).132
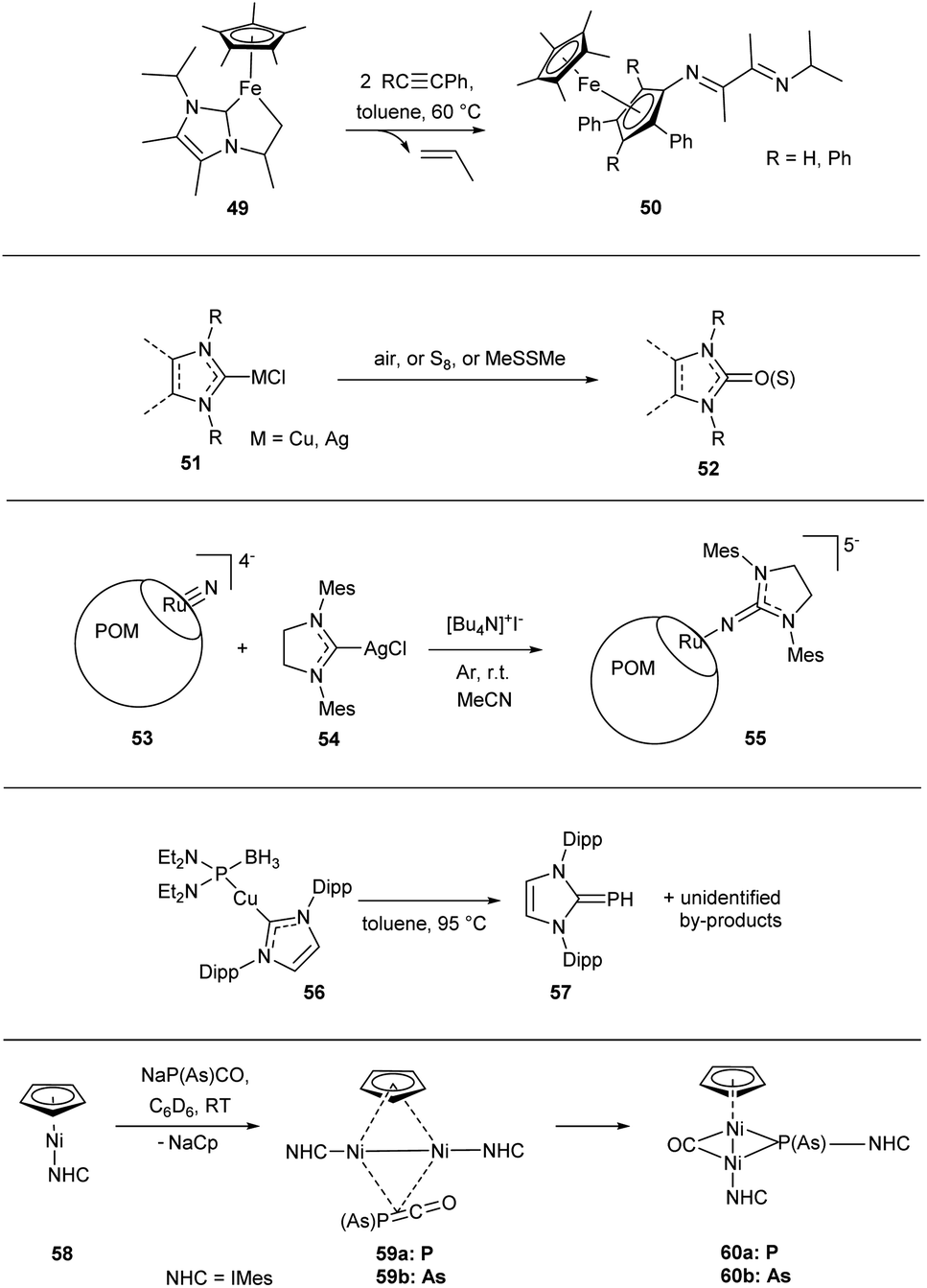 | ||
| Scheme 11 Representative examples of the C–NHC and X–NHC bond forming reactions with unclear mechanisms. | ||
Oxidation of M/NHC complexes 51, leading to formation of O–NHC bond and affording imidazoline-2-ones 52 or related substances, certainly deserves attention.172,181,182,222 For example, homogeneous aerobic oxidation of CuI/NHC complexes can be used for preparative synthesis of cyclic ureas (Scheme 11).181 The reaction is highly sensitive to the NHC steric bulkiness and possibly includes a reductive elimination step. Similar reactions of CuI/NHC and AgI/NHC complexes with sulfur afford azoline-2-thions.186–188,207 The mechanisms of these reactions remain unexplored.
Formation of N–NHC bond between ruthenium(VI) nitride-containing polyoxometalate [PW11O39RuVIN]4−53 and (NHC)AgCl complex 54 in the presence of iodide leads to compound 55 (Scheme 11).206 The presence of iodide is prerequisite; the authors suggest that iodide ensures reduction of RuIV to RuIII in the presence of Ag/NHC complex.
Formation of P–NHC and As–NHC bonds is an option as well (Scheme 11). For example, CuI/NHC complex 56 transforms into phosphaalkene 57 in good yields under heating in toluene at 95 °C.185 Interaction of NiI/NHC complex 58 with NaPCO or NaAsCO affords binuclear NiI complexes 60via intermediates 59.157 The mechanisms of these reactions remain unexplored.
4. Rational catalyst design for the tuning of M/NHC catalytic systems
As it follows on from the above discussion, M/NHC complexes can decompose via diverse range of M–NHC bond cleavage reactions, and this phenomenon is common for most of the metals and NHC ligands. To a greater or a lesser extent, R–NHC coupling inevitably takes place in catalytic systems and can be quite influential to take it into account.Given the possibility of M–NHC bond cleavage, what criteria should be used when selecting an M/NHC catalyst to ensure effective catalysis of a particular reaction? What changes in the structure of M/NHC complexes and catalytic conditions would enhance the efficacy of a catalytic system?
We believe that the type of catalytic mechanism is the primary thing to be taken into account. In particular, the NHC-connected and NHC-disconnected modes of catalysis may require quite different optimization approaches.
4.1. The NHC-connected catalysis
M–NHC bond cleavage may represent a serious obstacle for the classical, NHC-connected metal catalysis. This mode of catalysis is dependent on the M–NHC bond stability, as the M–NHC framework participates in catalytic cycle and notably in transition states of the catalyzed reaction. As some excellent reviews on the molecular M/NHC catalysis are available,7–12,18 we will only briefly consider the properties of catalytic systems that are relevant to the problem of M–NHC bond cleavage.Catalytic efficacy of M/NHC complexes is highly dependent on:
(i) Electronic and steric parameters of M/NHC complexes, especially the NHC ligands; (ii) the ease of M/NHC complexes activation; (iii) the M–NHC bond stability, both at the activation stage and during the catalytic cycle.
The great success of M/NHC complexes as catalysts is primarily due to the strong sigma electron-donating ability of NHC ligands, which ensures the strong metal–NHC bonding8,9 and usually accelerates the oxidative addition step. The electron-donating properties of NHCs are chiefly determined by heterocyclic moiety and to a smaller extent by substituents.8,19,217 The ligands with non-aromatic NHC core (especially the expanded ring NHCs, “abnormal” NHCs, or NHCs with electron-donating groups conjugated with the aromatic N-heterocycle) typically reveal higher sigma electron-donating ability.11,12,223 Nevertheless, even NHCs with electron-withdrawing groups are sufficiently rich in electron density, e.g. to ensure the activation of chloroarenes.8
Steric properties of NHCs are evidently more significant for the tuning of M/NHC catalysts in a variety of cross-coupling, addition and CH-activation reactions.8,9,224 High catalytic activity has been observed for mono-NHC complexes comprising NHC ligands with the bulky and flexible N-aryl or N-alkyl substituents like, for example, 2,6-diisopropylphenyl.8,9,18,50,225,226 Such “bulky-yet-flexible”49,226 NHC ligands typically provide a sufficiently high buried volume (Vbur),11,19,224 and their performance is commonly interpreted in terms of the “flexible steric bulk” concept.49,227 According to this concept, effective ligands should be electron-rich and “small enough to accept sterically hindered substrates yet sufficiently bulky to support mono-ligation and promote reductive elimination”,228 and the “flexible steric bulk” should allow “the ligands to adapt to the changing needs of the catalytic cycle”.229 In this line, bisoxazoline-derived N-heterocyclic carbene ligands were introduced (IBiox, Fig. 3).226,229 The IBiox ligands revealed excellent performance in Pd-catalyzed Suzuki–Miyaura coupling between sterically hindered aryl chlorides and boronic acids; the reaction provides access to tetra-ortho-substituted biaryls.229 Cyclic (alkyl)(amino)carbenes (CAACs) demonstrated good activity in Pd-catalyzed α-arylation of ketones with non-hindered arylchlorides.230 However, the most common recognition have been received by families of N,N′-bis-[2,6-(di-iso-propyl)phenyl]imidazol-2-ylidene (IPr) and its saturated analog 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene (SIPr) (Fig. 3).12,18,49,50,226 The examples include SINap,231,232 ITent (Tent seems to reflect tentacular structure),18,49,233 IPr*,50,234 IPentAn,235 IPr*An236 and other bulky-yet-flexible NHC ligands. Metal complexes with these ligands show superior catalytic activity in various cross-coupling reactions. Besides the N-substituents, heterocyclic core of NHCs also contributes to Vbur.224 For instance, the expanded ring NHCs237 usually have slightly higher Vbur compared to the corresponding imidazol-2-ylidene NHCs with the same N-substituents.224 Imidazol-2-ylidene ligands with Cl238 and N(Alk)2239–242 substituents at C4 and C5 of imidazole ring show enhanced activity in many cross-coupling reactions, apparently due to a buttressing effect of these groups on the N-substituents.238,242
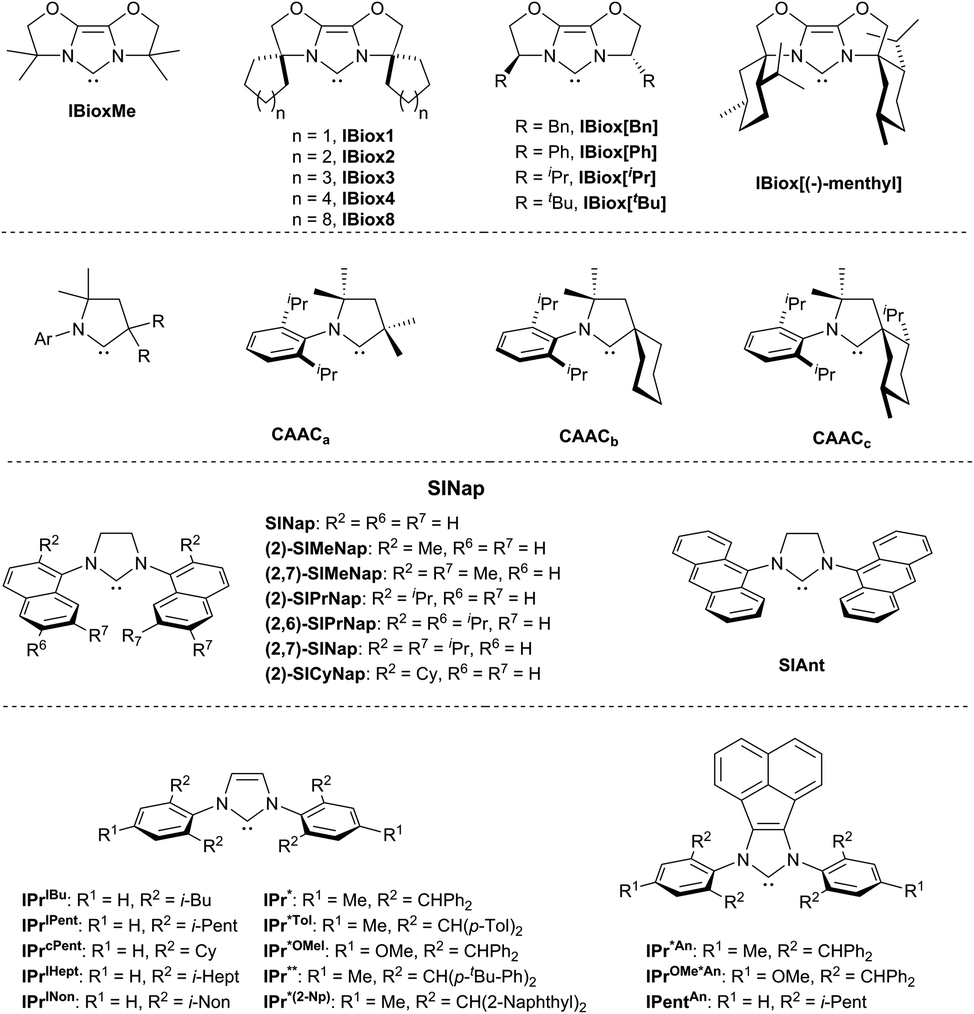 | ||
| Fig. 3 Major types of bulky-yet-flexible NHC ligands used in M/NHC catalysis. | ||
It has been commonly believed that steric bulkiness facilitates reductive elimination. However, DFT calculations show that IPr ligands can also facilitate oxidative addition of aryl halides, owing to favorable intramolecular π–π and C–H/π interactions between ArCl and bulky N-substituents (steric attraction) that decrease the activation barriers.243 Thus, bulky groups can accelerate not only the reductive elimination, but also the oxidative addition step.243 Moreover, it was revealed experimentally that bulky NHCs with aromatic N-substituents like IPr and IMes provide better activation of Pd nanoparticles in the Pd catalyzed hydrogenolysis of bromobenzene than NHCs with alkyl N-substituents (ICy, IMe).46 X-ray photoelectron spectra revealed that NHCs with aromatic N-substituents provide higher donation of electron density to Pd nanoparticles than NHCs with aliphatic N-substituents. The effect was explained by DFT calculations, which showed that NHCs like IPr and IMes bind to Pd nanoclusters not just by the carbene carbon but also by aromatic N-substituents via their delocalized π-orbitals.46 Such coordination facilitates the transfer of electron density from NHCs to Pd nanoclusters and lowers the activation barriers of aryl halide oxidative addition.46 Thus, NHC ligands with bulky aromatic N-substituents can also promote oxidative addition to the NHC-connected metal clusters and nanoparticles.
However, correlation between steric bulkiness and catalytic activity is not straightforward and optimum dependence is observed oftentimes.49,226 For example, in the Suzuki–Miyaura coupling of 2-chloromesitylene with 2,6-dimethylbenzene boronic acid activity of Pd complexes with ITent ligands increased significantly from IPr to IPent, then it decreased from IPent to IHept and INon. A similar effect, with the highest activity for IHept, was observed in Buchwald–Hartwig amination.49 In C–S cross-couplings between aryl halides and thiols, the excessively bulky IPr* and IPr*OMe ligands provide lower reaction rates but higher selectivity than IPr.54 In reactions of alkyne hydrothiolation, Pd and Ni complexes with IMes ligand show better catalytic performance than corresponding complexes with the bulkier IPr and SIPr ligands.100,244
The next important factor in the catalytic performance of M/NHC complexes is the ease of their activation under catalytic conditions.25 Well-defined and stable M/NHC precatalysts often require transformation into active form capable of catalysis. Activation can include only removing of throw-away ligand, for example chloride in (NHC)AuCl complexes, and may be facilitated by more bulky NHC and external activators like silver salts used for capture halide ions.25 In many cases the activation requires reduction of a stable Mn+2/NHC precatalyst to active Mn/NHC species that enter the oxidative addition step. For example, PdII/NHC complexes must be reduced to Pd0/NHC complexes which activate aryl halide and thus initiate C–S cross-coupling with thiols.98 Reduction of the metal is usually accomplished by using external reducing agents or sacrificial ligands. The ease of activation is essential for the catalytic output.55,98,245 Hardly activated PdII/NHC precatalysts are highly prone to formation of the catalytically inactive thiolate complexes and Pd–NHC bond breaking products associated with deactivation of the catalytic system.55,98
Stability of M–NHC bond is a very important point, both at the activation stage and during catalysis. Foremost, if metal can change its oxidation degree by 2 in catalytic conditions, activators and typical reagents can induce reductive elimination of NHC ligands from the M/NHC catalyst. C–NHC, H–NHC and X–NHC couplings are highly probable in such systems. For example, ESI-MS monitoring of various Pd/NHC complexes with IMes, IPr, SIpr and other typical ligands in DMF solution of iodobenzene at 100 °C revealed decomposition of the complexes and facile formation of the Ph–NHC coupling products.64 H–NHC coupling represents a problem during the activation of M/NHC complexes with hydride donors, e.g. alcohols or aliphatic amines.54,59,62 Strong bases, e.g. alkali metal alkoxides and hydroxides used in many catalytic systems, may trigger O–NHC coupling.97 The effect of O–NHC coupling on the catalytic performance of PEPPSI–IPr precatalyst was assessed in the reaction of acetophenone CH-arylation with chlorobenzene. Simple preheating of PEPPSI–IPr 2j with t-BuOK for 10 min and 3 h at 100 °C led to, respectively, 26% and 56% decrease in the yields of CH–arylation product (Scheme 12).97 It should be remembered that M/NHC complexes with metals in the highest oxidation states, e.g. NiIV and PdIV, are susceptible to the reductive elimination of NHC, and the use of such complexes in catalytic systems with assumed participation of MIV species may be inefficient due to their low stability.60 It should also be noted that activated M0/NHC complexes are usually susceptible to M–NHC bond dissociation and prone to ligand displacement and elimination, which may trigger formation of metal clusters and nanoparticles.25
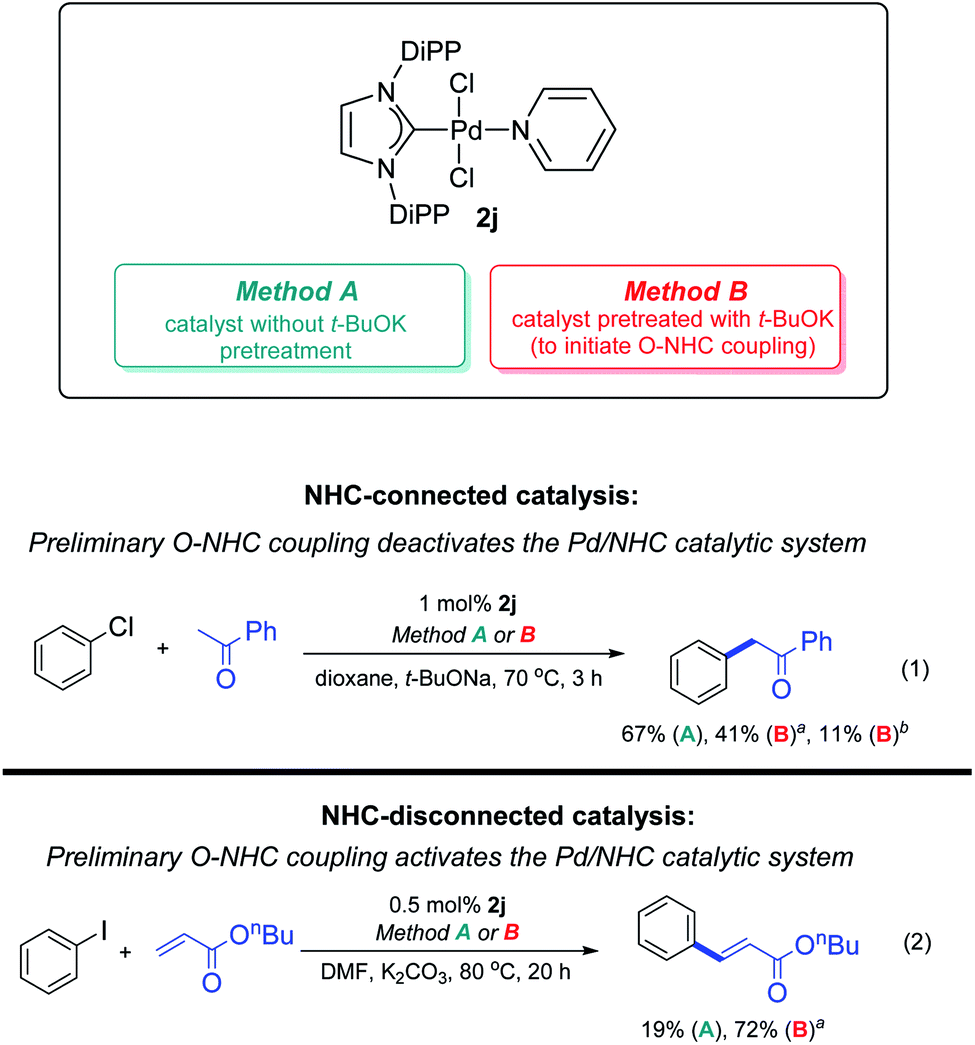 | ||
| Scheme 12 Possible impacts of O–NHC coupling on the Pd-catalyzed C–C coupling reactions. Method A: pretreatment of the catalyst with t-BuOK for 10 min at 100 °C. Method B: pretreatment of the catalyst with t-BuOK for 3 h at 100 °C. Adapted with permission.97 Copyright © 2018 Royal Society of Chemistry. | ||
Thus, suppression of the undesirable M–NHC bond cleavage is generally beneficial for the NHC-connected metal catalysis, and to protect this bond by rational catalyst design is challenging.
Reductive elimination of NHC ligands can be suppressed by building them up with bulky-yet-flexible moieties; this approach also helps to minimize formation of dimeric M–M species.25 However, bulky NHC ligands and strongly binding co-ligands can hinder reductive activation.25 In such cases, the use of ancillary sacrificial ligands as internal reductants for precatalyst activation is feasible. For example, allyl, cinnamyl and related anions are used as convenient sacrificial ligands which can activate PdII/NHC and NiII/NHC complexes under the action of alkoxide anions.57,246,247 The use of cinnamyl or η3-indenyl sacrificial ligands in combination with a bulky NHC ligand ensures facile precatalyst activation while suppressing the formation of PdI–PdI dimeric species.248,249
Morpholine as a sacrificial co-ligand for the base-induced activation of PdII/NHC complexes in C–S cross-coupling reactions was successfully used.55 Preparation of the aliphatic amine complexes in advance turned out to be redundant, as a variety of primary and secondary amines in combination with strong bases can be used directly for the activation of Pd–PEPPSI complexes in C–S cross-coupling reactions.54 Strong bases (e.g. potassium tert-butoxide) deprotonate the NH group of Pd-coordinated amine in the in situ formed amino complexes and facilitate β-hydride transfer from amine to Pd. Besides, the base accelerates reductive elimination of HX from the forming hydride intermediates (NHC)PdHXL thus decreasing their concentration and suppressing the undesirable H–NHC coupling.54
Undesirable formation of (NHC)PdHXL intermediates can be suppressed, in some reactions, by using special additives serving as reversibly coordinating ligands. For example, it was disclosed recently that deactivation of Pd/NHC catalysts in Negishi reaction of alkylzinc reagents can be effectively diminished by the addition of LiBr.165 Excess of bromide ions help to keep Pd atom coordinately saturated in (NHC)Pd(Alkyl)(Aryl)Br catalytic intermediates thus preventing β-hydride transfer from alkyl group to Pd leading to hydride complexes responsible for catalyst deactivation via H–NHC coupling.165
Stability of M/NHC complexes against reductive elimination of NHC ligands can be enhanced by the use of chelated NHC–ligands.250–252 For example, certain complexes with the tridentate pyridine-bridged bis-NHC ligands resist Me–NHC coupling even at 150 °C.250 However, stability of chelated NHC complexes significantly depends on steric factors and can be affected by bulky N-substituents (e.g.tBu).250 Moreover, stable tridentate ligands can hinder coordination of reagents to the metal by reducing the availability of vacant coordination sites.25 For this reason, bidentate chelated NHC ligands that contain a second donor functionality less strongly binding with the metal center and capable of reversible dissociation (oxygen, nitrogen, sulfur, phosphine, or other hemilabile group) may be a better option for certain catalytic systems.5,25,253–255
Overall, rational balance between steric bulkiness and flexibility of NHC ligand, combined with the ease of the throw-away ligands elimination and the use of effective activators, is a prerequisite for high efficacy of the NHC-connected metal catalysis.
4.2. The NHC-disconnected catalysis
One may doubt concerning the benefits of the use of M/NHC complexes in reactions catalyzed by NHC-disconnected metal species. Indeed, the NHC-disconnected mode implies the breakage of the M/NHC complex. Isn't it better to use some cheaper metal salts or complexes?Apparently, there are quite a few reactions proceeding in the mode of NHC-disconnected metal catalysis, in which the M/NHC complexes can be successfully replaced with cheaper metal compounds. For example, in the Pd catalyzed Mizoroki–Heck reaction of butyl acrylate with iodobenzene relatively cheap Pd(OAc)2 combined with a tetraalkylammonium salt [Bu4N]+Br− (Pd nanoparticle stabilizer) demonstrated efficacy no worse than Pd/NHC complexes in the same conditions.27 However, in certain catalytic systems operating by NHC-disconnected metal catalysis, the use of M/NHC precatalysts is reasonable, because of unique features provided by [NHC–R]+ counterions and [NHC–R]+X− salts stabilization mechanisms (Fig. 2C and D).
The NHC-disconnected metal catalysis can be conventionally described as ligandless.38–40,43,256 In this mode, performance of a catalytic system depends significantly on the rate of active metal species formation and their working concentration in solution.38,40,43,256 In the reactions driven by M0 active species, agglomeration of M0 nanoparticles into inactive metal precipitates poses a serious problem.39 A well-known example of this effect is formation of palladium black in Pd-catalyzed reactions. Actual concentrations of active M0 species in such systems depend on the rates of their formation from a precatalyst as related to their stability.38,40,43,256 In many cases, concentration of M0 active species significantly affects their stability, with higher concentrations enhancing the rates of agglomeration thus destabilizing the catalytic system.38,256
The use of M/NHC complexes allows fine tuning of the rates of M0 active species formation; at the same time, organic products of M/NHC decomposition may act as stabilizers for the active metal species despite the M–NHC bond absence (see Fig. 2 as an example).
The main impact of M/NHC precatalyst structure concerns the rate of formation of NHC-disconnected active metal species. This rate strongly depends on the rate of the M–NHC bond cleavage which, in turn, depends on the structure of the NHC ligand and co-ligands.27,54,58–60,62,63,97 M/NHC complexes with bulky NHC ligands (or bis-NHC complexes,59 especially chelated250) are usually more resistant to R–NHC coupling and therefore decompose slower than NHC complexes with non-bulky NHC ligands. For example, the rate of Mizoroki–Heck reactions shows inverse relationship with the stability of Pd/NHC precatalyst.27,250
Decomposition of Pd–PEPPSI–IPr precatalyst via O–NHC coupling by preheating with t-BuOK enhances its catalytic performance significantly (Scheme 12).97 Moreover, catalysis of Mizoroki–Heck reactions by Pd–PEPPSI complexes in the presence of aliphatic amine bases has been shown to proceed by a previously unknown mechanism of the active species generation that provides enhanced robustness of the formed catalytic systems (Scheme 13).59 Heated with tertiary aliphatic amines (e.g. triethylamine) under typical conditions of the Mizoroki–Heck reaction, Pd–PEPPSI complexes undergo the amine-induced H–NHC coupling. The reaction is channeled as follows: at first, Pd–PEPPSI precatalyst reacts with amine to give a primary pool of active metal clusters or nanoparticles while releasing a primary portion of NHC ligand in the form of azolium salt [NHC–H]+X− (Scheme 13). The released NHC reacts promptly with the Pd/NHC species in solution to produce a relatively stable bis-NHC complex Pd(NHC)2X2, this process can also occur due to Pd–NHC bond dissociation. The second channel involves sluggish decomposition of the formed bis-NHC complex for continuous production of the azolium salt and active Pd clusters or nanoparticles (Scheme 13). Thus, the bis-NHC complex acts as a molecular reservoir of active metal species. The observed combination of fast- and slow-release channels ensures prolonged performance of the catalytic system. The efficiency of this approach and the correctness of its mechanistic interpretation were confirmed experimentally; the setting included repeated cycles of filtration of the reaction mixture and reloading with fresh substrates.59
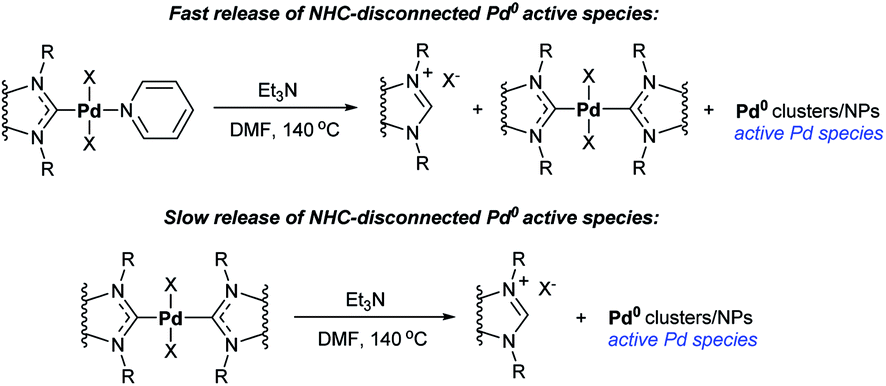 | ||
| Scheme 13 Reactions of fast and slow release of the NHC-disconnected Pd0 active species in the catalytic systems with Pd–PEPPSI precatalyst and triethylamine.59 | ||
As mentioned above, another benefit of using M/NHC precatalysts for the NHC-disconnected catalysis concerns stabilization of active metal species with azolium salts derived from NHC-ligands. The M–NHC bond cleavage via H–NHC coupling, protonolysis or C–NHC coupling produces azolium salts [NHC–R]+X−. Azolium salts are well-known stabilizers of metal nanoparticles.257,258 Imidazolium cations formed via C–NHC coupling provide enhanced stability against strong bases and are known as highly promising subclass of ionic liquids.72
Thus, active metal clusters and nanoparticles formed from M/NHC precatalysts can be effectively stabilized in situ by azolium salts. This mechanism was proposed for Pd/NHC catalysis in Mizoroki–Heck reaction27 and confirmed experimentally for Rh/NHC catalyzed hydrogenation of arenes. Under conditions of arene hydrogenation, by means of ex situ and operando XAFS studies, scanning transmission electron microscopy and IR spectroscopy, it was revealed that [(CAAC)Rh(COD)Cl] complexes form Rh nanoparticles stabilized with the cations of protonated CAAC (Fig. 4), which function as active centers for the arene hydrogenation.148,149 Moreover, CAAC-derived products adsorbed on Rh nanoparticles were shown to play a key role in providing high chemoselectivity of fluorinated arenes hydrogenation.259 Remarkably, azolium cations can effectively stabilize anionic [PdX3]− complexes released from Pd/NHC precatalysts in Mizoroki–Heck reactions at low Pd loadings (0.1 mM concentrations and below).62 Ionic complex ([NHC–Ph]+)2[Pd2X6]2−, formed by fast Ph–NHC coupling and isolated from the reaction mixture, was recognized as a new type of ionic palladium precatalysts for Mizoroki–Heck reaction. In this case, the [NHC–Ph]+ cation acts as ionic stabilizer for NHC-disconnected active species; the ionic state prevents aggregation and progressive deactivation of the catalyst in diluted solutions.62
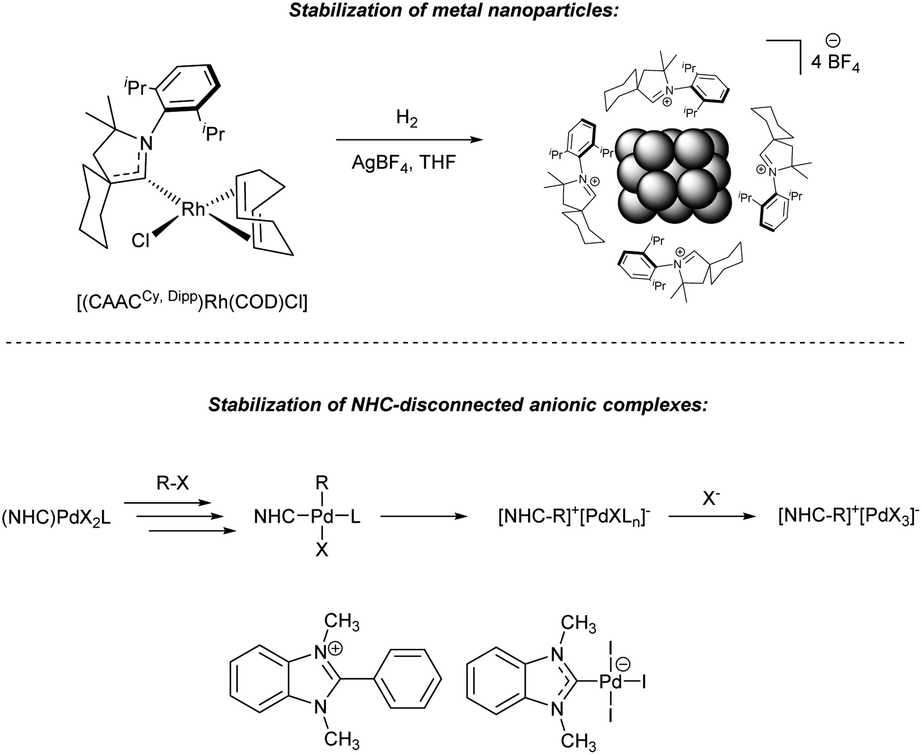 | ||
| Fig. 4 Stabilization of the NHC-disconnected active metal species with azolium salts formed from M/NHC precatalysts. (a) Rh nanoparticles in the [(CAAC)Rh(COD)Cl]-catalyzed arene hydrogenation;148,149 (b) anionic Pd complexes in Mizoroki–Heck reaction.62 | ||
Overall, in certain catalytic systems M/NHC precatalysts can provide high catalytic performance owing to the regulated release of NHC-disconnected M0 active species and their in situ stabilization by azolium salts and non-coordinative cations derived from the NHC ligands.
5. Recycling and sustainability aspects of M/NHC systems
Consideration of catalyst recycling highlights a long-standing contradiction in the area of M/NHC catalysis. On one hand M/NHC complexes were believed to be highly stable, while on the other hand the number of successful recycling experiments is drastically limited. In fact, molecular M/NHC complexes are not considered as easily recyclable catalysts.260–265 The topic disclosed in the present review sheds some light on this problem. Indeed, catalyst evolution readily takes place during M/NHC-catalyzed reactions and in many cases the complexes can not be recycled in initial state. It should be mentioned that supported heterogenized M/NHC catalysts were studied for several reactions to perform recycling,266–270 however it is a different approach which is not considered in details here.Among the three possible pathways for M/NHC catalysis, catalyst recycling in the NHC-connected molecular mode (Fig. 2B) is less studied. Many M/NHC complexes are stable under catalytic conditions and retain their activity due to the strong M–NHC bond. However, a few only can be recovered in their initial form after the reaction. (NHC)NiCl(Cp) complexes were used in a regioselective Markovnikov-type thiol-yne click reaction.100 (IMes)NiCl(Cp) complex was found to be the most active catalyst. However, its recovery, although technically possible, was profoundly inefficient. The losses were due to IMes-S coupling that acted as the major catalyst decomposition pathway during the reaction.
Of note, there is another possible option, which includes a different type of supported heterogenized NHC systems. Heterogenized systems have been reviewed previously and are not considered here (corresponding reviews can be found elsewhere269,270).
Recycling with NHC-disconnected molecular catalysis involving ionic species (Fig. 2C) is more feasible, although yet not explicitly explored. One of the major pathways to irreversibly eliminate NHC from the coordination sphere of palladium is Ar–NHC coupling. This process leads to ionic pair that can be stabilized in solution by either decreasing the concentration or increasing the ionic strength. Adjustment of these parameters allowed efficient isolation of the [NHC–Ph]+[NHC–PdI3]− (NHC = 1,3-dimethylbenzimidazol-2-ylidene) ionic complex from the reaction mixture; it was successfully reused for five consecutive cycles without loss of catalytic activity (Fig. 5).62
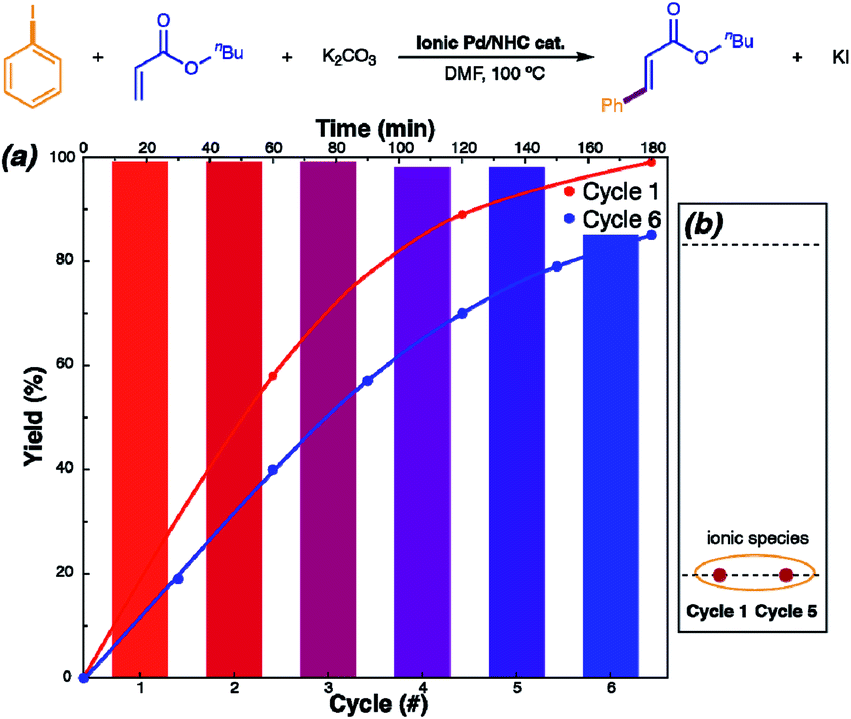 | ||
| Fig. 5 Ionic Pd/NHC-catalyzed Mizoroki–Heck reaction with counterion stabilization: mechanism, recycling, and scope. The reaction was examined for recovery of the ionic Pd/NHC complex. (a) Reaction profiles for a sequence of cycles, with cycle 6 performed after 3 week storage of the isolated catalyst. (b) Model TLC analysis of the ionic Pd/NHC complex catalyst recovered after cycles 1 and 5. Adapted with permission.62 Copyright ©2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
With increasing cycle number, the [NHC–Pd(I)3]− was progressively transformed into ligandless [PdI3]−, although the [NHC–Ph]+[PdI3]− became detectable by TLC, NMR and ESI-MS after the fifth cycle only. A solid dimer of this complex with [Pd2I6]2− anion readily dissociates in solution. In the studied Mizoroki–Heck reaction, H–NHC coupling acts as a reversible pathway to the molecular NHC–Pd bonded mode of catalysis, whereas Ph–NHC coupling promotes formation of a stabilizing non-coordinating cation to keep the metal in its active form.
Noteworthy, leaching driven formation of NHC-disconnected ionic systems might be a promising method to achieve catalyst recycling. C–C and C–H oxidative additions to palladium nanoparticles promote separation of individual metal complexes and their leaching into liquid phase. The heating of [NHC–Me]+ salt with aryl halides in the presence of palladium nanoparticles, copper or nickel salts leads to formation of [NHC–Ar]+X− salts, that is a vivid example of the C–C bond oxidative addition-driven leaching.61
In the meantime, high concentrations of precatalyst promote sintering of the NHC-disconnected palladium, and catalyst recycling in the metal cluster pathway (Fig. 2D) can be achieved. In general, it follows the more developed approach, available for cocktail-type systems.
Some NHC–Pd–R complexes are thermally labile but capable of forming water-resistant nanoparticles stabilized by NHC ligands. A protocol for obtaining Pd nanoparticles (NPs) from (NHC)2PdMe2 (NHC = 3-sodium sulfonatopropyl substituted imidazolylidene) was developed by water-mediated thermal decomposition of the complex at 80 °C (Scheme 14).166 This reaction yields both NHC–Me and NHC–H products (corresponding to about 60% and 40% of the conversion, respectively). The metal-free NHC species eventually bind to the forming Pd NPs, which makes them water-soluble. The resulting Pd NPs successfully catalyzed 10 consecutive cycles of styrene hydrogenation in water, and no degradation of the catalyst by precipitation of bulk palladium was observed during the reaction.
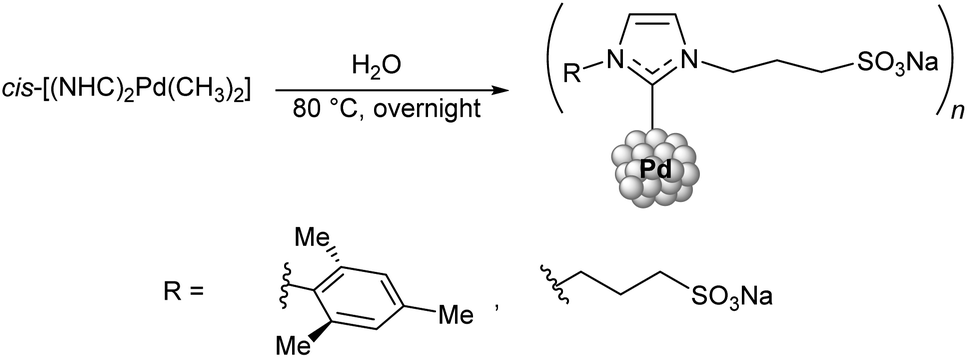 | ||
| Scheme 14 Synthesis of water-soluble Pd NPs by thermal decomposition of the dimethyl bis-NHC complexes.166 | ||
Although it is usually not accentuated, leaching that affects nanoparticle catalysts in the systems with NHC–H precursors might be classified as NHC ligand-assisted leaching due to activation of the C–H bond by either a base or direct M0 oxidative addition.
To summarize this section, recycling of M/NHC complexes for re-use in the further reaction is on the early stages of development. More in depth studies are required to develop efficient recycling protocols and solve sustainability problem. Revealing the nature of active species is the key-requirement for successful recycling.
6. Conclusions
M/NHC complexes have been long considered as robust homogeneous catalysts with catalytic performance substantially dependent on the enhanced strength of the M–CNHC bond. It is currently becoming clear that the M–NHC bonds are susceptible to facile cleavage even under mild conditions. The integrity of the M–NHC framework can be violated by reactions of reductive elimination, protonolysis and ligand displacement. The analysis carried out in this article shows that M–NHC bond cleavage phenomenon is typical for most of the metals and NHC ligands and may significantly affect the catalysis. Among the considered reactions (Table 1), the H–NHC bond formation may be a reversible step, while many cleavage reactions with the formation of C–NHC and X–NHC bonds may not be reversible under catalytic conditions.The impact of the M–NHC bond breakage on a catalytic system largely depends on the mode of its functioning (Fig. 2). Many M/NHC-catalyzed reactions proceed by NHC-connected mechanisms where the M–NHC framework directly participates in catalytic cycle, notably affecting transition states of the transformation. In such cases, the M–NHC bond cleavage is highly undesirable as it ultimately leads to deactivation of the catalytic system.
However, a number of reaction systems operate in the NHC-disconnected catalysis mode, with M/NHC complexes acting as precursors of active species. In such systems, M–NHC bond cleavage leads to the generation of active centers and, thus, represents a process of catalyst activation. Rational M/NHC catalyst design should therefore account for the type of catalytic mechanism.
In the systems that operate in the mode of NHC-connected metal catalysis, the structure of the M/NHC catalyst has to ensure a combination of high catalytic activity with sufficient stability of the M–NHC framework. At present, the most reliable approaches are based on the rational balance between the steric bulkiness and flexibility of NHC ligands combined with the use of easily eliminated throw-away co-ligands and effective activators. A new promising approach is to use chelating NHC ligands that, in addition to the strongly binding NHC carbon, contain a mild donor site capable of switchable coordination/decoordination with the metal.
In the systems that operate in the mode of NHC-disconnected metal catalysis, the M/NHC precatalyst should provide the optimal rate of M–NHC bond breaking to release the active metal species. At the same time, the products of M/NHC decomposition have to ensure effective stabilization of the active species (unless it is ensured by the use of ancillary stabilizers).
Processes of M–NHC bond cleavage can change molecular M/NHC catalytic system to nano-sized dimension. Nanoparticles bearing NHC frameworks are actively studied and found their applications in many fields, including materials science, which is not a subject of this review. Alongside, clusters being a transition between nanoparticles and molecular systems are of high importance. Many clusters were proposed to possess high catalytic properties, however studies of well-defined metal clusters with NHC ligands are yet very limited. Thus, pronounced developments in this area are anticipated in the near future.
We hope that this review will draw attention of researchers to the problem of M–NHC framework lability and promote the development of new effective approaches for the rational design of M/NHC catalytic systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Russian Science Foundation grant no. 19-73-20085 for the support of organometallic part and Russian Science Foundation grant no. 19-13-00460 for the support of dynamic catalysis part.Notes and references
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef CAS PubMed.
- R. H. Crabtree, New J. Chem., 2011, 35, 18–23 RSC.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed. Engl., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed. Engl., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed. Engl., 2007, 46, 2768–2813 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed. Engl., 2010, 49, 6940–6952 CrossRef PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- D. Janssen-Müller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- S. Budagumpi, R. S. Keri, G. Achar and K. N. Brinda, Adv. Synth. Catal., 2020, 362, 970–997 CrossRef CAS.
- C. Fliedel, A. Labande, E. Manoury and R. Poli, Coord. Chem. Rev., 2019, 394, 65–103 CrossRef CAS.
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244–2253 CrossRef CAS PubMed.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS.
- K. J. Cavell and D. S. McGuinness, Coord. Chem. Rev., 2004, 248, 671–681 CrossRef CAS.
- K. Cavell, Dalton Trans., 2008, 6676–6685 RSC.
- K. J. Cavell and A. T. Normand, in N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer Netherlands, Dordrecht, 2011, pp. 299–314 Search PubMed.
- B. R. M. Lake, M. R. Chapman and C. E. Willans, in Organometallic Chemistry, Royal Society of Chemistry, 2016, vol. 40, pp. 107–139 Search PubMed.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- D. J. Nelson, J. M. Praetorius and C. M. Crudden, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, London, UK, 2017, pp. 46-98 Search PubMed.
- D. S. McGuinness, M. J. Green, K. J. Cavell, B. W. Skelton and A. H. White, J. Organomet. Chem., 1998, 565, 165–178 CrossRef CAS.
- A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 36, 1981–1992 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- K. K. Hii and K. Hellgardt, Top. Organomet. Chem., 2016, 57, 249–262 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595–1604 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- A. S. Kashin and V. P. Ananikov, J. Org. Chem., 2013, 78, 11117–11125 CrossRef CAS PubMed.
- C. G. Baumann, S. De Ornellas, J. P. Reeds, T. E. Storr, T. J. Williams and I. J. S. Fairlamb, Tetrahedron, 2014, 70, 6174–6187 CrossRef CAS.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- A. F. Schmidt, A. A. Kurokhtina and E. V. Larina, Catal. Sci. Technol., 2014, 4, 3439–3457 RSC.
- J. F. Sonnenberg and R. H. Morris, Catal. Sci. Technol., 2014, 4, 3426–3438 RSC.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 909–933 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, in New Trends in Cross-Coupling: Theory and Applications, The Royal Society of Chemistry, 2015, pp. 355–478 Search PubMed.
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS PubMed.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS.
- A. M. Trzeciak and A. W. Augustyniak, Coord. Chem. Rev., 2019, 384, 1–20 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- J. B. Ernst, C. Schwermann, G.-i. Yokota, M. Tada, S. Muratsugu, N. L. Doltsinis and F. Glorius, J. Am. Chem. Soc., 2017, 139, 9144–9147 CrossRef CAS PubMed.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed. Engl., 2018, 57, 4981–4985 CrossRef CAS PubMed.
- M. A. Ortuño and N. López, Catal. Sci. Technol., 2019, 9, 5173–5185 RSC.
- S. Meiries, G. Le Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2013, 19, 17358–17368 CrossRef CAS PubMed.
- F. Izquierdo, S. Manzini and S. P. Nolan, Chem. Commun., 2014, 50, 14926–14937 RSC.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed. Engl., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- G. Bastug and S. P. Nolan, J. Org. Chem., 2013, 78, 9303–9308 CrossRef CAS PubMed.
- O. V. Khazipov, M. A. Shevchenko, D. V. Pasyukov, A. Y. Chernenko, A. V. Astakhov, V. A. Tafeenko, V. M. Chernyshev and V. P. Ananikov, Catal. Sci. Technol., 2020, 10, 1228–1247 RSC.
- J. L. Farmer, M. Pompeo, A. J. Lough and M. G. Organ, Chem.–Eur. J., 2014, 20, 15790–15798 CrossRef CAS PubMed.
- E. Marelli, M. Corpet, S. R. Davies and S. P. Nolan, Chem.–Eur. J., 2014, 20, 17272–17276 CrossRef CAS PubMed.
- J. A. Fernández-Salas, E. Marelli, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2015, 21, 3906–3909 CrossRef PubMed.
- E. G. Gordeev, D. B. Eremin, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2018, 37, 787–796 CrossRef CAS.
- O. V. Khazipov, M. A. Shevchenko, A. Y. Chernenko, A. V. Astakhov, D. V. Pasyukov, D. B. Eremin, Y. V. Zubavichus, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2018, 37, 1483–1492 CrossRef CAS.
- A. V. Astakhov, S. B. Soliev, E. G. Gordeev, V. M. Chernyshev and V. P. Ananikov, Dalton Trans., 2019, 48, 17052–17062 RSC.
- E. A. Denisova, D. B. Eremin, E. G. Gordeev, A. M. Tsedilin and V. P. Ananikov, Inorg. Chem., 2019, 58, 12218–12227 CrossRef CAS PubMed.
- D. B. Eremin, E. A. Denisova, A. Yu. Kostyukovich, J. Martens, G. Berden, J. Oomens, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Chem.–Eur. J., 2019, 25, 16564–16572 CrossRef CAS PubMed.
- E. G. Gordeev and V. P. Ananikov, J. Comput. Chem., 2019, 40, 191–199 CrossRef CAS PubMed.
- A. Y. Kostyukovich, A. M. Tsedilin, E. D. Sushchenko, D. B. Eremin, A. S. Kashin, M. A. Topchiy, A. F. Asachenko, M. S. Nechaev and V. P. Ananikov, Inorg. Chem. Front., 2019, 6, 482–492 RSC.
- L.-C. Campeau, P. Thansandote and K. Fagnou, Org. Lett., 2005, 7, 1857–1860 CrossRef CAS PubMed.
- S. B. Soliev, A. V. Astakhov, D. V. Pasyukov and V. M. Chernyshev, Russ. Chem. Bull., 2020, 69, 683–690 CrossRef CAS.
- D. S. McGuinness, W. Mueller, P. Wasserscheid, K. J. Cavell, B. W. Skelton, A. H. White and U. Englert, Organometallics, 2002, 21, 175–181 CrossRef CAS.
- P. Csabai, F. Joó, A. M. Trzeciak and J. J. Ziółkowski, J. Organomet. Chem., 2006, 691, 3371–3376 CrossRef CAS.
- D. McGuinness, Dalton Trans., 2009, 6915–6923 RSC.
- D. S. McGuinness and K. J. Cavell, in N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer Netherlands, Dordrecht, 2011, pp. 105–129 Search PubMed.
- T. J. Williams, J. T. W. Bray, B. R. M. Lake, C. E. Willans, N. A. Rajabi, A. Ariafard, C. Manzini, F. Bellina, A. C. Whitwood and I. J. S. Fairlamb, Organometallics, 2015, 34, 3497–3507 CrossRef CAS.
- N. K. T. Ho, B. Neumann, H.-G. Stammler, V. H. Menezes da Silva, D. G. Watanabe, A. A. C. Braga and R. S. Ghadwal, Dalton Trans., 2017, 46, 12027–12031 RSC.
- X. Xie and H. V. Huynh, Org. Chem. Front., 2015, 2, 1598–1603 RSC.
- R. Li, Y. Hu, R. Liu, R. Hu, B. Li and B. Wang, Adv. Synth. Catal., 2015, 357, 3885–3892 CrossRef CAS.
- D. Ghorai and J. Choudhury, Chem. Commun., 2014, 50, 15159–15162 RSC.
- R. Thenarukandiyil and J. Choudhury, Organometallics, 2015, 34, 1890–1897 CrossRef CAS.
- D. Ghorai, C. Dutta and J. Choudhury, ACS Catal., 2016, 6, 709–713 CrossRef CAS.
- R. Thenarukandiyil, H. Thrikkykkal and J. Choudhury, Organometallics, 2016, 35, 3007–3013 CrossRef CAS.
- Z. She, Y. Wang, D. Wang, Y. Zhao, T. Wang, X. Zheng, Z.-X. Yu, G. Gao and J. You, J. Am. Chem. Soc., 2018, 140, 12566–12573 CrossRef CAS PubMed.
- S. Haslinger, J. W. Kück, M. R. Anneser, M. Cokoja, A. Pöthig and F. E. Kühn, Chem.–Eur. J., 2015, 21, 17860–17869 CrossRef CAS PubMed.
- B. R. M. Lake, A. Ariafard and C. E. Willans, Chem.–Eur. J., 2014, 20, 12729–12733 CrossRef CAS PubMed.
- D. S. McGuinness, N. Saendig, B. F. Yates and K. J. Cavell, J. Am. Chem. Soc., 2001, 123, 4029–4040 CrossRef CAS PubMed.
- M. Heckenroth, A. Neels, M. G. Garnier, P. Aebi, A. W. Ehlers and M. Albrecht, Chem.–Eur. J., 2009, 15, 9375–9386 CrossRef CAS PubMed.
- K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 2685–2686 CrossRef CAS PubMed.
- K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 13964–13965 CrossRef CAS PubMed.
- K. Araki, S. Kuwata and T. Ikariya, Organometallics, 2008, 27, 2176–2178 CrossRef CAS.
- K. J. Hawkes, K. J. Cavell and B. F. Yates, Organometallics, 2008, 27, 4758–4771 CrossRef CAS.
- J. C. Lewis, A. M. Berman, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 2493–2500 CrossRef CAS PubMed.
- S. Kuwata and F. E. Hahn, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS PubMed.
- D. S. McGuinness, K. J. Cavell, B. F. Yates, B. W. Skelton and A. H. White, J. Am. Chem. Soc., 2001, 123, 8317–8328 CrossRef CAS PubMed.
- D. C. Graham, K. J. Cavell and B. F. Yates, Dalton Trans., 2007, 4650–4658 RSC.
- N. D. Clement and K. J. Cavell, Angew. Chem., Int. Ed. Engl., 2004, 43, 3845–3847 CrossRef CAS PubMed.
- T. Steinke, B. K. Shaw, H. Jong, B. O. Patrick and M. D. Fryzuk, Organometallics, 2009, 28, 2830–2836 CrossRef CAS.
- J. W. Sprengers, J. Wassenaar, N. D. Clement, K. J. Cavell and C. J. Elsevier, Angew. Chem., Int. Ed. Engl., 2005, 44, 2026–2029 CrossRef CAS PubMed.
- S. Fantasia, J. D. Egbert, V. Jurčík, C. S. J. Cazin, H. Jacobsen, L. Cavallo, D. M. Heinekey and S. P. Nolan, Angew. Chem., Int. Ed. Engl., 2009, 48, 5182–5186 CrossRef CAS PubMed.
- J. Broggi, V. Jurčík, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588–4591 CrossRef CAS PubMed.
- V. M. Chernyshev, O. V. Khazipov, M. A. Shevchenko, A. Y. Chernenko, A. V. Astakhov, D. B. Eremin, D. V. Pasyukov, A. S. Kashin and V. P. Ananikov, Chem. Sci., 2018, 9, 5564–5577 RSC.
- M. Sayah, A. J. Lough and M. G. Organ, Chem.–Eur. J., 2013, 19, 2749–2756 CrossRef CAS PubMed.
- J. Li, J. Morris, W. W. Brennessel and W. D. Jones, J. Chem. Crystallogr., 2014, 44, 15–19 CrossRef CAS.
- D. A. Malyshev, N. M. Scott, N. Marion, E. D. Stevens, V. P. Ananikov, I. P. Beletskaya and S. P. Nolan, Organometallics, 2006, 25, 4462–4470 CrossRef CAS.
- B.-L. Lin, P. Kang and T. D. P. Stack, Organometallics, 2010, 29, 3683–3685 CrossRef CAS PubMed.
- X. Hu and K. Meyer, J. Am. Chem. Soc., 2004, 126, 16322–16323 CrossRef CAS PubMed.
- K. Fauché, L. Nauton, L. Jouffret, F. Cisnetti and A. Gautier, Chem. Commun., 2017, 53, 2402–2405 RSC.
- K. Nomura, G. Nagai, A. Nasr, K. Tsutsumi, Y. Kawamoto, K. Koide and M. Tamm, Organometallics, 2019, 38, 3233–3244 CrossRef CAS.
- M. Uzelac, A. Hernán-Gómez, D. R. Armstrong, A. R. Kennedy and E. Hevia, Chem. Sci., 2015, 6, 5719–5728 RSC.
- M. Chen, Y. Wang, R. J. Gilliard, Jr., P. Wei, N. A. Schwartz and G. H. Robinson, Dalton Trans., 2014, 43, 14211–14214 CAS.
- R. S. Ghadwal, D. Rottschäfer, D. M. Andrada, G. Frenking, C. J. Schürmann and H.-G. Stammler, Dalton Trans., 2017, 46, 7791–7799 RSC.
- Y. Wang, M. Y. Abraham, R. J. Gilliard, P. Wei, J. C. Smith and G. H. Robinson, Organometallics, 2012, 31, 791–793 CrossRef CAS.
- Y. Wang, Y. Xie, P. Wei, H. F. Schaefer and G. H. Robinson, Dalton Trans., 2016, 45, 5941–5944 RSC.
- L. P. Ho, A. Nasr, P. G. Jones, A. Altun, F. Neese, G. Bistoni and M. Tamm, Chem.–Eur. J., 2018, 24, 18922–18932 CAS.
- A. Hernán-Gómez, M. Uzelac, S. E. Baillie, D. R. Armstrong, A. R. Kennedy, M. Á. Fuentes and E. Hevia, Chem.–Eur. J., 2018, 24, 10541–10549 CrossRef PubMed.
- Y. Wang, N. A. Maxi, Y. Xie, P. Wei, H. F. Schaefer and G. H. Robinson, Chem. Commun., 2019, 55, 8087–8089 RSC.
- T. Chu, S. F. Vyboishchikov, B. Gabidullin and G. I. Nikonov, Angew. Chem., Int. Ed. Engl., 2016, 55, 13306–13311 CrossRef CAS PubMed.
- C.-C. Tai, Y.-T. Chang, J.-H. Tsai, T. Jurca, G. P. A. Yap and T.-G. Ong, Organometallics, 2012, 31, 637–643 CrossRef CAS.
- A. L. Schmitt, G. Schnee, R. Welter and S. Dagorne, Chem. Commun., 2010, 46, 2480–2482 RSC.
- G. Schnee, O. Nieto Faza, D. Specklin, B. Jacques, L. Karmazin, R. Welter, C. Silva López and S. Dagorne, Chem.–Eur. J., 2015, 21, 17959–17972 CrossRef CAS PubMed.
- V. Dardun, L. Escomel, E. Jeanneau and C. Camp, Dalton Trans., 2018, 47, 10429–10433 RSC.
- A. Hock, H. Schneider, M. J. Krahfuß and U. Radius, Z. Anorg. Allg. Chem., 2018, 644, 1243–1251 CrossRef CAS.
- M. L. Cole, D. E. Hibbs, C. Jones, P. C. Junk and N. A. Smithies, Inorg. Chim. Acta, 2005, 358, 102–108 CrossRef CAS.
- W.-C. Shih, C.-H. Wang, Y.-T. Chang, G. P. A. Yap and T.-G. Ong, Organometallics, 2009, 28, 1060–1067 CrossRef CAS.
- T. Fukuda, H. Hashimoto and H. Tobita, J. Organomet. Chem., 2017, 848, 89–94 CrossRef CAS.
- M. M. D. Roy, S. Fujimori, M. J. Ferguson, R. McDonald, N. Tokitoh and E. Rivard, Chem.–Eur. J., 2018, 24, 14392–14399 CrossRef CAS PubMed.
- M. L. Cole, A. J. Davies and C. Jones, J. Chem. Soc., Dalton Trans., 2001, 2451–2452 RSC.
- P. L. Arnold, I. A. Marr, S. Zlatogorsky, R. Bellabarba and R. P. Tooze, Dalton Trans., 2014, 43, 34–37 RSC.
- P. L. Arnold, T. Cadenbach, I. H. Marr, A. A. Fyfe, N. L. Bell, R. Bellabarba, R. P. Tooze and J. B. Love, Dalton Trans., 2014, 43, 14346–14358 RSC.
- T. Simler, T. J. Feuerstein, R. Yadav, M. T. Gamer and P. W. Roesky, Chem. Commun., 2019, 55, 222–225 RSC.
- S.-T. Liu, R.-Z. Ku, C.-Y. Liu and F.-M. Kiang, J. Organomet. Chem., 1997, 543, 249–250 CrossRef CAS.
- S. Li, C. W. Kee, K.-W. Huang, T. S. A. Hor and J. Zhao, Organometallics, 2010, 29, 1924–1933 CrossRef CAS.
- R. Schowner, W. Frey and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2019, 2019, 1911–1922 CrossRef CAS.
- E. Mas-Marzá, P. M. Reis, E. Peris and B. Royo, J. Organomet. Chem., 2006, 691, 2708–2712 CrossRef.
- A. A. Grineva, O. A. Filippov, S. E. Nefedov, N. Lugan, V. César and D. A. Valyaev, Organometallics, 2019, 38, 2330–2337 CrossRef CAS.
- T. Hatanaka, Y. Ohki and K. Tatsumi, Angew. Chem., Int. Ed., 2014, 53, 2727–2729 CrossRef CAS PubMed.
- T. Pugh and R. A. Layfield, Dalton Trans., 2014, 43, 4251–4254 RSC.
- B. M. Day, T. Pugh, D. Hendriks, C. F. Guerra, D. J. Evans, F. M. Bickelhaupt and R. A. Layfield, J. Am. Chem. Soc., 2013, 135, 13338–13341 CrossRef CAS PubMed.
- Ö. Karaca, M. R. Anneser, J. W. Kück, A. C. Lindhorst, M. Cokoja and F. E. Kühn, J. Catal., 2016, 344, 213–220 CrossRef.
- J. Cheng, J. Liu, X. Leng, T. Lohmiller, A. Schnegg, E. Bill, S. Ye and L. Deng, Inorg. Chem., 2019, 58, 7634–7644 CrossRef CAS PubMed.
- J. J. Dunsford, I. A. Cade, K. L. Fillman, M. L. Neidig and M. J. Ingleson, Organometallics, 2014, 33, 370–377 CrossRef CAS.
- X. Wang, J. Zhang, L. Wang and L. Deng, Organometallics, 2015, 34, 2775–2782 CrossRef CAS.
- C. Ma, C. Ai, Z. Li, B. Li, H. Song, S. Xu and B. Wang, Organometallics, 2014, 33, 5164–5172 CrossRef CAS.
- R. C. da Costa, F. Hampel and J. A. Gladysz, Polyhedron, 2007, 26, 581–588 CrossRef.
- M. Rouen, P. Queval, L. Falivene, J. Allard, L. Toupet, C. Crévisy, F. Caijo, O. Baslé, L. Cavallo and M. Mauduit, Chem.–Eur. J., 2014, 20, 13716–13721 CrossRef CAS PubMed.
- H. Yoo and D. H. Berry, Inorg. Chem., 2014, 53, 11447–11456 CrossRef CAS PubMed.
- V. H. Mai, I. Korobkov and G. I. Nikonov, Organometallics, 2016, 35, 936–942 CrossRef CAS.
- B. M. Day, K. Pal, T. Pugh, J. Tuck and R. A. Layfield, Inorg. Chem., 2014, 53, 10578–10584 CrossRef CAS PubMed.
- C. B. Hansen, R. F. Jordan and G. L. Hillhouse, Inorg. Chem., 2015, 54, 4603–4610 CrossRef CAS PubMed.
- P. Karak, C. Dutta, T. Dutta, A. L. Koner and J. Choudhury, Chem. Commun., 2019, 55, 6791–6794 RSC.
- D. P. Allen, C. M. Crudden, L. A. Calhoun and R. Wang, J. Organomet. Chem., 2004, 689, 3203–3209 CrossRef CAS.
- B. L. Tran, J. L. Fulton, J. C. Linehan, M. Balasubramanian, J. A. Lercher and R. M. Bullock, ACS Catal., 2019, 9, 4106–4114 CrossRef CAS.
- B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441–8449 CrossRef CAS.
- C. Y. Tang, W. Smith, A. L. Thompson, D. Vidovic and S. Aldridge, Angew. Chem., Int. Ed. Engl., 2011, 50, 1359–1362 CrossRef CAS PubMed.
- T. Zell, P. Fischer, D. Schmidt and U. Radius, Organometallics, 2012, 31, 5065–5073 CrossRef CAS.
- B. R. Dible, M. S. Sigman and A. M. Arif, Inorg. Chem., 2005, 44, 3774–3776 CrossRef CAS PubMed.
- R. J. Hazlehurst, S. W. E. Hendriks, P. D. Boyle and J. M. Blacquiere, ChemistrySelect, 2017, 2, 6732–6737 CrossRef CAS.
- T. J. Hadlington, T. Szilvási and M. Driess, Angew. Chem., Int. Ed. Engl., 2017, 56, 7470–7474 CrossRef CAS PubMed.
- D. S. McGuinness, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 1596–1605 CrossRef CAS.
- A. T. Normand, A. Stasch, L.-L. Ooi and K. J. Cavell, Organometallics, 2008, 27, 6507–6520 CrossRef CAS.
- G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed. Engl., 2018, 57, 431–436 CrossRef CAS PubMed.
- S. Caddick, F. G. N. Cloke, P. B. Hitchcock, J. Leonard, A. K. d. K. Lewis, D. McKerrecher and L. R. Titcomb, Organometallics, 2002, 21, 4318–4319 CrossRef CAS.
- J.-F. Lefebvre, J.-F. Longevial, K. Molvinger, S. Clément and S. Richeter, C. R. Chim., 2016, 19, 94–102 CrossRef CAS.
- D. S. McGuinness and K. J. Cavell, Organometallics, 2000, 19, 4918–4920 CrossRef CAS.
- A. T. Normand, M. S. Nechaev and K. J. Cavell, Chem.–Eur. J., 2009, 15, 7063–7073 CrossRef CAS PubMed.
- E. P. Couzijn, E. Zocher, A. Bach and P. Chen, Chem.–Eur. J., 2010, 16, 5408–5415 CrossRef CAS PubMed.
- O. Esposito, P. M. P. Gois, A. K. de K. Lewis, S. Caddick, F. G. N. Cloke and P. B. Hitchcock, Organometallics, 2008, 27, 6411–6418 CrossRef CAS.
- W. J. Marshall and V. V. Grushin, Organometallics, 2003, 22, 1591–1593 CrossRef CAS.
- P. Eckert and M. G. Organ, Chem.–Eur. J., 2020, 26, 4861–4865 CrossRef CAS PubMed.
- J. M. Asensio, S. Tricard, Y. Coppel, R. Andrés, B. Chaudret and E. de Jesús, Chem.–Eur. J., 2017, 23, 13435–13444 CrossRef CAS PubMed.
- S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2018, 140, 3551–3554 CrossRef CAS PubMed.
- C.-F. Fu, C.-C. Lee, Y.-H. Liu, S.-M. Peng, S. Warsink, C. J. Elsevier, J.-T. Chen and S.-T. Liu, Inorg. Chem., 2010, 49, 3011–3018 CrossRef CAS PubMed.
- C.-Y. Wang, Y.-H. Liu, S.-M. Peng, J.-T. Chen and S.-T. Liu, J. Organomet. Chem., 2007, 692, 3976–3983 CrossRef CAS.
- A. Y. Chernenko, D. V. Pasyukov, A. V. Astakhov, V. A. Tafeenko and V. M. Chernyshev, Russ. Chem. Bull., 2018, 67, 1196–1201 CrossRef CAS.
- D. S. McGuinness, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 1596–1605 CrossRef CAS.
- E. Lee, D. Y. Bae, S. Park, A. G. Oliver, Y. Kim and D. V. Yandulov, Eur. J. Inorg. Chem., 2016, 2016, 4561–4564 CrossRef CAS.
- A. M. Magill, D. S. McGuinness, K. J. Cavell, G. J. P. Britovsek, V. C. Gibson, A. J. P. White, D. J. Williams, A. H. White and B. W. Skelton, J. Organomet. Chem., 2001, 617–618, 546–560 CrossRef CAS.
- X. Zhou and R. F. Jordan, Organometallics, 2011, 30, 4632–4642 CrossRef CAS.
- O. Esposito, D. E. Roberts, F. G. N. Cloke, S. Caddick, J. C. Green, N. Hazari and P. B. Hitchcock, Eur. J. Inorg. Chem., 2009, 2009, 1844–1850 CrossRef.
- D. Bacciu, K. J. Cavell, I. A. Fallis and L.-l. Ooi, Angew. Chem., Int. Ed. Engl., 2005, 44, 5282–5284 CrossRef CAS PubMed.
- S. Li, J. Tang, Y. Zhao, R. Jiang, T. Wang, G. Gao and J. You, Chem. Commun., 2017, 53, 3489–3492 RSC.
- E. D. Blue, T. B. Gunnoe, J. L. Petersen and P. D. Boyle, J. Organomet. Chem., 2006, 691, 5988–5993 CrossRef CAS.
- F. Lazreg and C. S. J. Cazin, Organometallics, 2018, 37, 679–683 CrossRef CAS.
- F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef CAS.
- D. Li and T. Ollevier, Org. Lett., 2019, 21, 3572–3575 CrossRef CAS PubMed.
- D. Li and T. Ollevier, J. Organomet. Chem., 2020, 906, 121025 CrossRef CAS.
- W. Zeng, E. Wang, R. Qiu, M. Sohail, S. Wu and F.-X. Chen, J. Organomet. Chem., 2013, 743, 44–48 CrossRef CAS.
- E. L. Kolychev, V. V. Shuntikov, V. N. Khrustalev, A. A. Bush and M. S. Nechaev, Dalton Trans., 2011, 40, 3074–3076 RSC.
- M. Blum, J. Kappler, S. H. Schlindwein, M. Nieger and D. Gudat, Dalton Trans., 2018, 47, 112–119 RSC.
- S. Groysman and R. H. Holm, Inorg. Chem., 2009, 48, 621–627 CrossRef CAS PubMed.
- M. Slivarichova, R. Correa da Costa, J. Nunn, R. Ahmad, M. F. Haddow, H. A. Sparkes, T. Gray and G. R. Owen, J. Organomet. Chem., 2017, 847, 224–233 CrossRef CAS.
- G. Venkatachalam, M. Heckenroth, A. Neels and M. Albrecht, Helv. Chim. Acta, 2009, 92, 1034–1045 CrossRef CAS.
- C. E. Cooke, M. C. Jennings, M. J. Katz, R. K. Pomeroy and J. A. C. Clyburne, Organometallics, 2008, 27, 5777–5799 CrossRef CAS.
- Y.-F. Han, G.-X. Jin and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 9263–9266 CrossRef CAS PubMed.
- J. Holmes, R. J. Kearsey, K. A. Paske, F. N. Singer, S. Atallah, C. M. Pask, R. M. Phillips and C. E. Willans, Organometallics, 2019, 38, 2530–2538 CrossRef CAS.
- P. Langer, L. Yang, C. R. Pfeiffer, W. Lewis and N. R. Champness, Dalton Trans., 2019, 48, 58–64 RSC.
- A. Poethig and T. Strassner, Organometallics, 2011, 30, 6674–6684 CrossRef CAS.
- G. Rodríguez-López, P. Montes-Tolentino, T. O. Villaseñor-Granados and A. Flores-Parra, J. Organomet. Chem., 2017, 848, 166–174 CrossRef.
- S. Simonovic, A. C. Whitwood, W. Clegg, R. W. Harrington, M. B. Hursthouse, L. Male and R. E. Douthwaite, Eur. J. Inorg. Chem., 2009, 2009, 1786–1795 CrossRef.
- C.-X. Wang, Y. Gao, Y.-X. Deng, Y.-J. Lin, Y.-F. Han and G.-X. Jin, Organometallics, 2015, 34, 5801–5806 CrossRef CAS.
- T. Yan, L.-Y. Sun, Y.-X. Deng, Y.-F. Han and G.-X. Jin, Chem.–Eur. J., 2015, 21, 17610–17613 CrossRef CAS PubMed.
- L. Zhang, R. Das, C.-T. Li, Y.-Y. Wang, F. E. Hahn, K. Hua, L.-Y. Sun and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 13360–13364 CrossRef CAS PubMed.
- L. Zhang and Y.-F. Han, Dalton Trans., 2018, 47, 4267–4272 RSC.
- Y. W. Zhang, R. Das, Y. Li, Y. Y. Wang and Y. F. Han, Chem.–Eur. J., 2019, 25, 5472–5479 CrossRef CAS PubMed.
- M. Marinelli, M. Pellei, C. Cimarelli, H. V. R. Dias, C. Marzano, F. Tisato, M. Porchia, V. Gandin and C. Santini, J. Organomet. Chem., 2016, 806, 45–53 CrossRef CAS.
- A. Juzgado, M. M. Lorenzo-Garcia, M. Barrejon, A. M. Rodriguez, J. Rodriguez-Lopez, S. Merino and J. Tejeda, Chem. Commun., 2014, 50, 15313–15315 RSC.
- F. Lazreg, M. Lesieur, A. J. Samson and C. S. J. Cazin, ChemCatChem, 2016, 8, 209–213 CrossRef CAS.
- M. Mechler, W. Frey and R. Peters, Organometallics, 2014, 33, 5492–5508 CrossRef CAS.
- S. Ono, T. Watanabe, Y. Nakamura, H. Sato, T. Hashimoto and Y. Yamaguchi, Polyhedron, 2017, 137, 296–305 CrossRef CAS.
- C. Besson, J.-H. Mirebeau, S. Renaudineau, S. Roland, S. Blanchard, H. Vezin, C. Courillon and A. Proust, Inorg. Chem., 2011, 50, 2501–2506 CrossRef CAS PubMed.
- M. Paas, B. Wibbeling, R. Fröhlich and F. E. Hahn, Eur. J. Inorg. Chem., 2006, 2006, 158–162 CrossRef.
- H. Türkmen, O. Şahin, O. Büyükgüngör and B. Çetinkaya, Eur. J. Inorg. Chem., 2006, 2006, 4915–4921 CrossRef.
- A. Nandy, T. Samanta, S. Mallick, P. Mitra, S. Kumar Seth, K. Das Saha, S. S. Al-Deyab and J. Dinda, New J. Chem., 2016, 40, 6289–6298 RSC.
- A. Rit, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed. Engl., 2013, 52, 4664–4667 CrossRef CAS PubMed.
- A. Rit, T. P. Spaniol, L. Maron and J. Okuda, Organometallics, 2014, 33, 2039–2047 CrossRef CAS.
- L. E. Lemmerz, T. P. Spaniol and J. Okuda, Z. Anorg. Allg. Chem., 2016, 642, 1269–1274 CrossRef CAS.
- K. Naktode, S. Anga, R. K. Kottalanka, H. P. Nayek and T. K. Panda, J. Coord. Chem., 2014, 67, 236–248 CrossRef CAS.
- A. V. Astakhov, O. V. Khazipov, E. S. Degtyareva, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2015, 34, 5759–5766 CrossRef CAS.
- O. Saker, M. F. Mahon, J. E. Warren and M. K. Whittlesey, Organometallics, 2009, 28, 1976–1979 CrossRef CAS.
- E. Lee, J. Lee and D. V. Yandulov, Eur. J. Inorg. Chem., 2017, 2017, 2058–2067 CrossRef CAS.
- L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407–5413 CrossRef CAS.
- L. R. Titcomb, S. Caddick, F. G. N. Cloke, D. J. Wilson and D. McKerrecher, Chem. Commun., 2001, 1388–1389 RSC.
- R. W. Simms, M. J. Drewitt and M. C. Baird, Organometallics, 2002, 21, 2958–2963 CrossRef CAS.
- C.-Y. Wang, Y.-H. Liu, S.-M. Peng and S.-T. Liu, J. Organomet. Chem., 2006, 691, 4012–4020 CrossRef CAS.
- V. M. Chernyshev, A. V. Astakhov, I. E. Chikunov, R. V. Tyurin, D. B. Eremin, G. S. Ranny, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2019, 9, 2984–2995 CrossRef CAS.
- W. Zeng, R. Qiu, E. Y. Wang and F. X. Chen, Adv. Mater. Res., 2013, 788, 164–167 Search PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef CAS.
- S. M. P. Vanden Broeck, F. Nahra and C. S. J. Cazin, Inorganics, 2019, 7, 78 CrossRef.
- G. Le Duc, S. Meiries and S. P. Nolan, Organometallics, 2013, 32, 7547–7551 CrossRef CAS.
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed. Engl., 2003, 42, 3690–3693 CrossRef CAS PubMed.
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed. Engl., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- X. Luan, R. Mariz, M. Gatti, C. Costabile, A. Poater, L. Cavallo, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 6848–6858 CrossRef CAS PubMed.
- L. Vieille-Petit, X. Luan, R. Mariz, S. Blumentritt, A. Linden and R. Dorta, Eur. J. Inorg. Chem., 2009, 2009, 1861–1870 CrossRef.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed. Engl., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- G. Berthon-Gelloz, M. A. Siegler, A. L. Spek, B. Tinant, J. N. H. Reek and I. E. Markó, Dalton Trans., 2010, 39, 1444–1446 RSC.
- D.-D. Lu, X.-X. He and F.-S. Liu, J. Org. Chem., 2017, 82, 10898–10911 CrossRef CAS PubMed.
- F.-Y. Zhang, X.-B. Lan, C. Xu, H.-G. Yao, T. Li and F.-S. Liu, Org. Chem. Front., 2019, 6, 3292–3299 RSC.
- A. Binobaid, M. Iglesias, D. J. Beetstra, B. Kariuki, A. Dervisi, I. A. Fallis and K. J. Cavell, Dalton Trans., 2009, 7099–7112 RSC.
- M. Pompeo, R. D. J. Froese, N. Hadei and M. G. Organ, Angew. Chem., Int. Ed. Engl., 2012, 51, 11354–11357 CrossRef CAS PubMed.
- Y. Zhang, V. Cesar, G. Storch, N. Lugan and G. Lavigne, Angew. Chem., Int. Ed. Engl., 2014, 53, 6482–6486 CrossRef CAS PubMed.
- Y. Zhang, V. César and G. Lavigne, Eur. J. Org. Chem., 2015, 2015, 2042–2050 CrossRef CAS.
- Y. Zhang, G. Lavigne and V. César, J. Org. Chem., 2015, 80, 7666–7673 CrossRef CAS PubMed.
- Y. Zhang, G. Lavigne, N. Lugan and V. Cesar, Chem.–Eur. J., 2017, 23, 13792–13801 CrossRef CAS PubMed.
- T. Szilvási and T. Veszprémi, ACS Catal., 2013, 3, 1984–1991 CrossRef.
- E. S. Degtyareva, J. V. Burykina, A. N. Fakhrutdinov, E. G. Gordeev, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2015, 5, 7208–7213 CrossRef CAS.
- M. Sayah and M. G. Organ, Chem.–Eur. J., 2013, 19, 16196–16199 CrossRef CAS PubMed.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- A. R. Martin, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2012, 8, 1637–1643 CrossRef CAS PubMed.
- P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596–5606 CrossRef CAS.
- D. Balcells and A. Nova, ACS Catal., 2018, 8, 3499–3515 CrossRef CAS.
- D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2006, 359, 1855–1869 CrossRef CAS.
- S. S. Subramanium and L. M. Slaughter, Dalton Trans., 2009, 6930–6933 RSC.
- J. Deng, H. Gao, F. Zhu and Q. Wu, Organometallics, 2013, 32, 4507–4515 CrossRef CAS.
- S. Hameury, P. de Fremont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC.
- C. Fliedel and P. Braunstein, J. Organomet. Chem., 2014, 751, 286–300 CrossRef CAS.
- K. J. Evans and S. M. Mansell, Chem.–Eur. J., 2020, 26, 5927–5941 CrossRef CAS PubMed.
- A. S. Sigeev, A. S. Peregudov, A. V. Cheprakov and I. P. Beletskaya, Adv. Synth. Catal., 2015, 357, 417–429 CrossRef CAS.
- J. Łuczak, M. Paszkiewicz, A. Krukowska, A. Malankowska and A. Zaleska-Medynska, Adv. Colloid Interface Sci., 2016, 230, 13–28 CrossRef PubMed.
- S. Wegner and C. Janiak, Top. Curr. Chem., 2017, 375, 65 CrossRef PubMed.
- D. Moock, M. P. Wiesenfeldt, M. Freitag, S. Muratsugu, S. Ikemoto, R. Knitsch, J. Schneidewind, W. Baumann, A. H. Schäfer, A. Timmer, M. Tada, M. R. Hansen and F. Glorius, ACS Catal., 2020, 6309–6317 CrossRef CAS PubMed.
- C. Diner and M. G. Organ, Organometallics, 2019, 38, 66–75 CrossRef CAS.
- J. D. Hayler, D. K. Leahy and E. M. Simmons, Organometallics, 2019, 38, 36–46 CrossRef CAS.
- M. Weck and C. W. Jones, Inorg. Chem., 2007, 46, 1865–1875 CrossRef CAS PubMed.
- R. Zhong, A. Pöthig, Y. Feng, K. Riener, W. A. Herrmann and F. E. Kühn, Green Chem., 2014, 16, 4955–4962 RSC.
- R. Bhaskar, A. K. Sharma and A. K. Singh, Organometallics, 2018, 37, 2669–2681 CrossRef CAS.
- A. Ortiz, P. Gómez-Sal, J. C. Flores and E. de Jesús, Organometallics, 2018, 37, 3598–3610 CrossRef CAS.
- S. Sabater, J. A. Mata and E. Peris, ACS Catal., 2014, 4, 2038–2047 CrossRef CAS.
- S. Ruiz-Botella and E. Peris, Chem.–Eur. J., 2015, 21, 15263–15271 CrossRef CAS PubMed.
- S. Ruiz-Botella and E. Peris, ChemCatChem, 2018, 10, 1874–1881 CrossRef CAS.
- R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS PubMed.
- W. Wang, L. Cui, P. Sun, L. Shi, C. Yue and F. Li, Chem. Rev., 2018, 118, 9843–9929 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |