
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-fused cyclobutanes via cycloisomerization of alkylidenecyclopropane acylsilanes†
Sarah
Eichenberger
 ,
Moritz
Hönig
,
Matthieu J. R.
Richter
,
Renana
Gershoni-Poranne
* and
Erick M.
Carreira
*
,
Moritz
Hönig
,
Matthieu J. R.
Richter
,
Renana
Gershoni-Poranne
* and
Erick M.
Carreira
*
Laboratorium für Organische Chemie, Eidgenössische Technische Hochschule Zürich, HCI H335, Vladimir-Prelog-Weg 3, 8093 Zürich, Switzerland. E-mail: carreira@org.chem.ethz.ch
First published on 4th May 2020
Abstract
A novel Lewis acid-catalyzed cycloisomerization of alkylidenecyclopropane acylsilanes is disclosed. The readily available starting materials participate in tandem Prins addition/ring expansion/1,2-silyl shift to grant access to bicyclo[4.2.0]octanes and bicyclo[3.2.0]heptanes, which are common motifs in terpenoid natural products. Notably, the transformation relies on the ability of acylsilanes to act sequentially as acceptors and donors on the same carbon atom.
Introduction
Bicyclo[4.2.0]octanes and bicyclo[3.2.0]heptanes are common motifs in natural products, such as those belonging to the protoilludane, sterpurane and punctaporonane families.1 Additionally, both hydrocarbon and heterocyclic analogues have found interest in the discovery and development of small molecule therapeutics.2 Herein, we report a novel Lewis acid-catalysed ring-expanding cycloisomerization of alkylidenecyclopropane acylsilanes 1. The transformation generates bicyclic products 2 that incorporate fused cyclobutanes with embedded quaternary centers (Fig. 1). Notably, this method offers high diastereocontrol over the newly formed quaternary stereocenters, and the resulting α-silyl ketones offer a key handle for divergent product functionalization.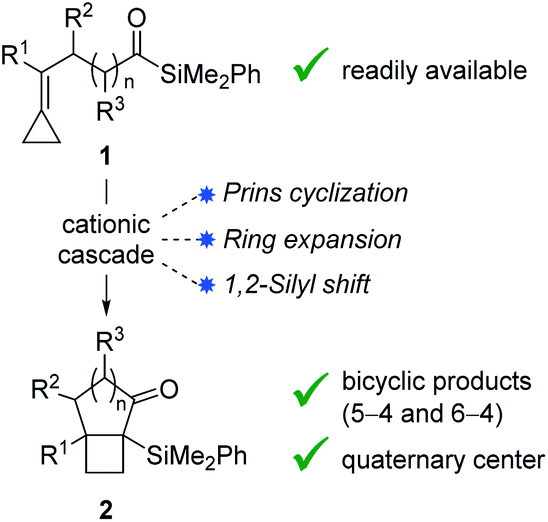 | ||
| Fig. 1 Cycloisomerization cascade. | ||
Sterpurene, repraesentin F, and illudiolone (Scheme 1a) exemplify natural products that include bicyclo[4.2.0]octane and bicyclo[3.2.0]heptane. The complex scaffolds along with the quaternary centers at the cyclobutane ring junction render these structures challenging targets for synthesis.3 Among the most widespread methods to access bicyclo[4.2.0]octane and bicyclo[3.2.0]heptane scaffolds are [2 + 2] photocycloaddition reactions involving cyclic enone acceptors and ethylene.4 However, experimental inconvenience associated with the use of ethylene limits the utility of such approaches. Alternative strategies towards these include cobalt-mediated [2 + 2 + 2] cycloaddition,5 cyclopropane ring expansion,6 and homo-Favorskii rearrangement,7 among others. However, cyclobutanes fused to either 5- or 6-membered rings that possess quaternary stereocenters at the ring junction continue to pose considerable challenges for synthesis. In a pioneering study, Fürstner reported the use of alkylidene cyclopropanes for the synthesis of cyclobutenes mediated by platinum-catalysis.8 The requisite starting materials were accessed in one step from aldehydes by use of the Julia–Kocienski reaction. Other convenient access routes to alkylidene cyclopropanes from aldehydes include Wittig9 and Petasis10 olefination approaches. Transformations exploiting alkylidenecyclopropanes or vinyl cyclopropanes as cyclobutane precursors have been developed employing π-acid catalysis.6b–g For example, acetylenic alkylidene cyclopropanes have been reported to undergo Au-catalyzed rearrangement to furnish cyclobutane-fused 1,3-cyclohexadienes 6 (Scheme 1b).6f We recently implemented this method in the first total synthesis and stereochemical revision of harziane diterpenoid 7, using a key gold-catalyzed cycloisomerization to access the natural product's cyclobutane core.11 Despite the power of the transformation involving alkylidene cyclopropanes, expansion to other reactive functionality, such as acylsilanes would considerably expand its utility.
