
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron/N-doped graphene nano-structured catalysts for general cyclopropanation of olefins†
Abhijnan
Sarkar
a,
Dario
Formenti
b,
Francesco
Ferretti
 a,
Carsten
Kreyenschulte
b,
Stephan
Bartling
b,
Kathrin
Junge
b,
Matthias
Beller
*b and
Fabio
Ragaini
*a
a,
Carsten
Kreyenschulte
b,
Stephan
Bartling
b,
Kathrin
Junge
b,
Matthias
Beller
*b and
Fabio
Ragaini
*a
aDipartimento di Chimica – Università degli Studi di Milano, Via C. Golgi 19, 20133 Milano, Italy. E-mail: fabio.ragaini@unimi.it
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
First published on 8th June 2020
Abstract
The first examples of heterogeneous Fe-catalysed cyclopropanation reactions are presented. Pyrolysis of in situ-generated iron/phenanthroline complexes in the presence of a carbonaceous material leads to specific supported nanosized iron particles, which are effective catalysts for carbene transfer reactions. Using olefins as substrates, cyclopropanes are obtained in high yields and moderate diastereoselectivities. The developed protocol is scalable and the activity of the recycled catalyst after deactivation can be effectively restored using an oxidative reactivation protocol under mild conditions.
Introduction
The use of available, environmentally compatible and inexpensive 3d-metals in catalysis is an hot topic in modern chemistry.1–4 Among the various transition metals, particularly Fe is a privileged element due to its abundance, very low price and negligible toxicity.5–7 In the past decade, a remarkable number of reports were disclosed concerning the use of specific well-defined iron homogeneous complexes, supported or unsupported Fe-based nanoparticles, or other kinds of aggregates (e.g. nanoclusters) in various catalytic transformations.6,8–15 In 2013, inspired by electrocatalysis, some of us reported the development of an iron-based heterogeneous catalyst prepared by the pyrolysis of Fe(OAc)2/Phen (Phen = 1,10-phenanthroline) impregnated onto Vulcan carbon.16 According to the most recent and accurate characterization, the active material Fe/Phen@C-800 is composed of Fe-based nanoparticles (NPs), small Fe clusters, and even single Fe atoms embedded in a nitrogen doped carbon matrix (see below for a more detailed description). Based on this work, a whole family of catalytically interesting materials has been prepared using different approaches.17,18 So far, they have been mainly used in reduction/oxidation reactions and no application in other advanced organic transformations has been reported.19 Clearly, the discovery of new reactivity for iron-based heterogeneous catalysts will give new impetus for 3d-metal catalysis in general, and probably facilitate their actual implementation in industry (Scheme 1). | ||
| Scheme 1 (A) Preparation of Fe/Phen@C-800, (B) selected known catalytic applications and (C) the present work: cyclopropanation reactions using Fe/Phen@C-800 as the catalyst. | ||
Inspired by the availability of different Fe oxidation states in such materials,20,21 we considered them also to be promising candidates for processes known to be catalysed by iron species in higher oxidation states. To prove this idea, the cyclopropanation of olefins using diazo compounds as carbene sources was chosen as a model reaction. Indeed, this transformation is efficiently catalysed by homogeneous Fe(II/III) complexes based on nitrogen-containing ligands such as porphyrins, phthalocyanines or other multidentate N- and N,O-ligands.22
Cyclopropanes constitute an important scaffold in natural products and many biologically active molecules including several pharmaceuticals and agrochemicals.23 Furthermore, their highly strained structure provides a surprising reactivity making them versatile reagents in synthetic organic chemistry.24 Thus, many homogeneous complexes based on Fe, Ru, Co, and Cu have been developed, which successfully catalyse this transformation.25,26 Along with homogeneous catalysts, immobilized metal complexes (mainly based on Cu and, to a less extent, on Ru and Rh) are also known to be active in cyclopropanations. However to date, very few reports describe the use of truly heterogeneous catalysts. After the pioneering work of Mayoral in 1997,27 Cu NPs on TiO2–Al2O3,28 Al2O3 (ref. 29) and core–shell Fe–FexOy (ref. 30) were reported as catalysts. More recently, the groups of Coleman and Mack described the use of silver foil as a recyclable catalyst under mechanochemical conditions.31,32
Here, we present for the first time a heterogeneous iron catalyst for such reactions, which can be conveniently used and professionally recycled.
Results and discussion
Optimization and scope of olefin cyclopropanation using the Fe/Phen@C-800 catalyst
Initially, we explored the activity of Fe/Phen@C-800 in the reaction of α-methylstyrene 1a with ethyl diazoacetate (EDA) as a model transformation (Scheme 2). To our delight, the Fe/Phen@C-800 catalyst is able to efficiently catalyse this reaction affording the cyclopropane 2a in very high to excellent yields with a moderate diastereoselectivity in favour of the trans isomer. Control experiments using materials prepared by the same procedure employed for Fe/Phen@C-800, but omitting either Fe(OAc)2 or Phen, or by using Fe(OAc)2 and Phen as such resulted in no detectable formation of the desired product (Table S2, entries 6–8†). | ||
| Scheme 2 Cyclopropanation of 1a using Fe/Phen@C-800 as the catalyst. | ||
Notably, the nature of the solvent has a minimal influence both on the reaction yield and diastereoselectivity making this reaction versatile in terms of the media profile (Table S2†). Applying 1,2-dimethoxyethane (DME) as solvent, 60 °C was identified as the optimal reaction temperature (Table S3†). Even though in most optimization experiments an excess of the olefin (5-fold amount) with respect to the diazo compound has been used, it is possible to decrease the amount of 1a (1.5 eq.) and still a very good yield of the cyclopropanes can be achieved (Table S4, entry 3†). This demonstrates the applicability of the procedure even to more expensive olefins. Under the latter conditions, diethyl fumarate and diethyl maleate derived from EDA homocoupling were also observed as side products. Advantageously, the catalyst is water tolerant and only a slightly decreased yield was obtained using a “wet” solvent (Table S4, entry 4†).
Given the good results obtained in the benchmark reaction and the apparent robustness of the catalyst, the substrate scope was investigated (Scheme 3).
 | ||
| Scheme 3 Substrate scope with respect to olefins. Reaction conditions: 0.50 mmol of EDA, 2.50 mmol of alkene, 18.5 mg of Fe/Phen@C-800 (corresponding to 2 mol% Fe), and 3 mL of DME at 60 °C for 4 h. Yields are based on the starting EDA and refer to the isolated compounds (sum of the trans and cis diastereoisomers). Diastereomeric trans/cis ratios (dr) are based on separated diastereomers. aIsolated yield of the major isomer in parentheses, dr measured by 1H NMR. bOnly major isomer isolated, dr measured by 1H NMR. c1.5 mmol of alkene was employed. dReaction time: 8 h. Isolated as an isomeric mixture. Where possible, dr are measured by 1H NMR. | ||
Styrene and related derivatives (1b–1e) gave the corresponding cyclopropanes in high yields regardless of the position of the substituent on the arene ring. Similarly, substrates with electron-donating groups (1f–1g) afforded the corresponding cyclopropanes in very high yields. Also halogen-containing substrates (1h–1j, 1m, 1p, 1q) were well tolerated, maintaining intact the C–X bond, which can allow further functionalization. To our surprise, pentafluoro styrene (1l) as an example of an electron-poor substrate afforded the corresponding product in a good yield. Notably, more challenging aliphatic substrates such as 1-octene (1t) and β-pinene (1v) gave the products in satisfactory yields. Using vinyl cyclohexene (1u) the cyclopropanation reaction occurred regioselectively on the terminal olefinic bond maintaining intact the internal one. The selectivity for the terminal double bond in 1u is explained by the lack of activity of the catalyst in the case of internal olefins (Fig. S1†), most likely due to a hindered approach of the substrate to the carbene formed on the surface of the catalyst.
Under the optimized conditions, the scope of the diazo compound was also examined (Scheme 4). Mono-substituted diazo compounds (ester or ketone) gave the corresponding cyclopropanes 2aa–2ab in excellent yields. Interestingly, the use of more sterically demanding diazo compounds has a dramatic positive effect on the diasteroselection, furnishing the trans isomer with >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. The use of di-substituted diazomethanes proved to be more challenging. Nevertheless, diphenyldiazomethane yielded the product 2ac in a moderate yield (100 °C, 8 h in toluene).
:1 dr. The use of di-substituted diazomethanes proved to be more challenging. Nevertheless, diphenyldiazomethane yielded the product 2ac in a moderate yield (100 °C, 8 h in toluene).
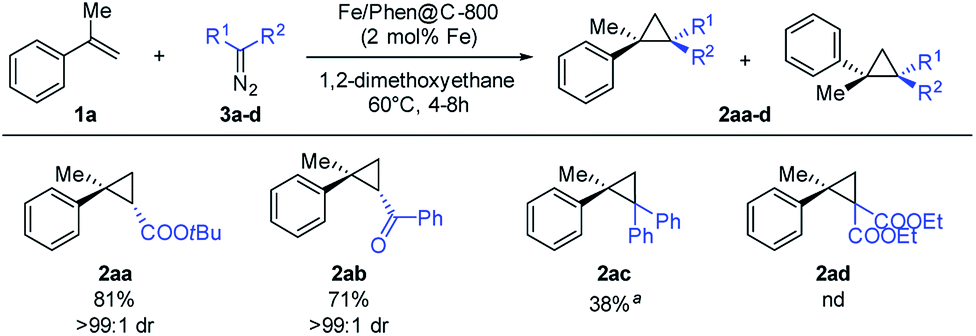 | ||
| Scheme 4 Substrate scope with respect to diazo compounds. Reaction conditions: 0.50 mmol of 3a–d, 2.50 mmol of alkene, 18.5 mg of Fe/Phen@C-800 (2 mol% Fe), and 3 mL of DME, at 60 °C for 4 h. Isolated yields are reported, based on the starting EDA; dr were measured by 1H NMR. aReaction conditions: 100 °C in toluene for 8 h. | ||
Catalyst reactivation and recycling
In order to demonstrate the practical utility of this protocol, the scalability and recyclability of the system were studied. The model reaction was successfully scaled-up to 15-fold without significant variations of yield and diasteroisomeric ratio compared to the small-scale run. Regarding the recyclability, a significant decrease of the activity was observed after the 4th run (D.1 in Fig. 1, cycles 1–5). ICP analysis of the solution showed a negligible (ca. 0.1%) loss of iron from the catalyst, so this cannot be the reason for deactivation.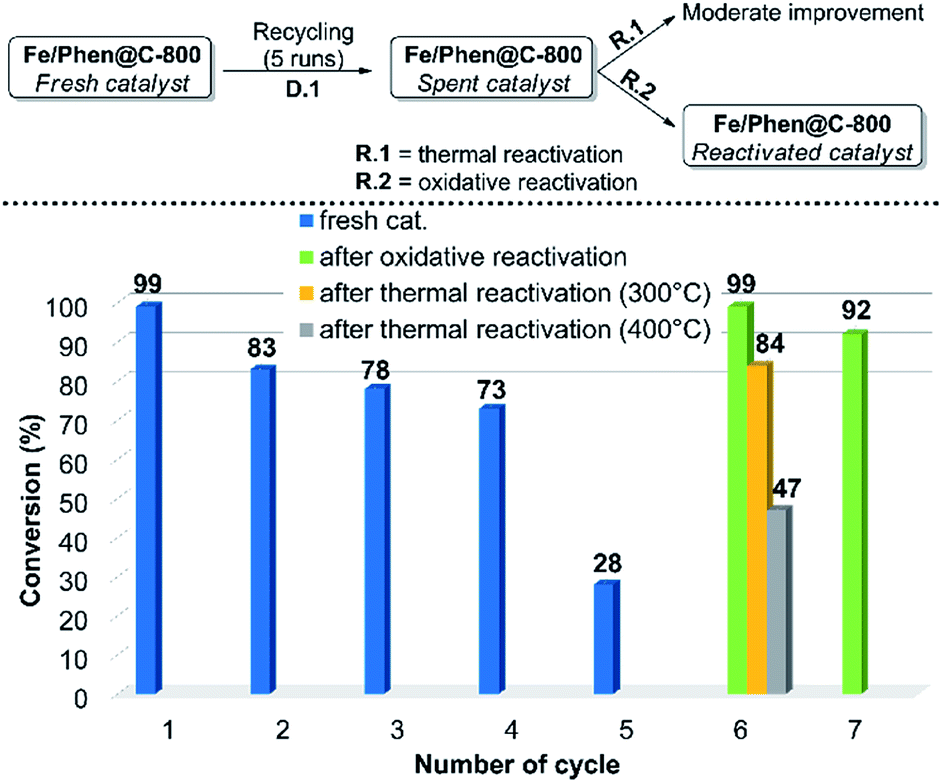 | ||
| Fig. 1 Recycling and reactivation steps. For experimental details regarding the recycling runs and the reactivation procedures see the ESI.† | ||
In order to make the whole process both efficient and effective, two routes of re-activation of the spent catalyst were explored. The material obtained after 5 runs (Fe/Phen@C-800_S) was thermally treated under an inert atmosphere (R.1 in Fig. 1). However, despite a partial reactivation was gained, still complete conversion of EDA could not be achieved. In contrast, by treating Fe/Phen@C-800_S with an aqueous solution of 3 v/v% H2O2, the initial catalyst activity was restored (R.2 in Fig. 1)! The reactivated catalyst, Fe/Phen@C-800_R, could be further recycled as the fresh one (Fig. 1, cycle 7). Such an oxidative regeneration is typical for catalysts that suffer from physicochemical deactivation (e.g. fouling or poisoning).33,34 Apparently, the olefinic substrates and in particular styrenes underwent to a small extent oligo- and/or polymerization, which deactivates the iron catalyst. Indeed, we verified that complete deactivation of the catalyst occurred even by treating the parent material only with styrene under the reaction conditions. Also in this case, the activity was restored by oxidative treatment (see the ESI† for experimental details).
Note that most oxidative reactivation processes are based on the use of air at high temperatures, but such a treatment would result in the complete burning of the carbon support of our catalyst. Hydrogen peroxide has been reported as a milder alternative in a few cases.35–37 Moreover, the Fen+/H2O2 system (known as Fenton's reagent) is also effective in these processes thanks to the generated HO˙ radicals.38 Thus, it appears that in our case both the direct action of H2O2 and related Fenton-like reactions are responsible for the removal of the oligomers/polymers from the spent catalyst.
Characterisation of fresh, spent and reactivated catalysts
To further elucidate the reason for the declined activity, the fresh, spent and regenerated catalysts were characterized by X-ray photoelectron spectroscopy (XPS) and scanning transmission electron microscopy (STEM). XPS N 1s analysis showed common peaks at around 399 eV, 400 eV, and 401 eV for all the materials (Fig. S2–S6†). These results are in agreement with three different nitrogen bonding situations, namely N bonded in residual organic matrices and/or Fe–Nx centers, pyrrolic-N, and graphitic-N, respectively.16,39,40 A minor peak at 398.1 eV ascribed to pyridinic-N has been detected in Fe/Phen@C-800_S. C 1s core levels displayed an analogous pattern for all the materials (Fig. S2†). Indeed, signals corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–H (284.8 eV), CN or C–O (approximately at 285 eV), and C–N or CO (285–291 eV) functionalities can be detected.41 Deconvolution of the Fe 2p3/2 region showed four pairs of peaks which can be attributed to various Fe states. Peaks at around 708 eV, 711 eV, and 714 eV in the region correspond to Fe(0), Fe(II) and Fe(III), respectively.42 Additionally, the peak at 710 eV suggests the presence of Fe–Nx bonds which is in agreement with the previous results from N 1s spectra.43
C/C–H (284.8 eV), CN or C–O (approximately at 285 eV), and C–N or CO (285–291 eV) functionalities can be detected.41 Deconvolution of the Fe 2p3/2 region showed four pairs of peaks which can be attributed to various Fe states. Peaks at around 708 eV, 711 eV, and 714 eV in the region correspond to Fe(0), Fe(II) and Fe(III), respectively.42 Additionally, the peak at 710 eV suggests the presence of Fe–Nx bonds which is in agreement with the previous results from N 1s spectra.43
Finally, the interpretation of the O 1s region is not trivial because of the large amount of oxygen functionalities both derived from oxygen groups present in the carbonaceous matrix44 and iron oxides.45 For this reason, unambiguous assignments are not possible here. In general, only small changes in the XPS data can be observed when comparing the fresh, spent, and regenerated catalysts. STEM images and analytical data of the three materials were then acquired. As depicted in Fig. 2, the annular dark field (ADF) STEM images and related elemental maps show the presence of Fe, N, and O on the carbon support. Nitrogen is mainly distributed in a phase also containing C, O and Fe on the surface of the support alongside Fe-based particles. In addition to the elemental maps, the different contrast visible in both the annular bright field (ABF) and high angle annular dark field (HAADF) images (Fig. S7†) confirmed the predominant presence of probably metallic Fe NPs in addition to Fe oxide NPs. Whereas layers of graphene covered the former, the latter are not covered thus indicating a possible protecting role of graphene towards oxidation. The nitrogen-doped amorphous carbonaceous matrix attached to the support showed the presence of dispersed Fe clusters. Their identity is also confirmed by electron energy loss spectra (see Fig. S10B†). The complex pattern revealed by Fe 2p XPS spectra reflects the variety of iron functionalities (metallic Fe, FeOx, and FeNx centres) in the material. The spent catalyst (Fe/Phen@C-800_S) showed similar structures: still defined Fe-based NPs (mainly metallic and to a less extent oxidic) enveloped in carbon shells and dispersed iron clusters can be clearly observed (Fig. 2B, S8 and S10C†). Finally, the reactivated catalyst Fe/Phen@C-800_R generally showed more extended oxidic particles, while maintaining intact small iron clusters (Fig. 2C, S9 and S10D†).
 | ||
| Fig. 2 ADF-STEM images and EELS elemental maps of Fe/Phen@C-800 (A), Fe/Phen@C-800_S (B) and Fe/Phen@C-800_R (C). | ||
The general morphology and distribution of Fe, N, O and C remained similar between the three different states of the catalyst as shown by the STEM data. Combined with the small changes in the electronic structure revealed by XPS, the data are consistent with the deactivation of the catalyst being due to fouling, which is removed by the H2O2 treatment, rather than by a structural change of the catalyst itself.
Conclusions
In conclusion, here we report the first iron-based heterogeneous catalyst for cyclopropanation reactions. While in the past, this class of non-noble metal catalysts was generally employed for reduction or oxidation reactions, they are indeed effective catalysts also for carbene transfer reactions. The developed protocol allows obtaining several cyclopropanes from aromatic and aliphatic olefins and different diazo compounds in a practical and efficient manner. The heterogeneous nature of the catalyst and its robustness make it also an ideal candidate for flow chemistry applications. The deactivation of the catalyst has also been studied and an oxidative regeneration protocol was effectively developed, which may be of more general use even for other reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Ministero dell'Università e della Ricerca (MIUR) (PRIN 20154X9ATP). Financial support of the BMBF is also gratefully acknowledged. F. F. thanks the Dipartimento di Chimica, Università degli Studi di Milano, for financial support (Piano di Sostegno alla Ricerca 2019). D. F. thanks LIKAT for a Post-Doctoral Fellowship. Dr Pavel Ryabchuk and Dr Annette-Enrica Surkus (both LIKAT, Germany) are gratefully acknowledged for fruitful discussions. We thank Prof. Dr Hansjörg Grützmacher (ETH, Switzerland) for suggesting us to test the use of H2O2 for the reactivation of the catalyst.References
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- M. S. Holzwarth and B. Plietker, ChemCatChem, 2013, 5, 1650–1679 CrossRef CAS.
- R. M. Bullock, Catalysis without precious metals, Wiley-VCH Verlag, 2010 Search PubMed.
- J. Maes, E. A. Mitchell and B. U. W. Maes, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, ed. L. Summerton, H. F. Sneddon, L. C. Jones and J. H. Clark, The Royal Society of Chemistry, London, 2016, pp. 192–202 Search PubMed.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- A. K. Srivastava, M. Ali, S. Siangwata, N. Satrawala, G. S. Smith and R. K. Joshi, Asian J. Org. Chem., 2020, 9, 377–384 CrossRef CAS.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed.
- J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- E. Bisz and M. Szostak, ChemSusChem, 2017, 10, 3964–3981 CrossRef CAS PubMed.
- A. Boudier, P.-A. R. Breuil, L. Magna, H. Olivier-Bourbigou and P. Braunstein, Chem. Commun., 2014, 50, 1398–1407 RSC.
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS.
- A. Li, S. A. Nicolae, M. Qiao, K. Preuss, P. A. Szilágyi, A. Moores and M.-M. Titirici, ChemCatChem, 2019, 11, 3602–3625 CrossRef CAS.
- D. Formenti, F. Ferretti, C. Topf, A.-E. Surkus, M.-M. Pohl, J. Radnik, M. Schneider, K. Junge, M. Beller and F. Ragaini, J. Catal., 2017, 351, 79–89 CrossRef CAS.
- D. Formenti, C. Topf, K. Junge, F. Ragaini and M. Beller, Catal. Sci. Technol., 2016, 6, 4473–4477 RSC.
- D. Intrieri, D. M. Carminati and E. Gallo, in Non-Noble Metal Catalysis, ed. R. J. M. K. Gebbink and M.-E. Moret, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2019, pp. 163–189 Search PubMed.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS PubMed.
- O. G. Kulinkovich, Cyclopropanes in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2015 Search PubMed.
- D. Intrieri, D. M. Carminati and E. Gallo, Dalton Trans., 2016, 45, 15746–15761 RSC.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- J. M. Fraile, B. García, J. I. García, J. A. Mayoral and F. Figueras, in Stud. Surf. Sci. Catal., ed. H. U. Blaser, A. Baiker and R. Prins, Elsevier, 1997, vol. 108, pp. 571–578 Search PubMed.
- X. Liu, Y. Liu, X. Li, S. Xiang, Y. Zhang, P. Ying, Z. Wei and C. Li, Appl. Catal., A, 2003, 239, 279–286 CrossRef CAS.
- M. Lakshmi Kantam, V. Swarna Jaya, M. Jaya Lakshmi, B. R. Reddy, B. M. Choudary and S. K. Bhargava, Catal. Commun., 2007, 8, 1963–1968 CrossRef.
- S. Ishikawa, R. Hudson, M. Masnadi, M. Bateman, A. Castonguay, N. Braidy, A. Moores and C.-J. Li, Tetrahedron, 2014, 70, 6162–6168 CrossRef CAS.
- L. Chen, D. Leslie, M. G. Coleman and J. Mack, Chem. Sci., 2018, 9, 4650–4661 RSC.
- L. Chen, M. O. Bovee, B. E. Lemma, K. S. M. Keithley, S. L. Pilson, M. G. Coleman and J. Mack, Angew. Chem., Int. Ed., 2015, 54, 11084–11087 CrossRef CAS PubMed.
- J. A. Moulijn, A. E. van Diepen and F. Kapteijn, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, K. Helmut, S. Ferdi and W. Jens, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 1829–1845 Search PubMed.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- C. A. Querini, Catal. Today, 2000, 62, 135–143 CrossRef CAS.
- G. F. Thiele and E. Roland, J. Mol. Catal. A: Chem., 1997, 117, 351–356 CrossRef CAS.
- J. C. Chen and J. J. Huang, in 4th International Conference on Environmental Science and Development-ICESD 2013, ed. Y. Dan, Elsevier Science BV, Amsterdam, 2013, vol. 5, pp. 107–111 Search PubMed.
- M. V. Morales, K. Gora-Marek, H. Musch, A. Pineda, B. Murray, S. Stefanidis, L. Falco, K. Tarach, E. Ponomareva, J. H. Marsman and I. Melian-Cabrera, Appl. Catal., A, 2018, 562, 215–222 CrossRef CAS.
- F. Jaouen, J. Herranz, M. Lefèvre, J.-P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CrossRef CAS PubMed.
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel, S. Krakert, A. Terfort and W. E. S. Unger, Surf. Sci., 2009, 603, 2849–2860 CrossRef CAS.
- Y. Su, Y. Zhu, H. Jiang, J. Shen, X. Yang, W. Zou, J. Chen and C. Li, Nanoscale, 2014, 6, 15080–15089 RSC.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- Z.-Y. Wu, X.-X. Xu, B.-C. Hu, H.-W. Liang, Y. Lin, L.-F. Chen and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, ACS Nano, 2014, 8, 6856–6862 CrossRef CAS PubMed.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, Chem. Mater., 1990, 2, 186–191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for catalyst preparation and catalytic reactions, additional catalytic results, catalyst characterisation, and cyclopropane characterisation. See DOI: 10.1039/d0sc01650k |
| This journal is © The Royal Society of Chemistry 2020 |