
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencetrans-Hydroboration–oxidation products in Δ5-steroids via a hydroboration-retro-hydroboration mechanism†
J. Ciciolil
Hilario-Martínez
 ab,
Fernando
Murillo
a,
Jair
García-Méndez
a,
Eugenia
Dzib
a,
Jesús
Sandoval-Ramírez
b,
Miguel Ángel
Muñoz-Hernández‡
c,
Sylvain
Bernès
d,
László
Kürti
e,
Fernanda
Duarte
f,
Gabriel
Merino
*a and
María A.
Fernández-Herrera
*a
ab,
Fernando
Murillo
a,
Jair
García-Méndez
a,
Eugenia
Dzib
a,
Jesús
Sandoval-Ramírez
b,
Miguel Ángel
Muñoz-Hernández‡
c,
Sylvain
Bernès
d,
László
Kürti
e,
Fernanda
Duarte
f,
Gabriel
Merino
*a and
María A.
Fernández-Herrera
*a
aDepartamento de Física Aplicada, Centro de Investigación y de Estudios Avanzados, Unidad Mérida. km 6 Antigua Carretera a Progreso. Apdo. Postal 73, Cordemex, 97310, Merida, Yuc., Mexico. E-mail: mfernandez@cinvestav.mx; gmerino@cinvestav.mx
bFacultad de Ciencias Químicas, Benemérita Universidad Autónoma de Puebla, Ciudad Universitaria, 72570, Puebla, Pue., Mexico
cCentro de Investigaciones Químicas, Universidad Autónoma del Estado de Morelos, Av. Universidad 1001, Cuernavaca, Mor. C. P. 62209, Mexico
dInstituto de Física, Benemérita Universidad Autónoma de Puebla, Ciudad Universitaria, 72570, Puebla, Pue., Mexico
eDepartment of Chemistry, Rice University, BioScience Research Collaborative, 6500 Main Street, Houston, Texas 77030, USA
fChemistry Research Laboratory, Oxford University, Mansfield Road, OX1 3TA Oxford, UK
First published on 14th September 2020
Abstract
Herein, we report for the first time a “trans-hydroboration–oxidation product” isolated and characterized under traditional hydroboration–oxidation conditions using cholesterol and diosgenin as substrates. These substrates are excellent starting materials because of the rigidity and different structural environments around the double bond. Further investigations based on experimental evidence, in conjunction with theoretical studies, indicate that the formation of this trans-species occurs via a retro-hydroboration of the major product to generate the corresponding Δ6-structure and the subsequent hydroboration by the β-face. Besides, the corresponding Markovnikov type products have been isolated in synthetically useful yields. The behavior of the reaction under a range of temperatures is also investigated.
Introduction
The addition of electron-deficient boranes to alkenes is one of the most common reactions to produce organoboranes.1,2 The resulting alkylboranes are easy-to-handle and highly versatile intermediates that participate in many synthetically useful transformations. Perhaps the most common reaction of organoboranes is their oxidation by hydrogen peroxide in an alkaline medium to furnish the corresponding alcohols regio- and stereospecifically.3 The hydroboration (HB) of alkenes is a traditional and well-known organic transformation where the governing stereochemical principle is the addition of hydrogen and BH2 to the same π-face (syn-addition). This reaction, in principle, proceeds via a four-membered transition state (Scheme 1).1,2 Most olefins readily undergo HB under the latter conditions, usually giving the corresponding anti-Markovnikov product, and the subsequent oxidation step with hydrogen peroxide proceeds with retention of configuration.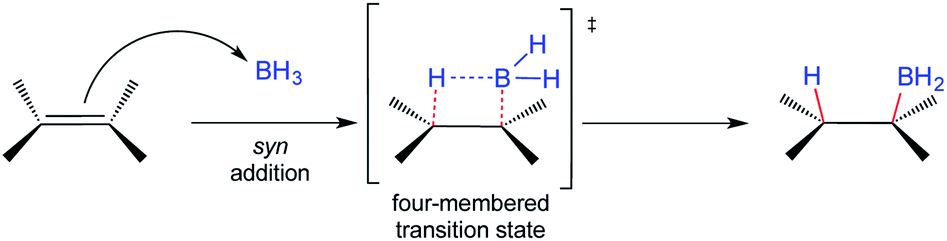 | ||
| Scheme 1 General mechanism for the hydroboration reaction of alkenes. | ||
These observations lead to the generalization that HB takes place via an anti-Markovnikov syn-addition from the less hindered side of the double bond. Even the HB of terminal alkynes occurs in an anti-Markovnikov stereospecific fashion, where the syn addition also results from HB on the same side of the alkyne.
After a plethora of literature reports on olefin HB reactions, the following question arises: is it possible to obtain a trans-hydroboration product? The answer is yes, but it has never been reported under traditional HB conditions. To the best of our knowledge, no examples of olefin trans-HB have been described in the literature. Herein, we report a trans-hydroboration–oxidation product from a hydroboration-retro-hydroboration pathway employing cholesterol (1) and diosgenin (2) as substrates. These outcomes violate, in principle, all HB reported mechanisms so far.
Results and discussion
The first application of the hydroboration–oxidation (HBO) procedure to the double bond in cholesterol (1, Fig. 1) was reported in 1959 simultaneously by Wechter4 and Sondheimer.5,6 Wechter reported that the HB reaction employing BH3·THF yielded a mixture of dialkyl boranes in 98% of isolated yield. Upon oxidation (H2O2/NaOH), the crude product was found to consist of 5α-cholestane-3β,6α-diol (1a, 78%), 5β-cholestane-3β,6β-diol (1c, traces), and a third compound reported as “not characterized” (see Fig. 1). In parallel, Sondheimer reported the use of BH3·OEt2 and cholesterol to deliver, after oxidation using H2O2 in ethanolic KOH, a mixture of compounds consisting of 1a (68%), 1c (20%), and recovering some 1 (9%). In general, most of the HBO studies on Δ5-steroids describe the generation of only one alcohol with the stereochemistry of 1a.7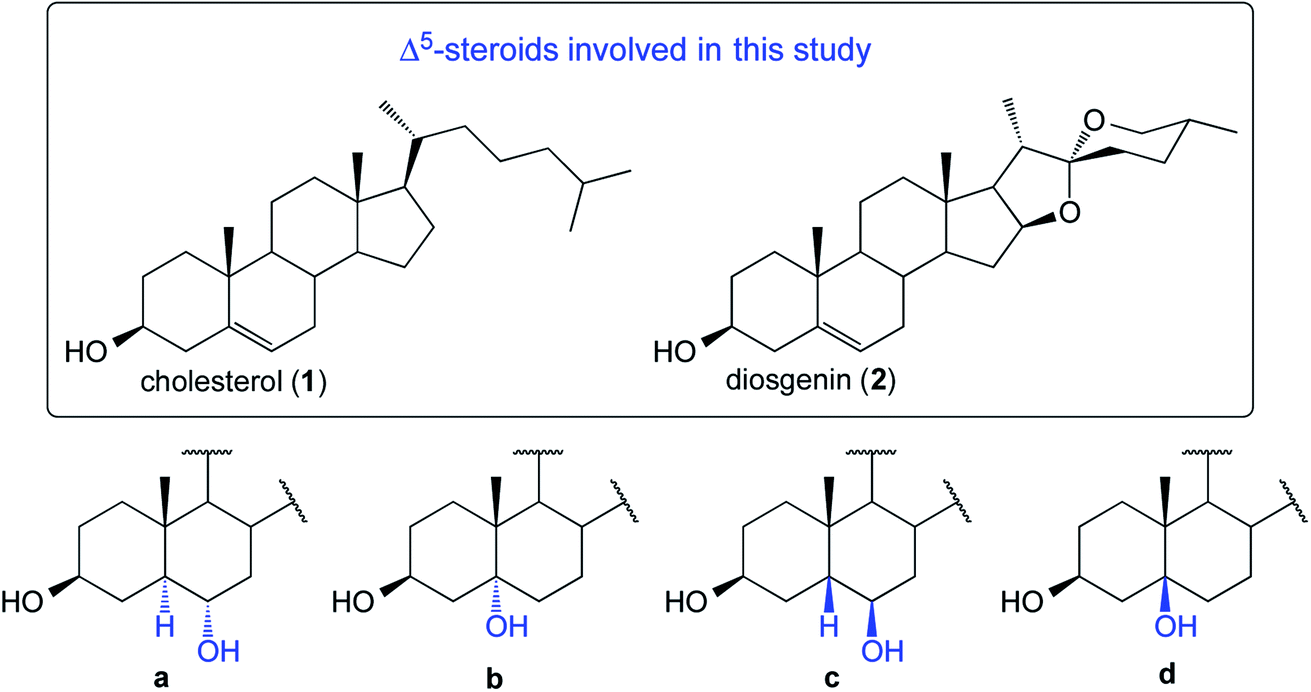 | ||
| Fig. 1 Structures of four possible products derived from the HBO reaction of substrates 1 and 2 (structures type a–d). | ||
We decided to perform the HBO of 1 and 2 (Fig. 1).8,9 The reactions were subjected to a thorough temperature study (from 20 °C to −20 °C). In our hands, for both substrates, the crude contained three main components and ultimately four products were isolated and fully characterized.
Due to the steric shielding exerted by Me-19, not surprisingly the major products (1a and 2a) arose via hydroboration of the α-steroidal face.10 It is remarkable that the lowering of the reaction temperature led to lower yields of compounds type a (see Table 1).11–13 Compounds 1b and 2b14 are the Markovnikov addition products. These compounds have been recently reported employing transition-metal-catalyzed asymmetric HB reactions.15,16 However, under the typical HB conditions (as in our case), they were unexpectedly obtained in synthetically meaningful yields (8–18%). It is also notable that their formation is favored with the decrease of temperature. The isolation of compounds type c was challenging. However, after several purifications by column chromatography, both 1c and 2c successfully crystallized from an enriched fraction of c. The full 1D and 2D NMR characterization data are detailed in the ESI.† Compounds of type d were not detectable, and the unreacted starting materials were not recoverable from any of the reactions.
| Diol type | Isolated yields at different temperatures (%) | ||||
|---|---|---|---|---|---|
| 20a °C | 10 °C | 0 °C | −10 °C | −20 °C | |
| a Reactions carried out at rt (25 °C) provided similar yields. | |||||
| Diols derived from cholesterol (1) | |||||
| 1a | 66 | 62 | 59 | 54 | 40 |
| 1b | 9 | 10 | 12 | 14 | 17 |
| 1c | Traces | Traces | Traces | Traces | Traces |
| 1e | 22 | 24 | 27 | 30 | 38 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Diols derived from diosgenin (2) | |||||
| 2a | 65 | 61 | 55 | 52 | 41 |
| 2b | 8 | 10 | 12 | 15 | 18 |
| 2c | Traces | Traces | Traces | Traces | Traces |
| 2e | 21 | 23 | 26 | 29 | 36 |
Unexpectedly, we also found the “trans”-HBO products, 1e and 2e,17 under classical HB conditions! The yields of these unexpected stereoisomers are 21–38%, surprisingly high when considering their nature. At room temperature, yields of products type e are about one-third of the HBO products type a. The 5α,6β-stereochemistry of 1e and 2e18 are confirmed by NMR; for further details, see the ESI.†
For structures b, c, and e, the observed stereochemistry in solution is supported by the single-crystal X-ray crystallography structure determinations of compounds 2b, 1c, 2c, and 1e, respectively (see ESI and CIF files deposited with the CCDC†). Of particular interest is the “trans” product 1e, in which the 6β-OH and 19-Me groups are assumed to induce steric repulsion (Fig. 2). Indeed, the O6⋯C19 separation, 3.029(4) Å, is shortened by 0.19 Å as compared to the van der Waals distance. However, since the OH group is free to rotate, there are no occurrences of destabilizing H⋯H contacts, and both 3β and 6β hydroxyl groups are engaged in classical intermolecular hydrogen bonds, to form supramolecular chains in the crystal. The same situation is observed for 1c and 2c.
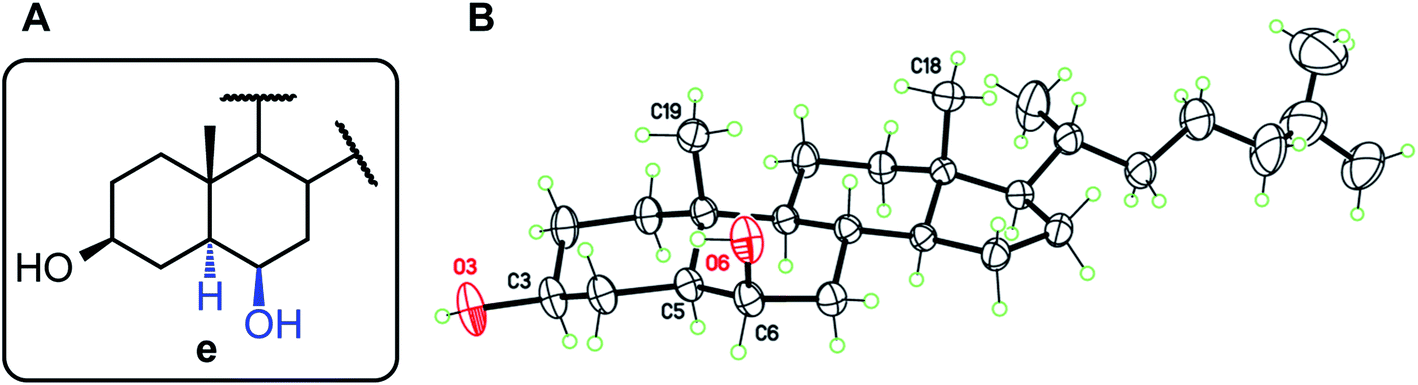 | ||
| Fig. 2 (A) Structure type e (derived from the HBO reaction of 1 and 2). (B) Molecular structure for compound 1e with displacement ellipsoids at 30% probability level. | ||
To elucidate the formation of the unexpected HBO products type e, we performed a series of quantum chemical computations at the DLPNO-CCSD(T)19–21/def2-TZVPP22 level by taking the PBE0 (ref. 23) structures from a model composed of two rings (a decalin framework, see Fig. 3), and considering the solvent effects (THF) via the SMD24 approximation. Entropic contributions and thermal corrections were computed at the SMD-PBE0-D3 (ref. 25)/def2-TZVP level at 273.15 K. Hydroboration has been theoretically studied by several groups.26–28 Validation of this methodology and further details are provided in the ESI.† All these computations were done in Gaussian 16 (ref. 29) and Orca 4.1.1.30 Considering that the alcohol is formed with retention of configuration in a subsequent oxidation step, we focus our analysis on the HB mechanism; thus, we label the alkylborane system as 1a′ that produces 1a after the oxidation.
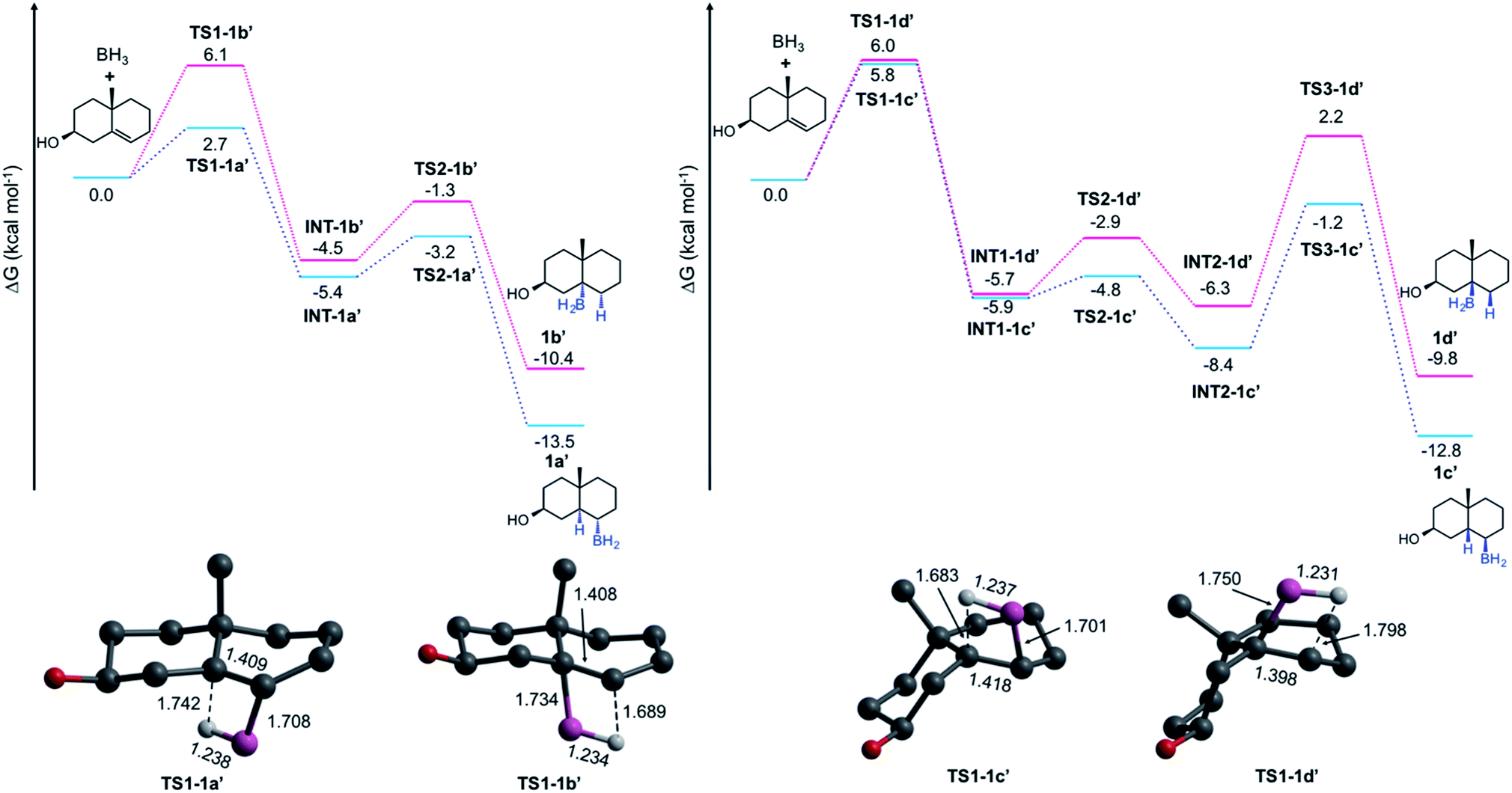 | ||
| Fig. 3 Gibbs energy profiles (in kcal mol−1) of hydroboration of the decalin model of 1 and transition state structures for the first steps. Bond distances are in Å. In the depicted structures, H atoms are omitted for clarity, except the H atom involved in the hydroboration. | ||
The Gibbs energy pathways for the suprafacial HB additions of 1 are illustrated in Fig. 3. The smallest barrier (ΔG≠ = 2.7 kcal mol−1) is obtained for one of the α-additions. It is easily explained due to the presence of Me-19. As is expected, the difference between anti-Markovnikov and Markovnikov barriers (ΔΔG≠ = 3.4 kcal mol−1) favors the former addition. Since the addition involves the bridgehead C-5, these transition states (TSs) are connected to twisted-chair conformations (INT-1a′ and INT-1b′), which are more stable than the reactants by 5.4 and 4.5 kcal mol−1, respectively. The barriers to obtain the final products are relatively low (2.2 and 3.2 kcal mol−1) and correspond to a conformational arrangement to form the final products with a trans-A/B rings fusion. The formation of both 1a′ (ΔGrxn = 13.5 kcal mol−1) and 1b′ (ΔGrxn = 10.4 kcal mol−1) is exergonic in nature. At this point, there are no surprises.
The formation of 1c′ and 1d′ involves an additional conformational change to provide the cis-A/B rings fusion. The barriers for HB additions at the β-face are similar to that computed for 1b′ (∼6 kcal mol−1). The first conformational barriers through TS2-1c′ and TS2-1d′ are small (1.1 and 2.8 kcal mol−1). However, the barriers for the final step are much higher (7.2 and 8.5 kcal mol−1) and become the rate-limiting steps. This explains why 1d is not formed, despite having an HB barrier similar to 1b, and why 1c is only isolated in traces.
The pathways discussed above are relevant to understand the HB by both α- and β-face, nevertheless, they do not explain the formation of the trans-HB product. We hypothesize that the formation of this product involves a retro-hydroboration to regenerate the double bond but at C-6 (Fig. 4). This leads to the formation of the Δ6-steroid 3 (5α-cholest-6-en-3β-ol), followed by typical HB on the β-face. Our computations indicate that 1 is more stable than 3 by 2.6 kcal mol−1. The formation of 3 from 1a′ is concerted viaTS1-1e′ (ΔG≠ = 18.0 kcal mol−1). The result is the π-complex INT-1e′, which is then converted to the trans-product (1e′) viaTS2-1e′ (ΔG≠ = 6.8 kcal mol−1) or to 1f′viaTS1-1f′ (7.1 kcal mol−1). After all the experimental and computational evidence, these results indicate that even though less preferred, a retro-hydroboration mechanism is viable under traditional experimental conditions, explaining the formation of 1e.
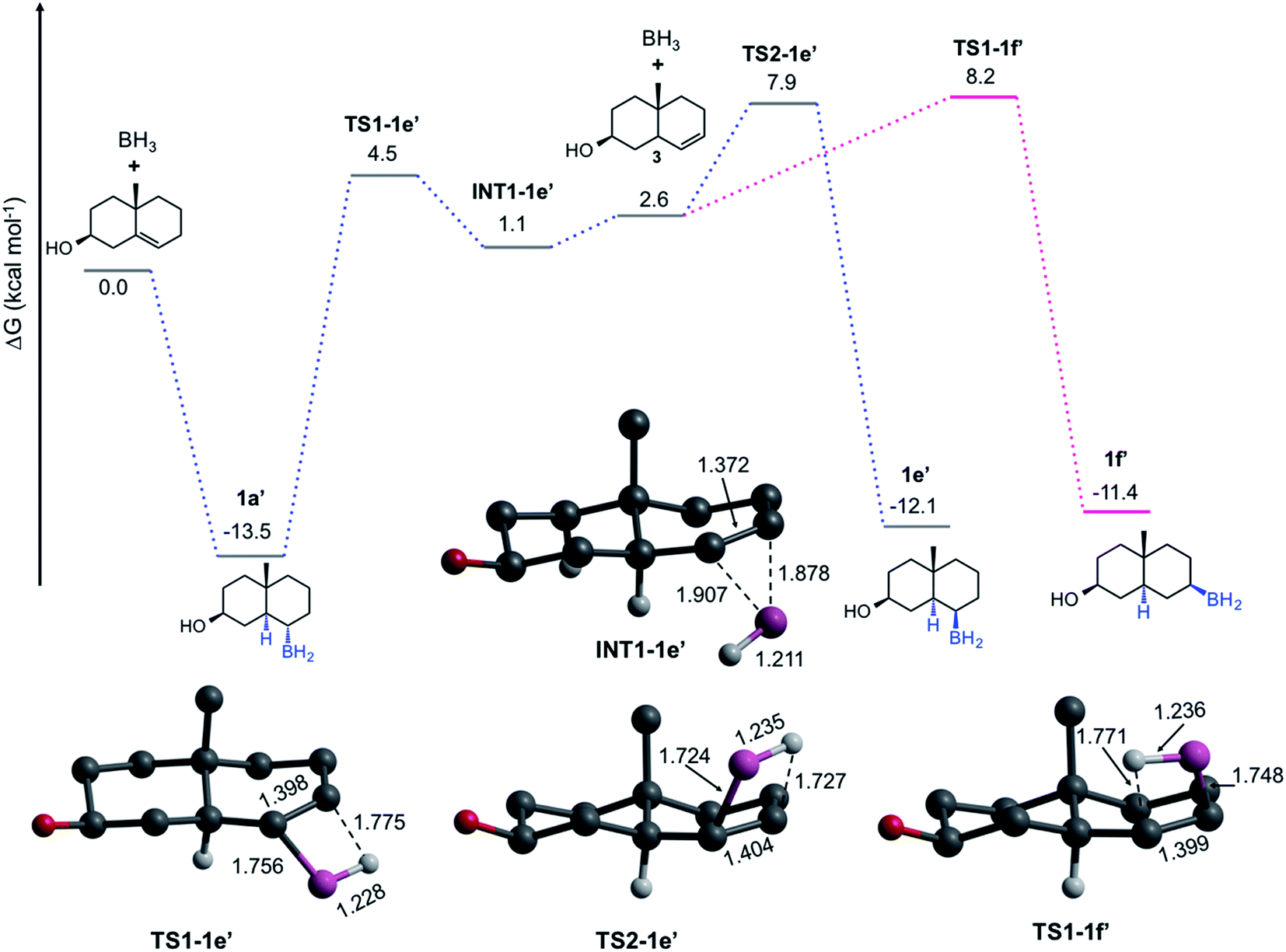 | ||
| Fig. 4 Gibbs energy profiles (in kcal mol−1) for the formation of 1e′ and 1f′ and transition state structures involved. Bond distances are in Å. In the depicted structures, H atoms are omitted for clarity, except the H atom involved in the hydroboration. | ||
Conclusions
In summary, we report the complete characterization of the hydroboration–oxidation products of cholesterol and diosgenin. Because of the steric effect exerted by Me-19, the most abundant products are the anti-Markovnikov ones by the α-steroidal face. Surprisingly, a trans-product is also obtained! This is the first time that such a type of structure is synthesized and characterized under typical hydroboration–oxidation conditions. The best way to explain the formation of this “trans-species” is via a retro-hydroboration of the major product (a-type products) to generate the corresponding Δ6-structure and the subsequent hydroboration by the β-face. We were lucky to select these steroids as substrates because of the rigidity and different structural environments around the double bond, which allowed us to isolate and characterize the trans-product.Experimental section
General procedure for the hydroboration of Δ5-steroids
To a solution of 1 or 2 (2 mmol) in dry THF (30 mL), NaBH4 (0.38 g, 10 mmol) was added. The system was kept sealed, under stirring, and under argon atmosphere. The reaction mixture was set at the temperature specified in Table 1 before the addition of BF3·OEt2 (1.65 mL, 13.4 mmol). After stirring for 2 h, the remaining pressure was released, and the work up was performed adding brine (10 mL) dropwise. The THF was evaporated under reduced pressure and the crude product was dissolved in CH2Cl2, washed with brine (1 × 50 mL) and water (3 × 50 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude mixture was immediately dissolved in a solution of 2% KOH/MeOH (50 mL) and then 5 mL of 35% H2O2 were added dropwise. After 1 h of stirring at rt, 200 mL of water were added, and the resulting precipitate was filtered off and dried under vacuum. The resultant white solids were purified by flash chromatography on a Combiflash apparatus using a gradient elution employing hexane/ethyl acetate (see specific details in the ESI†).Note: all procedures were carried out employing a commercial 1.0 M solution of BH3·THF and the results were the same.
Author contributions
JCHM, JGM, JSR, and MAFH carried out the experiments. FM, ED, FD, and GM performed the computational studies. MAMH and SB performed the X-ray determinations. All authors interpreted the results and LK, FD, GM, and MAFH prepared the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully thank Conacyt (Grants 254742, 254604, and 268178). JCHM, FM, and ED acknowledge Conacyt for PhD fellowships, and JGM for MSc fellowship.References
- H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1956, 78, 5694–5695 Search PubMed.
- H. Brown and B. C. Rao, J. Org. Chem., 1957, 22, 1135–1136 Search PubMed.
- P. Kaur, G. L. Khatik and S. K. Nayak, Curr. Org. Synth., 2017, 14, 665–682 Search PubMed.
- W. J. Wechter, Chem. Ind., 1959, 294–295 Search PubMed.
- S. Wolfe, M. Nussim, Y. Mazur and F. Sondheimer, J. Org. Chem., 1959, 24, 1034 Search PubMed.
- M. Nussim, Y. Mazur and F. Sondheimer, J. Org. Chem., 1964, 29, 1120–1131 Search PubMed.
- M. B. Smith, in Organic Synthesis, Academic Press, Oxford, 3rd edn, 2010, pp. 491–540 Search PubMed.
- R. Zeferino-Diaz, J. C. Hilario-Martinez, M. Rodriguez-Acosta, A. Carrasco-Carballo, M. G. Hernandez-Linares, J. Sandoval-Ramirez and M. A. Fernandez-Herrera, Steroids, 2017, 125, 20–26 Search PubMed.
- M. A. Fernandez-Herrera, J. Sandoval-Ramirez, H. Lopez-Munoz and L. Sanchez-Sanchez, Arkivoc, 2009, 170–184 Search PubMed.
- Compound 2a matched an authentic sample of α-chlorogenin.
- P. K. Agrawal, D. C. Jain, R. K. Gupta and R. S. Thakur, Phytochemistry, 1985, 24, 2479–2496 Search PubMed.
- S. C. Sharma and O. P. Sati, Phytochemistry, 1982, 21, 1820–1821 Search PubMed.
- M. E. Wall and H. A. Walens, J. Am. Chem. Soc., 1955, 77, 5661–5665 Search PubMed.
- H. Nawa, M. Uchibayashi, A. Okabori, K. Morita and T. Miki, Chem. Pharm. Bull., 1963, 11, 139–144 Search PubMed.
- H. Iwamoto, T. Imamoto and H. Ito, Nat. Commun., 2018, 9, 2290 Search PubMed.
- J. R. Smith, B. S. L. Collins, M. J. Hesse, M. A. Graham, E. L. Myers and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 9148–9151 Search PubMed.
- D. G. Chincharadze, A. N. Kel'ginbaev, M. B. Gorovits, L. I. Éristavi, T. T. Gorovits and N. K. Abubakirov, Chem. Nat. Compd., 1979, 15, 442–446 Search PubMed.
- Compound 2e matched an authentic sample of β-chlorogenin.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 Search PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 Search PubMed.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- N. J. R. V. E. Hommes and P. v. R. Schleyer, J. Org. Chem., 1991, 56(12), 4074–4076 Search PubMed.
- Y. Oyola and D. A. Singleton, J. Am. Chem. Soc., 2009, 131, 3130–3131 Search PubMed.
- J. O. Bailey and D. A. Singleton, J. Am. Chem. Soc., 2017, 139, 15710–15723 Search PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, 2016, https://gaussian.com/citation/ Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization of all the isolated compounds, computational details, and the Cartesian coordinates of all the stationary points. CCDC 1921283–1921286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01701a |
| ‡ Present address: Department of Chemistry, 310 President's Circle, Mississippi State University, MS State, Mississippi 39762, United States. |
| This journal is © The Royal Society of Chemistry 2020 |