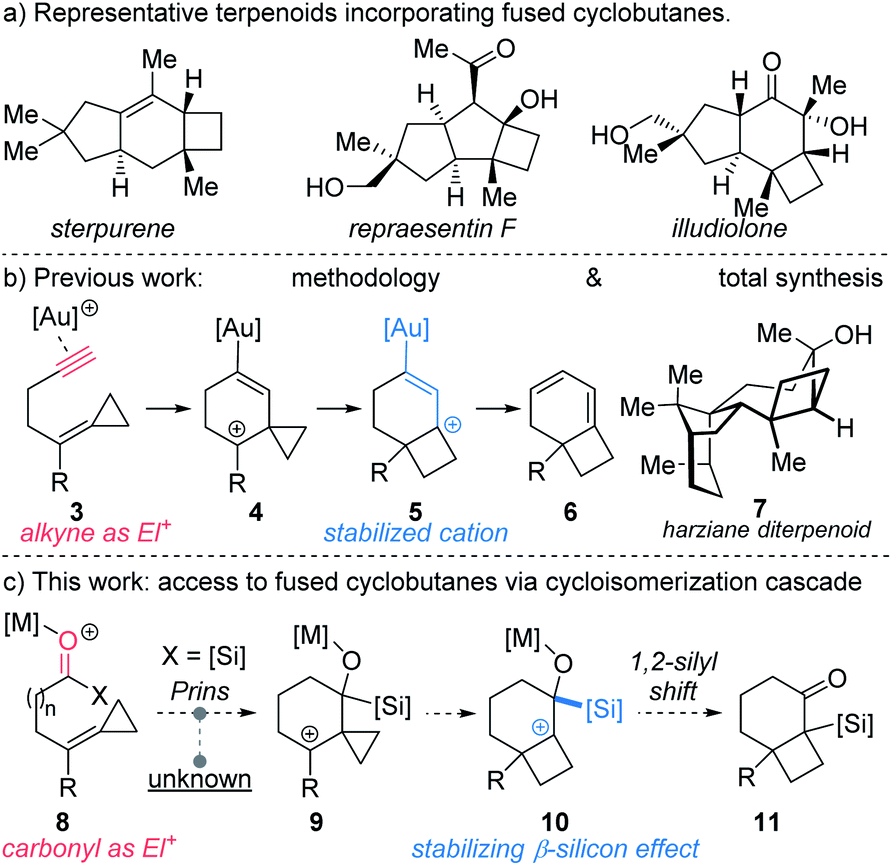 | ||
| Scheme 1 (a) Relevant natural products; (b) prior work; and (c) this work. | ||
Results and discussion
Analysis of the enyne isomerization over the course of the sequence of steps reveals that the alkyne and its derivatives display complementary reactivity patterns.12 Thus, in the conversion of 3 to 4 the coordinated alkyne is electrophilic; by contrast, the ensuing cation 5 enjoys stabilization by the electron-rich vinyl gold fragment (AuC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) derived from the alkyne. Accordingly, we became interested in identifying acetylene surrogates that would exhibit similar behavior and trigger analogous reaction cascades. Based on the pioneering work of Kuwajima,13 Reich,14 and, more recently, Johnson,15 we reasoned that acyl acylsilanes could serve as a suitable proxy for the alkyne in cycloisomerization reactions.16
C) derived from the alkyne. Accordingly, we became interested in identifying acetylene surrogates that would exhibit similar behavior and trigger analogous reaction cascades. Based on the pioneering work of Kuwajima,13 Reich,14 and, more recently, Johnson,15 we reasoned that acyl acylsilanes could serve as a suitable proxy for the alkyne in cycloisomerization reactions.16
Accordingly, we hypothesized that in the presence of a Lewis acid acylsilane 8 might trigger intramolecular Prins-type addition of alkylidenecyclopropane to generate cyclopropyl carbinyl cation 9 (Scheme 1c).17 Although addition of substituted vinylidene cyclopropanes to aldehydes have been reported,18 additions to acylsilanes are unprecedented. The formation of 9 might subsequently bring about ring expansion to form cyclobutyl cation 10,8,19 which following 1,2-silyl shift would lead to formation of α-silyl ketone 11.20
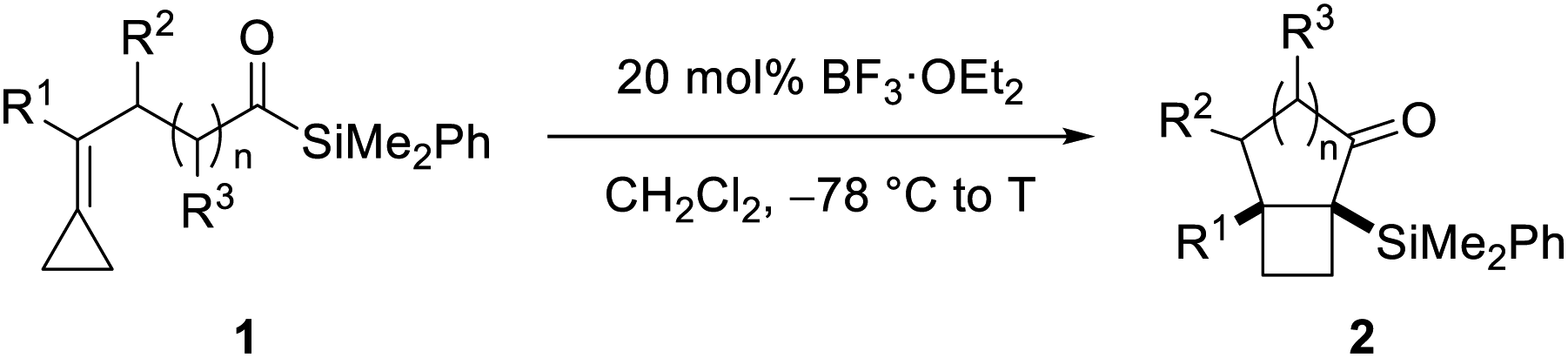
Our synthetic studies commenced with investigation of the cycloisomerization of acylsilane 1a containing a phenyl-substituted alkylidenecyclopropane (Scheme 2). This substrate was accessed from ketoamide 12 in two steps involving chemoselective olefination using Petasis' dicyclopropyl titanocene reagent,10 followed by addition of dimethylphenylsilyllithium.21 Treatment of acylsilane 1a with 20 mol% BF3·OEt2 in dichloromethane at −78 to −20 °C effected cycloisomerization and yielded bicyclo[3.2.0]heptane 2a in 83% yield and as a single diastereoisomer.22 We also examined other Lewis acids on related substrates, such as TMSOTf, EtAlCl2, In(OTf)3, but none gave the desired product. The relative configuration of ring fusion was initially, tentatively assigned to be cis based on the severe energetic penalty that would otherwise be associated with formation of the alternative trans fusion.
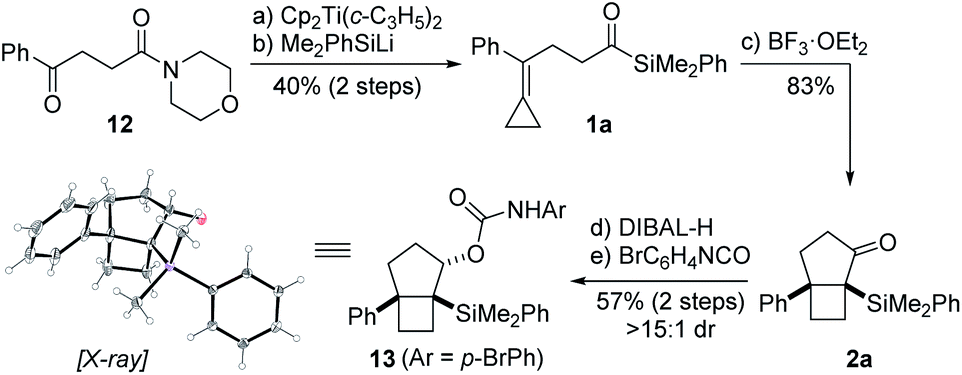 | ||
Scheme 2 Proof of concept. Reagents and conditions: (a) Cp2Ti(c-C3H5)2, PhMe, 50 °C; (b) Me2PhSiLi, THF, −78 °C, 40% (2 steps); (c) BF3·OEt2 (20 mol%), CH2Cl2, −78 to −20 °C, 83%; (d) DIBAL-H, CH2Cl2, −78 °C; (e) 4-bromophenyl isocyanate, Et3N, CH2Cl2, 37 °C, 57% (2 steps), >15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. The thermal ellipsoids in the X-ray structure of 13 are displayed at 50% probability level. Moreover, the carboxamide on the secondary alcohol was omitted for clarity. :1 dr. The thermal ellipsoids in the X-ray structure of 13 are displayed at 50% probability level. Moreover, the carboxamide on the secondary alcohol was omitted for clarity. | ||
This assignment was subsequently corroborated by X-ray crystallographic analysis of carbamate derivative 13, which was accessed from α-silyl ketone 2a by diastereoselective carbonyl reduction (DIBAL-H, >15:1 dr), followed by treatment of the resulting secondary alcohol with para-bromophenyl isocyanate. As a control experiment, it is noteworthy that when the corresponding aldehyde was used in lieu of the acyl silane, we did not observe product formation. Having demonstrated the viability of the reaction, we next investigated the scope of the novel cycloisomerization reaction (Table 1). The cycloisomerization proved suitable for the synthesis of fused 5–4 and 6–4 ring systems, as demonstrated by the formation of bicyclo[3.2.0]heptane 2a (83%) and bicyclo[4.2.0]octane 2b (84%). The transformation could furthermore be extended to substrates containing alkyl substituents (2c–2e). Among these, cyclopropyl-substituted cyclobutane 2e is an interesting case because it results from regioselective ring expansion of the spirocyclic cyclopropane ring of the putative intermediate cyclopropylcarbinyl cation 14. The observed preference for migration of a spirocyclic cyclopropane C–C bond may result from the greater stability of the ensuing tertiary cyclobutyl cation (15) over the alternative secondary cyclobutyl cation in spirocycle 16.
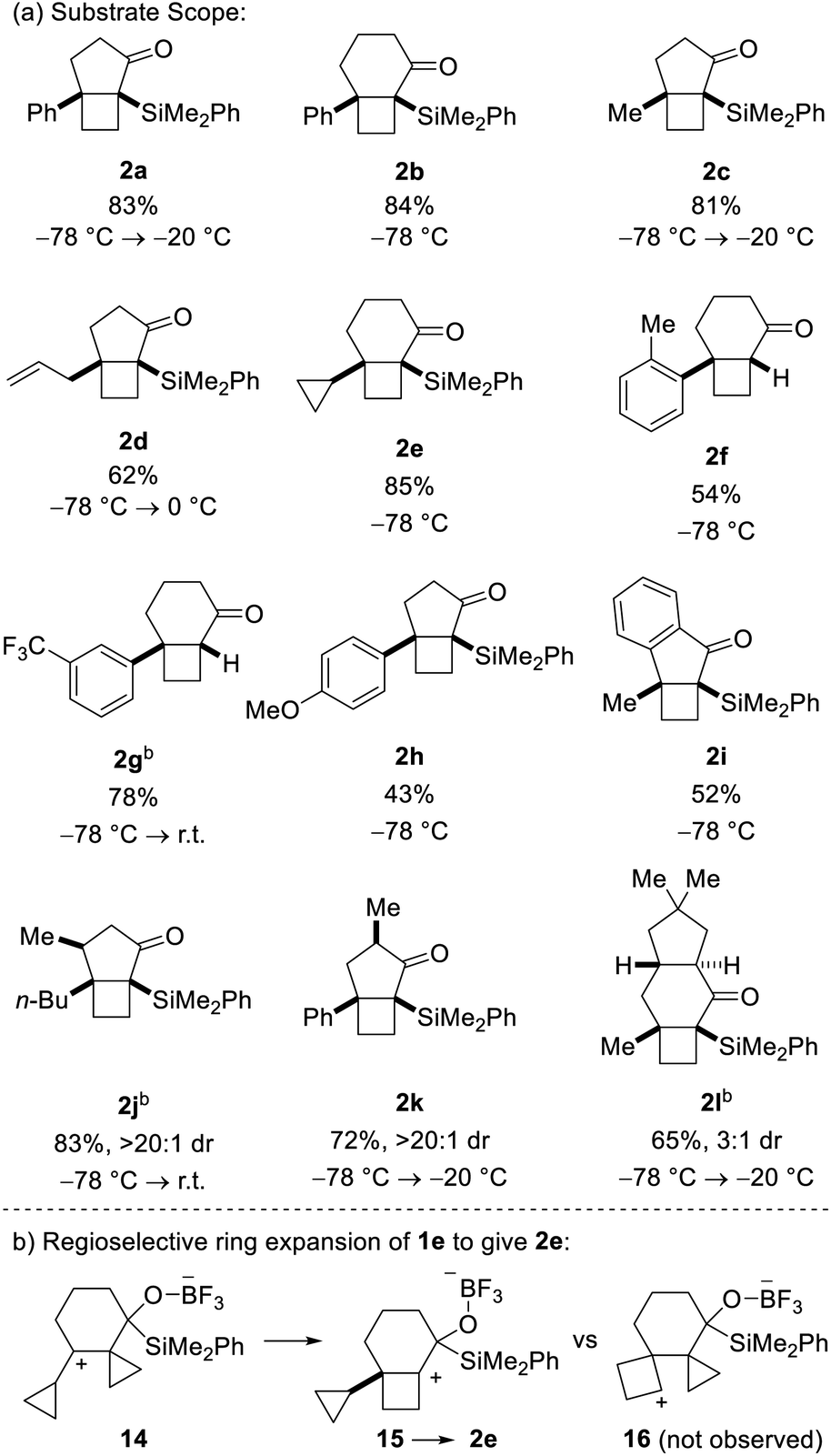
The substrate scope of the novel cycloisomerization reaction was found to extend to substrates incorporating aryl substituents bearing electron-donating and electron-withdrawing groups (2f–2h). Interestingly, cycloisomerization of arenes with ortho- or meta-substitution was accompanied by product lacking the silyl group (cf.2f and 2g). For these two cases, we hypothesize that the additional steric encumbrance exerted by the substituted arenes results in competing desilylation of intermediate 10 (Scheme 1) thereby producing directly an enolate, which is protonated upon work-up. Intriguingly, incorporation of various substitution patterns along the alkyl chain linking the alkylidene cyclopropanes and acyl silane led to formation of a range of substituted polycyclic products 2i–2l in moderate to high yield. It is notable that the cyclization takes place in a highly diastereoselective manner. Accordingly, incorporation of a methyl group at the allylic position or at Cα in the acylsilane led to highly diastereoselective formation of the all-cis isomers of bicyclo[3.2.0]heptanes 2j and 2k in >20:1 dr. Furthermore, incorporation of a dimethyl-substituted cyclopentane ring in cyclization precursor 1l led to generation of the corresponding tricyclic product 2l in 65% yield and 3:1 dr. Notably, 2l incorporates the characteristic carbon skeleton of sterpurane derived natural products, which underscores the significance of the novel cycloisomerization reaction to access complex terpenoids.
The α-silyl ketone functionality present in the cycloisomerization products provide a suitable handle for further functionalization, as demonstrated for bicyclo[4.2.0]octane 2b as a representative example (Scheme 3). For instance, protodesilylation of 2b was effected in high yield upon exposure to potassium fluoride in methanol at room temperature to give bicyclo[4.2.0]octane 17. Alternatively, heating of 2b in toluene at 90 °C promoted rearrangement to the isomeric silyl enol ether 18 in high yield.23 This silyl enol ether, featuring a strained alkylidenecyclobutane double bond, would be difficult to generate by regioselective enol ether formation of the parent ketone. Marek has recently demonstrated the ability of related tetrasubstituted phenyldimethylsilyl enol ethers to engage in aldol addition reactions for the formation of quaternary stereocenters.24 Consequently, this corroborates the synthetic utility associated with phenyldimethylsilyl enol ethers such as 18 and, by extension, their corresponding α-silyl ketones 2. The exclusive formation of α-silyl ketone products (2) from Lewis acid-induced cycloisomerization of acylsilanes 1 likely reflects kinetic preference for 1,2-silyl shift of the intermediate cyclobutyl cation 10 over competing Brook rearrangement to furnish silyl enol ether isomer.
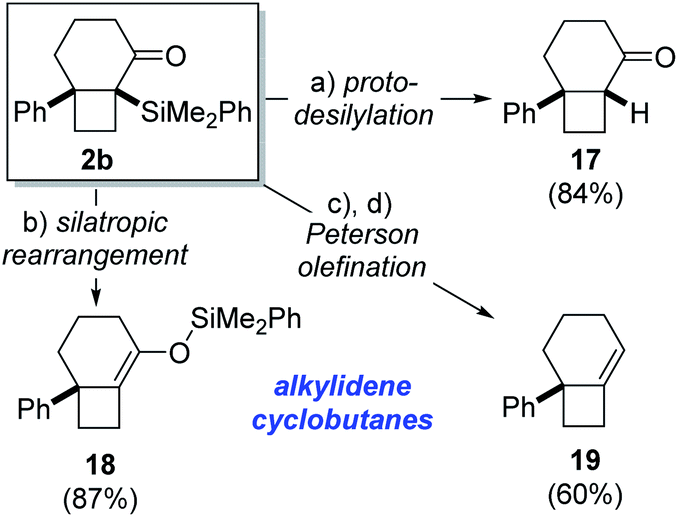 | ||
| Scheme 3 Product functionalization. Reagents and conditions: (a) KF, MeOH, r.t., 84%; (b) PhMe, 90 °C, 87%; (c) DIBAL-H, CH2Cl2, −78 °C; (d) BF3·OEt2, CH2Cl2, 0 °C, 60% (2 steps). | ||
The synthetic utility of the α-silyl ketone motif in the cycloisomerization product 2c is further demonstrated by a reduction/Peterson olefination25 sequence (Scheme 3). Accordingly, treatment of 2b with DIBAL-H led to formation of a diastereomeric mixture of secondary alcohols (dr 3:1), which underwent highly regioselective elimination to generate alkylidenecyclobutane 19 in 60% yield over 2 steps. It is notable that both diastereomeric alcohols proved competent substrates for elimination26 and did not lead to formation of regioisomeric olefin products. This demonstrates the strategic advantage associated with the novel cycloisomerization reaction in providing a handle for installation of strained alkylidenecyclobutane double bonds. Additionally, the ability to access cyclobutanes with exocyclic double bonds renders this method complementary to Fürstner's platinum-catalyzed isomerization of alkylidenecyclopropanes, which generates cyclobutenes containing endocyclic double bonds.7
Reflection on the transformation described prompts interesting questions concerning the mechanism. Rearrangement pathways for two limiting conformations that highlight the relationship between the C–Si and the cyclopropyl C–C are outlined in Scheme 4: C–Si and C–Cβanti-periplanar (20) along with C–Si and C–Cβ roughly syn-periplanar (20′). The former is expected to furnish the observed products (2) while products resulting from the latter are not observed because of poor alignment of C–Si relative to the migrating Cα cyclopropane carbon. To further probe the origin of the selectivity for the syn isomer, we performed a computational analysis.27,28
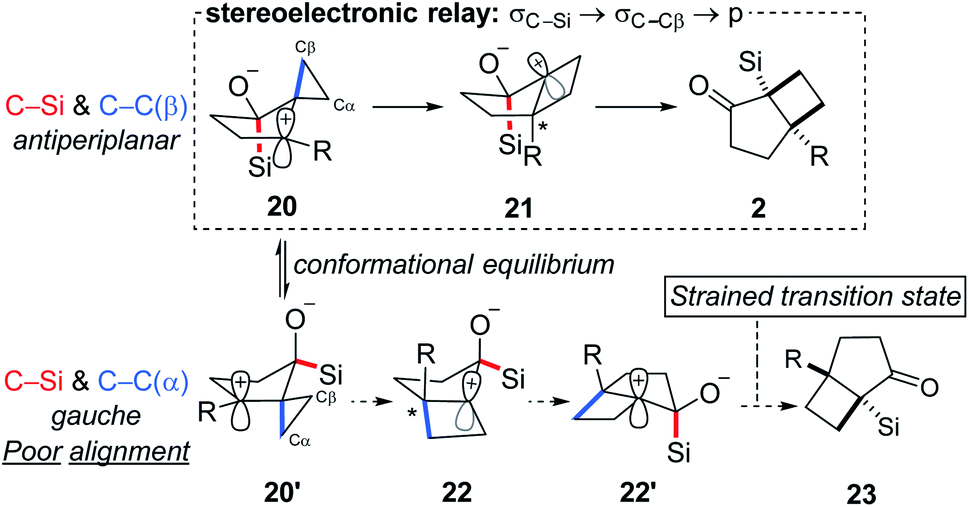 | ||
| Scheme 4 Stereochemical course of the cyclization reaction. | ||
Although 20′ was calculated to be favored thermodynamically by ∼1 kcal mol−1, 20 undergoes a facile migration with no meaningful energy barrier, leading to 21. Conversely, 20′ is unable to undergo an analogous migration, and attempts to obtain structure 22 as a local minimum failed, indicating that not only is the rearrangement disfavored, but the intermediate itself is not a stable compound. Analysis of the geometries of the conformers reveal the underlying reasons for this difference.
The calculated optimized geometry of 20′ shows the cyclopropyl bisected by the plane of the cyclopentane (ii in Scheme 5a). This allows for a stabilizing interaction between the Walsh-like cyclopropane orbital and the neighbouring carbocation. In contrast, the cyclopropyl ring in 20 is forced “upwards” from the plane of the cyclopentane ring, in the direction opposite the pseudo-axial silicon (Scheme 5a structure i). This distortion is likely triggered by the destabilizing out-of-phase interaction between the cyclopropyl Walsh-like orbital and the σC–Si orbital. This results in the orbital becoming more localized and positioned almost parallel to the empty p orbital (Scheme 5b20), allowing for facile migration of Cβ to the neighboring carbon. Importantly, because the cyclopropyl group is distorted as shown in 20, it is always Cβ that migrates, leading to the selectivity for the syn positioning of the R and Si groups. An analogous interaction with the σC–O orbital was not identified in 20′. Following the migration of Cβ in 20, the positive charge is located β to the Si group, leading to a rapid 1,2-silyl shift and resulting in the formation of the observed cis-fused product 2, which is found to be 29.5 kcal mol−1 lower in energy than 21. Additional calculations and visualizations of the molecular orbitals supporting this analysis are provided in the ESI.†
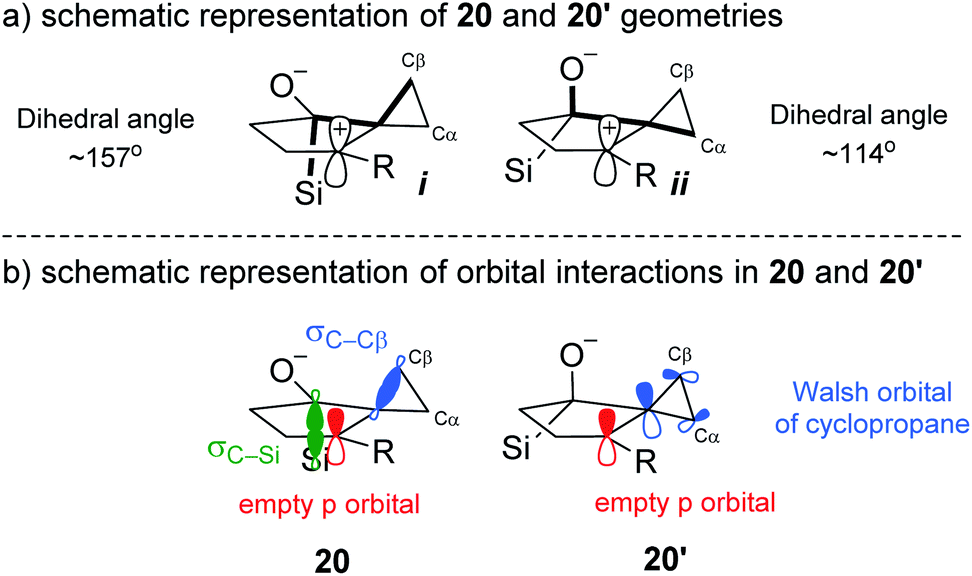 | ||
| Scheme 5 Geometrical and orbital analysis of the stereoelectronic relay based on computational results. | ||
Conclusions
We developed a highly diastereoselective ring-expanding cycloisomerization reaction of alkylidenecyclopropane acylsilanes to bicyclic α-silyl ketones. The products feature cyclobutane rings containing a quaternary stereocenter, which are challenging motifs for synthesis. The α-silyl ketone functionality present in the cyclization products provides a suitable handle for further functionalization of the cyclobutane ring. Notably, all cyclization precursors were synthesized in two steps from simple acyclic ketoamide precursors. More broadly, the cationic annulation of vinylidine cyclopropanes have now been expanded to include acylsilanes as reaction partners. Applications of this method in the context of total syntheses are currently ongoing and will be reported in due course.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. H. acknowledges a fellowship of the Stipendienfonds Schweizerische Chemische Industrie (SSCI) and the Swiss National Science Foundation (SNF 200020172516). RGP acknowledges the generous support of the Branco Weiss Fellowship and is grateful to Eno Paenurk for insightful discussions.Notes and references
- T. C. McMorris, A. Kashinatham, R. Lira, H. Rundgren, P. K. Gantzel, M. J. Kelner and R. Dawe, Phytochemistry, 2002, 61, 395 CrossRef CAS ; for a review on protoilludanes and related sesquiterpenoids, see: P. Siengalewicz, J. Mulzer and U. Rinner, Eur. J. Org. Chem., 2011, 7041 CrossRef.
- See, for example: R. Vorberg, N. Trapp, E. M. Carreira and K. Müller, Chem.–Eur. J., 2017, 23, 3126 CrossRef CAS PubMed , and references therein.
- For reviews on the synthesis of cyclobutanes, see: E. Lee-Ruff, Synthesis of Cyclobutanes, in PATAI's Chemistry of Functional Groups, ed. Z. Rappoport, John Wiley & Sons, Ltd., 2009 Search PubMed.
- Examples include: (a) T. Matsumoto, K. Miyano, S. Kagawa, S. Yu, J. Ogawa and A. Ichihara, Tetrahedron Lett., 1971, 12, 3521 CrossRef; (b) S. Kagawa, S. Matsumoto, S. Nishida, S. Yu, J. Morita, A. Ichihara, H. Shiraham and T. Matsumoto, Tetrahedron Lett., 1969, 10, 3913 CrossRef; (c) H. Takeshita, H. Iwabuchi, I. Kouno, M. Iino and D. Nomura, Chem. Lett., 1979, 649 CrossRef CAS; (d) H. Takeshita, I. Kouno, M. Iino, H. Iwabuchi and D. Nomura, Bull. Chem. Soc. Jpn., 1980, 53, 3641 CrossRef CAS; (e) J. Furukawa, N. Morisaki, H. Kobayashi, S. Iwasaki, S. Nozoe and S. Okuda, Chem. Pharm. Bull., 1985, 33, 440 CrossRef CAS; (f) E. J. Corey, H. Uda and R. B. Mitra, J. Am. Chem. Soc., 1963, 85, 362 CrossRef CAS; (g) E. J. Corey, R. B. Mitra and H. Uda, J. Am. Chem. Soc., 1964, 86, 485 CrossRef CAS; (h) G. Mehta, P. Ghosh and K. Sreenivas, ARKIVOC, 2003, 176 Search PubMed; for a related example of a [2 + 2] photocycloaddition towards a protoilludane sesquiterpene, see (i) A. Zech, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 14629 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Kögl, L. Brecker, R. Warrass and J. Mulzer, Angew. Chem., Int. Ed., 2007, 46, 9320 CrossRef PubMed; (b) M. Kögl, L. Brecker, R. Warrass and J. Mulzer, Eur. J. Org. Chem., 2008, 2714 CrossRef; for a related example, see: (c) E. P. Johnson and K. P. C. Vollhardt, J. Am. Chem. Soc., 1991, 113, 381 CrossRef CAS.
- For selected examples, see: (a) T. Fujita, T. Ohtsuka, H. Shirahama and T. Matsumoto, Tetrahedron Lett., 1982, 23, 4091 CrossRef CAS; (b) E. Jiménez-Núñez, C. K. Claverie, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5452 CrossRef PubMed; (c) A. Pitaval, D. Leboeuf, J. Ceccon and A. M. Echavarren, Org. Lett., 2013, 15, 4580 CrossRef CAS PubMed; (d) S. G. Sethofen, S. T. Staben, O. Y. Hung and F. D. Toste, Org. Lett., 2008, 10, 4315 CrossRef PubMed; (e) G. Li, X. Huang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 6944 CrossRef CAS PubMed; (f) H. Zheng, R. J. Felix and M. R. Gagné, Org. Lett., 2014, 16, 2272 CrossRef CAS; (g) S. Ferrer and A. M. Echavarren, Org. Lett., 2018, 20, 5784 CrossRef CAS PubMed.
- L. Zhang and M. Koreeda, Org. Lett., 2002, 4, 3755 CrossRef CAS PubMed.
- A. Fürstner and C. Aïssa, J. Am. Chem. Soc., 2006, 128, 6306 CrossRef CAS PubMed; A. Masarwa, A. Fürstner and I. Marek, Chem. Commun., 2009, 5760 RSC ; ref. 4 and references cited therein.
- T. Matsuda, M. Makino and M. Murakami, Angew. Chem., Int. Ed., 2005, 44, 4608 CrossRef CAS PubMed.
- N. A. Petasis and E. I. Bzowej, Tetrahedron Lett., 1993, 34, 943 CrossRef CAS.
- M. Hönig and E. M. Carreira, Angew. Chem., Int. Ed., 2020, 59, 1192 CrossRef PubMed.
- For a discussion of transformations, which invert the Lewis acid–base properties of a given carbon atom, see: D. A. Evans and G. C. Andrews, Acc. Chem. Res., 1974, 7, 147 CrossRef CAS.
- I. Kuwajima and M. Kato, Tetrahedron Lett., 1980, 21, 623 CrossRef CAS.
- H. J. Reich, E. K. Eisenhart, R. E. Olson and M. J. Kelly, J. Am. Chem. Soc., 1986, 108, 7791 CrossRef CAS PubMed.
- D. A. Nicewicz and J. S. Johnson, J. Am. Chem. Soc., 2005, 127, 6170 CrossRef CAS PubMed.
- For selected reviews on acylsilanes, see: (a) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540 RSC; (b) P. F. Cirillo and J. S. Panek, Org. Prep. Proced. Int., 1992, 24, 553 CrossRef CAS; (c) P. C. B. Page, S. S. Klair and S. Rosenthal, Chem. Soc. Rev., 1990, 19, 147 RSC; (d) A. Ricci and A. Degl'Innocenti, Synthesis, 1989, 9, 647 CrossRef. For a selected example, see: (e) T. Takatsu, S. Ito, T. Kan, H. Shirahama and S. Matsumoto, Synlett, 1989, 1, 40 Search PubMed.
- G. A. Olah, V. P. Reddy and G. K. S. Prakash, Chem. Rev., 1992, 92, 69 CrossRef CAS.
- K. Miura, M. Takasumi, T. Hondo, H. Saito and A. Hosomi, Tetrahedron Lett., 1997, 38, 4587 CrossRef CAS.
- For related examples of intermediate generation of cyclopropylcarbinyl cations undergoing ring expansion to form cyclobutane products, see: (a) S. G. Sethofer, S. T. Staben, O. Y. Hung and F. D. Toste, Org. Lett., 2008, 10, 4315 CrossRef CAS; (b) E. Jiménez-Núñez, C. K. Claverie, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5452 CrossRef PubMed; (c) J. P. Markham, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 9708 CrossRef CAS PubMed; (d) G. Li, X. Huang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 6944 CrossRef CAS PubMed; (e) F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178 CrossRef CAS PubMed.
- For a related example of a 1,2-silyl shift, see: R. L. Danheiser and D. M. Fink, Tetrahedron Lett., 1985, 26, 2513 CrossRef CAS.
- (a) C. T. Clark, B. C. Milgram and K. A. Scheidt, Org. Lett., 2004, 6, 3977 CrossRef CAS PubMed; (b) I. Fleming and U. Ghosh, J. Chem. Soc., Perkin Trans. 1, 1994, 3, 257 RSC.
- Treatment of related cycloisomerization precursors containing an aldehyde or ketone as acceptor instead of the acylsilane under identical reaction conditions did not lead to formation of bicyclic products arising from ring expansion of the cyclopropane (see ESI†). This demonstrates the importance of the acylsilane motif in enabling productive cyclization and ring expansion.
- A. G. Brook, D. M. MacRae and W. W. Limburg, J. Am. Chem. Soc., 1967, 89, 5493 CrossRef CAS.
- P.-Y. Wang, G. Duret and I. Marek, Angew. Chem., Int. Ed., 2019, 58, 14995 CrossRef CAS PubMed.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780 CrossRef CAS.
- The fact that both diastereomeric alcohols generated by treatment of 2b with DIBAL afford olefin 19 upon treatment with acid (Scheme 3) suggests an E1 mechanism. The mechanism for formation of olefins from vicinal silyl alcohols under basic conditions also appears to be proceed via multiple mechanistic pathways, see J.-N. Heo, E. B. Holson and W. R. Roush, Org. Lett., 2003, 5, 1697 CrossRef CAS.
- All structures were optimized with the M06-2X functional in combination with the def2-TZVP basis set, using the Gaussian suite of programs. Grimme's D3 dispersion correction and an implicit solvent model for CH2Cl2 were both employed. Frequencies analyses were performed to ensure true minima of all obtained species.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC Deposition number 1981477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02224a |
| This journal is © The Royal Society of Chemistry 2020 |