
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnodic oxidation triggered divergent 1,2- and 1,4-group transfer reactions of β-hydroxycarboxylic acids enabled by electrochemical regulation†
Zhenxing
Zhang‡
ab,
Lei
Zhang‡
a,
Xianhao
Zhang
c,
Jianxin
Yang
c,
Yunxing
Yin
c,
Yangye
Jiang
d,
Chengchu
Zeng
 d,
Gang
Lu
e,
Yang
Yang
f and
Fanyang
Mo
*ag
d,
Gang
Lu
e,
Yang
Yang
f and
Fanyang
Mo
*ag
aDepartment of Energy and Resources Engineering, College of Engineering, Peking University, Beijing 100871, China. E-mail: fmo@pku.edu.cn
bCollege of Chemistry and Chemical Engineering, Anyang Normal University, Anyang 455000, China
cWuXi AppTec (Tianjin) Co., Ltd, Tianjin 300457, P. R. China
dCollege of Life Science & Bioengineering, Beijing University of Technology, Beijing 100124, China
eSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, Shandong 250100, China
fDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA
gJiangsu Donghai Silicon Industry S&T Innovation Center, Jiangsu 222300, China
First published on 28th September 2020
Abstract
We report a set of electrochemically regulated protocols for the divergent synthesis of ketones and β-keto esters from the same β-hydroxycarboxylic acid starting materials. Enabled by electrochemical control, the anodic oxidation of carboxylic acids proceeded in either a one-electron or a two-electron pathway, leading to a 1,4-aryl transfer or a semipinacol-type 1,2-group transfer product with excellent chemoselectivity. The 1,4-aryl transfer represents an unprecedented example of carbon-to-oxygen group transfer proceeding via a radical mechanism. In contrast to previously reported radical group transfer reactions, this 1,4-group transfer process features the migration of electron-rich aryl substituents. Furthermore, with these chemoselective electrochemical oxidation protocols, a range of ketones and β-keto esters including those possessing a challenging-to-access medium-sized ring could be synthesized in excellent yields.
Introduction
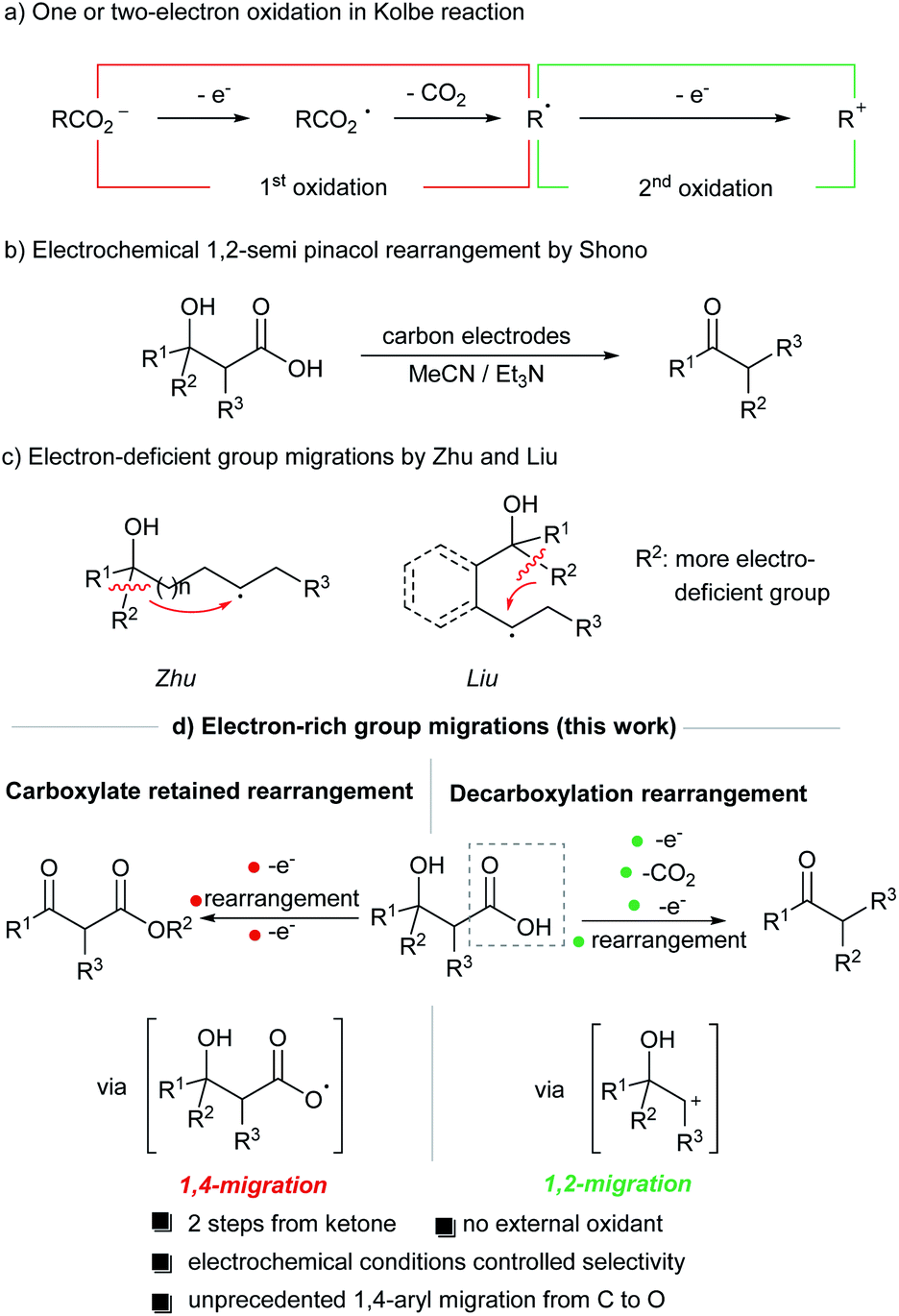
As an essential class of synthetically useful transformations, rearrangement reactions such as ring expansion, ring contraction and functional group transposition have received considerable attention due to their excellent atom economy, stereospecificity and efficiency.1 Among various rearrangement processes, radical and ionic group migrations represent two of the most commonly encountered mechanisms. In this context, it is of great interest if the same substrate can undergo either radical or ionic rearrangement when being subjected to different reaction conditions.Our laboratory has been interested in utilizing organic electrochemistry as a mild and efficient alternative to conventional chemical approaches for sustainable synthesis.2 One of the most appealing advantages of organic electrochemistry lies in its ability to exert exquisite control over the reaction outcome. Through the fine tuning of applied potential and other electrochemical parameters (e.g., choice of electrodes and electrolytes), common organic starting materials can selectively undergo one- or two-electron redox processes at the electrode and generate highly reactive intermediates.3 We surmise that the judicious selection of electrochemical conditions could be used to trigger mechanistically distinct rearrangement processes from the same substrate.
Kolbe electrolysis is a classical organic electrochemical reaction which provides an effective means to couple alkyl radicals generated from the anodic oxidation of carboxylic acids.4 In these anodic processes, the initial one-electron oxidation-triggered decarboxylation leads to the formation of an alkyl radical (Scheme 1a). This transient alkyl radical can also be further oxidized electrochemically, giving rise to a new reactive carbocation species. For example, this electrochemically generated carbocation has been utilized in semi-pinacol rearrangement by Shono (Scheme 1b).5 We postulated that this electrochemistry-enabled mechanistic dichotomy could be further leveraged to allow for divergent rearrangement processes from the same substrate.
 | ||
| Scheme 1 Electrochemical oxidation of alkyl carboxylic acids. | ||
We envisioned an electrochemically controlled product selectivity in the anodic oxidation induced rearrangement of β-hydroxycarboxylic acids (Scheme 1d). In this design, the two-electron oxidation of β-hydroxycarboxylic acids would lead to a carbocation intermediate at the β-position of the alcohol, thereby inducing the semipinacol-type ionic rearrangement. On the other hand, under appropriate electrochemical conditions, one-electron oxidation of the carboxylic acid would give rise to a highly reactive oxygen-centered radical, thereby allowing for novel functional group migration processes via a radical mechanism.6 More importantly, we demonstrate that the electron-rich aryl migration can be accomplished in this carbon-to-oxygen group transfer triggered by anodic one-electron oxidation of β-hydroxycarboxylic acids. We note that carbon to oxygen group transfer initiated by an oxygen-centered radical is unprecedented in the literature.6a
Radical-mediated functional group migration reactions have received considerable attention and a number of elegant protocols for carbon-group transfer processes have been recently reported by Zhu7 and Liu8 (Scheme 1c). In these previously reported radical aryl migration reactions, electron-deficient aryl group migrates preferentially over electron-rich ones due to the nucleophilic nature of the carbon-centered radical. The inverse reactivity pattern, namely the migration of electron-rich aryl groups, is seldom reported.
Results and discussion
We commenced our studies by investigating reaction conditions for the electrochemical oxidation induced rearrangement of 3-hydroxy-2-methyl-3,3-diphenylpropanoic acid 1a (Table 1). When glassy carbon was used as electrodes (anode and cathode), a mixture of products 2a and 3a was obtained with low chemoselectivity (entry 1). Similar to our previous work on intramolecular oxidative cyclization,2b a catalytic amount of NaOH could serve as the base and the supporting electrolyte in this reaction, and no additional electrolyte was needed for the electrochemical transformation. To improve the chemoselectivity of this reaction, we examined the use of a range of electrodes and solvents. With graphite as the anode and Pt plate as the cathode, using a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and pyridine as the solvent, 3a formed with excellent chemoselectivity (entry 3). Increasing the electric quantity from 2 F mol−1 to 2.3 F mol−1 further improved the yield of 3a to 74% (entry 4). On the other hand, the use of Pt anode in conjunction with a mixed MeCN/H2O solvent led to the selective formation of 2a (entries 6 and 7). Finally, no product was observed in the absence of electric current (entry 8).
:1 mixture of water and pyridine as the solvent, 3a formed with excellent chemoselectivity (entry 3). Increasing the electric quantity from 2 F mol−1 to 2.3 F mol−1 further improved the yield of 3a to 74% (entry 4). On the other hand, the use of Pt anode in conjunction with a mixed MeCN/H2O solvent led to the selective formation of 2a (entries 6 and 7). Finally, no product was observed in the absence of electric current (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Electrodes | Solvents (ratio) | 2a (%) | 3a (%) |
| a Entries 1–4 were performed on IKA Electrasyn 2.0 with its kit electrodes at room temperature. Entries 5–8 were performed in a test tube with Pt gauzes (52 mesh) as electrodes and IKA Electrasyn 2.0 as a power supply. Substrate 1a (0.3 mmol), solvents 4 mL, NaOH (10 mol%) for entries 1, 5–8, constant current (cc) 5 mA, 2 F mol−1. ND, not detected. b Yields were determined by 1H NMR using 1,3,5-trimethoxylbenzene as an internal standard. c 2.3 F mol−1. | ||||
| 1 | Glassy carbon (+/−) | MeOH/H2O (3:1) |
22 | 28 |
| 2 | Graphite (+/−) | Pyridine/H2O (1:3) |
3 | 46 |
| 3 | Graphite/Pt (+/−) | Pyridine/H2O (1:3) |
1 | 56 |
| 4c | Graphite/Pt (+/−) | Pyridine/H2O (1:3) |
1 | 74 |
| 5 | Pt (+/−) | MeOH/H2O (3:1) |
60 | 3 |
| 6 | Pt (+/−) | MeCN/H2O (3:1) |
73 | ND |
| 7c | Pt (+/−) | MeCN/H2O (3:1) |
78 | ND |
| 8 | Pt (+/−) | MeCN/H2O (3:1) |
ND | ND |
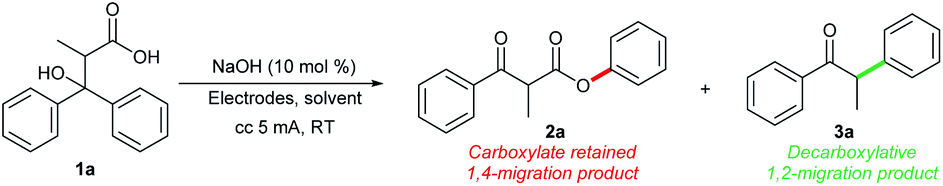
Based on previous study on Kolbe electrolysis9 and our own work, we reasoned that the choice of electrode plays a dominant role in determining the product selectivity.10 We observed that anode materials were key factor to the outcome of the reaction. Platinum electrodes favor 1,4-migration, whereas carbon-based electrodes favor 1,2-migration. For 1,4-aryl migration, the substrate undergoes a one-electron oxidation process to afford an oxygen-centered carboxylate radical. This triggers the 1,4-aryl migration followed by one-electron oxidation on the anode to deliver the final β-keto ester product (Scheme 2, left). For 1,2-group migration, a two-electron oxidation process takes place, resulting in the formation of a carbocation. The subsequent semi-pinacol rearrangement then furnishes the ketone product (Scheme 2, right).
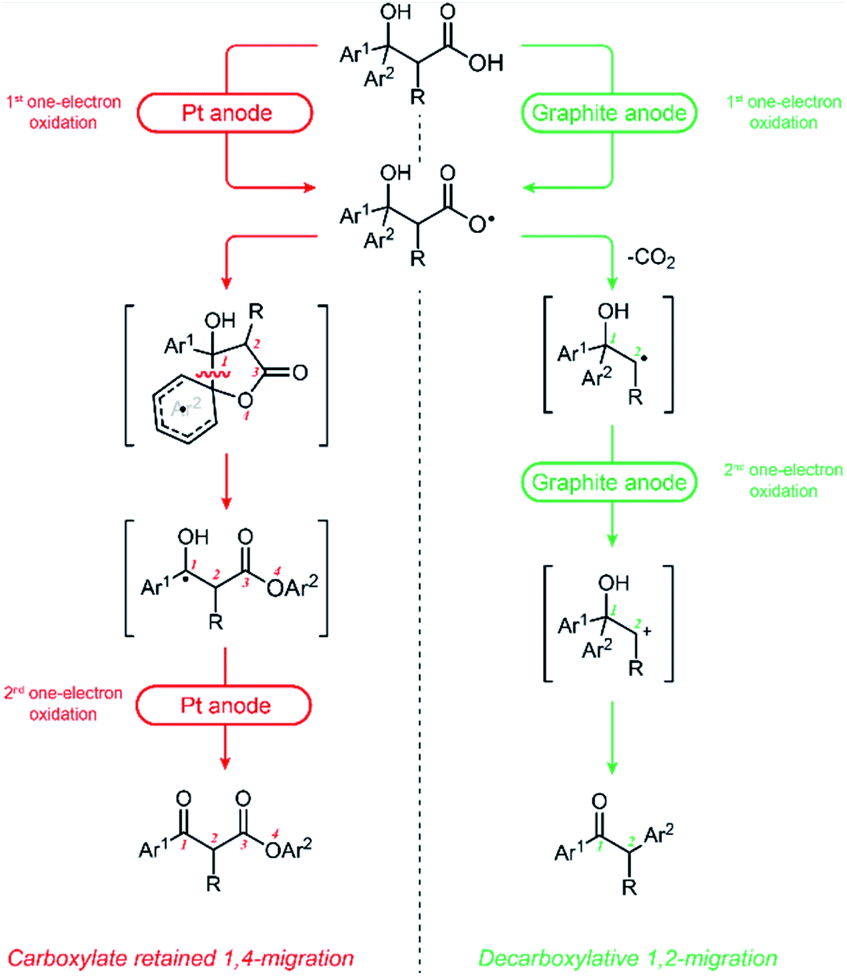 | ||
| Scheme 2 Rationale for migration selectivity. | ||
With the optimized reaction conditions in hand (Table 1, entries 4 and 7), we investigated the substrate scope of the one-electron oxidation induced 1,4-aryl migration (Table 2). A series of acyclic β,β-diaryl-β-hydroxycarboxylic acids 1a–1r were converted to the corresponding β-keto esters 2a–2r under electrochemical conditions in good to excellent yields. Some of these functionalized β-carbonyl esters require multistep synthetic procedure to synthesize using conventional methods. Therefore, this electricity-driven radical-mediated 1,4-aryl migration strategy provided an efficient access to these β-keto ester building blocks. Notably, in contrast to previously developed radical group transfer processes, when asymmetrical β,β-diaryl substituted β-hydroxycarboxylic acid 1i–1n was applied, the electron-rich aryl group migrated preferentially over the electron-neutral one. Additionally, we found that strong electron-withdrawing group substituted β,β-diaryl-β-hydroxycarboxylic acids had a poor reactivity toward 1,4-aryl migration, including cyan, amide and ester groups. We attribute this selectivity to the electrophilic nature of oxygen-centered radical generated from single-electron oxidation. From a mechanistic perspective, this rearrangement represents a selectivity reversal in radical migration reactions.6a Furthermore, a range of medium-sized lactones (2s–2u) that are challenging to synthesize11 could also be effectively prepared using this radical 1,4-aryl migration (n + 3 ring expansion), thus further demonstrating the synthetic utility of this electrochemically triggered group transfer process. We note that the current 1,4-aryl migration can only be applied to diaryl substituted β-hydroxycarboxylic acids. Retro-Reformatsky reaction took place when alkyl/aryl substituted β-hydroxycarboxylic acids were used.
| a Reaction conditions: substrate 1 (0.3 mmol), NaOH (10 mol%), MeCN 3 mL, H2O 1 mL, platinum gauzes as both anode and cathode, constant current (cc) 5 mA, 2.3 F mol−1. The reactions were performed in a test tube using Electrasyn 2.0 as power supply. |
|---|
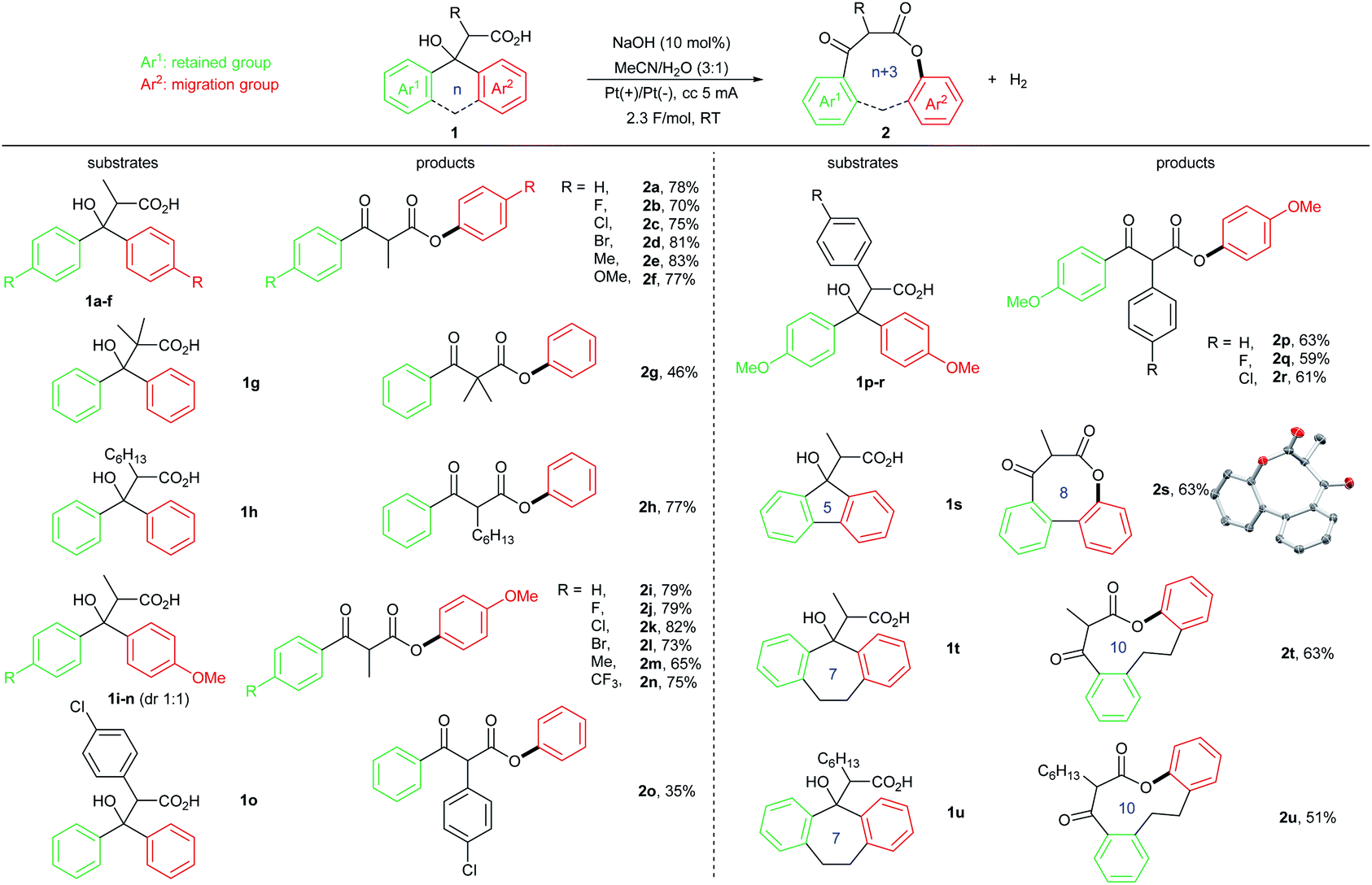
|
We next surveyed the substrate scope of the two-electron oxidation induced semi-pinacol rearrangement (1,2-aryl/alkyl migration) (Table 3). For substituted β-hydroxycarboxylic acids 1a–1e, both moderate electron-withdrawing and electron-donating groups were well tolerated, providing the corresponding products (3a–3e) in excellent yields (73–80%). However, substrates with strong electron-withdrawing group showed low reactivity (1v–1x). For β-alkyl-β-aryl-β-hydroxycarboxylic acid 1y, selective 1,2-aryl over 1,2-alkyl migration was observed, which is characteristic for semipinacol rearrangement involving a cationic intermediate.12 Additionally, a wide range of cyclic ketone-drived β-hydroxycarboxylic acids 1z–1ah were transformed into their corresponding n + 1 ring-expansion ketone products in moderate to good yields.
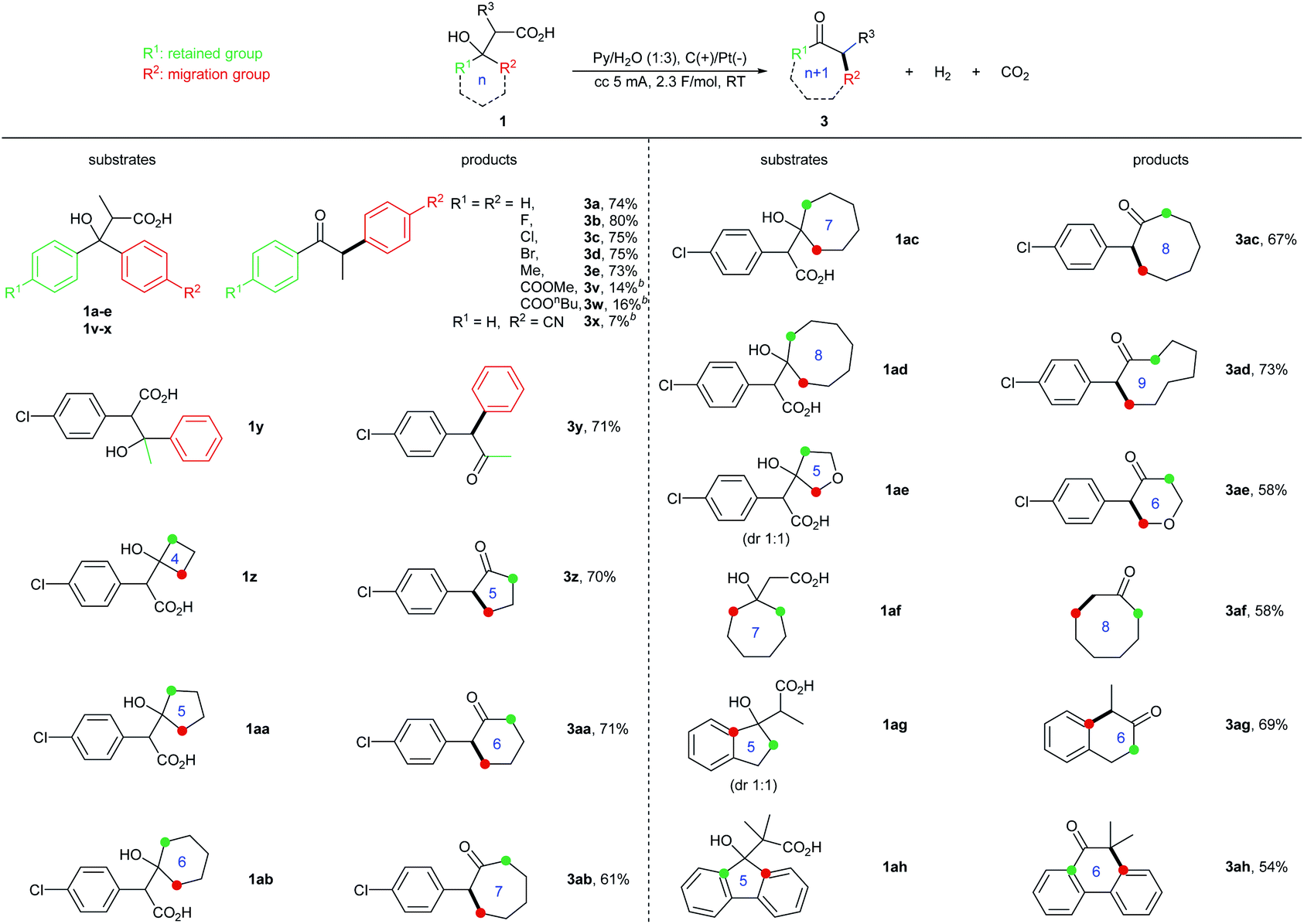
Thus, the electrochemical protocol detailed here allowed for a wide range of substrates including diaryl and aryl/alkyl substituted carboxylic acids to be effectively converted, thereby representing a more general process relative to the prior work described by Shono.5
This two-electron oxidation induced semipinacol rearrangement was also successfully applied in the construction of a biologically active polycyclic scaffold bicyclo[3.2.1]octadienone, which is found in plant-derived natural products including β-naphthocyclinone and engelharquinone.13 As shown in Scheme 3, the Diels–Alder reaction between anthrone 4 and benzyl acrylate 5 afforded bicyclo[2.2.2] 6, which was subsequently hydrogenated to compound 7 in 73% yield in two steps. Furthermore, 7 underwent the decarboxylative semi-pinacol rearrangement to afford the desired bicycle[3.2.1]octadienone 8 in good yield under the standard electrochemical conditions.
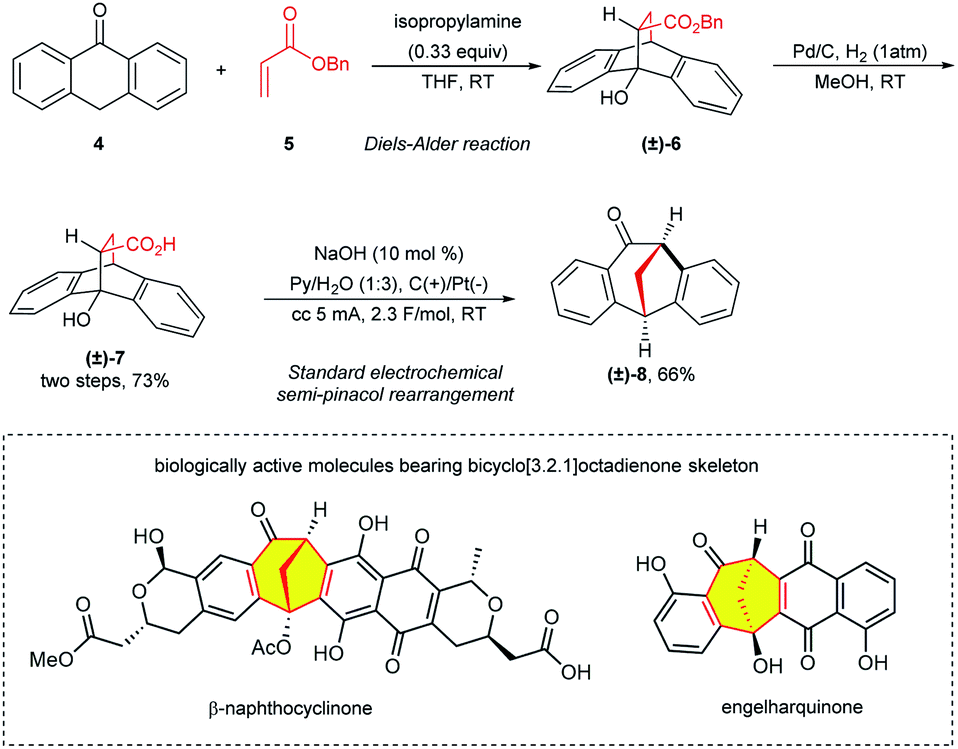 | ||
| Scheme 3 Synthesis of 8 and two biologically active molecules bearing bicycle[3.2.1]octadienone skeleton. | ||
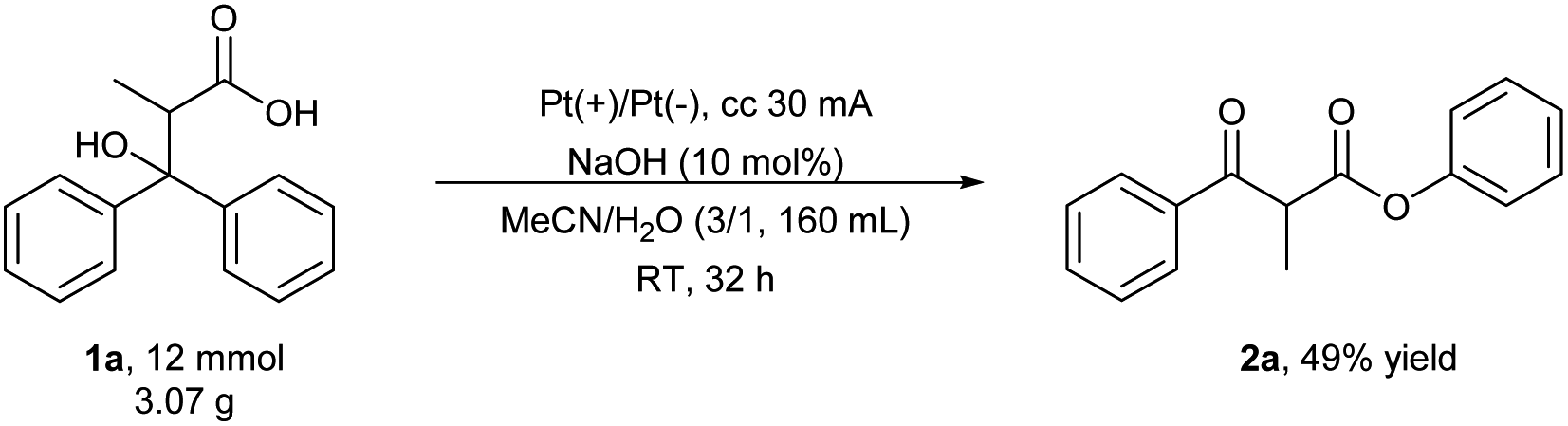
To further demonstrate the utility of this electrochemical method, a gram-scale synthesis of 2a was carried out under constant current electrolysis (eqn (1)). The 12 mmol-scale reaction furnished 2a in 49% yield, thereby demonstrating the potential of this method for the large-scale synthesis of valuable β-keto esters.
 | (1) |
Moreover, a number of derivatization transformations was carried out. Fluorination (9), chlorination (10), bromination (11), azidation (12) and sponification (13) of 2t were successfully executed, providing a range of synthetically useful building blocks (Scheme 4).
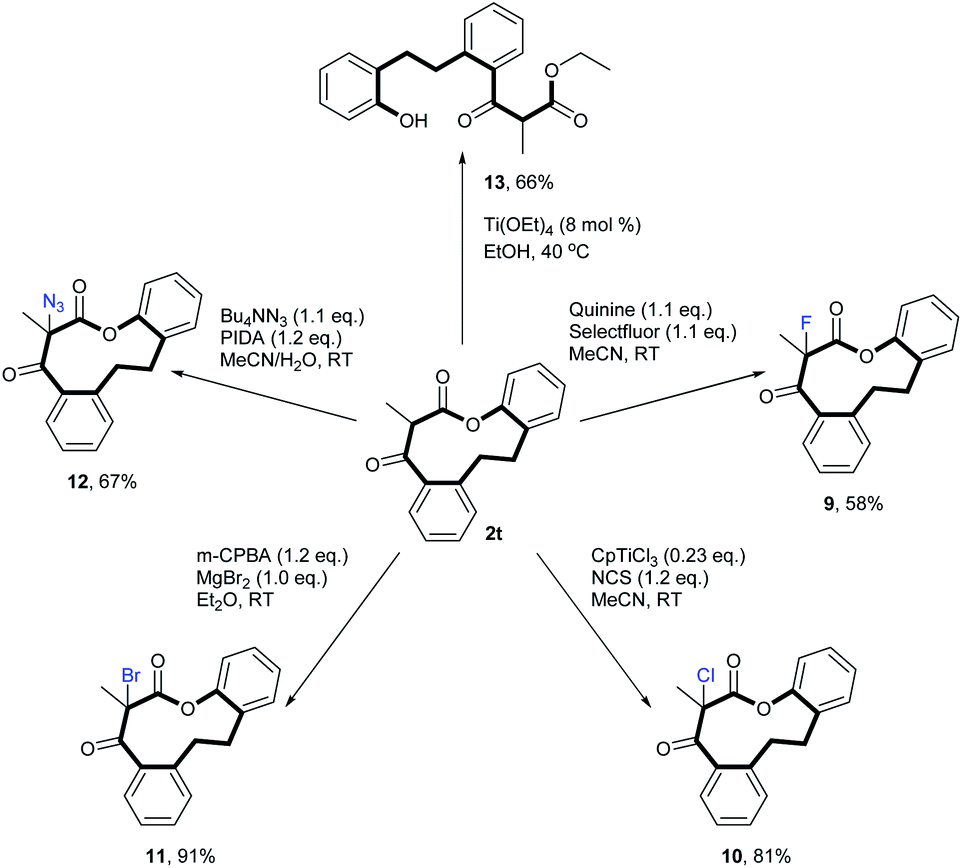 | ||
| Scheme 4 Versatile transformations of medium-sized lactone 2t. | ||
To revisit our original design for 1,4-migration reaction, we assumed that the carboxylate is oxidized first, followed by intramolecular addition of an oxygen-centered radical to the electron-rich arene (Scheme 5a). Nevertheless, there indeed exists a viable alternative mechanism in which the electron-rich arene is oxidized first, which then underwent intramolecular nucleophilic attack by the carboxylate (Scheme 5b).
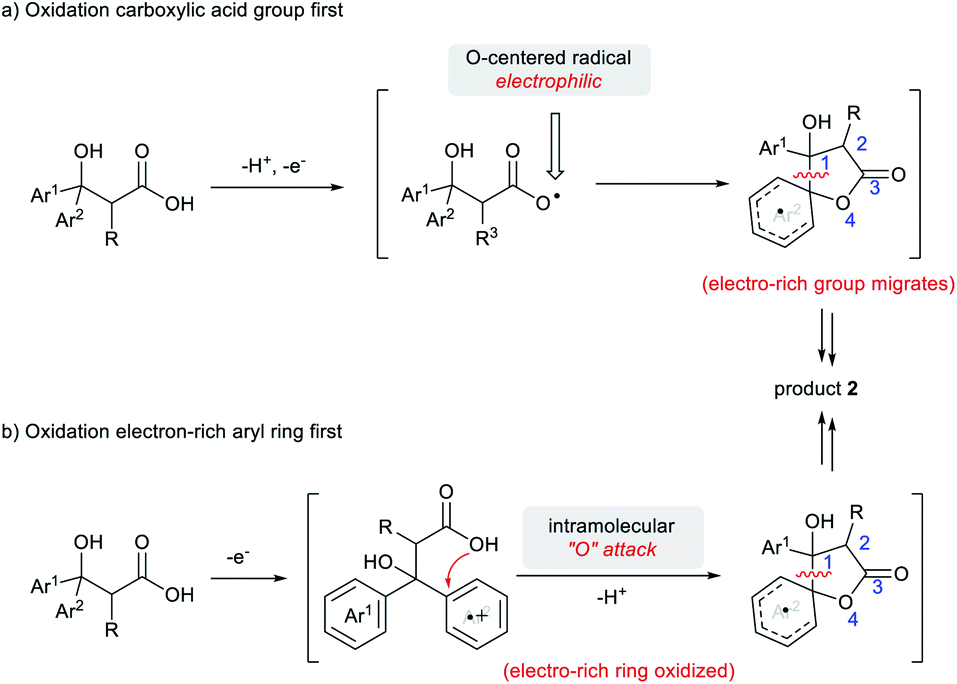 | ||
| Scheme 5 Two possible reaction mechanisms for 1,4-migration. | ||
To differentiate between these two mechanisms, cyclic voltammetry studies were carried out (Fig. 1). Our CV studies indicated that carboxylates undergo single electron oxidation at much lower potential relative to that of carboxylic acids (see the ESI† for details). For this reason, under electrolysis conditions, only the deprotonated substrate (i.e., the carboxylate) undergoes single electron oxidation. This deprotonation facilitated oxidation is common among Kolbe electrolysis. Thus, the redox potential difference between the carboxylate (1a′–1f′) and the arene (1a′′–1f′′) will dictate the functionality undergoing preferential oxidation. We found that the carboxylate form 1a′, 1c′ and 1f′ display a series of irreversible anodic events occurred with an onset potential of 0.73 V (Fig. 1b). In contrast, compounds 1a′′, 1c′′ and 1f′′, which contain no carboxylate functionality, showed no oxidation peaks at 0.7 V (Fig. 1c–e). As such, we assigned waves between 0.73 V and 0.78 V in Fig. 1b to the oxidation of carboxylate moiety in 1a′, 1c′ and 1f′. For compound 1f′′ containing the most electron-rich arene (anisole), an evident anodic wave appeared at 1.20 V. We attributed this wave to the oxidation of aryl ring, because the oxidation potential of tertiary alcohols is prohibitive according to literature report.14 By contrast, oxidation peaks for compounds 1a′′ and 1c′′ were insignificant, appearing as shoulder peaks at 1.35 V and 1.50 V, respectively. This is well in accord with that they possess electron-neutral or deficient aryl rings. We noticed that in the CV curve of compound 1f′, after the first oxidation peak at 0.78 V, another oxidation peak appeared at 1.12 V. We believe this second peak corresponds to arene oxidation in compound 1f′. Taken together, the data support the sequence of carboxylate oxidation prior to arene oxidation. DFT calculations were performed to investigate the energetics of reaction pathway involving carboxylate radical. The results show that the in situ formed carboxylate radical can easily undergo radical addition and C–C bond cleavage steps to realize an unusual carbon to oxygen 1,4-aryl migration. (See details in ESI†).
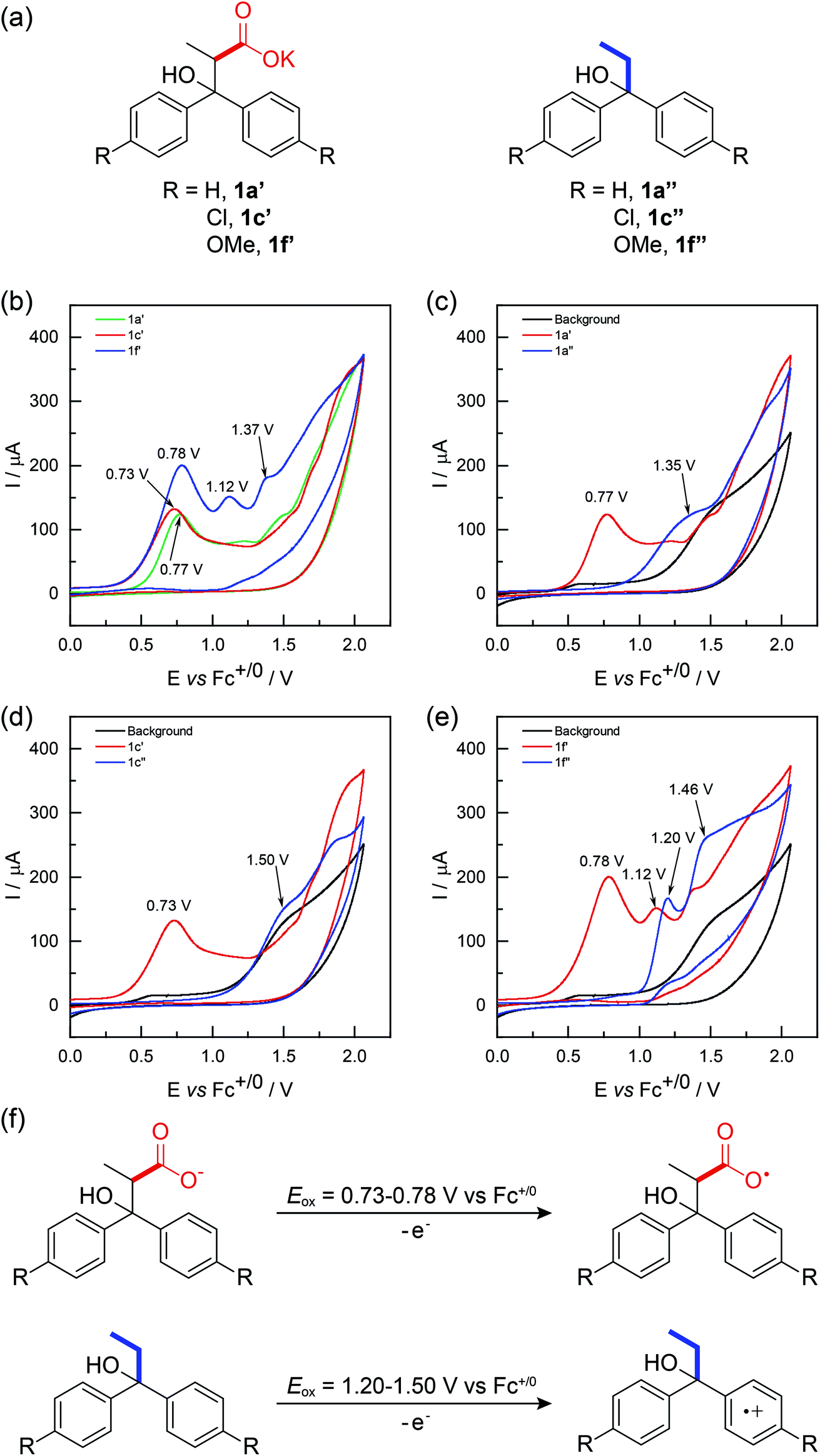 | ||
| Fig. 1 CV studies: working electrode, platinum disk (Φ 3 mm); counter electrode, silver wire; reference electrode, Ag/AgNO3 (0.1 M in MeCN); scan rate 0.1 V s−1; substrate concentration, 5 mM; background, 0.1 M LiClO4 in MeCN/H2O (3:1). | ||
To further verify this, two additional experiments were carried out (Scheme 6). Since the oxidation potential of carboxylate is lower than that of arene, we should be able to control the potential such that the carboxylate is more readily oxidized than the arene. Accordingly, a controlled potential electrolysis (CPE) was conducted using 1f as the substrate. The potential was controlled at 0.91 V vs. Fc+/0. After 8.18C of charge passed through the reaction mixture, desired product 2f was formed in 14% yield with 48% current efficiency (Scheme 6a). On the other hand, N-acyloxyphthalimides are widely used substrates for decarboxylative C–C bond-forming reactions under reductive conditions,15 which is different from most oxidizing decarboxylative reactions of carboxylic acids. Therefore, we hypothesized that N-hydroxyphthalimide derived from 1a might be transformed to 1,4-migration product 2a under redox neutral condition. To test our hypothesis, we first synthesized compound 14 from 1a and N-hydroxyphthalimide in one step. Next, an unoptimized conditions from literature15d was applied to substrate 14. Gratifyingly, 1,4-migration product 2a was formed in 10% yield upon irradiation (Scheme 6b). Mechanistically, a single-electron transfer from visible light-excited photocatalyst Ir(III)* to the N-acyloxyphthalimide 14 generates a radical anion that undergoes homolytic cleavage of the N–O bond to form the carboxylate radical and phthalimide anion. Final product 2a is produced via sequential radical carbon to oxygen 1,4-migration and oxidation by Ir(IV).
 | ||
| Scheme 6 Mechanistic studies on 1,4-migration. | ||
Conclusions
In summary, we have developed an orthogonal set of electrochemically controlled 1,4- and 1,2-group transfer reactions triggered by the oxidation of β-hydroxycarboxylic acids. Notably, through the judicious selection of electrochemical conditions, the same substrate could undergo either the 1,2-semipinacol rearrangement or the unprecedented carbon to oxygen 1,4-aryl migration with excellent chemoselectivity. This electrochemistry enabled chemoselective transformations provided a new method for the divergent transformation of easily available carboxylic acid derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the Natural Science Foundation of China (grants 21772003, 21933001, 22071004). This work was also partially funded by the BHP-PKU CCUS project supported by BHP Billiton.Notes and references
- C. M. Rojas, Molecular Rearrangements in Organic Synthesis, John Wiley & Sons, Inc., New Jersey, 2015 Search PubMed.
- (a) Q. Liu, B. Sun, Z. Liu, Y. Kao, B.-W. Dong, S.-D. Jiang, F. Li, G. Liu, Y. Yang and F. Mo, Chem. Sci., 2018, 9, 8731 RSC; (b) L. Zhang, Z. Zhang, J. Hong, J. Yu, J. Zhang and F. Mo, J. Org. Chem., 2018, 83, 3200 CrossRef CAS; (c) L. Zhang, Z. Zhang, J. Zhang, K. Li and F. Mo, Green Chem., 2018, 20, 3916 RSC; (d) J. Hong, M. Li, J. Zhang, B. Sun and F. Mo, ChemSusChem, 2019, 12, 6 CrossRef CAS; (e) Z. Zhang, L. Zhang, Y. Cao, F. Li, G. Bai, G. Liu, Y. Yang and F. Mo, Org. Lett., 2019, 21, 762 CrossRef CAS; (f) M. García-Melchor, A. A. C. Braga, A. Lledós, G. Ujaque and F. Maseras, Acc. Chem. Res., 2013, 46, 2626 CrossRef.
- (a) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS; (c) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS; (d) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27 CrossRef CAS; (e) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef; (f) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS; (g) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS; (h) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS; (i) K. D. Moeller, Tetrahedron, 2000, 56, 9527 CrossRef CAS.
- (a) M. Faraday, Ann. Phys., 1834, 108, 401 CrossRef; (b) H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257 CrossRef.
- T. Shono, J. Hayashi, H. Omoto and Y. Matsumura, Tetrahedron Lett., 1977, 18, 2667 CrossRef.
- (a) Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2015, 44, 5220 RSC; (b) A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649 CrossRef CAS.
- (a) X. Wu, M. Wang, L. Huan, D. Wang, J. Wang and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 1640 CrossRef CAS; (b) Z. Wu, R. Ren and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 10821 CrossRef CAS; (c) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388 CrossRef CAS; (d) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS.
- (a) L. Li, Z.-L. Li, Q.-S. Gu, N. Wang and X.-Y. Liu, Sci. Adv., 2017, 3, e1701487 CrossRef; (b) L. Li, Z.-L. Li, F.-L. Wang, Z. Guo, Y.-F. Cheng, N. Wang, X.-W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X.-Y. Liu, Nat. Commun., 2016, 7, 13852 CrossRef; (c) Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100 CrossRef CAS; (d) F. Mo, D. Qiu, Y. Zhang and J. Wang, Acc. Chem. Res., 2018, 51, 496 CrossRef CAS.
- (a) W. J. Koehl, J. Am. Chem. Soc., 1964, 86, 4686 CrossRef CAS; (b) S. D. Ross and M. Finkelstein, J. Org. Chem., 1969, 34, 2923 CrossRef CAS; (c) J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398 CrossRef CAS.
- (a) H.-J. Schäfer, Recent contributions of kolbe electrolysis to organic synthesis, Berlin, Heidelberg, 1990, DOI:10.1007/BFb0034365; (b) H. Tanaka, M. Kuroboshi and S. Torii, Organic Electrochemistry, CRC Press, 2015, p. 1265 Search PubMed.
- G. Rousseau, Tetrahedron, 1995, 51, 2777 CrossRef CAS.
- Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523 CrossRef CAS.
- (a) Y. Ando, S. Hori, T. Fukazawa, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2015, 54, 9650 CrossRef CAS; (b) T. Fukazawa, Y. Ando, K. Ohmori, T. Hayashi and K. Suzuki, Org. Lett., 2017, 19, 1470 CrossRef CAS; (c) C.-C. Wu, C.-F. Peng, I.-L. Tsai, M. H. Abd El-Razek, H.-S. Huang and I.-S. Chen, Phytochemistry, 2007, 68, 1338 CrossRef CAS.
- R. Francke, T. Quell, A. Wiebe and S. R. Waldvogel, Organic Electrochemistry, CRC Press, 2015, p. 983 Search PubMed.
- (a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS; (b) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440 CrossRef CAS; (c) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS; (d) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1987570. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02386h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |