
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient non-doped blue fluorescent OLEDs with low efficiency roll-off based on hybridized local and charge transfer excited state emitters†
Xianhao
Lv‡
a,
Mizhen
Sun‡
a,
Lei
Xu
a,
Runzhe
Wang
a,
Huayi
Zhou
a,
Yuyu
Pan
 b,
Shitong
Zhang
c,
Qikun
Sun
a,
Shanfeng
Xue
*a and
Wenjun
Yang
a
b,
Shitong
Zhang
c,
Qikun
Sun
a,
Shanfeng
Xue
*a and
Wenjun
Yang
a
aKey Laboratory of Rubber-Plastics of the Ministry of Education/Shandong Province (QUST), School of Polymer Science & Engineering, Qingdao University of Science &Technology, 53-Zhengzhou Road, Qingdao 266042, P. R. China. E-mail: sfxue@qust.edu.cn
bSchool of Petrochemical Engineering, Shenyang University of Technology, 30 Guanghua Street, Liaoyang, 111003, P. R. China
cState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun, 130012, P. R. China
First published on 28th April 2020
Abstract
Designing a donor–acceptor (D–A) molecule with a hybridized local and charge transfer (HLCT) excited state is a very effective strategy for producing an organic light-emitting diode (OLED) with a high exciton utilization efficiency and external quantum efficiency. Herein, a novel twisting D–π–A fluorescent molecule (triphenylamine–anthracene–phenanthroimidazole; TPAAnPI) is designed and synthesized. The excited state properties of the TPAAnPI investigated through photophysical experiments and density functional theory (DFT) analysis reveal that its fluorescence is due to the HLCT excited state. The optimized non-doped blue OLED using TPAAnPI as a light-emitting layer exhibits a novel blue emission with an electroluminescence (EL) peak at 470 nm, corresponding to the Commission International de L'Eclairage (CIE) coordinates of (0.15, 0.22). A fabricated device termed Device II exhibits a maximum current efficiency of 18.09 cd A−1, power efficiency of 12.35 lm W−1, luminescence of ≈29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 cd cm−2, and external quantum efficiency (EQE) of 11.47%, corresponding to a high exciton utilization efficiency of 91%. Its EQE remains as high as 9.70% at a luminescence of 1000 cd m−2 with a low efficiency roll-off of 15%. These results are among the best for HLCT blue-emitting materials involved in non-doped blue fluorescent OLEDs. The performance of Device II highlights a great industrial application potential for the TPAAnPI molecule.
900 cd cm−2, and external quantum efficiency (EQE) of 11.47%, corresponding to a high exciton utilization efficiency of 91%. Its EQE remains as high as 9.70% at a luminescence of 1000 cd m−2 with a low efficiency roll-off of 15%. These results are among the best for HLCT blue-emitting materials involved in non-doped blue fluorescent OLEDs. The performance of Device II highlights a great industrial application potential for the TPAAnPI molecule.
Introduction
Organic light-emitting diodes (OLEDs) exhibit impressive advantages in full colour flat-panel displays and solid-state luminescence due to their high luminous efficiency and brightness, wide field-of-view, flexibility, and low cost.1–4 Although red and green emitting materials exhibit satisfactory performances, the acquisition of high-performance blue-emitting materials involving high efficiency and novel colour purity remains a major challenge. This is due to the intrinsic wide band gaps of blue emitters, which generate a high charge-injection barrier and unbalanced charge-injection and transportation.5 Internal quantum efficiency (ηint) up to 100% can be obtained for some blue phosphorescent OLEDs (phOLEDs) based on Ir, Pt, and Os complexes. However, some distinct disadvantages such as introducing valuable metals, low photoluminescence quantum efficiency, and deficiency of excellent blue phosphorescent materials still exist. Also, phosphorescent materials necessitate dispersion into proper host materials with large band gaps to reduce the quenching effect of long-lived triplet excitons.6 Therefore, novel metal-free blue fluorescent materials suitable for producing low cost non-doped OLEDs with high efficiency are needed. Thermally activated delayed fluorescence (TADF)7 and hybridized local and charge transfer (HLCT)8 are two recently reported mechanisms that can yield 100% exciton utilization efficiency (EUE) in blue fluorescent OLEDs. In TADF emitters, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) separation is demonstrated by proper selection and linkage of donor (D) and acceptor (A) moieties to form a D–A molecular structure, with blue-emissive OLEDEQE capable of attaining 30%.9 However, like phosphorescent OLEDs, their EQEs are commonly affected by severe roll-off at a high current density due to cumulated triplet excited states. Therefore, TADF materials require doping into host materials with high triplet energy, which increases their fabrication cost.10Similar to the TADF mechanism, the CT excited state component in the HLCT excited state facilitates the efficient reverse intersystem crossing (RISC) process by the small ΔEST, accounting for the high EUE. However, unlike TADF emitters, the HLCT-type emitters display short excited lifetimes, which are favourable for fabricating cheap high-efficiency non-doped OLEDs. Especially, in some blue-emissive weekly coupled HLCT materials, the final emissive excited state is a nearly pure locally excited (LE) state rather than a CT excited state. This is beneficial for the blue-shift emission wavelength due to a shortened lifetime and satisfactory efficiency.11
In this study, we designed and synthesized a novel blue HLCT molecule, the TPAAnPI. In this molecule, triphenylamine (TPA) is a moderate electron-donating moiety with a propeller conformation, while phenanthroimidazole (PI) represents a highly efficient violet-blue chromophore with moderate electron-accepting ability. Anthracene acts as a conjugated core linking TPA and PI. Hence, the twisting D–π–A molecular conformation and the moderate electron donating and accepting ability can facilitate the partial separation of HOMO and LUMO, thereby improving the CT component in the molecule. The presence of the anthracene moiety increases the conjugation length and improves the LE component. Concurrently, the 9,10-substituted anthracene enhances twisting, which efficiently prevents the aggregation-caused quenching effect.
The photophysical experiments and density functional theory (DFT) calculations manifest that the emissive state of TPAAnPI is a HLCT state. The TPAAnPI exhibits a novel blue emission with a PL peak at 470 nm both in THF and solid film state, with a photo-luminescent quantum yield (PLQY) of TPAAnPI of 0.71 and 0.42, respectively. We fabricated the TPAAnPI non-doped Device I, which shows a novel blue emission with an electroluminescence (EL) emission peak at 470 nm, corresponding to the Commission International de L'Eclairage (CIE) coordinates of (0.15, 0.22). The maximum external quantum efficiency (EQE) is 8.00% at a luminance of 705 cd m−2. Although the efficiency roll-off is only of 0.75% at the luminance of 1000 cd m−2, the EQE remains unsatisfactory for industrial application. Further, the optimized non-doped Device II exhibited a maximum CE of 18.09 cd A−1, PE of 12.35 lm W−1, luminescence of ≈29900 cd cm−2, and EQE of 11.47%, corresponding to a high EUE of 91%. More importantly, the EQE remains as high as 9.70% at a luminescence of 1000 cd m−2. The efficiency roll-off is only 15% and the EL peak is still at 470 nm with CIE coordinates of (0.15, 0.22). We attribute the high performances of the non-doped device to the hybridized local and charge transfer (HLCT) excited state. These results are among the best involving HLCT blue-emissive materials for blue fluorescent OLEDs.
Results and discussion
Synthesis and characterization
The TPAAnPI synthesis route is illustrated in Scheme 1. PPI–Br was synthesized through a one-pot reaction of 9,10-phenanthraquinone, 4-aminobenzonitrile, ammonium acetate, and 4-bromobenzaldehyde. The corresponding boron ester, PPIB, was then immediately produced using the PPI–Br crude product. Subsequently, PIAn–Br was formed using a Suzuki coupling reaction between PPIB and 9,10-dibromoanthracene. Finally, the target product, TPAAnPI, was synthesized through a Suzuki coupling reaction between PIAn–Br and TPAB. Details of the synthesis are provided in the ESI,† with the 1H NMR spectra of the intermediate products displayed in Fig. S1–S3.† The purity and chemical structure of TPAAnPI are well characterized by 1H NMR (Fig. S4†), 13C NMR (Fig. S5†) and mass spectroscopy (Fig. S6†) and elemental analysis. | ||
| Scheme 1 (a) CH3COONH4,CH3COOH, 120 °C, N2 protection, 2 h. (b) Pd(dppf)Cl2, KOAc, 1,4-dioxane, 90 °C, N2 protection, overnight. (c) K2CO3, Pd(PPh3)4, THF & H2O, 70 °C, 24 h under N2 protection. | ||
Thermal properties
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed in a nitrogen atmosphere to evaluate the thermal properties of TPAAnPI. As depicted in Fig. S7,† the decomposition temperature (Td, corresponding to 5% weight loss) of TPAAnPI is 492 °C and the melting point (Tm) is at 366 °C, highlighting excellent thermal stability. Simultaneously, a glass-transition temperature (Tg) of 213 °C occurs on the DSC curve, indicating excellent morphological stability during heating in industrial production.Excitation-state properties
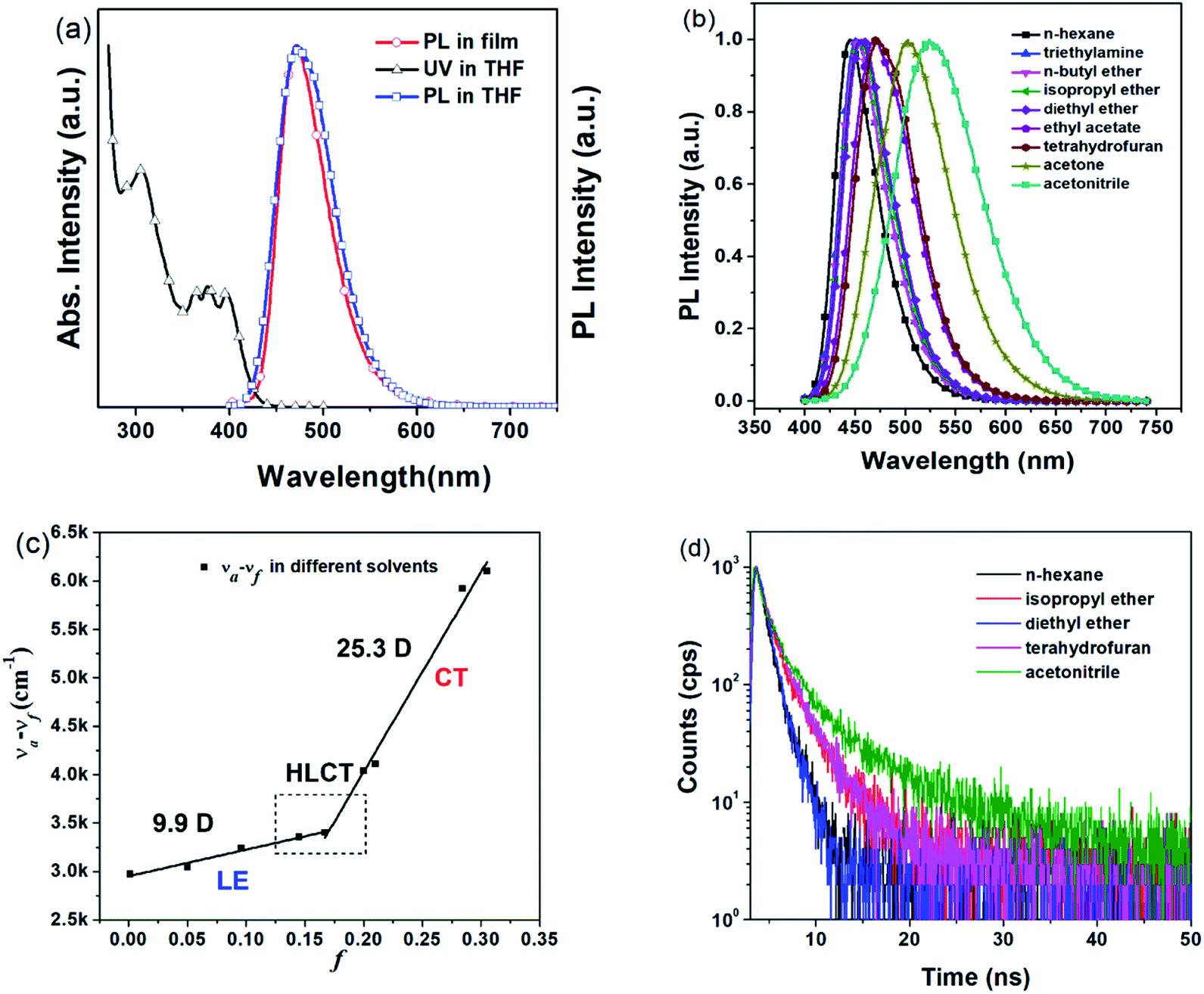 | ||
| Fig. 1 (a) Normalized UV-vis and PL spectra of TPAAnPI in the THF solution (10−5 M) and neat film. (b) Normalized PL spectra of TPAAnPI in nine different solvents with increasing polarity. (c) Linear fitting of the Lippert–Mataga model (the solid squares represent the Stocks shifts in different solvents, and the lines are fitted for the solvatochromic model), and the ground-state dipole moment (μg) was estimated to be 4.0332 D using DFT calculations at the level of M062X/6-31+G(d,p). (d) The transient PL decay spectra of TPAAnPI in different solvents and evaporated films. | ||
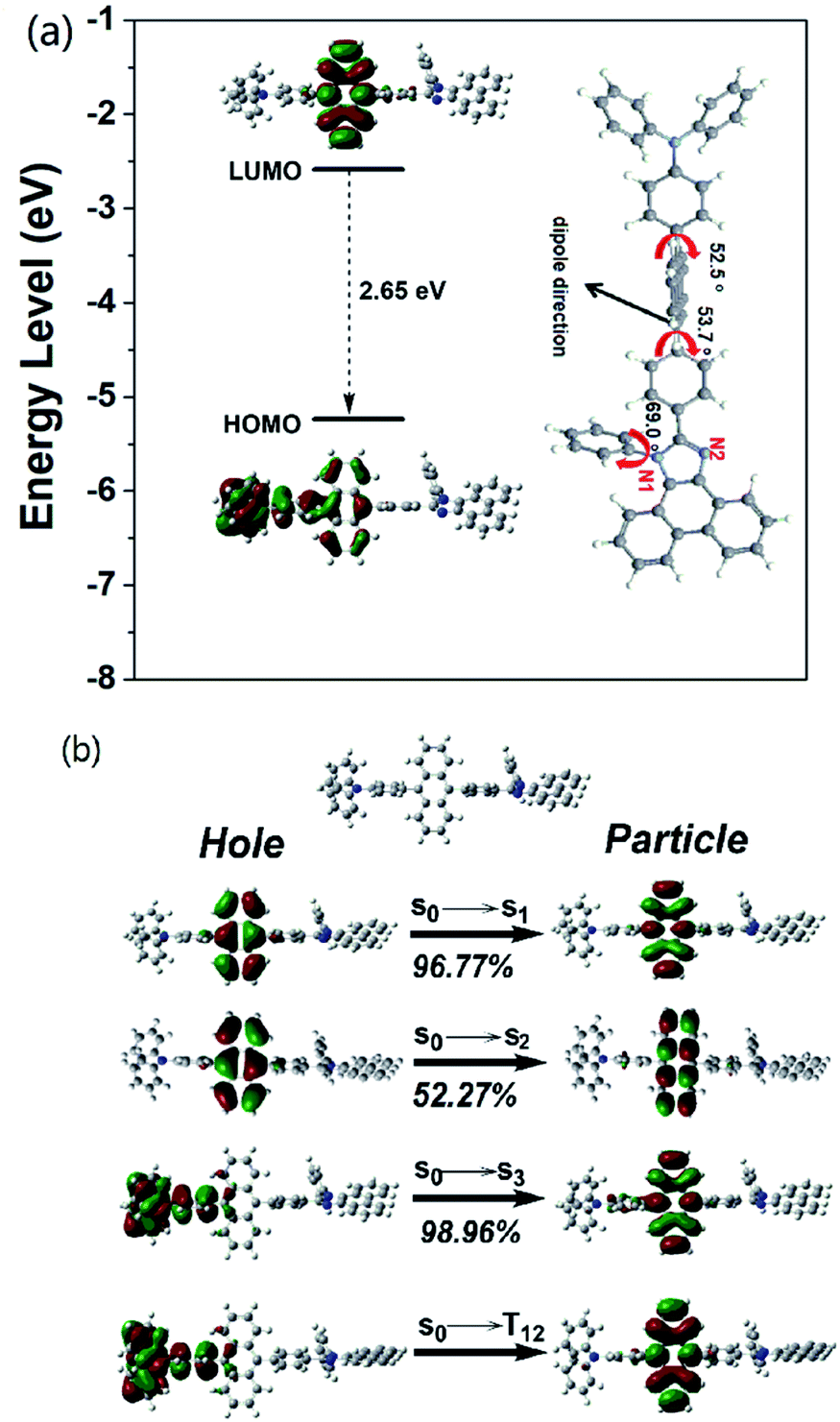 | ||
| Fig. 2 (a) The frontier molecular orbitals (highest occupied molecular orbital, HOMO and lowest unoccupied molecular orbital, LUMO) of TPAAnPI and its optimized molecular geometry of ground state (S0) at the M062X/6-31+G(d,p) level, (b) the NTOs of TPAAnPI for S0/S1, S0/S2, S0/S3 and S0/T12, S0: singlet ground state; S1: the lowest singlet excited state; S2: the second singlet excited state; S3: the third singlet excited state; T12: the twelfth triplet excited state. The percentage indicates the possibility of the transition. | ||
Theoretical calculations
Regarding the HLCT properties of TPAAnPI, we investigated its excited state using theoretical methods. First, the molecular configuration and frontier molecular orbital (FMO) of the TPAAnPI were optimized and calculated using the Gaussian 09 B.01 package at the level of M062X/6-31+G(d,p), as shown in Fig. 2a. As expected, in the 9,10-substituted anthracene derivate, the optimized structure of the TPAAnPI involves a twisted D–π–A molecular configuration with torsional angles of 52.5, 53.7, and 69.0°, respectively. Therefore, the calculated FMO exhibits partial separated characteristics. The highest occupied molecular orbital (HOMO) of the TPAAnPI is mainly localized on the TPA moiety but extends to the adjacent anthracene moiety. In contrast, the lowest unoccupied molecular orbital (LUMO) is fully distributed on the anthracene moiety, suggesting that the HOMO → LUMO transition involves an intercrossed CT and π–π* transition character, reflecting a typical HLCT character. Furthermore, NTO analysis was performed for the first 10 singlet and 15 triplet excited states based on the S0 state geometry using the time-dependent DFT (TDDFT) method at the same level as S0 (Fig. 2b and S9†). The S0 → S1, and S0 → S2 transitions of the TPAAnPI are the radiative π–π* and non-radiative n–π* transitions of anthracene, respectively, which are typical LE transitions. The oscillator strength of the S1 excited state is 0.5449 which is consistent with the PLQY in low-polarity solutions such as hexane. We also calculated the excited state dipole moment of the singlet excited state (Table S3†), obtaining values of 4.16 D and 27.39 D for the S1 and S3 states, respectively, which are also in accordance with the experimental results. Therefore, the emissive state of the TPAAnPI belongs to the S1 excited state, whereas the S3 excited state serves as the exciton utilization channel. Actually, the S3 excited state exhibits obvious CT character according to the NTO analysis. The NTO for S0 → S3 transition displays a total CT transition character, with a “hole” distributed on the TPA moiety and a “particle” distributed on the anthracene with a minor overlap on the adjacent phenyl moiety. The corresponding triplet CT excited state is the T12 excited state, and the S3–T12 energy gap is only 0.0013 eV. Therefore, this could afford efficiently increasing the RISC between S3 and T12 excited states, and eventually improving the EUE.Electrochemical properties
To understand the electrochemical properties of the TPAAnPI, the HOMO and LUMO energy levels were obtained from the electrochemical cyclic voltammetry measurements (CV) depicted in Fig. S10.† The onset oxidation and reduction potentials of the TPAAnPI are 0.67 and −2.14 V, with a corresponding HOMO and LUMO of −5.24 eV and −2.59 eV, respectively, according to Formula S3† (ferrocene was used as the internal standard, E+1/2 vs. Ag/Ag+ was measured as 0.23 V, and E−1/2 vs. Ag/Ag+ was 0.07 V). The calculated electrical band gap of 2.65 eV is blue-emissive (Table 1).| Compound | λ abs(s/f)a [nm] | λ PL(s/f)b [nm] | T g/Tmc [°C] | T d [°C] | HOMOe [eV] | LUMOe [eV] | E g [eV] | η PL(s/f)f [%] |
|---|---|---|---|---|---|---|---|---|
| a λ abs(s): maximum absorption wavelength in dilute THF (10−5 M); λabs(f): maximum absorption wavelength in the film. b λ PL(s): emission peak in dilute THF; λPL(f): emission peak in film. c T g: glass transition temperature; Tm: melting temperature. d T d: decomposition temperature. e Measured by cyclic voltammetry measurements. f η PL(s): the fluorescence quantum yield in dilute THF (10−5 M); ηPL(f): the fluorescence quantum yield in evaporated film. | ||||||||
| TPAAnPI | 395/404 | 470/470 | 213/366 | 492 | −5.24 | −2.59 | 2.65 | 71/42 |
Electroluminescence properties
To explore the potential application of the TPAAnPI as an electro-fluorescent material, a non-doped blue fluorescent OLED was fabricated. The optimized structures of the devices referred to as Device I and Device II contain the following: Device I: ITO/PEDOT:PSS (40 nm)/TAPC (5 nm)/TCTA (30 nm)/TPAAnPI (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm) and Device II: ITO/PEDOT:PSS (40 nm)/TCTA (30 nm)/TPAAnPI (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm).The conducting polymer polyethylene dioxythiophene/polystyrene sulfonate (PEDOT:PSS) served as the hole injecting layer, while di-(4-(N,N-ditolylamino)phenyl) cyclohexane (TAPC) acted as the hole transporting layer. 4,4′,4′′-tri(N-carbazolyl)triphenylamine (TCTA) was used as the hole transporting and electron blocking layer, whereas 1,3,5-tri(phenyl-2-benzimidazolyl)benzene (TPBi) represented an electron transporting and hole blocking layer and LiF the electron injecting layer. The summary properties of Device I and II are presented in Table 2.| Device | V turn-on [V] | CEmaxb [cd A−1] | PEmaxc [lm W−1] | EQEmax/1000/10000d [%] |
L max [cd m−2] | λ EL [nm] | V end [V] | CIEh [X, Y] | η roll-off [%] |
|---|---|---|---|---|---|---|---|---|---|
|
a Turn on voltage at the luminescence of 1 cd m−2.
b Maximal current efficiency.
c Maximal power efficiency.
d EQEmax: maximal external quantum efficiency; EQE1000: external quantum efficiency at the luminescence of 1000 cd m−2; EQE10000: external quantum efficiency at the luminescence of 10000 cd m−2.
e Maximum luminescence.
f Maximal EL peak value.
g End voltage at the luminescence of 1 cd m−2.
h Commission International de L'Eclairage (CIE) coordinates were obtained from EL spectra at the voltage of 6 V.
i Efficiency roll-off. Device I structure of: ITO/PEDOT:PSS (40 nm)/TAPC (5 nm)/TCTA (30 nm)/TPAAnPI (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm); Device II structure: ITO/PEDOT:PSS (40 nm)/TCTA (30 nm)/TPAAnPI (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm).
|
|||||||||
| I | 3.20 | 12.51 | 11.47 | 8.00/7.94/6.41 | 22489 |
470 | 7.80 | 0.15, 0.22 | 0.75 |
| II | 3.40 | 18.09 | 12.35 | 11.47/9.70/6.80 | 29900 |
470 | 7.00 | 0.15, 0.22 | 15 |
As shown in Fig. 3, Device I exhibited a novel blue emission, with the electroluminescence (EL) emission peak at 470 nm corresponding to the Commission International de L'Eclairage (CIE) coordinates of (0.15, 0.22) at 6 V (Fig. 3d). Its maximum current efficiency (CE) was 12.51 cd A−1 and the maximum power efficiency (PE) was 11.47 lm W−1. It also displayed a maximum luminescence of ≈22489 cd cm−2 and maximum external quantum efficiency (EQE) of only 8.00% at a luminescence of 705 cd m−2. Although the efficiency roll-off (ηroll-off) is only 0.75% (according to Formula S4†) at the luminance of 1000 cd m−2, the EQE remains unsatisfactory in industrial applications. Therefore, the non-doped Device II was fabricated to improve the performances. The optimized non-doped Device II exhibited a maximum CE of 18.09 cd A−1 and a maximum PE of 12.35 lm W−1. Its maximum luminescence is ≈29900 cd cm−2 and its maximum EQE is 11.47%, corresponding to the high EUE of 91%. More importantly, its EQE remains as high as 9.70% at the luminescence of 1000 cd m−2. Also the ηroll-off is only 15% and the EL peak is still at 470 nm with CIE coordinates of (0.15, 0.22) (Fig. 3d). Further the EL spectra are stable, with little vibronic features over the entire drive voltage range of 4 V to 8 V (Fig. S11†). The high EQE and the low ηroll-off of Device I and II further highlight a great potential of the TPAAnPI molecule for industrial applications.
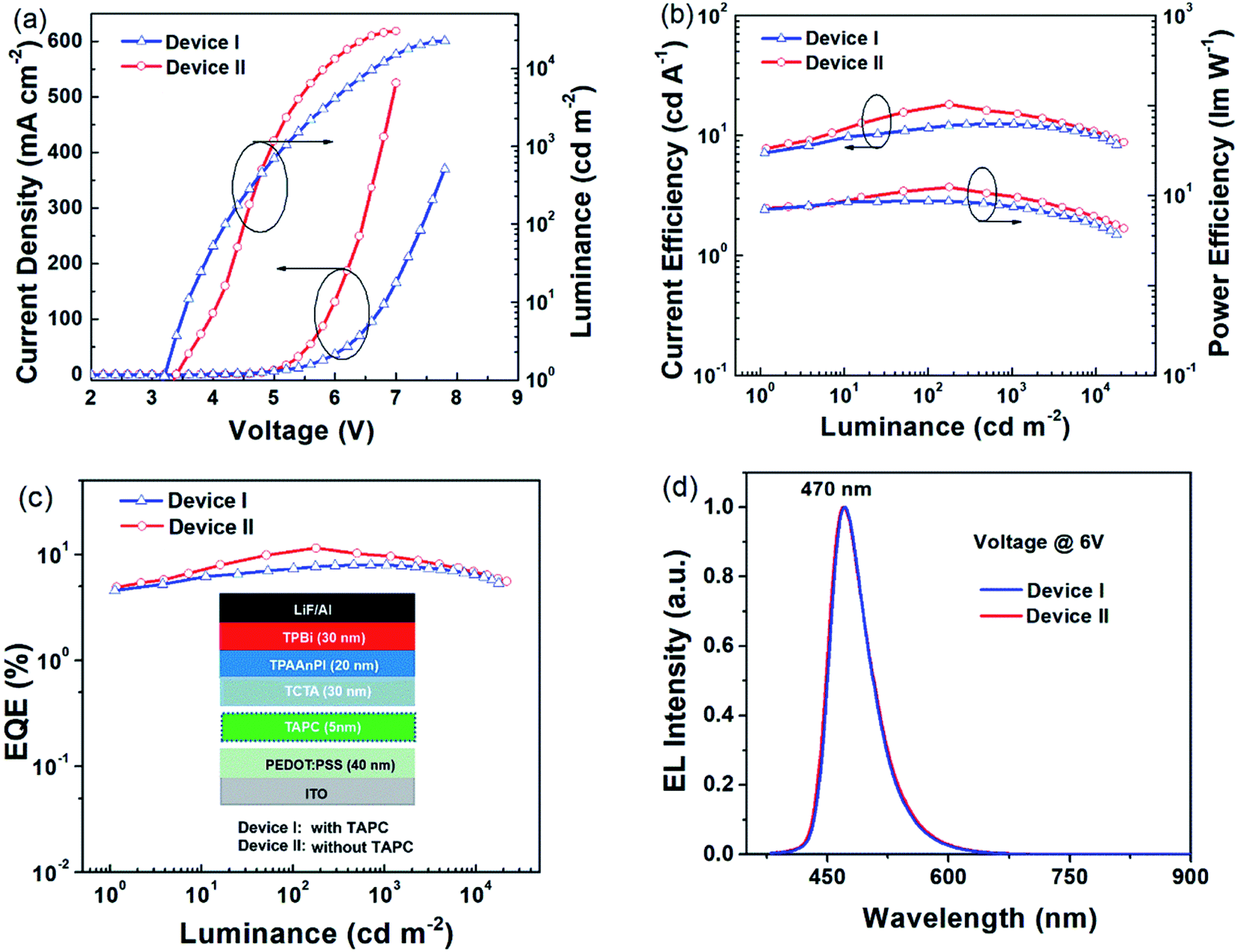 | ||
| Fig. 3 (a) The current density–voltage–luminance curve of Device I & II. (b) The current efficiency–luminance–power efficiency curves of Device I & II. (c) The external efficiency–luminance curves of Device I & II, the inset shows the device structure diagram. (d) Normalized EL spectra of Device I & II at 6 V. | ||
Then, the radiative exciton utilization efficiency of Device II was estimated according to the following equation: ηEL = ηrecηsηPLηout, where ηEL is the EQE of the device, ηrec denotes the recombination efficiency of the injected holes and electrons, ηPL is the PLQY of the emitter, ηs represents the EUE of device, and ηout is the light out-coupling efficiency. Assuming an ηout ≈ 0.3 for a glass substrate with a refraction index n of 1.5, ηrec is 100%, and ηPL of the TPAAnPI in solid film state is 42%. Therefore, the calculated ηs is 91%, breaking the limit of 25% singlet excitons of spin statistics rule (singlet/triplet: ∼1/3) in conventional fluorescent OLEDs,15 implying that almost all triplet excitons are converted to the singlet in the EL process. The recent non-doped blue OLEDs with CIEy ≈ 0.2 are listed in Table S4.†
The high ηs is attributed to the novel HLCT mechanism in the EL process. The small energy splitting between S3 and T12 (ΔEST ≈ 0) shown in Fig. 4 can create a potential RISC process from T12 to S3. In this way, the triplet excitons are converted to the singlet state in the EL process. According to the energy-gap law,16 the large energy gap between T2 and T1 (ΔET1T2= 1.07 eV) inhibits the internal conversion (IC) process, and the more competitive RISC process between T12 and S3 is prioritized because of the narrower energy gap (Fig. S12†). Therefore, a large fraction of the electro-generated triplet excitons can turn into singlet excitons through the T12 → S3 channel. Moreover, the large energy gap (ΔEST = 1.35 eV) between S1 and T1 prevents the ISC process from the low-lying S1 to T1 and excludes the existence of the TADF mechanism. More importantly, we measured the transient EL decay curves of Device II at different voltages (Fig. S13(a)†). The curves consisted of two components: a rapid EL decay originating from the promoted fluorescence of S1 and a delayed one. The delayed one is due to the collision recombination of previously injected holes and electrons which stayed in the emitter layer after the voltage was turned off.
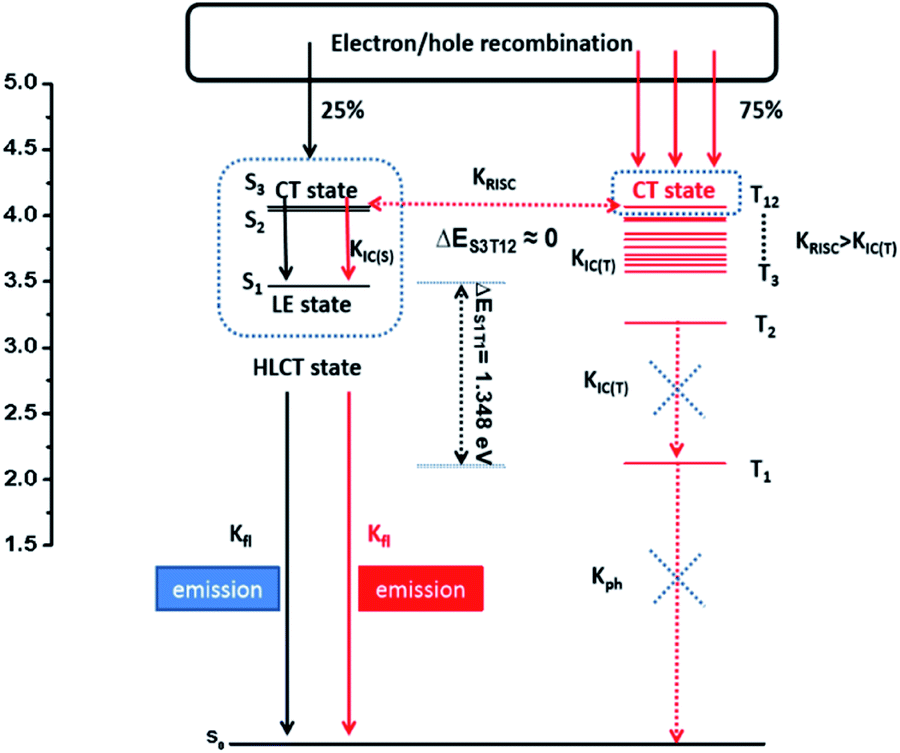 | ||
| Fig. 4 Model for exciton relaxation in the EL process. RISC: reverse intersystem crossing; IC(T): internal conversion between the triplet states; IC(S): internal conversion between the singlet states; fl: fluorescence; ph: phosphorescence. | ||
The amplified delayed component was obtained by taking the logarithm of EL intensity as Y and the logarithm of time as X, and the slope of the obtained curve is −0.28 in the initial time and −1.29 at the long lived EL transient (in Fig. S13(b)†). According to Monkman et al.,17 if the TTA mechanism is predominant in Device II, the delayed EL intensity should be proportional to t−2 at the long lived EL transient and the obtained slope should be −2 instead of −1.29. We further compared the transient EL decay curves at different driving voltages of Device II with a typical TTA device based on MADN (Fig. S13(c)†). The EL intensity of Device II decays much faster than that of the MADN-based device. As the driving voltage increased from 6 V to 8 V, the ratio of the delayed component of the MADN-based device decreased due to the enhanced triplet exciton quenching process. However, the delayed component of Device II exhibited a much shorter decay time and showed no obvious dependence on the driving voltage. The above results suggested that the contribution of the TTA is insignificant in the EL process. Therefore, the HLCT mechanism is likely responsible for the radiative exciton ratio above 25% in the non-doped OLED based on the TPAAnPI molecule.
Conclusions
In summary, a twisting novel blue D–π–A molecule, TPAAnPI, involving the HLCT excited state was designed and synthesized. The HLCT characteristic was demonstrated by photophysical experiments and DFT analysis. A fabricated optimized non-doped Device II based on TPAAnPI exhibited a maximum EQE of 11.47% and showed novel blue emission at 470 nm with CIE coordinates of (0.15, 0.22). More importantly, the EQE remained as high as 9.70% at the luminescence of 1000 cd m−2 with a ηroll-off of only 15%. The maximum EQE was only 8.00% for Device I, with an almost negligible ηroll-off of 0.75% at a luminance of 1000 cd m−2. These results are among the best for non-doped blue fluorescent OLEDs of HLCT blue-emissive materials. Our study therefore, proposed a molecular design method for a blue-emissive HLCT material characterized by a high radiative exciton ratio, high EQE, and low ηroll-off. These results are beneficial for further exploring molecular design and device optimization that can maximize triplet to singlet exciton up-conversion involving the HLCT mechanism. We believe that our results are valuable for improved practical applications in display and lighting.Experimental section
Materials
All raw materials were commercially purchased from Aldrich Chemical Co. or Energy Chemical Co., China and used without further purification unless otherwise noted. Tetrahydrofuran (THF) was dried and purified by fractional distillation over sodium/benzophenone before use.Measurements
The 1H NMR and 13C NMR spectra were recorded on a Bruker AC500 spectrometer at 500 and 125 MHz, respectively, using deuterated chloroform (CDCl3) or dimethyl sulphoxide (DMSO) as solvents. The chemical shift for each signal was reported in ppm units with tetramethylsilane (TMS) as a standard internal reference. The MALDI-TOF-MS mass spectra were recorded using an AXIMA-CFRTM instrument. The compounds were characterized by using a Flash EA 1112, CHN elemental analysis instrument.Device fabrication and characterization
The two typical device configurations fabricated by vacuum deposition were ITO/PEDOT:PSS (40 nm)/TCTA (30 nm)/TPAAnPI (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm) and ITO/PEDOT:PSS (40 nm)/TAPC (5 nm)/TCTA (30 nm)/TPAAnPI (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm). ITO-coated glass with a sheet resistance of 10 Ω per square was used as the substrate. The pre-treatment of ITO glass included routine chemical cleaning using detergent and alcohol in sequence and drying in an oven at 120 °C. After oxygen plasma cleaning it for 4 min it was finally transferred to a vacuum deposition system with a base pressure greater than 5 × 10−6 mbar for organics and metal deposition. The PEDOT (polyethylene dioxythiophene) and PSS (polystyrene sulfonate) mixture layer was deposited onto the ITO-coated substrate as the hole-injection layer. The cathode LiF was deposited by thermo-evaporation and followed by a thick Al capping layer. The current–voltage–luminance characteristics were measured by using a Keithley source measurement unit (Keithley 2400 and Keithley 2000) with a calibrated silicon photodiode. The electroluminescent (EL) spectra and Commission Internationale de l’Eclairage (CIE) coordinates of these devices were measured by using a PR650 spectroscan spectrometer. EQEs were calculated from the luminance, current density, and EL spectrum, assuming a Lambertian distribution (Formula S5†). All the results were measured in the forward-viewing direction without using any light outcoupling technique.Synthesis of TPAAnPI
A mixture of PIAn–Br (0.95 g, 1.52 mmol), N,N-diphenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (0.53 g, 1.83 mmol), K2CO3 (0.31 g, 2.24 mmol), tetrahydrofuran (THF, 50 ml) and deionized water (H2O, 10 ml), with Pd(PPh3)4 (0.05 g, 0.043 mmol) acting as a catalyst was refluxed at 70 °C for 24 h under nitrogen. After the mixture was cooled down, 50 ml H2O was added to the resulting solution and the mixture was extracted with dichloromethane several times. The organic phase was dried over MgSO4. After filtration and solvent evaporation, the given residue was purified through silica gel column chromatography using dichloromethane/petroleum ether (3:1; v/v) as an eluent to produce the product in the form of a light yellow solid (0.90 g, 75%). 1H NMR (500 MHz, chloroform-d) δ = 8.97 (d, J = 7.9 Hz, 1H), 8.81 (d, J = 8.4 Hz, 1H), 8.75 (d, J = 8.3 Hz, 1H), 7.85 (dd, J = 8.3, 5.9 Hz, 4H), 7.79 (t, J = 7.5 Hz, 1H), 7.73–7.64 (m, 8H), 7.54 (t, J = 7.7 Hz, 1H), 7.46–7.30 (m, 15H), 7.27 (d, J = 9.0 Hz, 6H), and 7.09 (t, J = 7.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ = 150.75, 147.78, 147.17, 139.81, 138.85, 137.56, 137.25, 136.08, 132.48, 132.08, 131.33, 130.23, 130.03, 129.94, 129.77, 129.38, 129.33, 129.22, 128.31, 127.32, 127.26, 127.10, 126.79, 126.31, 125.66, 125.11, 124.97, 124.69, 124.14, 123.08, 122.81, 120.91, 77.26, 77.01, 76.76, and 53.41. MALDI-TOF MS (mass m/z): 789.29 [M+]. Anal. calcd for C59H39N3: C, 89.70; H, 4.98; N, 5.32, found: C, 89.43; H, 5.00; N, 5.57.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Science Foundation of China (No. 51873095, 51673105 and 51803071), the Key Project of Higher Educational Science and Technology Program of Shandong Province of China (No. J18KZ001), and the Natural Science Foundation of Qingdao City of China (No. 16-5-1-89-jch). We thank the Open Project of the State Key Laboratory of Supramolecular Structure and Materials of Jilin University (SKLSSM-202032). We appreciate assistance from the State Key Laboratory of Luminescent Materials and Devices of the South China University of Technology for fabricating OLED devices.References
- (a) C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS; (b) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS; (c) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- (a) Y. Cao, I. D. Parker, G. Yu, C. Zhang and A. J. Heeger, Nature, 1999, 397, 414–417 CrossRef CAS PubMed; (b) S. Krotkus, D. Kasemann, S. Lenk, K. Leo and S. Reineke, Light: Sci. Appl., 2016, 5, e16121 CrossRef CAS PubMed; (c) J. Lee, M. Slootsky, K. Lee, Y. Zhang and S. R. Forrest, Light: Sci. Appl., 2014, 3, e181 CrossRef CAS; (d) W. H. Lee, Z. Zhao, Y. J. Cai, Z. Xu, Y. Yu, Y. Xiong, R. T. K. Kwok, Y. Chen, N. L. C. Leung, D. G. Ma, J. W. Y. Lam, A. J. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 6118–6125 RSC; (e) C. L. Li, J. B. Wei, X. X. Song, K. Q. Ye, H. Y. Zhang, J. Y. Zhang and Y. Wang, J. Mater. Chem. C, 2016, 4, 7013–7019 RSC; (f) C. L. Li, S. P. Wang, W. P. Chen, J. B. Wei, G. C. Yang, K. Q. Ye, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 10632–10635 RSC.
- (a) K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062–5066 CrossRef CAS PubMed; (b) J.-H. Lee, S.-H. Cheng, S.-J. Yoo, H. Shin, J.-H. Chang, C.-I. Wu, K.-T. Wong and J.-J. Kim, Adv. Funct. Mater., 2015, 25, 361–366 CrossRef CAS; (c) T. Suzuki, Y. Nonaka, T. Watabe, H. Nakashima, S. Seo, S. Shitagaki and S. Yamazaki, Jpn. J. Appl. Phys., 2014, 53, 052102 CrossRef; (d) T. B. Njuyan, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Mater., 2020, 1906614 Search PubMed; (e) L. Yu, Z. B. Wu, G. H. Xie, W. X. Zeng, D. G. Ma and C. L. Yang, Chem. Sci., 2018, 9, 1385–1391 RSC; (f) Z. Q. Li, C. L. Li, Y. C. Xu, N. Xie, X. C. Jiao and Y. Wang, J. Phys. Chem. Lett., 2019, 10, 842–847 CrossRef CAS PubMed.
- (a) Y. L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hanisch, S. Reineke, J. Fan and L. S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef CAS PubMed; (b) Q. Wang and D. G. Ma, Chem. Soc. Rev., 2010, 39, 2387–2398 RSC; (c) H.-H. Chou, Y.-H. Chen, H.-P. Hsu, W.-H. Chang, Y.-H. Chen and C.-H. Cheng, Adv. Mater., 2012, 24, 5867–5871 CrossRef CAS PubMed; (d) N. Sharma, E. Spuling, C. M. Mattern, W. B. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Brase, I. D. W. Samuel and E. Z. Colman, Chem. Sci., 2019, 10, 6689–6696 RSC; (e) C. L. Li, Z. Q. Li, X. J. Yan, Y. W. Zhang, Z. L. Zhang and Y. Wang, J. Mater. Chem. C, 2017, 5, 1973–1980 RSC; (f) C. L. Li, J. B. Wei, J. X. Han, Z. Q. Li, X. X. Song, Z. L. Zhang, J. J. Zhang and Y. Wang, J. Mater. Chem. C, 2016, 4, 10120–10129 RSC.
- (a) H. Sasabe, J. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Lange, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003–5007 CrossRef CAS; (b) T. Fleetham, G. J. Li, L. L. Wen and J. Li, Adv. Mater., 2014, 26, 7116–7121 CrossRef CAS PubMed; (c) C. L. Li, Z. Q. Li, J. X. Liang, H. Luo, Y. Liu, J. B. Wei and Y. Wang, J. Mater. Chem. C, 2018, 6, 12888–12895 RSC; (d) Z. Q. Li, Z. Cheng, J. Y. Lin, N. Xie, C. L. Li, G. C. Yang and Y. Wang, J. Mater. Chem. C, 2019, 7, 13486–13492 RSC.
- (a) S. Kappaun, C. Slugovc and J. W. List, Int. J. Mol. Sci., 2008, 9, 1527–1547 CrossRef CAS PubMed; (b) G. Cheng, S. C. F. Kui, W.-H. Ang, M.-Y. Ko, P.-K. Chow, C.-L. Kwong, C.-C. Kwok, C. S. Ma, X. G. Guan, K.-H. Low, S.-J. Su and C.-M. Che, Chem. Sci., 2014, 5, 4819–4830 RSC; (c) Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef; (d) V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. P. Loebl, V. van Elsbergen, H. Boerner, U. Scherf and M. Kreyenschmidt, J. Fluorine Chem., 2009, 130, 640–649 CrossRef CAS; (e) K. Klimes, Z. Q. Zhu and J. Li, Adv. Funct. Mater., 2019, 29, 1903068 CrossRef.
- (a) Q. S. Zhang, B. Li, S. P. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS; (b) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed; (c) X. K. Liu, Z. Chen, J. Qing, W. J. Zhang, B. Wu, H. L. Tam, F. R. Zhu, X. H. Zhang and C. S. Lee, Adv. Mater., 2015, 27, 7079–7085 CrossRef CAS PubMed; (d) D. X. Ding, Z. C. Wang, C. Y. Li, J. Zhang, C. B. Duan, Y. Wei and H. Xu, Adv. Mater., 2020, 1906950 CrossRef CAS; (e) I. S. Park, H. Komiyama and T. Yasuda, Chem. Sci., 2017, 8, 953–960 RSC.
- (a) W. J. Li, D. D. Liu, F. Z. Shen, D. G. Ma, Z. M. Wang, T. Feng, Y. X. Xu, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS; (b) S. T. Zhang, W. J. Li, L. Yao, Y. Y. Pan, F. Z. Shen, R. Xiao, B. Yang and Y. G. Ma, Chem. Commun., 2013, 49, 11302–11304 RSC; (c) S. T. Zhang, L. Yao, Q. M. Peng, W. J. Li, Y. Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. M. Wang, P. Lu, F. Li, S. J. Su, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS; (d) C. J. Zhou, D. L. Cong, Y. Gao, H. C. Liu, J. Y. Li, S. T. Zhang, Q. Su, Q. L. Wu and B. Yang, J. Phys. Chem. C, 2018, 122, 18376–18382 CrossRef CAS; (e) S. F. Xue, X. Qiu, S. A. Ying, Y. S. Lu, Y. Y. Pan, Q. K. Sun, C. Gu and W. J. Yang, Adv. Opt. Mater., 2017, 5, 1700747 CrossRef; (f) J. J. Shi, Q. Ding, L. Xu, X. H. Lv, Z. W. Liu, Q. K. Sun, Y. Y. Pan, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2018, 6, 11063–11070 RSC.
- (a) D. R. Lee, M. Kim, S. K. Jeon, S. H. Hwang, C. W. Lee and J. Y. Lee, Adv. Mater., 2015, 27, 5861–5867 CrossRef CAS PubMed; (b) M. Kim, S. K. Jeon, S.-H. Hwang, S.-S. Lee, E. Yu and J. Y. Lee, J. Phys. Chem. C, 2016, 120, 2485–2493 CrossRef CAS.
- (a) D. D. Zhang, M. H. Cai, Y. G. Zhang, D. Q. Zhang and L. Duan, ACS Appl. Mater. Interfaces, 2015, 7, 28693–28700 CrossRef CAS PubMed; (b) D. D. Zhang, L. Duan, C. Li, Y. L. Li, H. Y. Li, D. Q. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed; (c) H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- (a) W. J. Li, Y. Y. Pan, R. Xiao, Q. Peng, S. T. Zhang, D. G. Ma, F. Li, F. Z. Shen, Y. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS; (b) H. C. Liu, Q. Bai, W. J. Li, Y. C. Guo, L. Yao, Y. Gao, J. Y. Li, P. Lu, B. Yang and Y. G. Ma, RSC Adv., 2016, 6, 70085–70090 RSC; (c) X. Y. Tang, Q. Bai, Q. M. Peng, Y. Gao, J. Y. Li, Y. L. Liu, L. Yao, P. Lu, B. Yang and Y. G. Ma, Chem. Mater., 2015, 27, 7050–7057 CrossRef CAS; (d) H. C. Liu, Q. Bai, L. Yao, H. Y. Zhang, W. J. Li, Y. Gao, J. Y. Li, P. Lu, H. Y. Wang, B. Yang and Y. G. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC.
- R. N. Jones, Chem. Rev., 1947, 41, 353–371 CrossRef CAS PubMed.
- Z. Gao, Y. L. Liu, Z. M. Wang, F. Z. Shen, H. Liu, G. N. Sun, L. Yao, Y. Lv, P. Lu and Y. G. Ma, Chem.–Eur. J., 2013, 19, 2602–2605 CrossRef CAS PubMed.
- S. Tang, W. J. Li, F. Z. Shen, D. D. Liu, B. Yang and Y. G. Ma, J. Mater. Chem., 2012, 22, 4401–4408 RSC.
- S. J. Su, E. Gonmori, H. Sarabe and J. Kido, Adv. Mater., 2008, 20, 4189–4194 CAS.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, USA, 1991 Search PubMed.
- C. Rothe and A. P. Monkman, Phys. Rev. B, 2003, 68, 075208 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterization, general methods, detailed PL and EL performance and detailed TDDFT calculations, DSC curves, TGA curves, CV curves and the transient EL decay curves of the device. See DOI: 10.1039/d0sc01341b |
| ‡ Xianhao Lv and Mizhen Sun contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |