
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed synthesis of β-hydroxy compounds via a strained 6,4-palladacycle from directed C–H activation of anilines and C–O insertion of epoxides†
Raju S.
Thombal
 a,
Taisiia
Feoktistova
b,
Gisela A.
González-Montiel
b,
Paul H.-Y.
Cheong
*b and
Yong Rok
Lee
*a
a,
Taisiia
Feoktistova
b,
Gisela A.
González-Montiel
b,
Paul H.-Y.
Cheong
*b and
Yong Rok
Lee
*a
aSchool of Chemical Engineering, Yeungnam University, Gyeongsan 38541, Republic of Korea. E-mail: yrlee@yu.ac.kr
bDepartment of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, OR 97331, USA
First published on 22nd June 2020
Abstract
A palladium-catalyzed C–H activation of acetylated anilines (acetanilides, 1,1-dimethyl-3-phenylurea, 1-phenylpyrrolidin-2-one, and 1-(indolin-1-yl)ethan-1-one) with epoxides using O-coordinating directing groups was accomplished. This C–H alkylation reaction proceeds via formation of a previously unknown 6,4-palladacycle intermediate and provides rapid access to regioselectively functionalized β-hydroxy products. Notably, this catalytic system is applicable for the gram scale mono-functionalization of acetanilide in good yields. The palladium-catalyzed coupling reaction of the ortho-C(sp2) atom of O-coordinating directing groups with a C(sp3) carbon of chiral epoxides offers diverse substrate scope in good to excellent yields. In addition, further transformations of the synthesized compound led to biologically important heterocycles. Density functional theory reveals that the 6,4-palladacycle leveraged in this work is significantly more strained (>10 kcal mol−1) than the literature known 5,4 palladacycles.
Introduction
Transformative transition metal catalyzed coupling reactions concomitant with C–H functionalization and epoxide C–O insertion have recently been reported in the literature.1–3 The key enabling structural feature in these reactions is the use of strongly coordinating or bi-dentate directing groups. These pioneering reports all hinge on the use of 5,4-metallacycles (Scheme 1).1–4 Surprisingly, functionalization via 6,4-metallacycles is unknown in the literature. We reveal the first examples of successfully accessing and leveraging 6,4-palladacycles.5 Specifically, we report a palladium-catalyzed regioselective ortho-functionalization of anilines with O-coordinating directing groups, such as acetanilides, 1-phenylpyrrolidin-2-one, and 1-(indolin-1-yl)ethan-1-one, with epoxides (Scheme 1). We report the density functional theory study of the complete catalytic cycle and also reveal that the 6,4-palladacycle is more strained than the literature-known 5,4 palladacycles. Importantly, the synthesized β-hydroxy compounds are versatile synthons and can be readily converted into biologically important heterocycles such as indoles.6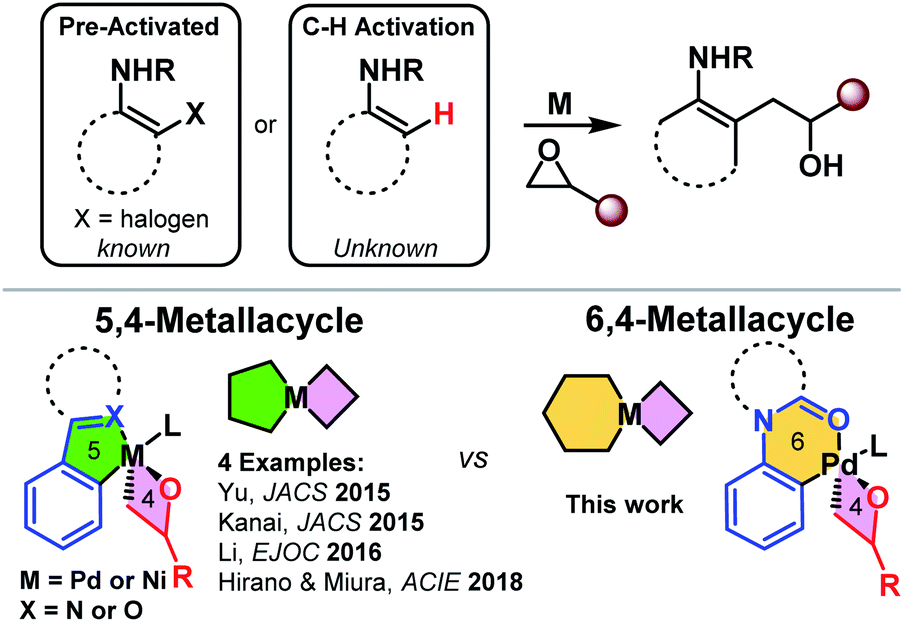 | ||
| Scheme 1 Transition metal catalyzed C–H/X functionalization and alkylation with epoxides (top) and known 5,4-metallacycle vs. the current strategy – 6,4-metallacycle (bottom). | ||
The C–H functionalization of arenes with various coupling partners has become a promising tool for synthetic chemists.7 Particularly, epoxides are widely used as alkylating reagents and building blocks in the construction of organic molecules via C–H bond functionalization.8 In the past few decades, significant progress has been made on the regioselective ring-opening of epoxides using a variety of nucleophiles on different catalytic platforms.9 The construction of C–C bonds by the opening of epoxides with aryl nucleophiles has played an important role in organic synthesis.10 Generally, the significant ring strain of epoxides enhances their susceptibility to ring-opening with a wide array of electron-rich species.11 In this context, several enantioselective and regioselective methods have been developed with the nucleophilic attack of –N, –O, and –halo nucleophiles using the stoichiometric amount of promoters. Therefore, the development of a catalytic version of these types of reactions for the functionalization at a specific position is still highly desirable.
Transition-metal-catalyzed direct utilization of epoxides has received much attention in organic synthesis in the past few decades.12–15 The reported representative modifications include the nickel-catalyzed opening of epoxides with aryl halides,16 and Li/Cu-catalyzed synthesis of enantiopure starting materials (Scheme 1).17
Results and discussion
To optimize the reaction conditions, we first examined the reaction between N-phenylacetamide 1a and 2-(phenoxymethyl)oxirane 2a using several catalysts and additives (Table 1). The initial attempts in the presence of 10 mol% of Ni(COD)2, Rh2(OAc)4, Co(OAc)2, [RuCl2(p-cymene)]2, and Cu(OAc)2 and 1 equiv. of AcOH in HFIP at 60 °C for 24 h did not provide any products (entries 1–5, Table 1). We also attempted the reaction using several palladium catalysts such as Pd(TFA)2, PdCl2, Pd(OAc)2, and PdCl2(PPh3)4 using HFIP as solvent (entries 6–9, Table 1). Among them, the 10 mol% of the Pd(OAc)2 catalyst provided the highest yield (64%). Several solvents were next screened. The reactions using 1,2-dichloroethane, CH3CN, trifluoroethanol, and tetrahydrofuran as solvents, afforded the desired product 3a in lower yields, while AcOH failed to produce the desired product (entries 10–14, Table 1). With 1 equiv. of acidic additives PivOH and 1-AdCO2H, 3a was produced in 31% and 27% yields, respectively (entries 15 and 16, Table 1). With 1 equiv. of basic additives NaOAc or Cs2CO3, 3a was not produced at all (entries 17 and 18, Table 1). Interestingly, increasing the loading of AcOH to 3 equiv. produced the best yield (79%, entry 19, Table 1). However, decreasing the amount of AcOH to 0.5 equiv. provided 3a in a lower yield (41%, entry 20, Table 1). Overall, acidic additives were found to be superior to basic additives, probably due to their participation in the C–H activation step. Increasing or decreasing the amount of the Pd(OAc)2 catalyst did not improve the yield of 3a (entries 21 and 22, Table 1). Importantly, the control reaction without the Pd(OAc)2 catalyst failed to produce 3a. The structure of synthesized compound 3a was identified by 1H NMR – there is a characteristic singlet signal of the NH-proton of the acetanilide amide at δ 9.23 ppm and newly generated methine proton at δ 4.25 ppm. The 13C NMR spectrum showed a characteristic signal of the amide carbonyl at δ 168.9 ppm. The structure was further confirmed by single-crystal X-ray diffraction analysis of the structurally related compound 6b-S.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additives (equiv.) | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (1 mmol), 2a (1 mmol), catalyst and additives in solvent (3.0 mL) for 24 h. b Isolated yields. | ||||||
| 1 | Ni(COD)2 (10) | AcOH (1) | HFIP | 60 | 24 | 00 |
| 2 | Rh2(OAc)4 (10) | AcOH (1) | HFIP | 60 | 24 | 00 |
| 3 | Co(OAc)2 (10) | AcOH (1) | HFIP | 60 | 24 | 00 |
| 4 | [RuCl2(p-cym)]2 (10) | AcOH (1) | HFIP | 60 | 24 | 00 |
| 5 | Co(OAc)2 (10) | AcOH (1) | HFIP | 60 | 24 | 00 |
| 6 | Pd(TFA)2 (10) | AcOH (1) | HFIP | 60 | 24 | 42 |
| 7 | PdCl2 (10) | AcOH (1) | HFIP | 60 | 24 | 38 |
| 8 | Pd(OAc)2 (10) | AcOH (1) | HFIP | 60 | 24 | 64 |
| 9 | PdCl2(PPh3)4 (10) | AcOH (1) | HFIP | 60 | 24 | 54 |
| 10 | Pd(OAc)2 (10) | AcOH (1) | DCE | 80 | 48 | 10 |
| 11 | Pd(OAc)2 (10) | AcOH (1) | CH3CN | 70 | 48 | 08 |
| 12 | Pd(OAc)2 (10) | AcOH (1) | TFE | 70 | 48 | 10 |
| 13 | Pd(OAc)2 (10) | — | AcOH | 60 | 48 | 00 |
| 14 | Pd(OAc)2 (10) | AcOH (1) | THF | 60 | 48 | 15 |
| 15 | Pd(OAc)2 (10) | PivOH (1) | HFIP | 60 | 24 | 31 |
| 16 | Pd(OAc)2 (10) | 1-AdCO2H (1) | HFIP | 60 | 24 | 27 |
| 17 | Pd(OAc)2 (10) | NaOAc (1) | HFIP | 60 | 24 | 00 |
| 18 | Pd(OAc)2 (10) | Cs2CO3 (1) | HFIP | 60 | 24 | 00 |
| 19 | Pd(OAc) 2 (10) | AcOH (3) | HFIP | 60 | 24 | 79 |
| 20 | Pd(OAc)2 (10) | AcOH (0.5) | HFIP | 60 | 24 | 41 |
| 21 | Pd(OAc)2 (20) | AcOH (3) | HFIP | 60 | 24 | 79 |
| 22 | Pd(OAc)2 (05) | AcOH (3) | HFIP | 60 | 24 | 66 |
| 23 | — | AcOH (3) | HFIP | 60 | 24 | 00 |
In order to explore the mechanism of this reaction, we computed the entire catalytic cycle using DFT – ωB97XD18/6-31G* (ref. 19) & LANL2DZ20/PCM21 (acetone)22 at 60 °C with Gaussian 16. All possible ligand coordination spheres around Pd, involving acetate, acetic acid, acetanilide 1a, and methyl epoxide for all pathways were considered. This reaction can first proceed via Pd(OAc)2 C–H activation of acetanilide 1a or Pd(OAc)2 C–O insertion into methyl epoxide. All possible routes towards the product were computed,23 and the proposed catalytic cycle with the resulting minimum energy reaction coordinate is shown in Fig. 1A.24 The most favored pathway involved first C–H activation of 1a, followed by C–O insertion of methyl epoxide, and finally reductive elimination to release the product as shown in the reaction coordinate diagram in Fig. 1B.
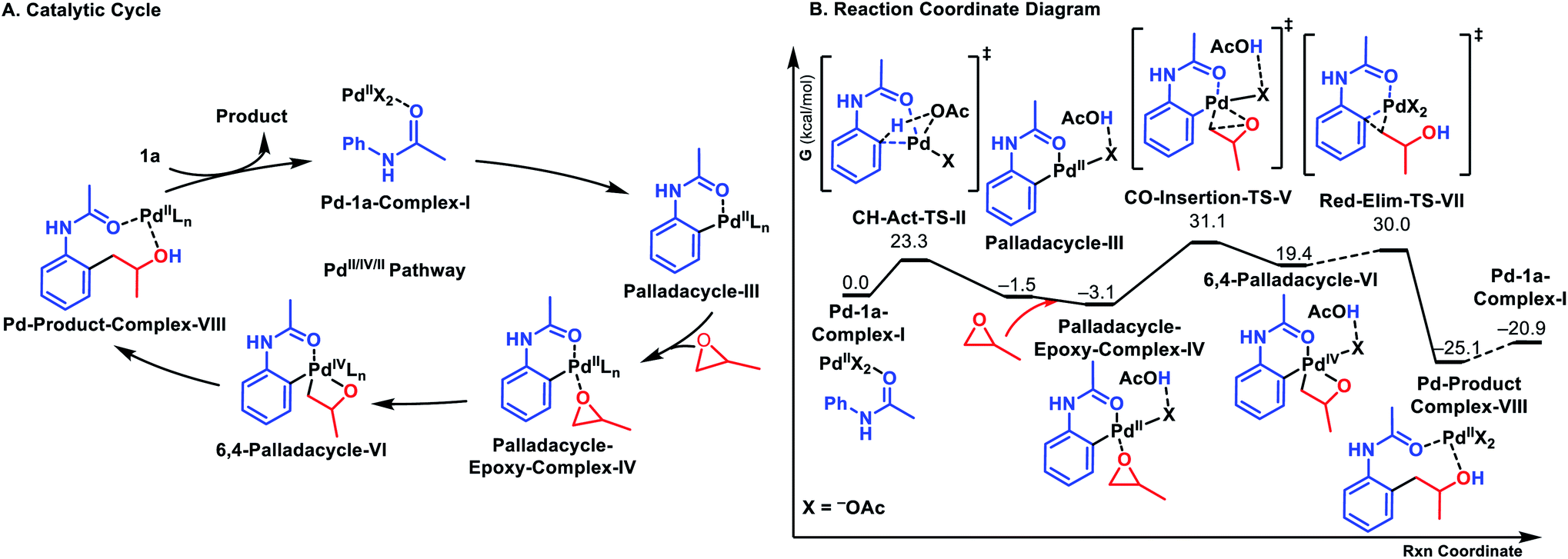 | ||
| Fig. 1 (A) Proposed catalytic cycle. (B) DFT reaction coordinate diagram for the title reaction. Energies in kcal mol−1. | ||
The catalytic cycle begins with Pd(OAc)2 coordinating to the carbonyl oxygen of the amide 1a (Pd-1a-Complex-I). Subsequent C–H activation (ΔG‡ = 23.3 kcal mol−1) leads to the formation of 6-membered Palladacycle-III (ΔG = −1.5 kcal mol−1). The C–H activation occurs via deprotonation of an ortho-hydrogen of 1a by a Pd-bound acetate (CH-Act-TS-II), rather than direct insertion of Pd into the C–H bond. Epoxide coordination leads to Palladacycle-Epoxy-Complex-IV (ΔG = −3.1 kcal mol−1). Oxidative addition into the C–O bond (CO-Insertion-TS-V, ΔG‡ = 31.1 kcal mol−1) leads to the formation of 6,4-Palladacycle-VI (ΔG = 19.4 kcal mol−1).25 Reductive elimination (Red-Elim-TS-VII, ΔG‡ = 30.0) leads to the Pd-Prod-Complex-VIII (ΔG = −25.1 kcal mol−1). The transfer of the palladium catalyst to a new acetanilide substrate is endergonic by 4.2 kcal mol−1.
One of the key features of the title reaction is the expansion into accessing and leveraging 6,4-palladacycles in reactions over the 5,4-palladacycles known in the literature (Scheme 1). We therefore explored the stability of the 6,4-Palladacycle-VIvs. the analogous 5,4-palladacycle derived from 2-phenylpyridine in an isodesmic reaction (Scheme 2).3 The DFT results reveal that the 6,4-palladacycle is higher in energy and more strained than the literature known 5,4 palladacycles by >10 kcal mol−1. This is in line with the slightly elevated temperatures required compared to the Kanai conditions involving the 2-phenylpyridine substrate.3a
 | ||
| Scheme 2 Isodesmic reaction of 6,4-Palladacycle-VI and the analogous 5,4-palladacycle derived from 2-phenylpyridine. | ||
It is noteworthy that given the significantly greater strain of the 6,4-palladacycle we were able to successfully engage it productively in the current methodology. We hypothesize that the greater strain of this palladacycle contributes to significantly elevating the barrier of the CO-Insertion-TS-V, in contrast to other reactions involving the 5,4-palladacycle where C–H activation is considered the rate-determining step (Fig. 2).3
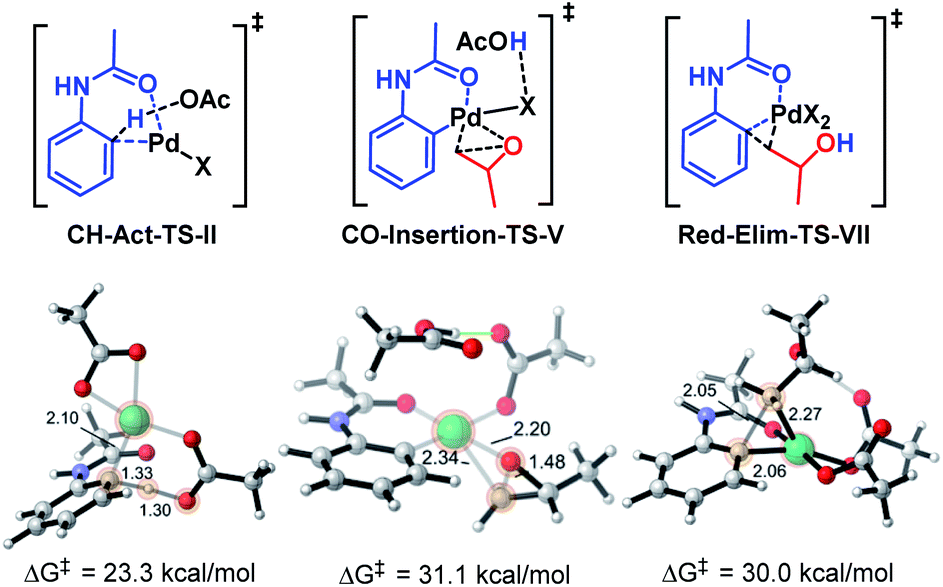 | ||
| Fig. 2 C–H activation, C–O insertion, and reductive elimination transition structures. | ||
We extensively investigated the possibility of a Pd(II)-dimer mechanism, as has been reported by Ritter and Sanford among others (for details see the ESI†).26 Rather, we discovered that the dimer mechanism proceeds via a concerted C–O bond breaking and C–C bond forming reductive elimination processes. Despite extensive efforts to locate a stepwise pathway, the dimeric stepwise transition structures were not stationary points on the potential energy surface. The lowest energy concerted Pd dimer reductive elimination barrier was 44.5 kcal mol−1, which was ∼15 kcal mol−1 higher than the monomer rate-determining step barrier.27
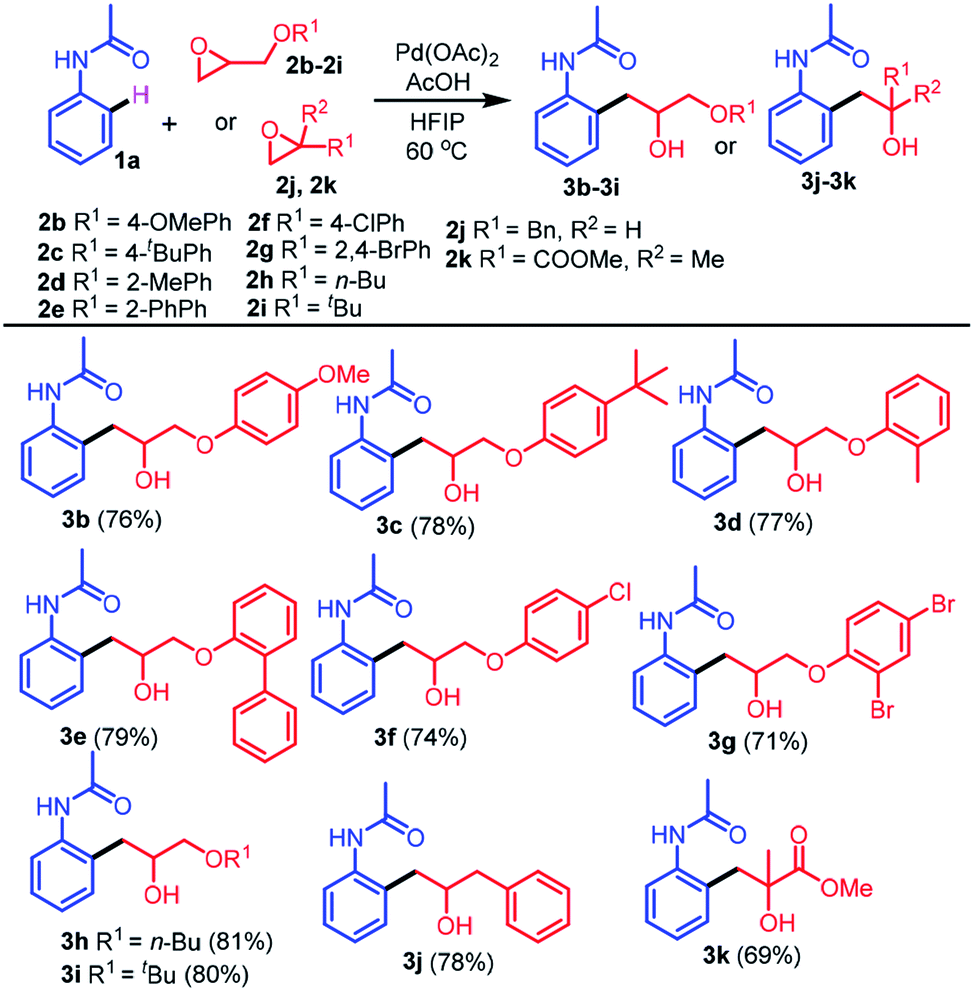
To explore the generality of this protocol, further reactions of different epoxides 2b–2k with 1a were next examined (Table 2). Treatment of 1a with several epoxides 2b–2e bearing electron-donating groups on the benzene ring, such as 4-methoxy, 4-t-butyl, 2-methyl, and 2-phenyl under optimized conditions provided the desired products 3b–3e in excellent yields (76–79%). Similarly, treatment of 1a with epoxides 2f or 2g bearing electron-withdrawing groups 4-Cl and 2,4-Br provided the desired products 3f and 3g in 74% and 71% yields, respectively. The reactions of epoxides bearing aliphatic substituents were also tolerated (3h and 3i in 81% and 80% yields, respectively). The reactions of epoxides bearing aliphatic substituents afforded the desired products in higher yields compared to the epoxides bearing aromatic substituents. The reaction of epoxide 2j bearing a benzyl substituent with 1a provided the desired alkylated product 3j in 78% yield. Importantly, the reaction of di-substituted epoxide 2k with 1a provided the desired alkylated product 3k containing a quaternary carbon in good yield (69%).
|
|
|---|
Next, the scope of this reaction was further explored using other substituted acetanilides 1b–1j bearing various electron-donating and -withdrawing groups on the benzene ring (Table 3). For example, the reaction of 1b–1e bearing electron-donating groups on the benzene ring at the 2 or 4-position with 2a or 2f afforded the expected products 4a–4d in 71–79% yields. Similarly, the reaction of 1f–1h bearing para-F, -Cl, or -Br groups with epoxide 2a led to the desired products 4e–4g in 70–74% yields. However, with 1i bearing a strong para electron-withdrawing nitro group, the desired product 4h was not isolated, probably due to strong deactivation of the meta-position of the nitro group on 1i. Interestingly, the reaction of 1j bearing a meta-Cl group with 2a regioselectively afforded the product 4i in 68% yield.
|
|
|---|

We then investigated the possibility of using 1,1-dimethyl-3-phenylurea 1k, 1-phenylpyrrolidin-2-one 1l, and 1-(indolin-1-yl)ethan-1-one 1m (Scheme 3). Interestingly, the reaction of the different directing groups 1k–1m with 2a or 2c regioselectively afforded the desired products in 64–80% yields.
 | ||
| Scheme 3 Reactions of various directing groups 1k–1m with epoxides 2a or 2c. | ||
To further explore the usefulness and broadness of this reaction, we carried out additional reactions using chiral epoxides as coupling partners (Scheme 4). A combination of (R)-2-(phenoxymethyl)oxirane (2a-R) with 1a afforded the product 6a-R in 75% yield (96% ee), whereas that of (S)-2-(phenoxymethyl)oxirane (2a-S) with 1a resulted in product 6b-S in 76% yield (98% ee). Further, the optical rotation study confirms the retention of the stereochemistry in the obtained products. The observed stereochemistry of the compound 6b-S was confirmed by single-crystal X-ray diffraction analysis.
 | ||
| Scheme 4 Reactions of chiral epoxides 2a-R and 2a-S with 1a. | ||
This new reaction can be carried out on a large scale (for details see the ESI†). To investigate the application of the novel protocol described herein, the conversion of the synthesized compounds 3a and 3g into new molecules was attempted (Scheme 5). Surprisingly, pyridinium chlorochromate (PCC) oxidation of 3a and 3g led to the direct formation of indoles 7 and 8 in 86 and 92% yields, respectively.
 | ||
| Scheme 5 Gram scale experiment and further transformations of the synthesized compounds 3a, and 3g to indoles. | ||
Conclusions
In summary, we have developed palladium-catalyzed regioselective C–H functionalization of O-coordinating directing groups with epoxides via Pd(II)/(IV)/(II) pathways. This reaction efficiently tolerates chiral epoxides, providing step-economical access to chiral alcohols on the benzene ring with high levels of regio- and stereo-selective control. Other directing groups such as 1,1-dimethyl-3-phenylurea, 1-phenylpyrrolidin-2-one, and 1-(indolin-1-yl)ethan-1-one were also successfully utilized in this transformation. This protocol also provides easy access to biologically important heterocycles with the further transformation of the synthesized compounds. The density functional theory study reveals that the new 6,4-palladacycles engaged in this work are significantly more strained than the literature known 5,4-palladacycles and their successful engagement in productive synthetic processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2018R1A2B2004432). This research was supported by the Nano Material Technology Development Program of the Korean National Research Foundation (NRF) funded by the Korean Ministry of Education, Science, and Technology (2012M3A7B4049675). PHYC is the Bert and Emelyn Christensen professor, and gratefully acknowledges financial support from the Vicki & Patrick F. Stone family and the National Science Foundation (NSF, CHE-1352663).Notes and references
- S.-B. Xu, K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2018, 57, 11797–11801 CrossRef CAS PubMed.
- G. Cheng, T.-J. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 10950–10953 CrossRef CAS PubMed.
- (a) Z. Wang, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 6140–6143 CrossRef CAS PubMed; (b) D.-D. Li, L.-F. Niu, Z.-Y. Ju, Z. Xu and C. Wu, Eur. J. Org. Chem., 2016, 18, 3090–3096 CrossRef.
- B. Lian, L. Zhang, S.-J. Li, L.-L. Zhang and D.-C. Fang, J. Org. Chem., 2018, 83, 3142–3148 CrossRef CAS PubMed.
- Recently, the acetanilides have been widely utilized in the C–H activation reactions. (a) X. Wan, Z. Ma, B. Li, K. Zhang, S. Cao, S. Zhang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 7416–7417 CrossRef CAS PubMed; (b) C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331–336 RSC; (c) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686–693 CrossRef CAS PubMed; (d) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728–731 CrossRef CAS PubMed; (e) G. E. M. Crisenza, O. O. Sokolova and J. F. Bower, Angew. Chem., Int. Ed., 2015, 54, 14866–14870 CrossRef CAS PubMed; (f) S. Grélaud, P. Cooper, L. J. Feron and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 9351–9356 CrossRef PubMed; (g) R. S. Thombal and Y. R. Lee, Org. Lett., 2020, 22, 3397–3401 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- (a) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed; (b) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304–8329 CrossRef CAS PubMed; (c) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (d) N. Thrimurtulu, A. Dey, A. Singh, K. Pal, D. Maiti and C. M. R. Volla, Adv. Synth. Catal., 2019, 361, 1441–1446 CrossRef CAS; (e) H. Kim, R. S. Thombal, H. D. Khanal and Y. R. Lee, Chem. Commun., 2019, 55, 13402–13405 RSC; (f) O. Baudoin, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202001224.
- (a) C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed; (b) R. E. Parker and N. S. Isaacs, Chem. Rev., 1959, 59, 737–799 CrossRef CAS; (c) I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250–5281 CrossRef CAS PubMed; (d) C. J. Morten, J. A. Byers, A. R. Van Dyke, I. Vilotijevic and T. F. Jamison, Chem. Soc. Rev., 2009, 38, 3175–3192 RSC; (e) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- C. Wang, L. Luo and H. Yamamoto, Acc. Chem. Res., 2016, 49, 193–204 CrossRef CAS PubMed.
- M. Alam, C. Wise, C. A. Baxter, E. Cleator and A. Walkinshaw, Org. Process Res. Dev., 2012, 16, 435–441 CrossRef CAS.
- R. M. Hanson, Chem. Rev., 1991, 91, 437–478 CrossRef CAS.
- Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed.
- C. Molinaro and T. F. Jamison, Angew. Chem., Int. Ed., 2005, 44, 129–132 CrossRef CAS PubMed.
- (a) S. Teng, M. E. Tessensohn, R. D. Webster and J. S. Zhou, ACS Catal., 2018, 8, 7439–7444 CrossRef CAS; (b) Y. Ikeda, H. Yorimitsu, H. Shinokubo and K. Oshima, Adv. Synth. Catal., 2004, 346, 1631–1634 CrossRef CAS.
- K. M. Miller, T. Luanphaisarnnont, C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2004, 126, 4130–4131 CrossRef CAS PubMed.
- Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2014, 136, 48–51 CrossRef CAS PubMed.
- P. T. Marcyk, L. R. Jefferies, D. I. AbuSalim, M. Pink, M. Baik and S. P. Cook, Angew. Chem., Int. Ed., 2019, 58, 1727–1731 CrossRef CAS PubMed.
- J. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 10, 6615–6620 CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef; (b) S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef; (c) J. L. Pascual-Ahuir, E. Silla and I. Tuñón, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- HFIP solvent effects have been successfully modeled using PCM(acetone) and PCM(TFE): (a) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed; (b) E. Erbing, A. Sanz-Marco, A. Vázquez-Romero, J. Malmberg, M. J. Johansson, E. Gómez-Bengoa and B. Martín-Matute, ACS Catal., 2018, 8, 920–925 CrossRef CAS.
- See the ESI† for other pathways considered..
- Structures involving the acetanilide amide-tautomer were also explored; in B3LYP, the C–H activation transition states of the tautomer were higher in energy by >13 kcal mol−1 than the non-tautomer. We were unable to locate a first order saddle point for the C–H activation transition state using wB97XD, but the intermediates prior to the C–H activation are also consistent with the tautomeric form being higher in energy (>9 kcal mol−1). See the ESI.†.
- We also examined the addition of an aryl-Pd nucleophile to the unopened epoxide, which would directly lead to the product catalyst complex without going through the C–O insertion of the epoxide and reductive elimination. The computed barrier for this process is 41.1 kcal mol−1, which is 10 kcal mol−1 higher in energy than that of the stepwise process shown in Fig. 1..
- (a) M. A. Gutierrez, G. R. Newkome and J. Selbin, J. Organomet. Chem., 1980, 202, 341–350 CrossRef CAS; (b) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234–11241 CrossRef CAS PubMed; (c) D. C. Powers, M. A. Geibel, J. E. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050–17051 CrossRef CAS PubMed; (d) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed; (e) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234–11241 CrossRef CAS PubMed.
- Various Pd dimer arrangements as well as syn- and anti-arrangements of the amides and HFIP as well as acetic acid coordination to the forming alkoxide in the concerted reductive elimination transition structures were computed. Alternate arrangements of the dimer species that are not analogous to the literature precedents were also considered, and they were even higher in energy.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, computational details and X-ray crystallographic structure and data for 6b-S. CCDC 1981280. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01462a |
| This journal is © The Royal Society of Chemistry 2020 |