
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dark side of disulfide-based dynamic combinatorial chemistry†
Mélissa
Dumartin
 a,
Jean
Septavaux
ab,
Marion
Donnier-Maréchal
a,
Emeric
Jeamet
a,
Elise
Dumont
cd,
Florent
Perret
*a,
Laurent
Vial
*a and
Julien
Leclaire
*a
a,
Jean
Septavaux
ab,
Marion
Donnier-Maréchal
a,
Emeric
Jeamet
a,
Elise
Dumont
cd,
Florent
Perret
*a,
Laurent
Vial
*a and
Julien
Leclaire
*a
aUniv. Lyon, Univ. Lyon 1, CNRS, INSA, CPE, ICBMS, F-69622 Lyon, France. E-mail: florent.perret@univ-lyon1.fr; laurent.vial@univ-lyon1.fr; julien.leclaire@univ-lyon1.fr
bSecoya Technologies, Louvain-La-Neuve, 1348 Belgium
cENS Lyon, Univ. Lyon 1, CNRS, Laboratoire de Chimie, F-69364, France
dInstitut Universitaire de France, 5 rue Descartes, 75005 Paris, France
First published on 17th July 2020
Abstract
During the last two decades, disulfide-based dynamic combinatorial chemistry has been extensively used in the field of molecular recognition to deliver artificial receptors for molecules of biological interest. Commonly, the nature of library members and their relative amounts are provided from HPLC-MS analysis of the libraries, allowing the identification of potential binders for a target (bio)molecule. By re-investigating dynamic combinatorial libraries generated from a simple 2,5-dicarboxy-1,4-dithiophenol building block in water, we herein demonstrated that multiple analytical tools were actually necessary in order to comprehensively describe the libraries in terms of size, stereochemistry, affinity, selectivity, and finally to get a true grasp on the different phenomena at work within dynamic combinatorial systems.
Introduction
Since its discovery, Dynamic Combinatorial Chemistry (DCC) has grown into a powerful strategy to produce, at a low synthetic cost, self-assembled architectures displaying complex topologies and/or innovative functions.1 DCC from disulfide-based building blocks has been particularly used in the field of molecular recognition to deliver tailored receptors for target (bio)molecules.2 DCC simultaneously allows the generation of a library of potential receptors, to screen their affinity for a targeted partner, to detect the best lead, and possibly to prepare this species by optimized scaled-up procedures. DCC relies on the amplification phenomenon, which is the increase in the amount of an assembled library member upon introduction of the targeted partner called the template. Amplification generally transcribes a stable and preferential association between both partners. A bottleneck of the DCC strategy is sometimes the difficulty to accurately quantify the amount of each library member within mixtures, and very frequently the impossibility to selectively extract the best binders from the mixtures. Commonly, only the nature of the members and their relative amounts are provided from HPLC-MS analysis of the libraries. These relative amounts and, more specifically, the amplification factor (i.e., the ratio between the amount of potential binder in the presence and absence of the template) are then used to estimate the binding constant rather than measure it experimentally through titration between the isolated partners.3,4During the last five years, we extensively re-investigated the dynamic combinatorial libraries generated from the single 2,5-dicarboxy-1,4-dithiophenol monomer M upon slow aerobic thiol oxidation and simultaneous fast disulfide exchange at physiological pH in water (Fig. 1A).5 Such libraries were first described in 2006 by some of us to quantitatively deliver the racemic homochiral tetramer of (pS)4/(pR)4 configuration M4-a upon incubation with the biogenic polyamine spermine (Fig. 1A and B).6 This collection of readily accessible constitutionally dynamic analogues of pillararenes, to which M4-a belongs, was enlarged in 2016 and named dyn[n]arenes.5d A landmark was the straightforward purification procedure of these new cyclophanes by selective precipitation and separation from the buffer and template. Herein, by extending this procedure to entire libraries instead of single members, we thoroughly investigated the impact of the degree of molecular information (i.e., the number of positive charges) borne by homologous templates (from spermine T4 to ammonium acetate T1, through cadaverine T3 and butylamine T2) on the (stereo)chemical composition of libraries made from monomer M (Fig. 1A).
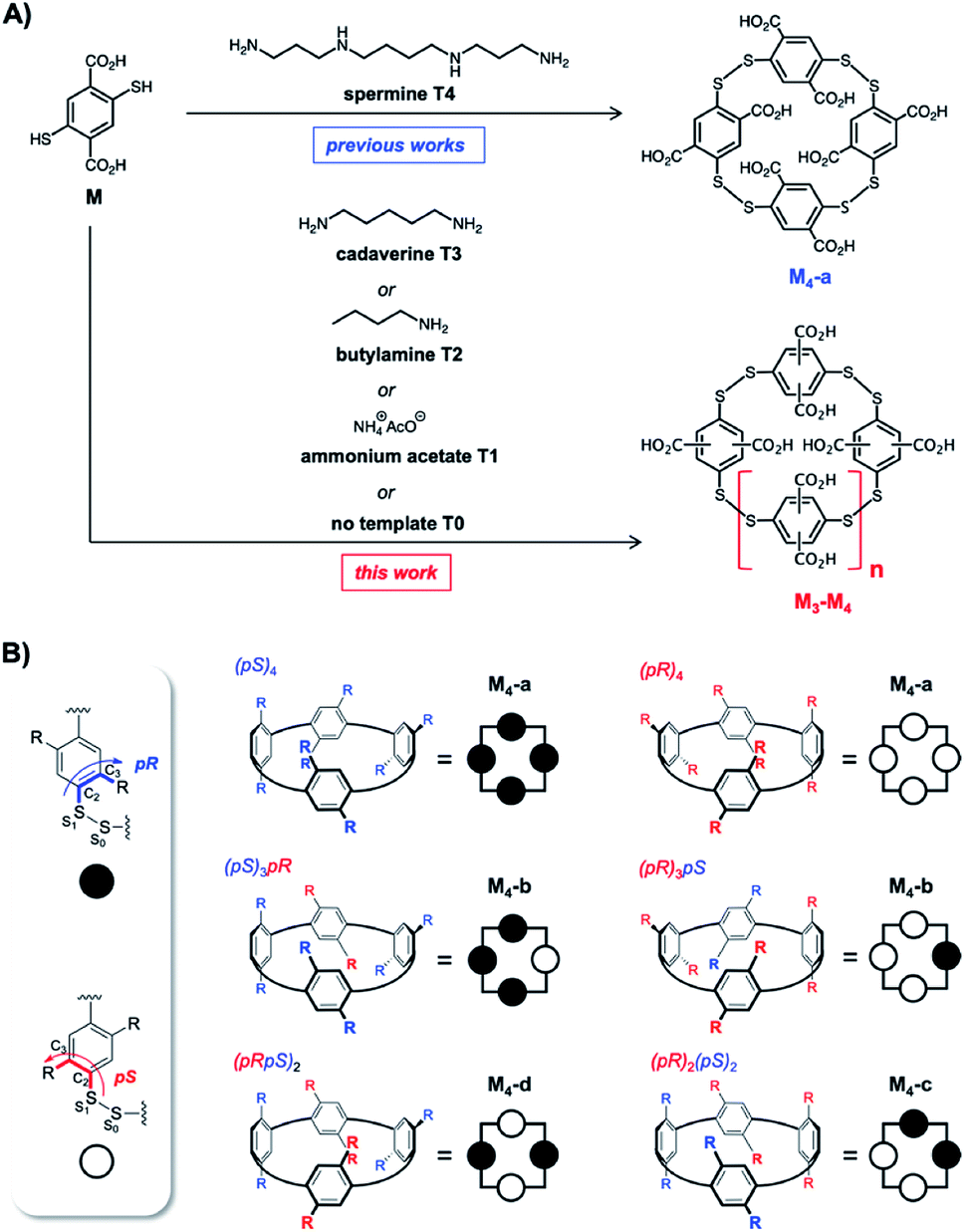 | ||
| Fig. 1 Previously reported synthesis of homochiral dyn[4]arene M4-a using spermine as the template, and current investigations involving truncated analogues of spermine (A), and dyn[4]arene's virtual configurational landscape arising from the rotation of phenyl units yielding hosts M4-a to M4-d (B). | ||
Results and discussion
When building block M undergoes spontaneous disulfide bridge formation and exchange in buffered water, the HPLC-MS analysis of the resulting libraries L0–L4 after 24 hours of incubation showed that – regardless of the template (T1–T4) used – the racemic homochiral tetramer M4-a of (pS)4/(pR)4 configuration was the main species produced (in blue in Fig. 2A). In comparison, HPLC analysis of the reference untemplated library L0 showed almost no detectable peak after one day. Comparison between the peak area corresponding to (pS)4/(pR)4M4-a in the various templated libraries L1–L4 revealed that, although this library member appeared at first sight to be the main or even sole species obtained, its true conversion ranged from 23% to 100% (Fig. 2A). The 100% reference was attributed to the peak area obtained with spermine T4 (i.e., corresponding to 100% of the disulfide-based material), as this template was previously shown by NMR monitoring to quantitatively deliver the homochiral dyn[4]arene M4-a. The fluctuation in the total amount of disulfide-based material within the various libraries L0–L4 suggested that a number of species failed to be detected by HPLC, and that complementary analytical tools would be required to properly and fully assess the template effects and provide a clear compositional view of the libraries.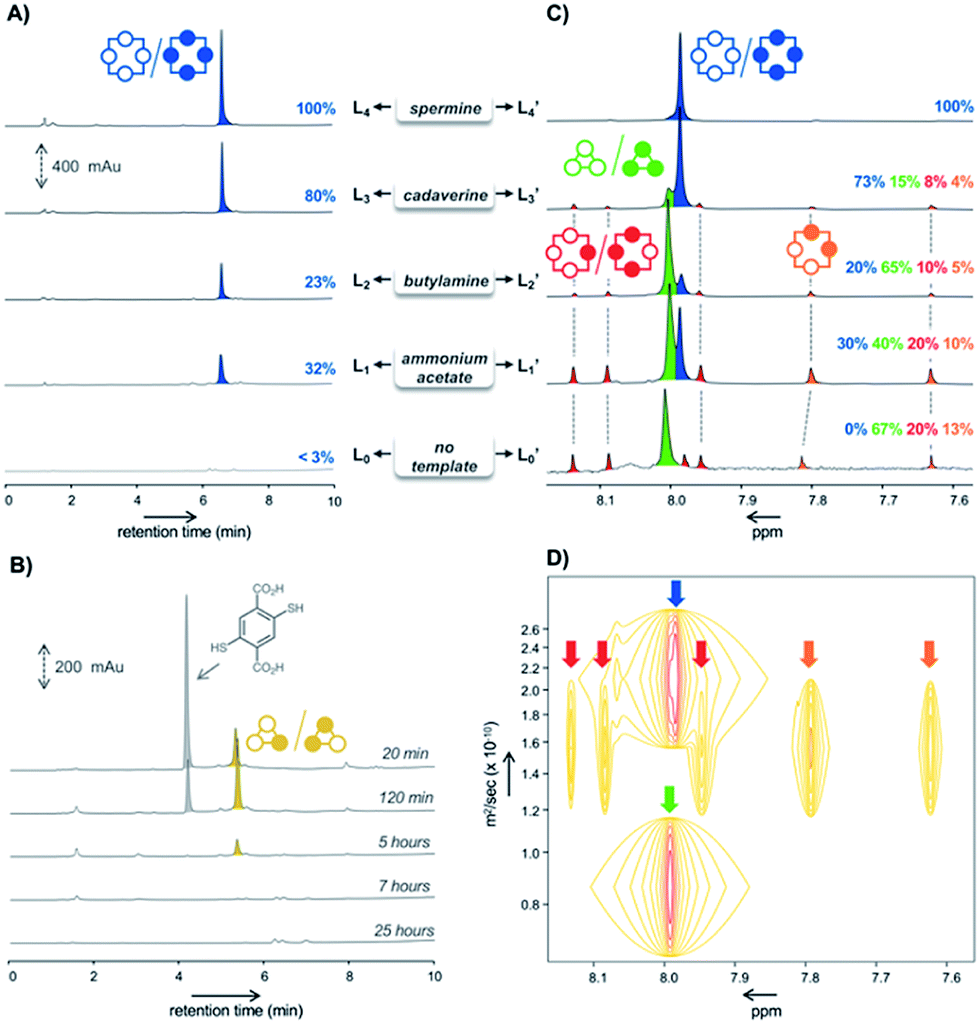 | ||
| Fig. 2 HPLC analyses of the libraries L1–L4 made from monomer M (4 mM) in the presence of the templates (A), and L0 in absence of any template (B); 1H NMR analyses of the corresponding libraries L0′–L4′ (C), and DOSY NMR analysis of the library L1 made from monomer M in presence of ammonium acetate T1 as the template (D). | ||
After template and buffer removal by selective precipitation of the library members upon neutralization of their anionic charges using trifluoroacetic acid, the (stereo)chemical composition of the corresponding static template-free libraries L0′–L4′ was assessed by ESI-MS and NMR spectroscopy analyses at pH = 7.4 (Fig. 2C). The former revealed that the unidentified species were either dyn[3]arenes or dyn[4]arenes (see Fig. S2†). The latter indicated that: (i) library L4′ generated with spermine as a template expectedly displayed a unique singlet at 7.98 ppm (in blue in Fig. 2C), which was formerly assigned to the homochiral dyn[4]arene M4-a of (pS)4/(pR)4 configuration,5d and that (ii) other templated libraries L1′–L3′ also showed the presence of (pS)4/(pR)4M4-a, along with at least six other visible singlets of varying areas depending on the template used, and (iii) that the homochiral tetramer M4-a was indeed absent from the untemplated system L0′. According to DOSY NMR experiments, these additional singlets corresponded to species displaying diffusion coefficients 1.6 to 3 times lower than the (pS)4/(pR)4 homochiral cyclophane M4-a (Fig. 2D). From these observations, it seemed legitimate to conclude that the trimers M3 and tetramers M4, which failed to be detected by HPLC-UV but could be quantified by 1H NMR spectroscopy, form (sub)nanoscopic aggregates in solution. This hypothesis was supported by the good agreement between the percentages of homochiral M4-a obtained by its integration in the NMR spectra (libraries Li′, Fig. 2C) and the relative peak area measured on each of the corresponding HPLC chromatograms (library Li, Fig. 2A). It also demonstrated that the composition of the system was unaltered by the precipitation process, i.e. that static template-free systems Li′ were true reflections of the dynamic templated mixtures Li.
Among the remaining 1H NMR signals, it was possible to unambiguously assign the singlet at 8.01 ppm (in green in Fig. 2C) to the homochiral trimer M3-a of (pS)3/(pR)3 configuration, as this dyn[3]arene had been previously isolated by us and fully characterized by NMR spectroscopy.5b We also ruled out the presence in the libraries of the heterochiral trimer M3-b of (pS)2(pR)/(pR)2(pS) configuration. Indeed, the early stage HPLC monitoring of the untemplated library L0 revealed that M3-b is a kinetic intermediate. While it appeared to be the major (detectable) library members during the first hours of incubation, it rapidly disappeared after a few additional hours (in yellow in Fig. 2B). HPLC-MS confirmed that this species was a trimer, which has to be the heterochiral stereoisomer since the homochiral counterpart M3-a yielded a flat HPLC-UV chromatogram. The remaining unassigned species present in L0′–L3′ could only be heterochiral tetramers M4-x (x = b–d, Fig. 1B), whose potential configuration may either be (pSpR)2, (pS)2(pR)2, or (pS)(pR)3/(pR)(pS)3. They correspond to C4, C2, and C1 symmetries and should consequently display a single, two, and four singlets in 1H NMR spectroscopy, respectively.
In order to assign these remaining unknown species, we performed an NMR titration on the static library of hosts L1′ generated with ammonium acetate as a template (corresponding to the templated library displaying the highest amount of unassigned species) using increasing amounts of cadaverine T3 as a guest molecule (see Fig. S3†). Interestingly, normalizing the chemical shifts' variation for each signal provided a supervised procedure for peak assignment within a complex mixture of homologous species. Not only does it validate the attribution of each C–H proton peak monitored throughout the titration, preventing accidental swapping, but it also allows the gathering of signals corresponding to the same host species on the basis of their evolution profile, even when splitting occurs during titration (Fig. 3A). Upon addition of cadaverine T3, the first binding event detected involves M4-a. As previously reported, it corresponds to the formation of a pseudorotaxane-type complex between both partners (T3 ⊂ M4-a). This binding event is slow at the NMR time scale: upon guest addition, the singlet corresponding to the free host disappears while the singlet corresponding to the complex appears further downfield. Additional amounts of guest T3 triggered simultaneously all other binding events, which appeared to be fast at the NMR time scale. Three distinct sets of signals could be identified: two singlets belonging to the meso (pS)2(pR)2 species (M4-c, in orange in Fig. 2B), and four singlets belonging to the heterochiral dyn[4]arene of (pS)(pR)3/(pR)(pS)3 configuration (M4-b, in red in Fig. 2B). Among all possible stereoisomers of dyn[4]arene M4, the meso alternate (pSpR)2 stereoisomer M4-d was missing in every library. This may be imputed to “head-to-head” repulsive coulombic interactions between adjacent aromatic units, which represent up to 4 unfavorable contacts within this meso species while only 2 can be found within the macrocycles M4-b and M4-c of (pS)2(pR)2 and (pS)(pR)3/(pR)(pS)3 configuration respectively, and none in the homochiral dyn[4]arene M4-a. To further confirm our assignment, the NMR titration of L2′ was also conducted with T2, which led to the same conclusions (see Fig. S4†). This thorough compositional and stereochemical analysis dramatically changed the picture of the dynamic libraries L0–L4 generated from 2,5-dicarboxy-1,4-dithiophenol monomer M, a widely used ingredient in disulfide based DCC.5–7 Relative quantification of the eluted library members by HPLC-UV/MS, which only displayed a minor portion of the system (the “bright side” of the libraries), would in fact lead to the following erroneous conclusions: (i) that templates T1–T4 stabilized the homochiral tetramer more or less to the same extent through its binding, (ii) that the untemplated system L0 presumably evolved toward the formation of non-eluting polymeric and polydisperse material, and (iii) that the heterochiral tetramers and the homochiral trimer, due to carboxylate–carboxylate contacts and/or ring constraints, were intrinsically too unstable to form and be observed. In fact, this portion of the system (the “dark side” of the libraries), which is not disclosed by conventional monitoring, represents not only most, if not all, the material content, but also most of its constitutional and stereochemical diversity. Unintuitively, this dark portion is stabilized by self-aggregation. From the experimental distribution of these cyclophanes measured in the reference system L0, and assuming that the limit of detection by 1H NMR spectroscopy of the homochiral tetramer M4-a is around 1% (with a signal/noise ratio superior to 450),8 it appeared that members of the dark side of the DCL are around 10 kJ mol−1 more stable than M4-a. It translates into an interconversion constant Kappiso in the range of 10 to 102 M−1 (Fig. 5A).9 This provides an order of magnitude of the counter-driving force that opposes the templated formation of the homochiral tetramer M4-a, which eventually appeared to be the one of the most unstable architectures within small size cyclo-oligomers. Yet, the fluctuations in the relative amounts of these “invisible” library members (all but the homochiral M4-a) between the different templated systems L1′–L4′ (Fig. 2B) indicated that the production of these species from M was guest-dependent, i.e. that each of them may display weak yet contrasted affinities toward each guest.
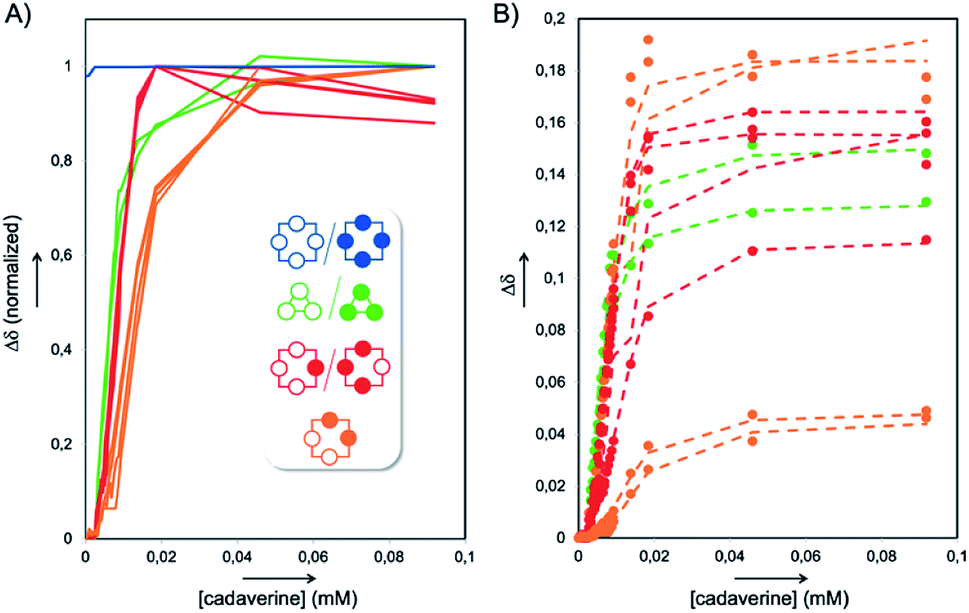 | ||
| Fig. 3 (A) Normalized chemical shifts' evolution of the various NMR signals from the titration of the static library L1′ with cadaverine T3, providing full host assignment and (B) corresponding concomitant fitting (dotted lines) of the experimental chemical shifts (circles). | ||
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding constant between the homochiral dyn[4]arene M4-a and cadaverine T3 was previously determined by ITC to be 3.11 × 107 M−1 in water at physiological pH.5a The apparent binding constants Kappbind between T3 and the dyn[n]arenes M3-a, M4-b and M4-c could be extracted from the abovementioned NMR titration by simultaneous curve fitting on every series of downfield displacements for each of the proton NMR signals of the hosts, as the concentration of the guest increased (Fig. 3A and B). To put it simply, the concomitant fitting was performed using homemade MATLAB scripts.10 The set of equations describing associations and mass balance could be solved using the fsolve MATLAB function to deliver the speciation of the system at equilibrium for a defined set of association constants (for the set of equations used, see the ESI†). The lsqcurvefit MATLAB function was then used to adjust the association constants by fitting the speciation to the NMR data. This methodology was applied for different binding stoichiometries, and a highly satisfactory average correlation coefficient R2 = 0.991 was obtained for binary complexes in the case of the homochiral trimer M3-a and ternary complex in the case of M4-b and M4-c.
:1 binding constant between the homochiral dyn[4]arene M4-a and cadaverine T3 was previously determined by ITC to be 3.11 × 107 M−1 in water at physiological pH.5a The apparent binding constants Kappbind between T3 and the dyn[n]arenes M3-a, M4-b and M4-c could be extracted from the abovementioned NMR titration by simultaneous curve fitting on every series of downfield displacements for each of the proton NMR signals of the hosts, as the concentration of the guest increased (Fig. 3A and B). To put it simply, the concomitant fitting was performed using homemade MATLAB scripts.10 The set of equations describing associations and mass balance could be solved using the fsolve MATLAB function to deliver the speciation of the system at equilibrium for a defined set of association constants (for the set of equations used, see the ESI†). The lsqcurvefit MATLAB function was then used to adjust the association constants by fitting the speciation to the NMR data. This methodology was applied for different binding stoichiometries, and a highly satisfactory average correlation coefficient R2 = 0.991 was obtained for binary complexes in the case of the homochiral trimer M3-a and ternary complex in the case of M4-b and M4-c.
As the full array of spectra corresponding to the titration revealed it, guest complexation with some of the dyn[n]arenes also induced a symmetric breaking of the hosts (see Fig. 3 and S3†). As a consequence, some C–H proton signals progressively split into minor (individual) and major (isochronous) peaks (see Fig. S2†). We therefore challenged the robustness of our procedure by fitting not only one series of signals for each species but all of them alternatively, where minor peaks were tested one-by-one while keeping major signals for other species. As mentioned earlier, this extended fitting allows the detection and self-correction of any assignment error on complex mixtures. Following the same procedure, apparent binding constants displayed individually by every library member contained in L2′ toward T2 were simultaneously extracted by the same concomitant fitting of the corresponding titration (see Fig. S5†).
From these sets of binding data, it appears that the apparent affinity of cadaverine T3 for dyn[n]arenes of various configurations spans five orders of magnitude (Fig. 4) with a selectivity factor (defined as the ratio of the binding constants) reaching up to 103 in favour of the template-responsive homochiral host M4-a. The same general trend was observed for T2: the binding constants, one or two orders of magnitude weaker than those displayed by cadaverine T3, span four orders of magnitude, with a marked selectivity in favour of M4-a by at least a factor 103.
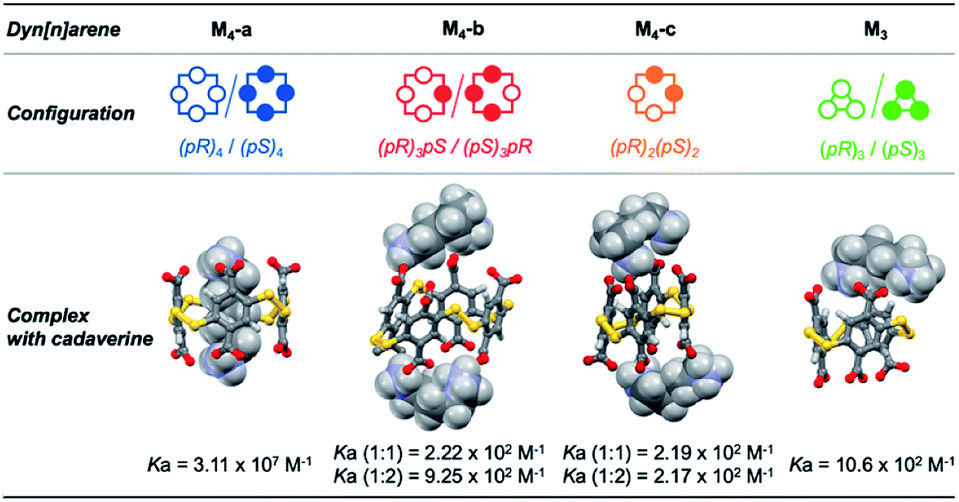 | ||
| Fig. 4 3D structures of the complexes between cadaverine and dyn[n]arenes M3 and M4 of various configurations obtained by MD simulations, and respective affinities between the partners measured by ITC and NMR titrations. | ||
To confirm the experimental trends, Molecular Dynamics (MD) simulations were performed and allowed to probe the three-dimensional structure of the corresponding complexes. MD simulations of 100 ns at 300 K with constant pressure in a truncated octahedral TIP3P water box were performed with the Amber 12 software package (see the ESI† for full computational details).11 The most representative structures were extracted after cluster analysis (Fig. 4). Modelling also confirms that the energetically favored association process displayed by the homochiral M4-a corresponds to molecular recognition through inclusion of the guest into the cavity of the host. Contrary to surface binding, inclusion is accompanied by an enthalpic gain originating from the optimization of the number of electrostatic contacts between partner-borne charges (i.e., the lock-and-key paradigm), as well as from the possible expulsion of high-energy water molecules from the cavity (i.e., the non-classical hydrophobic effect).
This picture of the full network of the affinities and selectivities between hosts and guests within the dynamic system can obviously not be apprehended from the fitted HPLC-UV based apparent amplifications. It underlines the true benefit of analyzing a static system Li′ derived from the parent DCL Li by NMR spectroscopy. In particular, 1H and DOSY NMR analysis of the untemplated library L0′ provided the last piece to fully apprehend the various noncovalent interactions and forces simultaneously at work within the dynamic system (Fig. 5A).
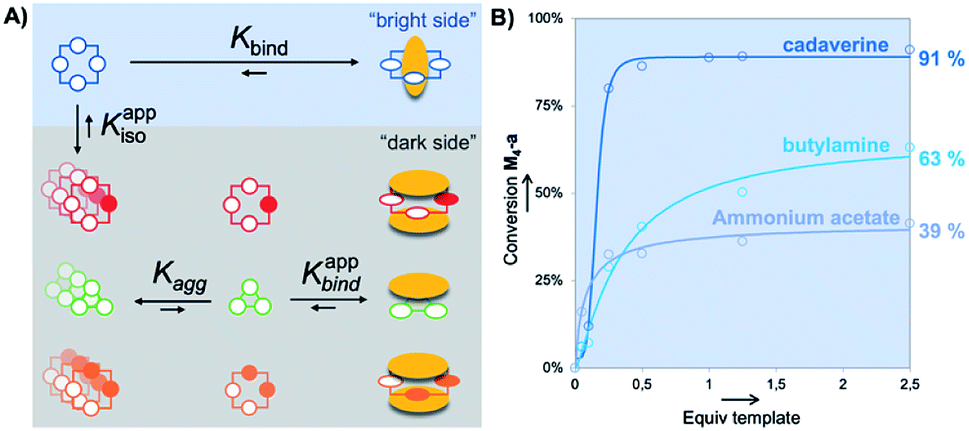 | ||
| Fig. 5 Merging the bright and the dark side: (A) schematic overview of the supramolecular processes (binding vs. aggregation) operating within the full DCL based on monomer M (Kbind: discrete host–guest binding constant; Kagg: host self-aggregation constant; Kappbind: apparent guest binding constant from aggregated host; Kappiso: apparent host isomerization constant from initial aggregated state); (B) conversions in M4-a from quantitative HPLC-UV monitoring with increasing amounts of templates T1–T3; x-axis is rescaled by a factor 1/5 for T1. | ||
As mentioned earlier, the untemplated library gathers self-aggregated cyclophanes (the “dark side”, Fig. 5A), which thereby become substantially more stable than the homochiral cyclotetramer M4-a (the “bright side”). Guest binding by these species formally required their partial or total disaggregation. This revealed that NMR titration delivers in fact the apparent binding constant Kappbind, which is related to the true binding constant Kbind through the relationship:
| Kbind = Kappbind × Kagg |
This led to the conclusion that the true selectivity (defined as the ratio of binding constant Kbind) displayed by each guest for M4-a with respect to any member of the dark side of the library was in reality one or two order of magnitudes lower (w.r.t. Kisobind in the range of 10 to 102 M−1) than the values obtained from the apparent binding constant. Another way to put it is that, within static and dynamic systems Li′ and Li, aggregation competes with binding and thereby reinforces an intrinsically modest supramolecular selectivity of each guest for the series of host molecules (which appears total and completely overestimated by HPLC).
Finally, we addressed the following question: can the quality (or amount) of molecular information contained by a guest be compensated (in terms of template effect on a host) by its absolute amount? Experimentally, we screened the impact (at fixed pH and ionic strength) of increasing amounts of templates T1–T3 on the absolute conversion (quantified by the peak area of M4-a measured in HPLC-UV with respect to the area obtained with 0.25 eq. of T4) of M4-a obtained from building block M (Fig. S1†). The results, summarized in Fig. 5B, indicate that cadaverine T3 was in fact the minimal truncated version of spermine, whose lack of molecular information can be compensated by a stoichiometric excess. In other words, almost quantitative amplification of M4-a could still be induced by equilibrium displacement provided that a strong excess of the α,ω-diamine core is used (91 ± 3% vs. 100% with spermine T4). In contrast, more truncated versions such as T1 (AcONH4) and T2 could only afford maximal conversions of respectively 39 ± 5 and 63 ± 6% in M4-a. This was corroborated by non-linear square fitting of the experimental data which, whatever the model used, confirmed that these maximal values were already reached experimentally with respectively 2.5 and 50 equivalents of each template.
Conclusions
We have shown here that exploring dynamic combinatorial libraries using a single, yet widely used analytical tool can unfortunately lead to a considerable underestimation of the diversity of mixtures in terms of size and stereochemistry, where the identity of the main species can even be misassigned. Quantifying the various host–guest affinities and apprehending the main underpinning supramolecular phenomena (binding, folding, aggregation) at work within a dynamic combinatorial system requires complementary procedures and techniques. In this perspective, we have extended our straightforward protocol of selective template and buffer removal, which herein allowed the preservation of the original composition of the dynamic libraries and provided robust static mixtures eligible for structural analyses and titrations. The propensity of some library members to form aggregates, which affected their detectability by HPLC and participation in the overall thermodynamic equilibrium of libraries, could easily be characterized by routine NMR spectroscopy experiments. Combined with a series of titrations on static mixtures of hosts, this analysis delivered the full array of binding affinities between templates and cyclophanes. It eventually provided the global view of the dark and bright side of this simple DCL, wherein intrinsic selectivity between hosts of varying sizes and configuration is modest but reinforced by self-aggregation processes. These two antagonistic forces operate both in the present static and dynamic parent mixtures, and presumably in many other water-soluble dynamic combinatorial libraries. Numerical simulations have previously shown that supramolecular interactions between library members could affect both degree and selectivity of the response of the library when a template molecule is added.12 Herein, we report – for the first time – an experimental illustration of this anticipated scenario. Our final recommendation, to complement HPLC screening by in situ and ex situ NMR spectroscopy is easy to implement and relies on readily accessible analytical techniques. Such guidelines should allow the community of system chemists to easily apprehend the interactions at work and to generate valuable knowledge on mixtures of increasing complexity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the LABEX iMUST (ANR-10-LABX-0064) of the Université de Lyon within the program “Investissements d'Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR). We are very grateful to A. Baudouin (CCRMN) for her help with the NMR measurements.References
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- For selected publications, see: (a) B. C. Peacor, C. M. Ramsay and M. L. Waters, Chem. Sci., 2017, 8, 1422–1428 RSC; (b) I. N. Gober and M. L. Waters, Org. Biomol. Chem., 2017, 15, 7789–7795 RSC; (c) P. Nowak, M. Colomb-Delsuc, S. Otto and J. Li, J. Am. Chem. Soc., 2015, 137, 10965–10969 CrossRef CAS PubMed; (d) J. Li, P. Nowak and S. Otto, Angew. Chem., Int. Ed., 2015, 54, 833–837 CrossRef CAS PubMed; (e) N. K. Pinkin and M. L. Waters, Org. Biomol. Chem., 2014, 12, 7059–7067 RSC; (f) S. Hamieh, R. F. Ludlow, O. Perraud, K. R. West, E. Mattia and S. Otto, Org. Lett., 2012, 14, 5404–5407 CrossRef CAS PubMed; (g) L. A. Ingerman, M. E. Cuellar and M. L. Waters, Chem. Commun., 2010, 46, 1839–1841 RSC; (h) A. R. Stefankiewicz, M. R. Sambrook and J. K. M. Sanders, Chem. Sci., 2012, 3, 2326–2329 RSC; (i) Z. Rodriguez-Docampo, E. Eugenieva-Ilieva, C. Reyheller, A. M. Belenguer, S. Kubik and S. Otto, Chem. Commun., 2011, 47, 9798–9800 RSC; (j) R. F. Ludlow and S. Otto, J. Am. Chem. Soc., 2008, 130, 12218–12219 CrossRef CAS PubMed.
- P. T. Corbett, J. K. M. Sanders and S. Otto, Chem.–Eur. J., 2008, 14, 2153–2166 CrossRef CAS PubMed.
- R. F. Ludlow, J. Liu, H. Li, S. L. Roberts, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2007, 46, 5762–5764 CrossRef CAS PubMed.
- (a) E. Jeamet, J. Septavaux, A. Héloin, M. Donnier-Maréchal, M. Dumartin, B. Ourri, P. Mandal, I. Huc, E. Bignon, E. Dumont, E. Bignon, E. Dumont, C. Morell, J.-P. Francoia, F. Perret, L. Vial and J. Leclaire, Chem. Sci., 2019, 10, 277–283 RSC; (b) M. Donnier-Maréchal, J. Septavaux, E. Jeamet, A. Héloin, F. Perret, E. Dumont, J.-C. Rossi, F. Ziarelli, J. Leclaire and L. Vial, Org. Lett., 2018, 20, 2420–2423 CrossRef PubMed; (c) L. Vial, M. Dumartin, M. Donnier-Maréchal, F. Perret, J.-P. Francoia and J. Leclaire, Chem. Commun., 2016, 52, 14219–14221 RSC; (d) P.-T. Skowron, M. Dumartin, E. Jeamet, F. Perret, C. Gourlaouen, A. Baudouin, B. Fenet, J.-V. Naubron, F. Fotiadu, L. Vial and J. Leclaire, J. Org. Chem., 2016, 81, 654–661 CrossRef CAS PubMed.
- L. Vial, R. F. Ludlow, J. Leclaire, R. Pérez-Fernández and S. Otto, J. Am. Chem. Soc., 2006, 128, 10253–10257 CrossRef CAS PubMed.
- (a) J. E. Beaver, B. C. Peacor, J. V. Bain, L. I. James and M. L. Waters, Org. Biomol. Chem., 2015, 13, 3220–3226 RSC; (b) S. Hamieh, V. Saggiomo, P. Nowak, E. Mattia, R. F. Ludlow and S. Otto, Angew. Chem., Int. Ed., 2013, 52, 12368–12372 CrossRef CAS PubMed; (c) P. Besenius, P. A. G. Cormack, R. F. Ludlow, S. Otto and D. C. Sherrington, Org. Biomol. Chem., 2010, 8, 2414–2418 RSC; (d) R. F. Ludlow and S. Otto, J. Am. Chem. Soc., 2008, 130, 12218–12219 CrossRef CAS PubMed.
- Good signal to noise is essential for accurate integration, and for integration errors <1%, a S/N of at least 250:1 is required. See: http://nmrweb.chem.ox.ac.uk/Data/Sites/70/userfiles/pdfs/quantitative-nmr.pdf.
- J. Clayden, N. Greeves and S. Warren, in Organic chemistry, Oxford University press, Oxford, 2nd edn, 2012, ch. 12, pp. 240–267 Search PubMed.
- MATLAB scripts are available at https://github.com/lovial/ChemSci.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- A. G. Orrillo and R. L. E. Furlan, J. Org. Chem., 2010, 75, 211–214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and computational details. See DOI: 10.1039/d0sc02399j |
| This journal is © The Royal Society of Chemistry 2020 |