
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantio- and diastereoselective conjugate borylation/Mannich cyclization†
Egor M.
Larin
 ,
Joachim
Loup
,
Iuliia
Polishchuk
,
Rachel J.
Ross
,
Andrew
Whyte
and
Mark
Lautens
*
,
Joachim
Loup
,
Iuliia
Polishchuk
,
Rachel J.
Ross
,
Andrew
Whyte
and
Mark
Lautens
*
Davenport Laboratories, Department of Chemistry, University of Toronto, 80 St. George St., Toronto, Ontario M5S 3H6, Canada. E-mail: mark.lautens@utoronto.ca
First published on 18th May 2020
Abstract
Strategies to capitalize on enolate intermediates generated from stereoselective conjugate borylation to α,β-unsaturated carbonyl systems are surprisingly rare despite the ubiquity of Michael acceptors, and the potential to generate valuable scaffolds bearing multiple stereocenters. Herein, we report a mild and stereoselective copper-catalyzed conjugate borylation/Mannich cyclization reaction. This strategy is feasible with a broad range of Michael acceptors, and can be leveraged to generate versatile borylated tetrahydroquinoline scaffolds bearing three contiguous stereocenters. The synthetic potential of these complex heterocycles has been explored through a series of derivatization studies.
Introduction
Copper-catalyzed asymmetric borylative 1,2-bisfunctionalizations of alkenes and other π-systems have been intensely investigated,1 with a wide variety of intramolecular cyclizations reported.2 These are attractive strategies for generating C–B bonds, which can be readily converted into C–C, C–O or C–N bonds (Scheme 1a).3 However, methods for stereoselective cyclization, following β-borylation of Michael acceptors, have remained rare. Existing reports are largely limited to either intermolecular or non-enantioselective variants.4 In 2006, Yun demonstrated that β-borylation of Michael acceptors could be performed enantioselectively,5 but only two enantioselective Cu-catalyzed domino 1,4-borylation/1,2-addition processes have been reported to date (Scheme 1b).6 Moreover, as these studies are focused largely on borylative aldol cyclizations of enones, the enantioselective construction of heterocycles through 1,4-borylation remains a worthwhile goal.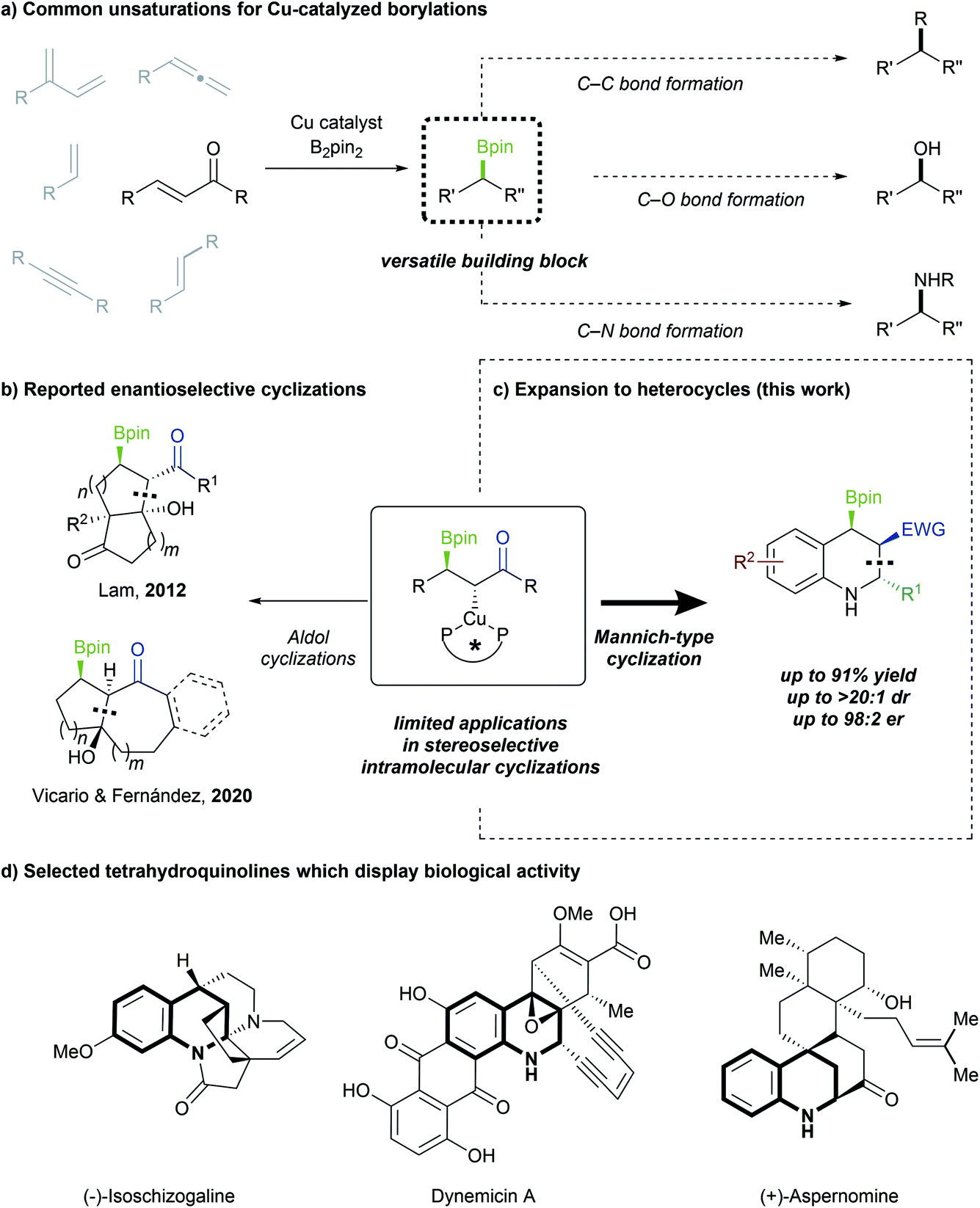 | ||
| Scheme 1 (a) Borylation strategies and applications thereof. (b) Reported enantioselective 1,4-borylation/cyclizations. (c) Enantio- and diastereoselective 1,4-borylation/Mannich cyclization. (d) Bioactive molecules containing the tetrahydroquinoline moiety. | ||
α,β-Unsaturated π-systems are potent electrophiles that allow for facile installation of a Bpin group at the β-position, as first disclosed by Miyaura7 and Hosomi.8 However, these systems are distinct from styrenes and other acceptors in terms of reactivity, mechanistic details and the resulting products. Notably, 1,4-borylation of Michael acceptors results in the in situ generation of a chiral enolate, which can be further utilized to conveniently build molecular complexity. The groups of Yun and Xiong have reported borylative enantioselective Mannich-type cyclizations using styrenes,2d,e but to date no complementary enantioselective process has yet been applied to more activated Michael acceptors. Considering that more than half of all known active pharmaceutical ingredients (APIs) contain at least one nitrogen heterocycle,9 and approximately half of all APIs are chiral,10 it is noteworthy that a 1,4-borylation/cyclization cascade strategy towards enantioenriched N-heterocycles has not been explored. Herein, we report an enantio- and diastereoselective Cu-catalyzed conjugate borylation/cyclization as a route to tetrahydroquinoline scaffolds, a pharmaceutically valuable and synthetically versatile boryl-functionalized N-heterocycle (Scheme 1c).
Shen and Xu had previously reported the synthesis of racemic borylated tetrahydroquinolines and notably obtained the all-cis isomer.4f Our choice of a related model substrate allowed us to generate enantioenriched tetrahydroquinoline structures with three contiguous stereocenters, and with complementary diastereoselectivity to that reported by Shen and Xu. The prevalence of the chiral tetrahydroquinoline moiety in natural products and bioactive molecules highlights the necessity of developing synthetic pathways towards this important scaffold (Scheme 1d).11 Additionally, the incorporation of a boryl group is advantageous as it enables further downstream functionalization.
Results and discussions
Reaction optimization
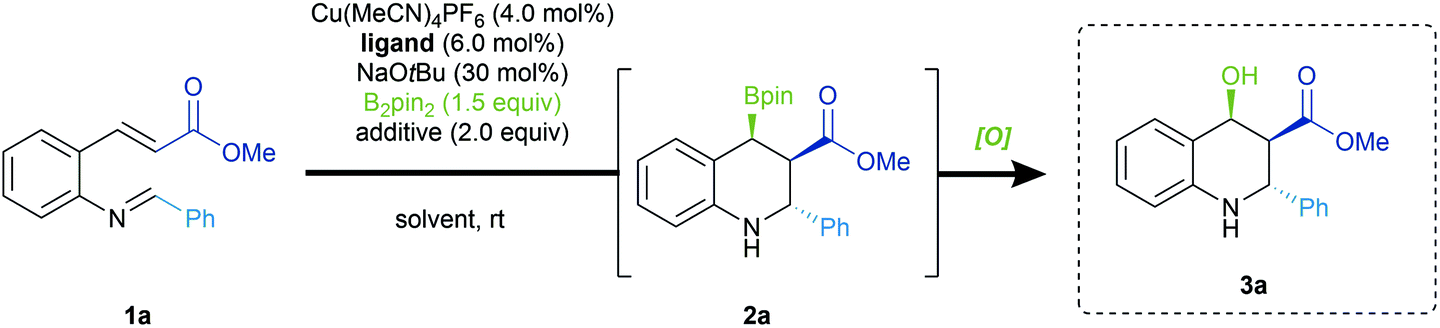
At the outset of our studies, we observed that the desired β-borylated products exhibited only moderate stability during purification via silica gel column chromatography. Isolated yields were significantly lower after chromatography than those measured through 1H NMR. This issue was previously observed by other groups for similar scaffolds.6b,12 In light of this obstacle, the borylated heterocycles were directly subjected to oxidation in order to simplify product isolation and characterization.13 The oxidation proceeds in a stereoretentive fashion, and affords the corresponding alcohol in quantitative yields (vide infra).Initial studies began employing (R)-BINAP (L1) as ligand in the presence of a substoichiometric amount of NaOtBu in THF. Under these conditions (Table 1, Entry 1), borylative cyclization of 1a was achieved with excellent diastereoselectivity as well as promising yields and enantioselectivities. Further screening of other chiral biaryl bisphosphine ligands such as (R)-Tol-BINAP (L2) (Entry 2) and (R)-SEGPHOS (L3) (Entry 3) did not lead to improved results. The less structurally constrained (S,S)-Ph-BPE (L4) (Entry 4), as well as monophosphine ligands (see ESI†), significantly eroded the selectivity of this domino reaction. However, Josiphos ligand (L5) provided the best enantioinduction accompanied by excellent diastereoselectivity (Entry 5). The use of bases other than NaOtBu led to inferior results in terms of selectivity (Entry 6). A significant proportion of the mass balance arose from protonation of the copper enolate instead of the desired cyclization into the imine. Nonetheless, the inclusion of an alcohol additive was essential to ensure high selectivity (Entry 7). While a bulkier alcohol such as tAmOH maintained high reaction selectivities, it suppressed the parasitic protonation of the copper enolate, with a significant amount of unreacted 1a (Entry 8). Finally, we noted a significant increase in product yields when the reaction was performed in diethyl ether with a more hindered alcohol additive (Entry 9). Presumably, the use of the relatively less polar and less coordinating solvent destabilized the enolate, thus facilitating the subsequent cyclization and securing our optimized reaction conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Additive | Yieldb (%) | drb | erc |
a Unless specified otherwise, reactions were carried out on 0.2 mmol scale of 1a in 4.0 mL of THF. Oxidation was performed with NaBO3·4H2O (4.0 equiv.) in THF/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 4 h.
b Yields and dr determined by 1H NMR analysis of the crude reaction mixture after oxidation using 1,3,5-trimethoxybenzene as the internal standard. Yield of isolated product given in parenthesis.
c Determined by HPLC analysis using a chiral stationary phase.
d NaOMe was used as the base.
e 27% of 1a remained.
f Reaction was carried out using Et2O as the solvent. :1), 4 h.
b Yields and dr determined by 1H NMR analysis of the crude reaction mixture after oxidation using 1,3,5-trimethoxybenzene as the internal standard. Yield of isolated product given in parenthesis.
c Determined by HPLC analysis using a chiral stationary phase.
d NaOMe was used as the base.
e 27% of 1a remained.
f Reaction was carried out using Et2O as the solvent.
|
|||||
| 1 | L1 | MeOH | 74 | >20:1 |
12.5:87.5 |
| 2 | L2 | MeOH | 47 | >20:1 |
16.5:83.5 |
| 3 | L3 | MeOH | 33 | >20:1 |
19:81 |
| 4 | L4 | MeOH | 13 | 1:1 |
48:52 |
| 5 | L5 | MeOH | 57 | >20:1 |
93:7 |
| 6d | L5 | MeOH | 55 | 7.8:1 |
88:12 |
| 7 | L5 | None | 53 | 6.6:1 |
85:15 |
| 8 | L5 | tAmOH | 54e | >20:1 |
92.5:7.5 |
| 9 | L5 | t AmOH | 89 (91) |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
94.5:5.5
|
|
|||||
Proposed stereochemical model
Based on our studies, we did not observe any major epimerization at the α-position of the ester. The observed minor diastereoisomer is believed to result from the disfavoured enolate cyclization with the imine, resulting in an all-cis configuration of the Bpin motif, ester and pendant phenyl group. Importantly, we found that the product dr ratio remained consistent during the reaction, and we observed no change of dr with respect to reaction time.These results prompted us to propose the following mechanism for this transformation. In the first step, the reaction proceeds through a syn-addition of the boronate and copper on the same face of the conjugated double bond.14 Due to the oxophilic nature of boron, a rotation around the sigma bond would stabilize the resulting enolate through the formation of a five-membered ring.15 The formation of this 5-membered cycle locks the ester cis to the Bpin group (Scheme 2a).
 | ||
| Scheme 2 Proposed stereochemical model. | ||
Since the ligand had a pronounced effect on the diastereoselectivity of the reaction, we believe the copper catalyst is likely substrate-bound during the second step of the reaction. The diastereoselectivity could arise from the interaction of the catalyst with the pendant aryl group of the imine (Scheme 2b), and the conformation of the cyclohexene-like transition state during the cyclization, although other mechanisms for diastereoinduction could not be excluded. The major diastereomer would then result from the carbon-bound14 copper enolate adding into the imine (IIA) through a half chair transition state. Conversely, attack of the enolate (IIB) through a boat transition state would generate the observed minor diastereomer; presumably, the latter pathway is disfavored due to the pendant aryl group's positioning in a sterically congested environment with the carbon-bound copper catalyst. Conversely, large ester groups may sterically clash with the pendant arene, thus favoring IIB and the pathway towards the all-cis product. Moreover, less structurally constrained or smaller ligands may also favor IIB, which would be in accordance with the all-cis selectivity of monophosphines and bisphosphines such as dppb, as was demonstrated by Shen and Xu.4f In the cases of other Michael acceptor groups, there may be more significant electronic effects that could alter the diastereoselectivity of the cyclization.
Substrate scope
We examined the versatility of this methodology by first assessing a variety of pendant aryl groups (Scheme 3). The reaction was successful with an ortho-substituted pendant arene group (3b, 3c). Moreover, halogen-containing imines were also tolerated (3c–3e). The reaction did prove sensitive to the electronics of the terminal electrophile, with electron-deficient imines (3f) cyclizing with less efficiency than electron-rich ones (3g). Further, thiophenyl (3h) and ferrocenyl (3i) containing substrates could be obtained in good to excellent enantioselectivity and diastereoselectivity. The absolute configuration of 3i was unambiguously determined through X-ray crystallography, and the configuration of other products was assumed by analogy.16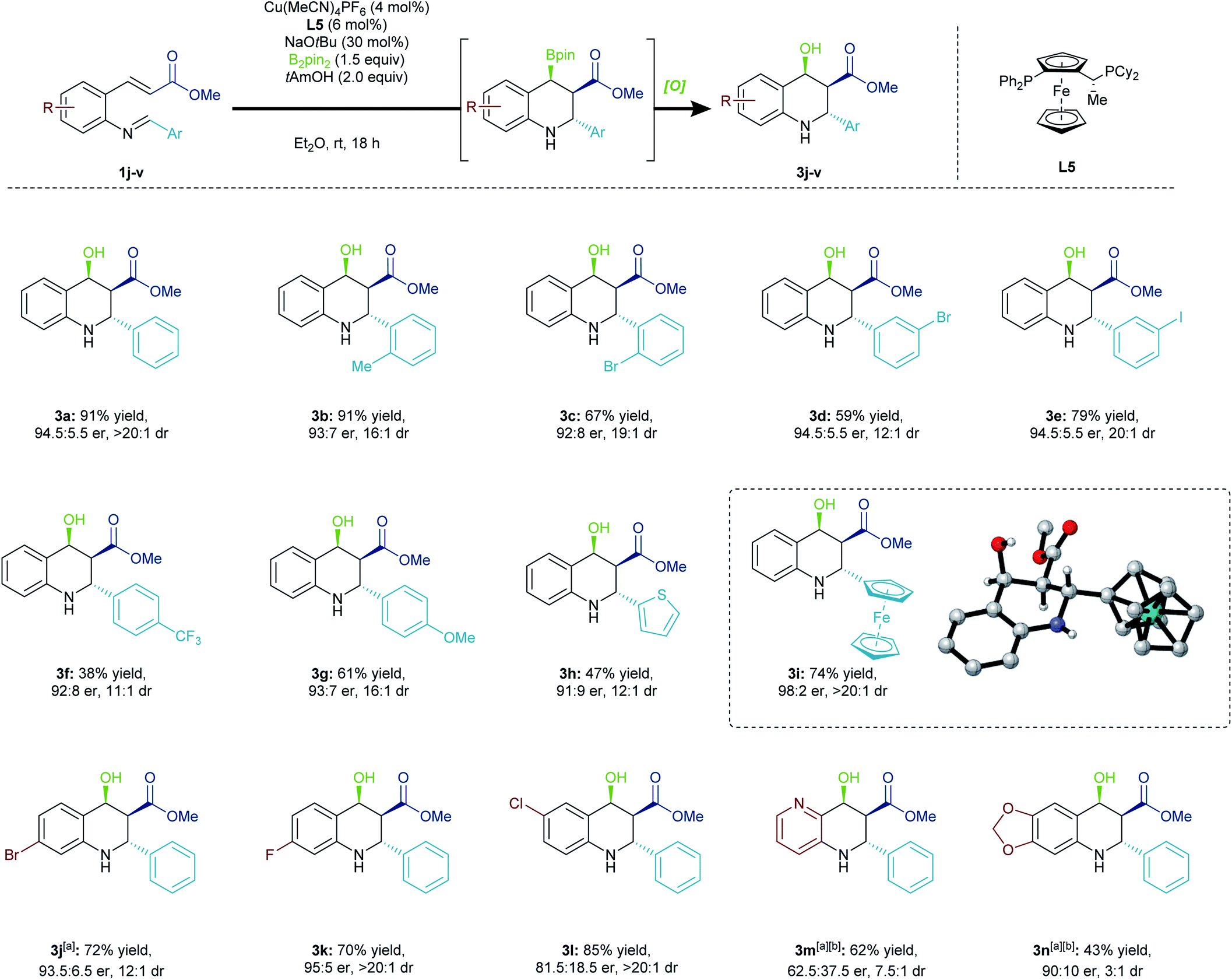 | ||
| Scheme 3 Sequential 1,4-borylation/Mannich cyclization. Reaction was conducted on 0.2 mmol scale. Oxidation was performed with NaBO3·4H2O (4.0 equiv.) in THF/H2O (2:1), 4 h. Yields reported are of the isolated major diastereomer. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixture. Enantiomeric ratios were assessed by HPLC analysis using a chiral stationary phase. [a] Combined isolated yield. [b] Reaction was performed over 6 h, subsequent oxidation over 4 h. | ||
Substituents on the aryl backbone were also investigated. Halogen-containing substrates participated (3j–3l), and the methodology could be expanded to include a pyridine heterocycle (3m). Unsurprisingly, the pyridine group significantly diminished the enantioselectivity of the initial borylation step, and this outcome could be attributed to the change in the electronic properties of the conjugated π-system. The product 3n, bearing a strongly electron-donating dioxolane substituent, was generated in low yield with poor selectivity. This compound suffered from a lack of stability due to its increased propensity to eliminate the hydroxyl group, leading to decomposition. Moreover, due to the products containing a Bpin or hydroxyl group that are both benzylic and β to a carbonyl, this methodology is limited by substrates with strongly electron-donating groups in the backbone. Electron rich backbone-bearing substrates exhibited a propensity towards elimination.
Michael acceptors
We were interested in assessing various Michael acceptor groups in the 1,4-borylation/Mannich cyclization reaction (Scheme 4). Notably, we observed lower selectivities upon increasing the size of the ester group (3a, 3o–3q), in accordance with our stereochemical model. The reaction conditions were amenable to α,β-unsaturated ketones (3r, 3s), with the methyl ketone derived tetrahydroquinoline product (3s) formed with excellent enantioselectivity (97.5:2.5 er). We also found that the α,β-unsaturated nitrile (3t) could be used in the conjugative borylation/Mannich cyclization cascade with the product being formed in high enantioenrichment (97:3 er). However, in the case of α,β-unsaturated amide, we were surprised to see a reversal of diastereoselectivity (3u, 3u′), with the all-cis diastereomer (3u′) being the major product of the reaction. Unfortunately, the reported conditions fail with sulfone or phosphonate ester acceptors (3v, 3w).
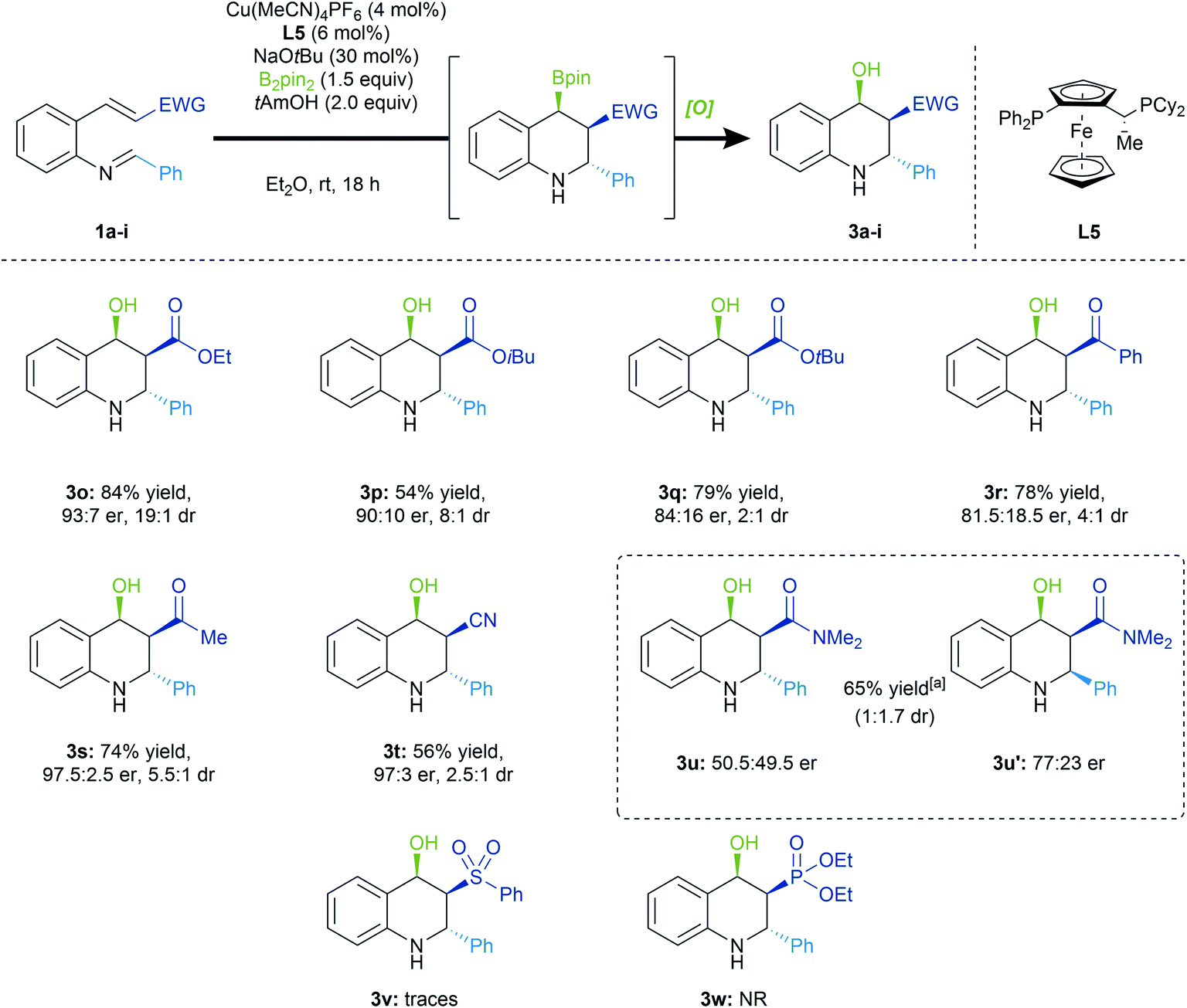 | ||
| Scheme 4 Feasibility of other Michael acceptors in the sequential 1,4-borylation/Mannich cyclization. Reaction was conducted on 0.2 mmol scale. Oxidation was performed with NaBO3·4H2O (4.0 equiv.) in THF/H2O (2:1), 4 h. Combined isolated yields. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixture. Enantiomeric ratios were assessed by HPLC analysis using a chiral stationary phase. [a] Combined NMR yield of both diastereomers. | ||
Product derivatizations
The reaction was performed on 3 mmol of the starting material 1a to ascertain reaction scalability. Some decrease in the yield of 2a was observed, albeit with limited impact on the enantio- and diastereoselectivity of the conjugate borylation/Mannich addition process (Scheme 5a). As inferred previously, the stereoretentive oxidation of the Bpin handle enables generation of the more easily handled hydroxy ester 3a in quantitative yield and complete retention of enantio- and diastereoselectivity (Scheme 5b).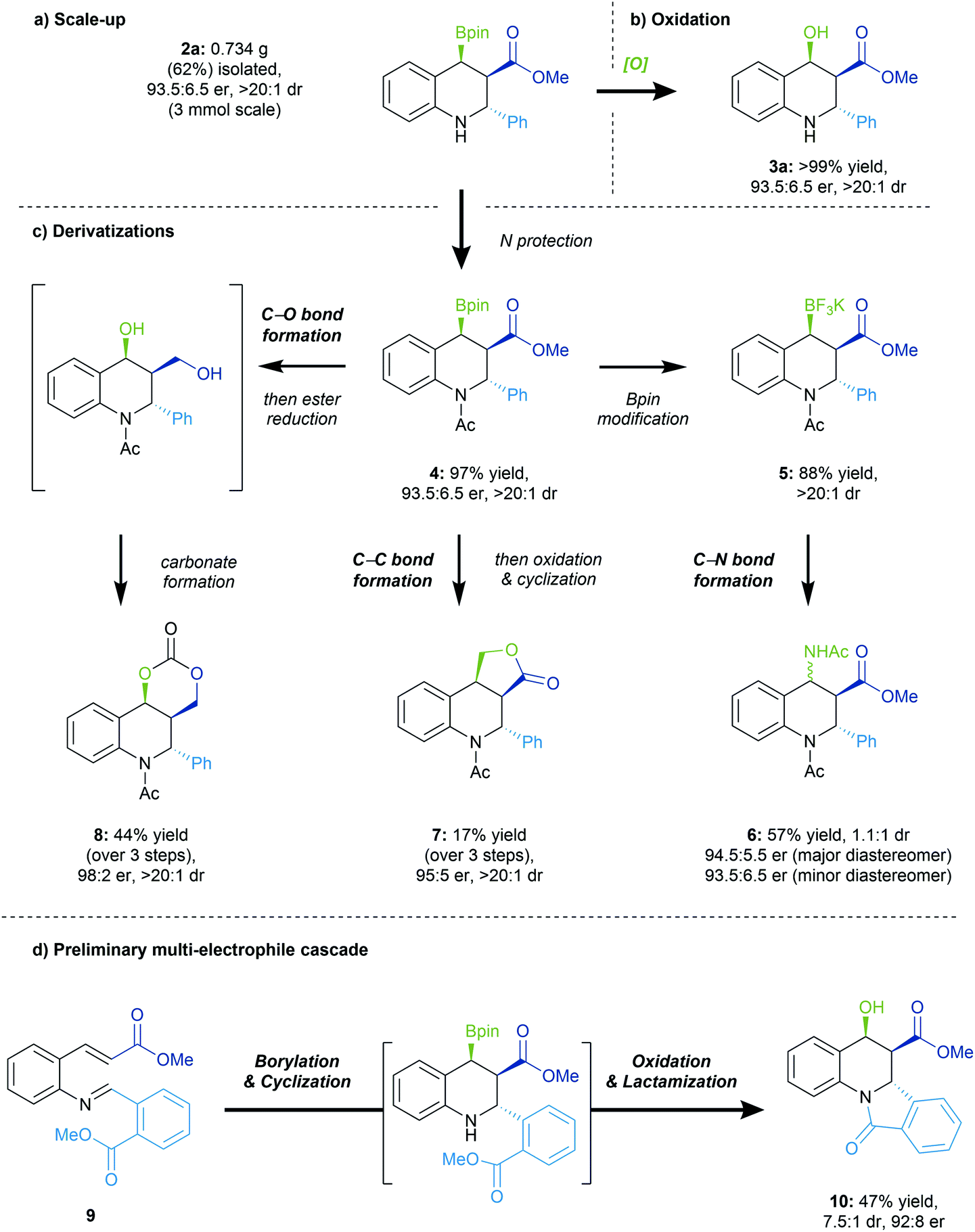 | ||
| Scheme 5 Reaction scale-up and subsequent elaborations. See ESI† for reaction details. | ||
Due to the Bpin group being both benzylic and β to a carbonyl, the borylated tetrahydroquinoline scaffold is very sensitive. Nonetheless, we wanted to capitalize on its high reactivity in order to demonstrate structural elaborations on the valuable tetrahydroquinoline scaffold (Scheme 5c). Initial protection of the free amine generated the N-acetyl protected 4 which was readily obtained in a high yield, with no impact on the dr or er. Subsequent formation of the respective trifluoroborate salt 5 enabled a successive C–N bond formation to furnish product 6 unfortunately as a mixture of two diastereomers in moderate yield, but preserving the other stereocenters. A Matteson homologation could be performed, followed by oxidation of the boryl group and cyclization to obtain the lactone product 7 as a single diastereomer with complete enantioretention. Finally, concomitant reduction of the ester and oxidation of the boryl group in 4 furnished a diol that subsequently formed dioxalanone 8 in moderate yield, as a single diastereomer.
We set out to demonstrate that the secondary amine generated in the conjugate borylation/cyclization reaction could be utilized as a nucleophile in a secondary intramolecular process (Scheme 5d). We thus designed substrate 9, bearing an ester group in the ortho position of the imine, and were pleased to see formation of the tetracyclic scaffold 10 with good enantioselectivity. This example highlights the applicability of the reactive amine in subsequent transformations to access more complex structures.
Conclusions
In summary, we identified a novel method to synthesize enantioenriched N-heterocycles through a copper-catalyzed conjugate borylation/Mannich cyclization. This reaction is a rare example of a copper-catalyzed borylation of an 1,4-unsaturated Michael system with subsequent cyclization, and the first to terminate into imines with high enantioselectively. The reported protocol is mild, amenable to scale-up and generates complex N-heterocycles with three contiguous stereocenters, with capacity for further structural elaborations. Moreover, a screen of various Michael acceptors demonstrates that this process is broadly applicable and we envision it can be utilized in other complex synthetic pathways.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Science and Engineering Research Council (NSERC), the University of Toronto (U of T), Alphora/Eurofins Scientific and Kennarshore Inc. for financial support. E.M.L. and R.J.R. thank NSERC for a postgraduate scholarship (CGS-D). J.L. thanks the Swiss National Science Foundation (SNSF) for a postdoctoral fellowship (Grant P2SKP2_187649). A.W. thanks the Walter C. Sumner Memorial Fellowship, Province of Ontario (OGS). We thank Dr I. Franzoni (NuChem Therapeutics), Dr D. A. Petrone (U of T), and Dr Y. Lebedev (U of T) for helpful discussions. Dr Alan Lough (U of T) is thanked for single-crystal X-ray structural analysis of 3i.References
- For select examples of borylative bisfunctionalizations see (a) H. Ito, S. Ito, Y. Sasaki, K. Matsuura and M. Sawamura, J. Am. Chem. Soc., 2007, 129, 14856 CrossRef CAS PubMed; (b) Y. Lee, H. Jang and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 18234 CrossRef CAS PubMed; (c) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894 CrossRef CAS PubMed; (d) F. Meng, X. Li, S. Torker, Y. Shi, X. Shen and A. H. Hoveyda, Nature, 2016, 537, 387 CrossRef CAS PubMed; (e) K. Yeung, R. E. Ruscoe, J. Rae, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 11912 CrossRef CAS PubMed; (f) C. Jarava-Barrera, A. Parra, A. López, F. Cruz-Acosta, D. Collado-Sanz, D. J. Cárdenas and M. Tortosa, ACS Catal., 2016, 6, 442 CrossRef CAS PubMed; (g) Y. Liu, D. Fiorito and C. Mazet, Chem. Sci., 2018, 9, 5284 RSC; (h) D. Fiorito, Y. Liu, C. Besnard and C. Mazet, J. Am. Chem. Soc., 2020, 142, 623 CrossRef CAS PubMed.
- For select examples of borylative cyclization see (a) H. Ito, T. Toyoda and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 5990 CrossRef CAS PubMed; (b) P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700 CrossRef CAS PubMed; (c) H. Iwamoto, Y. Ozawa, K. Kubota and H. Ito, J. Org. Chem., 2017, 82, 10563 CrossRef CAS PubMed; (d) D. Li, J. Kim, J. W. Yang and J. Yun, Chem.–Asian J., 2018, 13, 2365 CrossRef CAS PubMed; (e) G. Zhang, A. Cang, Y. Wang, Y. Li, G. Xu, Q. Zhang, T. Xiong and Q. Zhang, Org. Lett., 2018, 20, 1798 CrossRef CAS PubMed; (f) H.-M. Wang, H. Zhou, Q.-S. Xu, T.-S. Liu, C.-L. Zhuang, M.-H. Shen and H.-D. Xu, Org. Lett., 2018, 20, 1777 CrossRef CAS PubMed; (g) Y. Ozawa, H. Iwamoto and H. Ito, Chem. Commun., 2018, 54, 4991 RSC; (h) D. Li, J. Kim, J. W. Yang and J. Yun, Chem.–Asian J., 2018, 13, 2365 CrossRef CAS PubMed; (i) A. Whyte, K. I. Burton, J. Zhang and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 13927 CrossRef CAS PubMed; (j) A. Whyte, A. Torelli, B. Mirabi and M. Lautens, Org. Lett., 2019, 21, 8373 CrossRef CAS PubMed; (k) J. M. Zanghi, S. Liu and S. J. Meek, Org. Lett., 2019, 21, 5172 CrossRef CAS PubMed; (l) A. Whyte, B. Mirabi, A. Torelli, L. Prieto, J. Bajohr and M. Lautens, ACS Catal., 2019, 9, 9253 CrossRef CAS.
- (a) H. C. Brown and B. Singaram, Acc. Chem. Res., 1988, 21, 287 CrossRef CAS; (b) D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef CAS PubMed; (c) S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Chem. Rec., 2009, 9, 24 CrossRef CAS PubMed; (d) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481 RSC; (e) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- (a) I.-H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS PubMed; (b) M. Tobisu, H. Fujihara, K. Koh and N. Chatani, J. Org. Chem., 2010, 75, 4841 CrossRef CAS PubMed; (c) A. Welle, V. Cirriez and O. Riant, Tetrahedron, 2011, 68, 3435 CrossRef; (d) Y.-J. Zuo, X.-T. Chang, Z.-M. Hao and C.-M. Zhong, Org. Biomol. Chem., 2017, 15, 6323 RSC; (e) Y.-J. Zuo, Z. Zhong, Y. Fan, X. Li, X. Chen, Y. Chang, R. Song, X. Fu, A. Zhang and C.-M. Zhong, Org. Biomol. Chem., 2018, 16, 9237 RSC; (f) Y.-P. Bi, H.-M. Wang, H.-Y. Qu, X.-C. Liang, Y. Zhou, X.-Y. Li, D. Xu, M.-H. Shen and H.-D. Xu, Org. Biomol. Chem., 2019, 17, 1542 RSC.
- (a) S. Mun, J.-E. Lee and J. Yun, Org. Lett., 2006, 8, 4887 CrossRef CAS PubMed; (b) J.-E. Lee and J. Yun, Angew. Chem., Int. Ed., 2008, 47, 145 CrossRef CAS PubMed.
- (a) A. R. Burns, J. S. González and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 10827 CrossRef CAS PubMed; (b) J. Sendra, R. Manzano, E. Reyes, J. L. Vicario and E. Fernández, Angew. Chem., Int. Ed., 2020, 59, 2100 CrossRef CAS PubMed.
- (a) K. Takahashi, T. Ishiyama and N. Miyaura, Chem. Lett., 2000, 982 CrossRef CAS; (b) K. Takahashi, T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2001, 625, 47 CrossRef CAS.
- H. Ito, H. Yamanaka, J.-I. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821 CrossRef CAS.
- (a) J. S. Carey and D. Laffan, Pharmaceutical Process Development: Current Chemical and Engineering Challenges, ed. J. A. Blacker and M. T. Williams, Royal Society of Chemistry, 2011, pp. 39–65 Search PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- R. P. Jumde, F. Lanza, M. J. Veenstra and S. R. Harutyunyan, Science, 2016, 352, 433 CrossRef CAS PubMed.
- (a) I. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119, 5057 CrossRef CAS PubMed; (b) Y. Tokiwa, M. Miyoshi-Saitoh, H. Kobayashi, R. Sunaga, M. Konishi, T. Oki and S. Iwasaki, J. Am. Chem. Soc., 1992, 114, 4107 CrossRef CAS; (c) R. M. Kariba, P. J. Houghton and A. Yenesew, J. Nat. Prod., 2002, 65, 566 CrossRef CAS PubMed; (d) G. M. Staub, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Am. Chem. Soc., 1992, 114, 1015 CrossRef CAS.
- (a) K. Kubota, K. Hayama, H. Iwamoto and H. Ito, Angew. Chem., Int. Ed., 2015, 54, 8809 CrossRef CAS PubMed; (b) L.-J. Cheng and N. P. Mankad, Angew. Chem., Int. Ed., 2018, 57, 10328 CrossRef CAS PubMed.
- (a) M. V. Joannou, B. S. Moyer and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6176 CrossRef CAS PubMed; (b) Z. Niu, J. Chen, Z. Chen, M. Ma, C. Song and Y. Ma, J. Org. Chem., 2015, 80, 602 CrossRef CAS PubMed.
- (a) L. Dang, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 4443 CrossRef CAS; (b) L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987 RSC.
- (a) A. Whiting, Tetrahedron Lett., 1991, 32, 1503 CrossRef CAS; (b) R. J. Mears and A. Whiting, Tetrahedron, 1993, 49, 177 CrossRef CAS; (c) G. Conole, R. J. Mears, H. De Silva and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1995, 1825 RSC; (d) R. J. Mears, H. E. Sailes, J. P. Watts and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 2000, 3250 RSC; (e) H. E. Sailes, J. P. Watts and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 2000, 3362–3374 RSC.
- Due to the difficulties in isolation of sufficient quantitites or purification of N-alkylidene imine starting materials, we were unable to assess the efficiency of this reaction class..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1999811. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02421j |
| This journal is © The Royal Society of Chemistry 2020 |