
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formicamycin biosynthesis involves a unique reductive ring contraction†
Zhiwei
Qin‡
 a,
Rebecca
Devine‡
b,
Thomas J.
Booth
a,
Elliot H. E.
Farrar
c,
Matthew N.
Grayson
c,
Matthew I.
Hutchings
*ab and
Barrie
Wilkinson
*a
a,
Rebecca
Devine‡
b,
Thomas J.
Booth
a,
Elliot H. E.
Farrar
c,
Matthew N.
Grayson
c,
Matthew I.
Hutchings
*ab and
Barrie
Wilkinson
*a
aDepartment of Molecular Microbiology, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK. E-mail: barrie.wilkinson@jic.ac.uk
bSchool of Biological Sciences, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: matt.hutchings@jic.ac.uk
cDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 16th June 2020
Abstract
Fasamycin natural products are biosynthetic precursors of the formicamycins. Both groups of compounds are polyketide natural products that exhibit potent antibacterial activity despite displaying different three-dimensional topologies. We show here that transformation of fasamycin into formicamycin metabolites requires two gene products and occurs via a novel two-step ring expansion-ring contraction pathway. Deletion of forX, encoding a flavin dependent monooxygenase, abolished formicamycin production and leads to accumulation of fasamycin E. Deletion of the adjacent gene forY, encoding a flavin dependent oxidoreductase, also abolished formicamycin biosynthesis and led to the accumulation of new lactone metabolites that represent Baeyer–Villiger oxidation products of the fasamycins. These results identify ForX as a Baeyer–Villiger monooxygenase capable of dearomatizing ring C of the fasamycins. Through in vivo cross feeding and biomimetic semi-synthesis experiments we showed that these lactone products represent biosynthetic intermediates that are reduced to formicamycins in a unique reductive ring contraction reaction catalyzed by ForY.
Introduction
Bacterial aromatic polyketide natural products comprise an extensive family of biosynthetically related molecules that encompass great structural variety and often display potent biological activity.1 They include several important clinical agents, for example the tetracycline antibiotics and anthracycline antineoplastic drugs. The carbon skeletons of aromatic polyketides are invariably derived from the function of dissociated type II polyketide synthases (PKSs), and additional structural diversity is introduced by tailoring enzymes following polyketide biosynthesis.2,3 Such post-PKS transformations include acylation, alkylation, glycosylation and halogenation, as well as redox changes. Further structural complexity can be introduced via carbon–carbon bond cleavage and rearrangements in processes commonly catalyzed by oxidative enzymes. Illustrative examples include the reactions catalyzed by flavin-dependent Baeyer–Villiger (BV) monooxygenase enzymes4 during the biosynthesis of the jadomycins,5 gilvocarcins5 and chartreusins6 amongst others.We recently described the formicamycins, poly-halogenated tridecaketides that are co-products, along with the fasamycins, of the Streptomyces formicae KY5 for biosynthetic gene cluster (BGC; Fig. 1 and S1†) encoding a type II PKS.7 The fasamycins exhibit potent antibacterial activity and congeners were reported from heterologous expression of environmental DNA and were encoded by a clone expressing a type II PKS BGC, but no co-produced formicamycins were reported.8 Despite their common biosynthetic origin and carbon backbone connectivity, the fasamycins and formicamycins have very different three-dimensional structures as determined from NOESY NMR, ECD spectroscopy and computational modelling.7 Moreover, the formicamycins displayed more potent antibacterial activity and a barrier for the selection of resistant isolates.
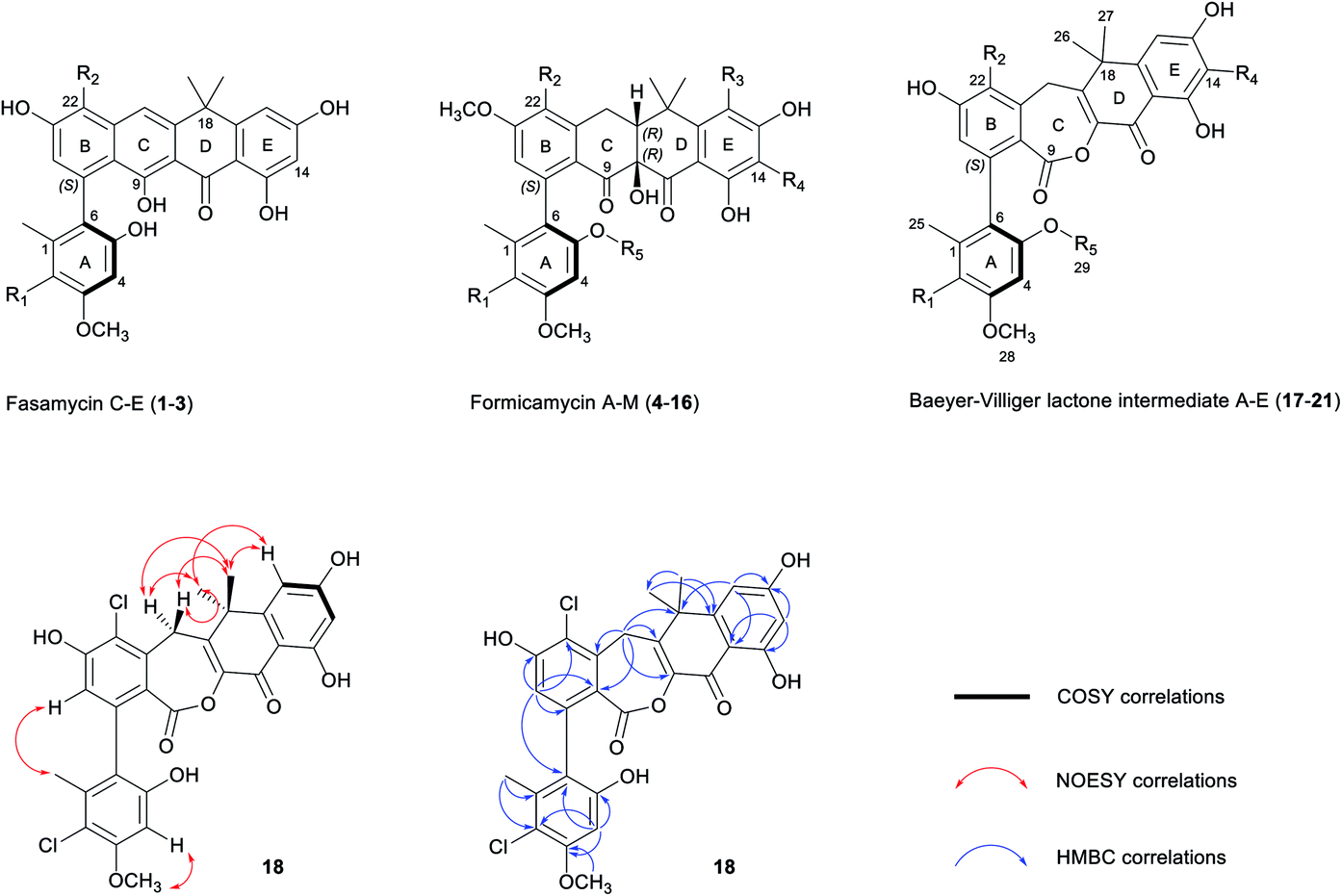 | ||
| Fig. 1 Chemical skeletons of fasamycins (1–3) and formicamycins (4–16) isolated from S. formicae KY5, and Baeyer–Villiger lactone intermediates (17–21) isolated from the S. formicae ΔforY mutant in this study (for detailed substituent variations including halogenation or O-methylation, see Fig. S1†). The 2D NMR structure determination of 18 is shown by COSY (bold), NOESY (red double-head arrow), and selected HMBC (blue single-head arrow) correlations respectively. R1–4 = H or Cl; R5 = H or CH3. | ||
Mutational analysis of the formicamycin (for) BGC identified a single flavin dependent halogenase (ForV) that can presumably catalyze multiple halogenation events.7 Deletion of forV abolished the production of all halogenated congeners and the ΔforV mutant produced only a single compound fasamycin C 1 but no formicamycins (Fig. 2b); the deletion could be complemented by ectopic expression of forV.7 This suggests a gatekeeper function for ForV, meaning the enzymes which convert fasamycins to formicamycins operate on halogenated intermediates, confirming that the fasamycins are biosynthetic precursors of the formicamycins. Structural comparison of the two skeletons indicates that unusual biosynthetic transformations are required to enable their interconversion, involving a dearomatization of fasamycin ring C via a formal tautomerization and two-electron reduction, plus the introduction of a tertiary hydroxyl group at C10. This suggests the requirement for a reductive enzyme in addition to a monooxygenase. These intriguing biosynthetic observations, in concert with their antibacterial properties, prompted us to study the biosynthetic steps linking the two compound families.
 | ||
| Fig. 2 Mutational analysis of the tailoring enzyme encoding genes involved in formicamycin biosynthesis. Deletion of forX abolishes the production of formicamycins but leads to accumulation of fasamycin E (3). Deletion of forY abolishes the production of formicamycins and leads to accumulation of six biosynthetic intermediates (17–22). Reconstituted HPLC traces (UV = 285 nm) showing: (a) S. formicae wild type; (b) S. formicae ΔforV (for details see ref. 7); (c) S. formicae ΔforX; (d) S. formicae ΔforY. | ||
Results
Bioinformatics analysis
The for BGC was sequenced previously7 and comparison to the originally reported fasamycin BGC8 did not identify any additional genes obviously responsible for formicamycin biosynthesis. However, comparison to a for-like BGC identified in a 140 kbp region of the Streptomyces kanamyceticus genome that is syntenic with S. formicae KY5 (Genbank ID LIQU00000000.1), identified a set of four grouped genes forX–forAA which are not present in S. kanamyceticus (Fig. 3). A similar observation was made when we compared the for BGC to the recently reported BGC encoding production of accramycin A, a new fasamycin congener.9 Three of these gene products (ForY, ForZ and ForAA) show high homology to sequences from Actinomadura species, suggesting they may have been acquired via horizontal gene transfer. The gene product ForX is a putative flavin-dependent monooxygenase, and both sequence homology and predicted structural homology10 searches indicated the most closely related characterized homologue is aklavinone 12-hydroxylase (RdmE) (Table 1), a Group A flavin-dependent monooxygenase.11,12 Group A monooxygenases are reported to catalyze hydroxylation or sulfoxidation. Sequence alignments of ForX and RdmE showed an overall identity of 41%, with three noticeable regions of low homology between residues 70–119, 212–245 and 365–393 in the RdmE structure (31HG), corresponding to the substrate binding pocket. Structural homology searches predicted ForY to be a TIM beta/alpha barrel oxidoreductase (Table 1), homologues of which typically utilize flavin cofactors to perform redox chemistry.13–17 The genes forZ and forAA encode a MarR family transcriptional regulator and an MFS family transporter respectively.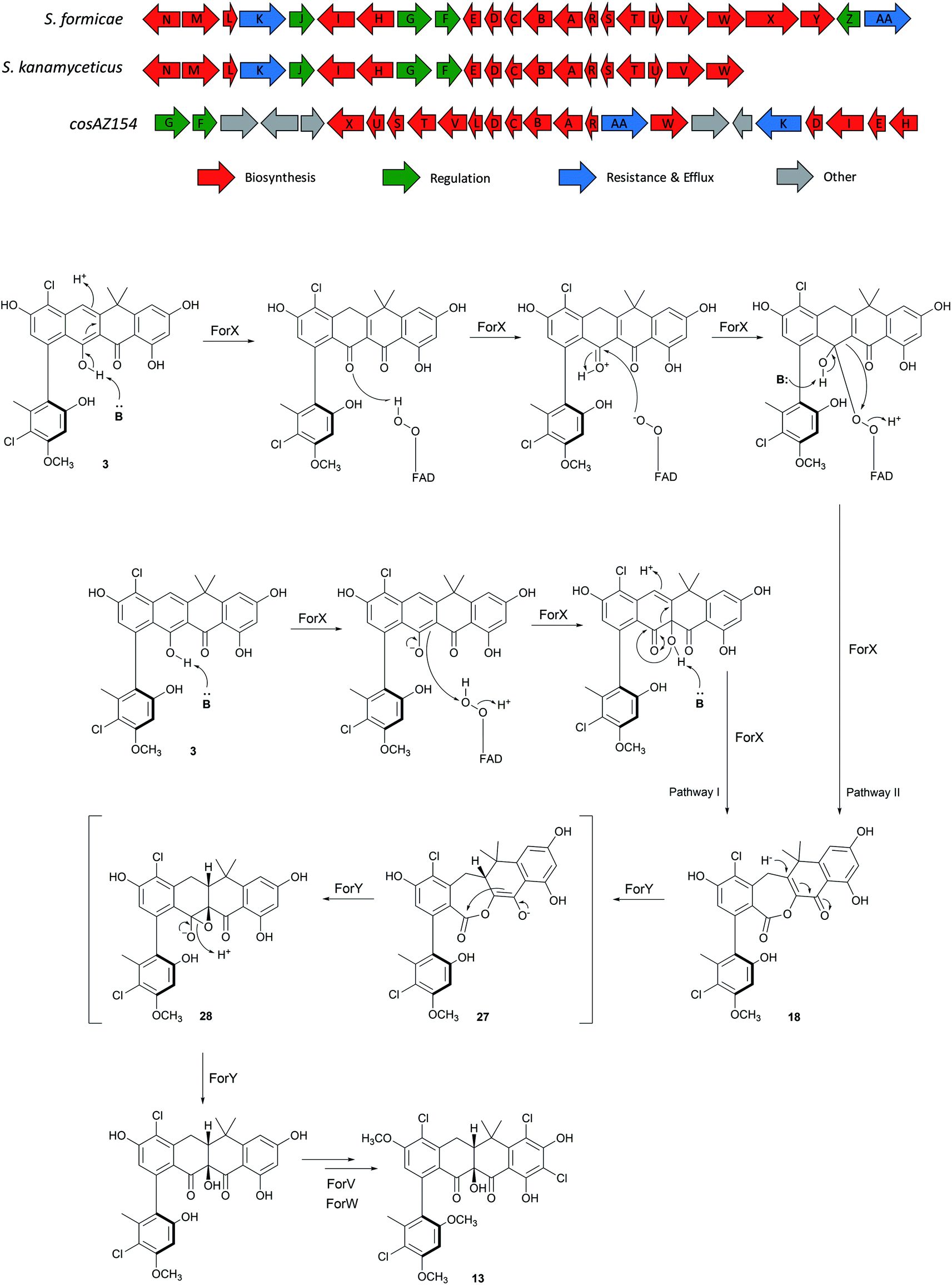 | ||
| Fig. 3 Comparison of the fasamycin/formicamycin biosynthetic gene clusters (top) and the alternate pathways for each step of the proposed two-step ring expansion-contraction mechanism of formicamycin biosynthesis from fasamycin E (3) (bottom). | ||
| Name | Accession number | Top hit | % coverage | % identity |
|---|---|---|---|---|
| ForX | WP_049717083.1 | Hypothetical protein, Streptomyces caatingaensis | 94 | 50 |
| ForY | WP_089326038.1 | LLM class flavin-dependent oxidoreductase, Actinomadura meyerae | 87 | 60 |
| ForZ | WP_121435158.1 | MarR transcriptional regulator, Actinomadura pelletieri | 84 | 42 |
| ForAA | WP_122199503.1 | DHA2 family efflux MFS transporter permease subunit, Actinomadura sp. NEAU-Ht49 | 96 | 57 |
Mutational analysis identifies a two-step biosynthetic transformation
Guided by our bioinformatic observations we made individual in-frame deletions in forX and forY using Cas9 mediated gene editing. The resulting mutants failed to produce formicamycins, but this was restored after ectopic expression of the deleted genes (Fig. S2†). Despite producing no formicamycins, S. formicae ΔforX accumulated the previously isolated fasamycin E (3) (Fig. 2c) which was confirmed by isolation and NMR characterization (Fig. S3 and S4†). This is consistent with the hypothesis that ForX catalyzes an oxidation required for formicamycin biosynthesis, and that it functions on halogenated fasamycin substrates. In contrast, S. formicae ΔforY accumulated six new compounds 17–22 (Fig. 2d). After scaling up fermentation (4 L) and solvent extraction, a small amount of each of the congeners 17–21 (5–13 mg) was isolated and subjected to structure determination using 2D NMR and MS. Compound 22 could not be isolated but analytical MS and UV data were consistent with it being a related biosynthetic congener. We first determined the structure of 18 because high-resolution ESI-MS predicted a molecular formula of C28H22O8Cl2 indicating one more oxygen atom compared with 3, but with a distinct UV spectrum (λmax 248 and 300 nm vs. 249, 289, 355, and 415 nm for 3) indicating a major structural change. 1D and 2D NMR experiments showed a striking low- to high-field chemical shift for the carbonyl group (C11) in comparison to 3, in addition to the appearance of new methylene and lactone carbonyl moieties. Based on these observations, and other connectivity, we assigned the structure of 18 as a Baeyer–Villiger lactone derivative of 3 (Fig. 1). Further analysis identified 19 as the C5-O-methyl congener of 18, and 17 as lacking a chlorine atom at C2 when compared to 18. The remaining two congeners 20 and 21 were determined to contain further variations in chlorination and O-methylation (Fig. S1†).These results are consistent with a two-step ring expansion-ring contraction pathway for the conversion of fasamycins into formicamycins with ForX catalyzing a Baeyer–Villiger reaction on fasamycin substrates, and ForY catalyzing the two-electron reduction of the Baeyer–Villiger intermediates with concomitant ring contraction via hydride addition at C19. To provide support for our hypothesis we attempted to express and isolate both proteins for biochemical analysis but, despite extensive experimentation, we were unable to obtain soluble protein. Undeterred, we instead utilized biomimetic chemical synthesis to investigate the biosynthetic pathway (see below), but first verified the intermediacy of the isolated lactones through a cross feeding experiment. Exogenous 18 (isolated from S. formicae ΔforY) was added to growing cultures of the S. formicae ΔforX biosynthetic mutant and after nine days incubation LCMS analysis of an ethyl acetate extract of the culture broth indicated that 18 was converted efficiently into the previously isolated formicamycin congeners 5, 8, 9, and 13 plus the O-methylated lactone 19 (Fig. S5†). This is consistent with 18 being a direct biosynthetic intermediate of the formicamycins and a putative substrate of ForY.
Biomimetic semi-synthesis of the formicamycins
To mimic the ForY catalyzed biosynthetic transformation we subjected an analytical sample (∼0.1 mg) of the lactone 19 to sodium borohydride reduction (Fig. 4a). This yielded three new products with a mass increase of 2 Da, plus an additional compound with a mass decrease of 14 Da (Fig. 4b). Notably, and in contrast to 19, the products 23 and 24 (0.97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) displayed formicamycin like UV spectra (Fig. 4c). The reaction was then scaled up (using 4 mg of 19) and the products 23 (∼0.4 mg) and 24 (∼0.2 mg) were purified by HPLC. 2D NMR experiments indicated that these both contained the formicamycin ring structure, consistent with the proposed reduction and ring contraction mechanism (Fig. S59†). Chemical shift comparisons to biosynthetic congeners, in addition to 1D NOE data, confirmed that 23 corresponds to the natural (10RS,19RS) diastereoisomer and 24 to the (10RS,19SR) diastereoisomer. The product 25 (∼0.4 mg) was isolated with 24 (1:1 mixture) and NMR analysis clearly showed 25 to be a lactone in which the C10–C19 double bond was reduced but for which rearrangement via ring contraction had not occurred. NMR analysis then showed that 25 corresponds to a single diastereoisomer and the NOESY spectrum showed a strong correlation between H-10 and H-19 indicating the (10RS,19RS) configuration. The NMR and MS data for the remaining −14 Da component is consistent with deoxygenation at C11 as shown for 26 (Fig. 4). The biomimetic reaction was then repeated using lactone congener 17 (5.4 mg) as the starting material and analysis of the products showed an almost identical outcome to that for sodium borohydride reduction of 19 (Fig. S6†).
:1 ratio) displayed formicamycin like UV spectra (Fig. 4c). The reaction was then scaled up (using 4 mg of 19) and the products 23 (∼0.4 mg) and 24 (∼0.2 mg) were purified by HPLC. 2D NMR experiments indicated that these both contained the formicamycin ring structure, consistent with the proposed reduction and ring contraction mechanism (Fig. S59†). Chemical shift comparisons to biosynthetic congeners, in addition to 1D NOE data, confirmed that 23 corresponds to the natural (10RS,19RS) diastereoisomer and 24 to the (10RS,19SR) diastereoisomer. The product 25 (∼0.4 mg) was isolated with 24 (1:1 mixture) and NMR analysis clearly showed 25 to be a lactone in which the C10–C19 double bond was reduced but for which rearrangement via ring contraction had not occurred. NMR analysis then showed that 25 corresponds to a single diastereoisomer and the NOESY spectrum showed a strong correlation between H-10 and H-19 indicating the (10RS,19RS) configuration. The NMR and MS data for the remaining −14 Da component is consistent with deoxygenation at C11 as shown for 26 (Fig. 4). The biomimetic reaction was then repeated using lactone congener 17 (5.4 mg) as the starting material and analysis of the products showed an almost identical outcome to that for sodium borohydride reduction of 19 (Fig. S6†).
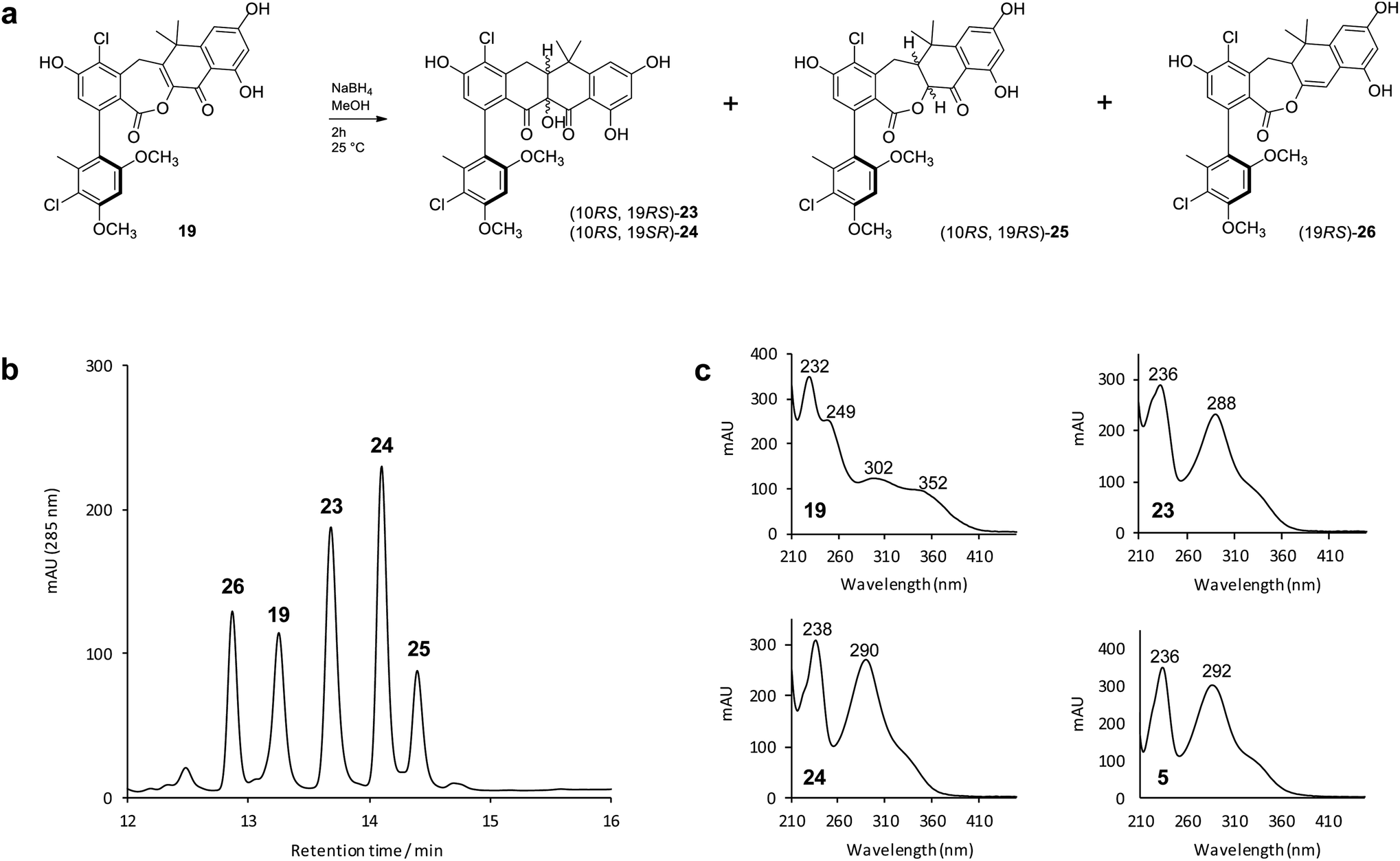 | ||
| Fig. 4 Biomimetic reduction of the biosynthetic intermediate 19 and isolated products. (b) HPLC-UV (285 nm) trace of the initial reaction products from (a); (c) UV spectra of the reaction substrate (19), two main products of interest (23 and 24), plus formicamycin B (5) as a standard for comparison. | ||
To better understand their stereochemical composition, we determined the [α]D20 values of 23 and 24 to be +68° and −73° respectively indicating that the reductive transformation had occurred with induction of chirality, most probably due to the Sa-atropoisomeric axis of the C6–C7 bond deriving from the fasamycin precursors. The fixed atropoisomeric axis was confirmed by determining the rotational energy barrier for the C6–C7 bond of fasamycin C (1) using DFT calculations (see ESI†). This yielded a value of ΔE = 156.1 kJ mol−1 indicating that little to no axial rotation should be expected about this bond, with the frequency of rotation on the order of years.18 The positive sign of the [α]D20 for 23 is consistent with all previous naturally occurring formicamycins which have [α]D20 values in the range +162° to +469°. Unfortunately, we had not isolated the formicamycin congener 23 in our previous work and therefore could not assign the enantiomeric excess of this molecule based on optical rotation comparison to a standard. While the isolated sample of 23 is a mixture of two enantiomers at C10/C19, given the essentially fixed atropoisomeric axis at C6–C7, these two molecules should be diastereotopic with respect to each other. However, the 1H NMR spectrum for this sample did not indicate split chemical shifts at any position and the initial HPLC analysis showed a single peak. We thus repeated the chromatographic analysis using the higher resolving power of UPLC, and varied the elution conditions, and were able to resolve the isolated sample of 23 into two peaks of the same mass and with the same UV spectra (Fig. 5) consistent with their diastereotopic relationship. Integration of these two peaks indicated a diastereomeric excess (% de) of 41.3%. The equivalent analysis of isolated 24 indicated a % de of 40.5%. Mixing the isolated samples of 23 and 24 and repeating the UPLC analysis confirmed the presence of 4 peaks, whereas reanalysis of this mixture using the original HPLC conditions showed only 2 peaks, consistent with no change to the sample following purification. UPLC analysis of formicamycins isolated from S. formicae KY5 indicated all congeners were single diastereoisomers.
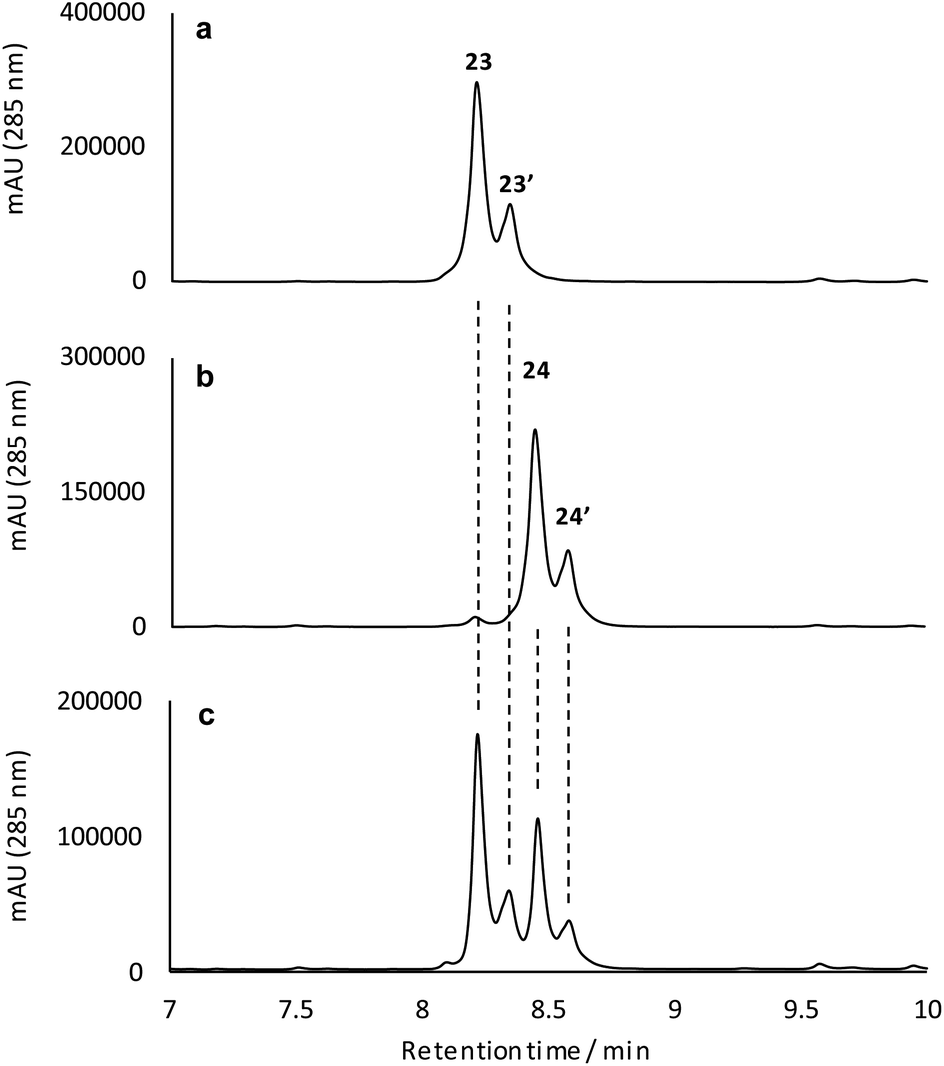 | ||
| Fig. 5 Reconstituted UPLC-UV (285 nm) analysis for the isolated reduction products 23 (a), 24 (b), and a mixture of the isolated products (c) indicating the diastereomeric composition of these samples. | ||
Conclusions
The two-step pathway described here explains the significant structural changes that must occur to transform the almost flat fasamycin skeleton into the twisted chair-like configuration of the formicamycins. The dearomatization of aromatic systems as a prelude to structural diversification has been described for several natural products including, for example, recent mutational analysis of the agnestins19 and cryptosporioptides20 where deletion of genes encoding putative Baeyer–Villigerases led to accumulation of the anticipated aromatic precursors. For the Baeyer–Villiger activity of ForX, two alternative mechanisms can be invoked for reaction between the fasamycin substrate, reduced flavin cofactor and oxygen. In the first pathway (Pathway I) the peroxyflavin species acts as an electrophile to catalyze oxygen introduction as a tertiary hydroxyl group at C10 following deprotonation of the adjacent phenol (Fig. 3). Subsequent rearrangement then occurs with bond migration and protonation at C20. Alternatively, (Pathway II) ForX could first induce tautomerization of ring C, possibly through substrate binding, followed by donation of a proton from the peroxyflavin cofactor to the resulting C9 ketone generating an oxonium intermediate. The resulting flavoperoxy anion can then act as a nucleophile in a canonical BV-like reaction to generate the observed lactone intermediate. The tautomerization required in the first step of Pathway II has precedent in the reaction of dehydrorabelomycin and congeners during the biosynthesis of gilvocarcins and jadomycins.5 However, we favor Pathway I which is reminiscent of that by which a nucleophilic chloride ion is converted into a highly reactive electrophilic species via the intermediacy of hypochlorous acid by flavin-dependent halogenases.21 This is consistent with our observation that ForX groups phylogenetically with the Group A monooxygenases that typically catalyze hydroxylation.We propose that ForY then acts as a flavin-dependent reductase catalyzing two-electron reduction of the intermediate lactone. This reaction most likely proceeds via 1,4-conjugate attack of a hydride ion at C19 to form the corresponding enolate 27. Collapse of the enolate can then occur with formation of the new C9–C10 sigma bond and formation of an epoxide intermediate with subsequent rearrangement to yield the C10-tertiary hydroxyl group with overall ring contraction in a process reminiscent of a Favorskii reaction. This pathway (Fig. 3) is consistent with the side products observed in the sodium borohydride biomimetic reduction, and with the steric requirements for orbital overlap during C9–C10 bond formation. Although rare in nature a Favorskii-type reaction has been demonstrated to occur during biosynthesis of the polyketide natural product enterocin produced by Streptomyces maritimus.22 This pathway involves oxidative activation of the preceding intermediate via a dual 4-electron oxidation. The reaction is catalyzed by the unique flavin dependent monooxygenase EncM which utilizes a flavin-N5-oxide, rather than a peroxyflavin intermediate, to catalyze substrate oxidation and trigger a Favorskii-type reaction.23 Oxidation promotes further aldol and heterocylization reactions which are also mediated by EncM.22,23 Based on the outcome of careful stable isotope labeling experiments, a Favorskii-like reaction has also been proposed to occur during biosynthesis of the fungal polyketide metabolite roussoellatide.24
The production of a single steric isomer during the biosynthesis of formicamycins and the lack of any conjugate reduction side products as are observed during the sodium borohydride biomimetic reduction of isolated lactone biosynthetic intermediates indicates that ForY must control both the stereochemical progress of this reaction and limit alternative reaction coordinates. As described, the proposed reductive ring contraction catalyzed by ForY is unique in polyketide biosynthesis and future work will seek to understand this novel biochemical process in greater detail. A search of all available microbial genome sequences did not identify any additional close homologues. Further searches using MultiGeneBlast25 with sequences for the minimal PKS genes plus forX, with and without forY, failed to identify any additional candidate BGCs that might encode pathways that operate as described here suggesting that this pathway is unique to formicamycin biosynthesis.
Materials and methods
For details regarding experimental procedures, microbiology and molecular biology procedures, spectroscopic data, see the ESI.†Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) via Institute Strategic Program Project BBS/E/J/000PR9790 to the John Innes Centre (ZQ), by BBSRC responsive mode grants BB/S009000/1 (to BW) and BB/S00811X/1 (to MIH), by a Norwich Research Park BBSRC Postdoctoral Training Program Studentship BB/M011216/1 (RD and TB), by the Engineering and Physical Sciences Research Council studentship (EHEF), and the University of Bath (EHEF and MNG). We thank Dr Edward Hems and Dr Martin Rejzek (JIC) for their comments on the chemical synthesis. We are also grateful to Dr Sergey Nepogodiev (JIC) for excellent NMR support. This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath.Notes and references
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed
.
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162 RSC
- S. C. Tsai, Annu. Rev. Biochem., 2018, 87, 503 CrossRef CAS PubMed
- H. Leisch, K. Morley and P. C. Lau, Chem. Rev., 2011, 111, 4165 CrossRef CAS PubMed
- N. Tibrewal, P. Pahari, G. Wang, M. K. Kharel, C. Morris, T. Downey, Y. Hou, T. S. Bugni and J. Rohr, J. Am. Chem. Soc., 2012, 134, 18181 CrossRef CAS PubMed
- Z. Xu, K. Jakobi, K. Welzel and C. Hertweck, Chem. Biol., 2005, 12, 579 CrossRef CAS PubMed
- Z. Qin, J. T. Munnoch, R. Devine, N. A. Holmes, R. F. Seipke, K. A. Wilkinson, B. Wilkinson and M. I. Hutchings, Chem. Sci., 2017, 8, 3218 RSC
- Z. Feng, D. Kallifidas and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12629 CrossRef CAS PubMed
- F. Maglangit, Q. Fang, V. Leman, S. Soldatou, R. Ebel, K. Kyeremeh and H. Deng, Molecules, 2019, 24, E3384 CrossRef PubMed
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. Sternberg, Nat. Protoc., 2015, 10, 845 CrossRef CAS PubMed
- Y. Lindqvist, H. Koskniemi, A. Jansson, T. Sandalova, R. Schnell, Z. Liu, P. Mantsala, J. Niemi and G. Schneider, J. Mol. Biol., 2009, 393, 966 CrossRef CAS PubMed
- M. M. E. Huijbers, S. Montersino, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Arch. Biochem. Biophys., 2014, 544, 2 CrossRef CAS PubMed
- S. W. Aufhammer, E. Warkentin, U. Ermler, C. H. Hagemeier, R. K. Thauer and S. Shima, Protein Sci., 2005, 14, 1840 CrossRef CAS PubMed
- T. Mukherjee, Y. Zhang, S. Abdelwahed, S. E. Ealick and T. P. Begley, J. Am. Chem. Soc., 2010, 132, 5550 CrossRef CAS PubMed
- M. N. Isupov, E. Schröder, R. P. Gibson, J. Beecher, G. Donadio, V. Saneei, S. A. Dcunha, E. J. McGhie, C. Sayer, C. F. Davenport, P. C. Lau, Y. Hasegawa, H. Iwaki, M. Kadow, K. Balke, U. T. Bornscheuer, G. Bourenkov and J. A. Littlechild, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 2344 CrossRef CAS PubMed
- S.-Y. Jun, K. M. Lewis, B. Youn, L. Xun and C. Kang, Mol. Microbiol., 2016, 100, 989 CrossRef CAS PubMed
- M. Okai, W. C. Lee, L. J. Guan, T. Ohshiro, Y. Izumi and M. Tanokura, Proteins: Struct., Funct., Bioinf., 2017, 85, 1171 CrossRef CAS PubMed
- S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505 CrossRef CAS PubMed
- A. J. Szwalbe, K. Williams, Z. Song, K. de Mattos-Shipley, J. L. Vincent, A. M. Bailey, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 233 RSC
- C. Greco, K. de Mattos-Shipley, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 2930 RSC
- C. Dong, S. Flecks, S. Unversucht, C. Haupt, K.-H. van Pee and J. H. Naismith, Science, 2005, 309, 2216 CrossRef CAS PubMed
- L. Xiang, J. A. Kalaitzis and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15609 CrossRef CAS PubMed
- R. Teufel, A. Miyanaga, Q. Michaudel, F. Stull, G. Louie, J. P. Noel, P. S. Baran, B. Palfey and B. S. Moore, Nature, 2013, 503, 552 CrossRef CAS PubMed
- E. L. F. Ferreira, D. E. Williams, L. P. Ióca, R. P. Morais-Urano, M. F. C. Santos, B. O. Patrick, L. M. Elias, S. P. Lira, A. G. Ferreira, M. R. Z. Passarini, L. D. Sette, R. J. Andersen and R. G. S. Berlinck, Org. Lett., 2015, 17, 5152 CrossRef CAS PubMed
- M. H. Medema, E. Takano and R. Breitling, Mol. Biol. Evol., 2013, 30, 1218 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: General remarks; full experimental details; Fig. S1–S60 and Tables S1–S5. See DOI: 10.1039/d0sc01712d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |