
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free electrochemical fluorodecarboxylation of aryloxyacetic acids to fluoromethyl aryl ethers†
Michael
Berger‡
a,
John D.
Herszman‡
a,
Yuji
Kurimoto
ab,
Goswinus H. M.
de Kruijff
a,
Aaron
Schüll
ac,
Sven
Ruf
c and
Siegfried R.
Waldvogel
 *a
*a
aDepartment of Chemistry, Johannes Gutenberg University, Duesbergweg 10–14, 55128 Mainz, Germany. E-mail: waldvogel@uni-mainz.de; Web: http://www.chemie.uni-mainz.de/OC/AK-Waldvogel/
bGraduate School of Natural Science and Technology, Okayama University, 700–8530 Okayama, Japan
cSanofi-Aventis Deutschland GmbH, 65926 Frankfurt am Main, Germany
First published on 1st June 2020
Abstract
Electrochemical decarboxylation of aryloxyacetic acids followed by fluorination provides easy access to fluoromethyl aryl ethers. This electrochemical fluorodecarboxylation offers a sustainable approach with electric current as traceless oxidant. Using Et3N·5HF as fluoride source and as supporting electrolyte, this simple electrosynthesis affords various fluoromethoxyarenes in yields up to 85%.
Introduction
Fluoromethyl aryl ethers have intriguing properties1 and show emerging importance in agrochemical and pharmaceutical applications.2 Replacing a hydrogen atom by fluorine as a bioisostere for example in methyl groups has advanced to a common method in molecular editing of drugs. The incorporation of fluorine can improve the metabolic stability of a drug and increase its potency.3 Furthermore, fluoromethyl aryl ethers feature a striking shift in volatility and scent compared to their non-fluorinated analogues, making them possible candidates for pheromones or fragrances.4 Therefore, widely applicable methods installing fluorine selectively into a molecule are of high interest.The first synthetic method for the formation of fluoromethyl aryl ethers involves an electrophilic monofluoromethylation of a phenol under basic conditions using FCH2Cl with chloride as leaving group.5 Furthermore, direct nucleophilic monofluoro-methylation of phenols and thiophenes with monofluoro-methyl-substituted sulfonium ylides has been reported.6 Although their scope shows wide applicability, firstly, the monofluoromethylating agent is considered as an impactful green-house gas (CH2FCl) and secondly the reagent has to be prepared by a tedious synthesis including several steps.7
In a much less troublesome approach, phenoxyacetic acids are used in decarboxylation reactions followed by the introduction of fluorine. One of the earliest methods for this reaction type involves the use of XeF2 for a radical fluorodecarboxylation,8,9 but this reagent is expensive and difficult to handle due to its enormous reactivity. In the recent years, several methods for decarboxylative fluorinations have been reported. For example, MacMillan showed the decarboxylative fluorination of aliphatic carboxylic acids using photoredox catalysis in combination with Selectfluor™.10 Furthermore, Sammis and Hartwig reported on Hunsdiecker-type fluorodecarboxylation reactions11,12 using AgF2 starting either from phenoxyacetic acids, α-fluoro- or α,α-difluorocarboxylates to obtain the corresponding mono-, di- and trifluoromethyl aryl ethers. Additionally, Tang showed the decarboxylative fluorination of electron-rich heteroaromatic carboxylic acids using Selectfluor™ in combination with KF in dichloroethane/H2O mixtures.13 However, these methods require either a photocatalyst or an excess of oxidizing and fluorinating agents like Selectfluor/NFSI,11,14 XeF2 or AgF2 which can be very powerful,8,12 but have many drawbacks like their hazardousness and high costs for the reagents and in particular the ruthenium based catalyst.
Thus, an electrochemical approach for fluorodecarboxylation reactions could tweak these findings, since electric current can be used as a green oxidant to generate reactive intermediates in situ, like the first electrosynthetic decarboxylation reaction shown by Kolbe in 1849.15 The well-known Kolbe-electrolysis gives the dimer of two aliphatic carboxylic acids by decarboxylation.15,16
Organic electrochemistry offers many advantages over traditional, reagent-based reactions, because usual reagents are often toxic, costly and generate a lot of reagent waste. It solely depends on electric current as a renewable, inexpensive and inherently safe reagent.17
Therefore, organic electrochemistry attracted a lot of attention in the recent years,18 and especially electrochemical fluorination reactions proved to be powerful tools for organofluorine synthesis.19
Recently, we worked on the electrochemical synthesis of aryl methoxymethyl ethers by electrochemical decarboxylation of phenoxyacetic acids (Scheme 1).20 Furthermore, Baran could demonstrate trapping carbocations generated by electrochemical decarboxylation of tertiary carboxylic acids. This way, fluorine has been introduced in few examples using KF/18-crown-6 and AgClO4 as sacrificial oxidant.21 That fluorodecarboxylation relies on silver(I) salts, which are known for Hunsdiecker-type decarboxylations (see Scheme 1).12 In contrast, we present a novel metal-free electrochemical fluorodecarboxylation of simply accessible aryloxyacetic acids22 to fluoromethyl aryl ethers by a pseudo-Kolbe pathway.
 | ||
| Scheme 1 Conventional vs. electrochemical fluorodecarboxylation of aryloxyacetic acids. | ||
Results and discussion
The screening experiments were conducted in undivided cells equipped with a simple two-electrode arrangement using constant current conditions.23 Electrolyses were performed at isostatic graphite electrodes (Cgr), a common anode material for pseudo-Kolbe electrolyses. The conversion of 4-tert-butyl-phenoxyacetic acid (1a) in dichloromethane with a substrate concentration of 0.1 mol L−1 served as benchmark reaction for optimisation studies. Various parameters and their influence on the reaction outcome like current density, applied charge, different fluoride sources and electrode materials were investigated. Since the electrolyte system turned out to be the most critical parameter, a selection of screening experiments is shown in Table 1, additional screening experiments are summarized in the ESI.† The yield of the optimisation experiments was determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard (Table 1). With an applied charge of 3 F, a current density of 5.5 mA cm−2, potassium fluoride (3.0 equiv.), 18-crown-6 (3.0 equiv.), 2,4,6-collidine (3.0 equiv.) in a 0.1 M NBu4PF6 in dichloromethane (5 mL) it was possible to generate the desired fluorinated product 2a in 30% yield (Table 1, entry 1). The control experiment without applied current (no conversion of starting material) indicated, that the anodic oxidation of the carboxylic acid is crucial for the conversion of 1a. Furthermore, a control experiment without adding a base demonstrated its significance (no product formation observed) for the reaction. Higher quantities of KF did not increase the yield (Table 1, entry 2). Replacing 2,4,6-collidine with DBU as base resulted in no product formation, while the starting material was consumed during electrolysis. However, the formation of Kolbe-type products was observed as side reaction when KF was used as fluoride source. Therefore, other fluoride sources such as amine fluorides were tested. Using Py·9HF (Table 1, entry 3) as fluoride source, the Kolbe reaction was completely suppressed, but the yield of 2a dropped as well. With the use of triethylamine trihydrofluoride (Et3N·3HF) also no product formation was observed. Yet, with triethylamine pentahydrofluoride (Et3N·5HF) as fluoride source, fluoromethyl aryl ether 2a was obtained in 58% yield with the starting material being completely consumed (Table 1, entry 5). Therefore, the following experiments were conducted with Et3N·5HF as fluoride source. Neither higher Et3N·5HF (CH2Cl2/Et3N·5HF, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) nor lower amounts (9:1) increased the yield of 2a. When the amount of amine-HF was raised even higher (CH2Cl2/Et3N·5HF, 2:3), the graphite anode degraded during electrolysis. Raising the fluoride concentration by adding KF also did not improve the yield, indicating that larger quantities of fluoride do not benefit the reaction outcome (Table 1, entry 8). In further optimisation studies, different current densities, amounts of charge and anode materials were tested. Both, with lower (2.8 mA cm−2) and higher current densities (11 mA cm−2) the yield of the fluorinated product 2a (47% and 0%) decreased. For the oxidation of the carboxylic acid 1a a theoretical charge of 2.0 F is needed, but lowering the applied charge from 3.0 F to 2.5 F led to lower yields (Table 1, entry 9). Lower quantities of 2,4,6-collidine decreased the yield substantially (Table 1, entry 10). The use of platinum or boron-doped diamond (BDD) as anode material lead to almost no formation of product 2a (Table 1, entries 11 and 12). Also using glassy carbon, another carbon allotrope close to graphite, as anode did not give the desired product with almost half of the starting material remaining non-converted upon electrolysis (Table 1, entry 13). In contrast to that, graphite foil as anode afforded the fluorinated product 2a in 54% yield but did not exceed previous results (Table 1, entry 14).
:2) nor lower amounts (9:1) increased the yield of 2a. When the amount of amine-HF was raised even higher (CH2Cl2/Et3N·5HF, 2:3), the graphite anode degraded during electrolysis. Raising the fluoride concentration by adding KF also did not improve the yield, indicating that larger quantities of fluoride do not benefit the reaction outcome (Table 1, entry 8). In further optimisation studies, different current densities, amounts of charge and anode materials were tested. Both, with lower (2.8 mA cm−2) and higher current densities (11 mA cm−2) the yield of the fluorinated product 2a (47% and 0%) decreased. For the oxidation of the carboxylic acid 1a a theoretical charge of 2.0 F is needed, but lowering the applied charge from 3.0 F to 2.5 F led to lower yields (Table 1, entry 9). Lower quantities of 2,4,6-collidine decreased the yield substantially (Table 1, entry 10). The use of platinum or boron-doped diamond (BDD) as anode material lead to almost no formation of product 2a (Table 1, entries 11 and 12). Also using glassy carbon, another carbon allotrope close to graphite, as anode did not give the desired product with almost half of the starting material remaining non-converted upon electrolysis (Table 1, entry 13). In contrast to that, graphite foil as anode afforded the fluorinated product 2a in 54% yield but did not exceed previous results (Table 1, entry 14).
|
|
|||
|---|---|---|---|
| Entry | Fluoride source | 1a (%) | 2a (%) |
| a Reaction conditions: undivided cell, graphite electrodes, 4-tert-butylphenoxy-acetic acid (0.5 mmol, 104 mg), CH2Cl2 (5 mL), 2,4,6-collidine (3.0 equiv.), NBu4PF6 (0.1 M) as supporting electrolyte, j = 5.5 mA cm−2, Q = 3 F, T = rt. b Determined by 1H NMR using 1,3,5-trimethoxybenzene (1.0 equiv.) as internal standard. c Q = 2.5 F. d 2,4,6-Collidine (1.5 equiv.). e Anode material: Pt. f BDD. g Glassy carbon. h Graphite foil. No supporting electrolyte necessary with amine-HF as fluoride source. | |||
| 1 | KF + 18-crown-6 (3.0 equiv.) | 0 | 30 |
| 2 | KF + 18-crown-6 (5.0 equiv.) | 0 | 26 |
| 3 | CH2Cl2/Py·9HF (4:1) |
0 | 7 |
| 4 | CH2Cl2/Et3N·3HF (4:1) |
32 | 0 |
| 5 |
CH
2
Cl
2
/Et
3
N·5HF (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
0 | 58 |
| 6 | CH2Cl2/Et3N·5HF (3:2) |
26 | 0 |
| 7 | CH2Cl2/Et3N·5HF (9:1) |
0 | 52 |
| 8 | 5 + KF + 18-crown-6 (1.0 equiv.) | 4 | 53 |
| 9c | CH2Cl2/Et3N·5HF (4:1) |
6 | 50 |
| 10d | CH2Cl2/Et3N·5HF (4:1) |
33 | 7 |
| 11e | CH2Cl2/Et3N·5HF (4:1) |
25 | 4 |
| 12f | CH2Cl2/Et3N·5HF (4:1) |
46 | 0 |
| 13g | CH2Cl2/Et3N·5HF (4:1) |
45 | 0 |
| 14h | CH2Cl2/Et3N·5HF (4:1) |
0 | 54 |

Finally, with an optimised electrolysis protocol, we explored the scope of this reaction with various aryl moieties (Scheme 2). The electrochemical fluorodecarboxylation of phenoxyacetic acid 1a at optimised conditions gave product 2a in 50% isolated yield. An experiment with the α,α-dimethylated homologue of 2a did not provide the tertiary fluoride, though it was anticipated that the resulting tertiary carbocation would stabilize the oxocarbenium ion. Tertiary fluorides might not be electrochemically stable (too ionizable) or undergo elimination reactions. An additional methyl substituent in ortho-position afforded the fluorinated product 2b in 61% yield. Starting from the para-nitro-substituted substrate gave fluoromethoxy-benzene 2c in 35% yield although nitro groups often tend to cathodic side reactions. With another methyl group in ortho-position to the nitro group product 2d was obtained in 31% yield.
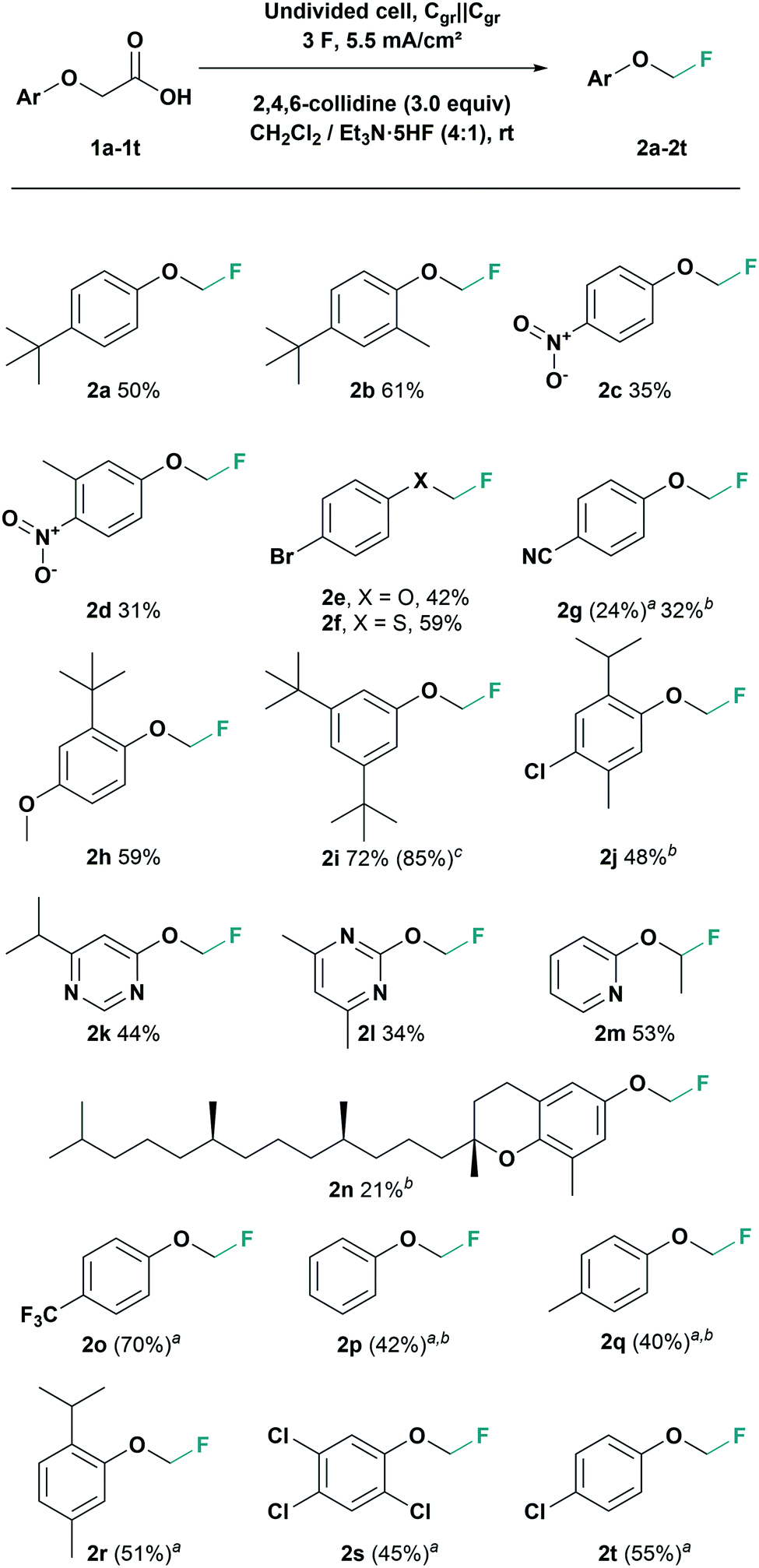 | ||
| Scheme 2 Synthesis of monofluoromethoxy arene derivatives by electrochemical fluorodecarboxylation. Standard reaction conditions: undivided cell, graphite electrodes, aryloxyacetic acid (0.5 mmol), CH2Cl2 (4 mL), Et3N·5HF (1 mL), 2,4,6-collidine (3.0 equiv), Q = 3 F, j = 5.5 mA cm−2, T = rt. Yields refer to isolated product. a Yield determined by 1H NMR using 1,3,5-trimethoxybenzene (1.0 equiv.) as internal standard. b Electrolysis conducted with graphite foil as electrode material. c Electrolysis conducted on 2.5 mmol scale. | ||
Furthermore, the 4-bromo-substituted phenoxyacetic acid gave the fluorinated product 2e in 42%. The corresponding thiophenoxyacetic acid gave an even higher yield with 59% of fluoromethyl aryl thioether 2f. With 4-cyano-phenoxyacetic acid only 24% of the fluorinated product 2g was obtained under standard conditions. However, using graphite foil the yield of fluoromethyl aryl ether 2g could be increased to 32%. In contrast, 4-methoxyphenoxyacetic acid did not show any product formation either with isostatic graphite (Cgr) or with graphite foil as electrodes. For the mechanism of this fluorodecarboxylation process we postulate a pseudo-Kolbe pathway24 (see ESI† for more information). Electron rich derivatives are therefore prone to possible side reactions at accessible ortho-positions. Blocking these positions should therefore prevent side reactions. Accordingly, with an additional sterically demanding tert-butyl-substituent in ortho-position, the fluorodecarboxylation gave 59% yield of derivative 2h. Further, a phenoxyacetic acid bearing two tert-butyl substituents in meta-position was tested giving 72% yield of fluoromethyl aryl ether 2i. To demonstrate the scalability of our method, that electrosynthesis was also performed on a 2.5 mmol scale and gave the fluorinated product 2i in 85% yield. Moreover, derivatives based on natural products like chloro-substituted thymol and δ-tocopherol (vitamin E) were tested. In both cases, the electrolysis with graphite foil was superior to isostatic graphite, giving the fluorinated thymol derivative 2j in 48% and the fluorinated δ-tocopherol 2n in 21% yield. Even with nitrogen heterocycles the fluorodecarboxylation was successful. The fluoromethoxy pyrimidine 2l was afforded in 44% and pyrimidine 2m with a 2-fluoromethoxy group in 34% yield, respectively. It was also possible to generate fluoro-ethoxypyridine 2n from the corresponding propionic acid in 53% yield.
In addition to that, for fluoromethoxyarenes 2o–2t NMR yields up to 70% were achieved. However, due to their enhanced volatility, these products are difficult to isolate, something that has been reported previously11 (see ESI† for more detailed information).
Conclusions
In summary, we established the first synthesis of fluoromethoxy aryl ethers by electrochemical fluorodecarboxylation of aryloxy-acetic acids using electric current as traceless oxidant. It enabled access towards a variety of fluoromethoxyarenes. This electrochemical protocol is very easy to conduct with a simple setup in undivided cells at constant current conditions with different aryl and heterocyclic moieties and substitution patterns in yields up to 85%. The successful fluorodecarboxylation of natural products and heterocycles demonstrated the broad applicability of this method even to demanding substrates. Besides, this electro-conversion is readily scalable with an increase in yield expected in the distillative solvent removal from the volatile fluoromethyl ethers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. H. acknowledges the Friedrich Ebert Foundation for granting a fellowship and S. R. W. thanks the Carl Zeiss Foundation for the research network ELYSION.References
- L. Xing, D. C. Blakemore, A. Narayanan, R. Unwalla, F. Lovering, R. A. Denny, H. Zhou and M. E. Bunnage, ChemMedChem, 2015, 10, 715 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827 CrossRef PubMed; B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140 CrossRef; P. Jeschke, ChemBioChem, 2004, 5, 571 CrossRef PubMed; Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed; S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; C. Fäh, R. Mathys, L. A. Hardegger, S. Meyer, D. Bur and F. Diederich, Eur. J. Org. Chem., 2010, 4617 CrossRef; W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef PubMed.
- P. T. Lowe and D. O'Hagan, J. Fluorine Chem., 2020, 230, 109420 CrossRef CAS.
- T. G. Miller and J. W. Thanassi, J. Org. Chem., 1960, 25, 2009 CrossRef CAS.
- X. Shen, M. Zhou, C. Ni, W. Zhang and J. Hu, Chem. Sci., 2014, 5, 117 RSC; Y. Nomura, E. Tokunaga and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 1885 CrossRef CAS PubMed; Y. Liu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2017, 56, 9930 CrossRef PubMed.
- R. Kowalczyk, A. J. F. Edmunds, R. G. Hall and C. Bolm, Org. Lett., 2011, 13, 768 CrossRef CAS PubMed; C. R. Johnson, R. A. Kirchhoff, R. J. Reischer and G. F. Katekar, J. Am. Chem. Soc., 1973, 95, 4287 CrossRef.
- T. B. Patrick, K. K. Johri and D. H. White, J. Org. Chem., 1983, 48, 4158 CrossRef CAS.
- Q. Lu, T. Benneche, J. Nielsen, C. Christophersen, R. Gawinecki, G. Häfelinger, M. N. Homsi, F. K. H. Kuske, M. Haugg, N. Trabesinger-Rüf and E. G. Weinhold, Acta Chem. Scand., 1996, 50, 850 CrossRef CAS.
- S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654 CrossRef CAS PubMed; B. Pieber, M. Shalom, M. Antonietti, P. H. Seeberger and K. Gilmore, Angew. Chem., Int. Ed., 2018, 57, 9976 CrossRef PubMed.
- M. Rueda-Becerril, O. Mahe, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J.-F. Paquin, J. Am. Chem. Soc., 2014, 136, 2637 CrossRef CAS PubMed.
- Q.-W. Zhang, A. T. Brusoe, V. Mascitti, K. D. Hesp, D. C. Blakemore, J. T. Kohrt and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 9758 CrossRef CAS PubMed.
- X. Yuan, J.-F. Yao and Z.-Y. Tang, Org. Lett., 2017, 19, 1410 CrossRef CAS PubMed.
- J. C. T. Leung and G. M. Sammis, Eur. J. Org. Chem., 2015, 2197 CrossRef CAS.
- H. Kolbe, Ann. Chem. Pharm., 1849, 69, 257 CrossRef.
- H.-J. Schäfer, Top. Curr. Chem., 1990, 152, 91 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef PubMed.
- T. Broese and R. Francke, Org. Lett., 2016, 18, 5896 CrossRef CAS PubMed; E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77 CrossRef PubMed; S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 13325 CrossRef PubMed; S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed; J. L. Röckl, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2019, 315 Search PubMed; E. Rodrigo, H. Baunis, E. Suna and S. R. Waldvogel, Chem. Commun., 2019, 55, 12255 RSC; M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC; S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef PubMed.
- T. Fuchigami and S. Inagi, Chem. Commun., 2011, 47, 10211 RSC; J. D. Haupt, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 242 CrossRef CAS PubMed; G. Laudadio, A. d. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. de Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832 CrossRef PubMed; J. D. Herszman, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 7893 CrossRef PubMed; Y. Takahira, M. Chen, Y. Kawamata, P. Mykhailiuk, H. Nakamura, B. K. Peters, S. H. Reisberg, C. Li, L. Chen, T. Hoshikawa, T. Shibuguchi and P. S. Baran, Synlett, 2019, 30, 1178 CrossRef; S. Doobary, A. T. Sedikides, H. P. Caldora, D. L. Poole and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 1155 CrossRef PubMed.
- X. Luo, X. Ma, F. Lebreux, I. E. Markó and K. Lam, Chem. Commun., 2018, 54, 9969 RSC; G. H. M. de Kruijff and S. R. Waldvogel, ChemElectroChem, 2019, 6, 4180 CrossRef CAS.
- J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398 CrossRef CAS PubMed.
- D.-W. Wang, H.-Y. Lin, B. He, F.-X. Wu, T. Chen, Q. Chen, W.-C. Yang and G.-F. Yang, J. Agric. Food Chem., 2016, 64, 8986 CrossRef CAS PubMed.
- C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26 CrossRef.
- J. P. Coleman, R. Lines, J. H. P. Utley and B. C. L. Weedon, J. Chem. Soc., Perkin Trans. 2, 1974, 9, 1064 RSC; M. Galicia, M. A. González-Fuentes, D. P. Valencia and F. J. González, J. Electroanal. Chem., 2012, 672, 28 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02417a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |