
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of primary amines catalyzed by thermotolerant fungal reductive aminases†
Juan
Mangas-Sanchez
 a,
Mahima
Sharma
b,
Sebastian C.
Cosgrove
a,
Jeremy I.
Ramsden
a,
James R.
Marshall
a,
Thomas W.
Thorpe
a,
Ryan B.
Palmer
a,
Gideon
Grogan
*b and
Nicholas J.
Turner
*a
a,
Mahima
Sharma
b,
Sebastian C.
Cosgrove
a,
Jeremy I.
Ramsden
a,
James R.
Marshall
a,
Thomas W.
Thorpe
a,
Ryan B.
Palmer
a,
Gideon
Grogan
*b and
Nicholas J.
Turner
*a
aSchool of Chemistry, University of Manchester, Manchester Institute of Biotechnology, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: nicholas.turner@manchester.ac.uk
bYork Structural Biology Laboratory, Department of Chemistry, University of York, YO10 5DD York, UK
First published on 5th May 2020
Abstract
Chiral primary amines are important intermediates in the synthesis of pharmaceutical compounds. Fungal reductive aminases (RedAms) are NADPH-dependent dehydrogenases that catalyse reductive amination of a range of ketones with short-chain primary amines supplied in an equimolar ratio to give corresponding secondary amines. Herein we describe structural and biochemical characterisation as well as synthetic applications of two RedAms from Neosartorya spp. (NfRedAm and NfisRedAm) that display a distinctive activity amongst fungal RedAms, namely a superior ability to use ammonia as the amine partner. Using these enzymes, we demonstrate the synthesis of a broad range of primary amines, with conversions up to >97% and excellent enantiomeric excess. Temperature dependent studies showed that these homologues also possess greater thermal stability compared to other enzymes within this family. Their synthetic applicability is further demonstrated by the production of several primary and secondary amines with turnover numbers (TN) up to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 as well as continous flow reactions, obtaining chiral amines such as (R)-2-aminohexane in space time yields up to 8.1 g L−1 h−1. The remarkable features of NfRedAm and NfisRedAm highlight their potential for wider synthetic application as well as expanding the biocatalytic toolbox available for chiral amine synthesis.
000 as well as continous flow reactions, obtaining chiral amines such as (R)-2-aminohexane in space time yields up to 8.1 g L−1 h−1. The remarkable features of NfRedAm and NfisRedAm highlight their potential for wider synthetic application as well as expanding the biocatalytic toolbox available for chiral amine synthesis.
Introduction
Chiral amines are a privileged functional group, ubiquitous in natural products, pharmaceuticals, agrochemicals and fine chemicals.1 Consequently, the discovery and development of more efficient ways to synthesise chiral amines continues to be a major research topic in synthetic chemistry. In this context, biocatalysis offers an alternative to conventional chemical strategies that often rely on the use of hazardous reagents and reaction conditions. Over the last 20 years, a substantial effort has been made in the field and a significant number of biocatalysts are now available for this task.2–6 Among these enzymes, imine reductases (IREDs), which catalyze the NAD(P)H-dependent reduction of prochiral imines to chiral amines, have assumed an important role within the biocatalytic toolbox for chiral amine synthesis, following their discovery by Mitsukura et al.7–9 A sub-group of IREDs, termed reductive aminases (RedAms), possesses additionally the distinctive ability to carry out reductive aminations in water, when presented with a range of carbonyl compounds and small amine donors in equimolar quantities.10 We have recently demonstrated that these enzymes catalyze the condensation of cyclic and aliphatic ketones with short-chain amines as well as the subsequent reduction of the imine intermediate,11 although the substrate scope has until now been mainly limited to the preparation of secondary and tertiary amines. The asymmetric synthesis of chiral primary amines, synthesized via reductive amination of ketones using inexpensive ammonia, continues to be a challenge in synthetic chemistry.12 Biocatalytic strategies for the asymmetric synthesis of chiral primary amines include the application of transaminases (TAs), which catalyse the pyridoxal phosphate-dependent amination of ketone at the expense of an ammonia donor,3,13 and amine dehydrogenases (AmDHs),5,14–16 which catalyse the NAD(P)H-dependent reductive amination of ketones to primary amines (Fig. 1). Nevertheless, these two families of enzymes present some difficulties with respect to their application. In TA-catalysed processes, in addition to the need of an amine donor in high concentration, such as alanine or isopropylamine, equilibrium issues make the use of enzymatic cascades or product removal strategies mandatory to achieve high yields.17–21 AmDHs are engineered amino acid dehydrogenases (AADHs), which use ammonium salts as amine partners and constitute an emerging source of biocatalysts, however their substrate scope is still limited.14,15,22–25 A new family of natural AmDHs (nat-AmDHs) has been recently described that will bring diversity and therefore enable the expansion of the substrate scope although to-date it still remains rather limited.26,27 IREDs have also been used to access primary amines,28 although poor conversions were obtained for the amination of prochiral ketones. In this context, and given that we have previously observed that, by forcing reaction conditions, RedAms can also perform the reductive amination of a wider range of ketones and amines,10 we envisaged that homologues within this family could also accept a broader range of carbonyl compounds as well as ammonia to access primary amines. In a recent report, one of our groups described the ability of two of these homologues – NfRedAm from Neosartorya fumigata and NfisRedAm from Neosartorya fischeri – for the reduction of the imine formed between acetophenone and ammonia, albeit at concentrations of 5 mM and 250 mM respectively, to give conversions to the primary amine product which, although modest, were superior to previously described RedAms such as AdRedAm from Ajellomyces dermatitidis.29 In this study we exploit this initial observation in the characterization of the activity of NfRedAm and NfisRedAm and their application to the scalable synthesis of primary and secondary amines using ammonia.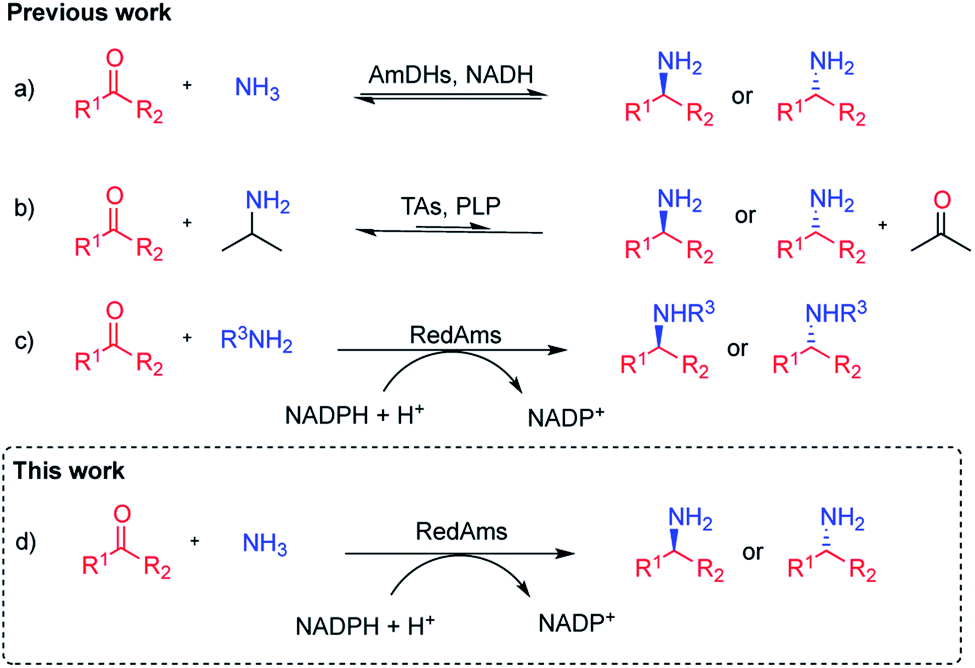 | ||
| Fig. 1 Biocatalytic reductive amination of ketones. (a) Reductive aminations mediated by amine dehydrogenases (AmDHs) using ammonia. (b) Formal reductive amination catalysed by transaminases (TAs) using isopropylamine as amine donor. (c) Reductive aminase (RedAm) catalysed reductive aminations with primary amines. (d) RedAm catalysed reductive aminations with ammonia. | ||
Results and discussion
The two target enzymes in this study, NfRedAm from Neosartorya fumigatus and NfisRedAm from Neosartorya fischeri, each displayed excellent levels of expression in E. coli BL21(DE3). The initial report, suggesting that these enzymes may display some activity for the reduction of the imine resulting from the ambient reaction of acetophenone and ammonia, prompted an in-depth investigation of the amination of ketones using NfRedAm and NfisRedAm for the synthesis of primary amines.Reductive amination with primary amines
The substrate scope of NfRedAm and NfisRedAm was initially assessed by combining a series of carbonyl compounds (1–7) with 1–5 equivalents of different primary amines (Fig. S4 in the ESI†). As previously observed with other fungal RedAms, the best results were observed after combining ketoesters, cyclic and aliphatic ketones such as ethyl levulinate (1), cyclohexanone (3) or 2-hexanone (4), with short-chain amines with certain π-character such as propargylamine (a), allylamine (b) and cyclopropylamine (c). This permits access to a wide range of chiral secondary amines, lactams and amino esters in good to excellent conversions (70% to >97%) and moderate to good enantiomeric excess and at low equivalents of the amine donor. Secondary amines derived from 2-octanone (6), 2-decanone (7) and 4-phenyl-2-butanone (5) can also be synthesised in this manner as well as δ-amino esters (2a,b) in moderate to good conversions (45–70%). The acceptance of benzylamine, which has previously been reported for an imine reductase homologue from Myxococcus stipitatus,30 was also explored. Remarkably, one equivalent of benzylamine (d) with 3 yielded 85% of the corresponding secondary amine 3d in 85% conversion for NfRedAm, whereas only traces of the product were observed for NfisRedAm. NfRedAm also outperforms other RedAms such as AdRedAm, where only 26% conversion was obtained.11 In terms of stereoselectivity, enantiomeric excesses ranged from poor to excellent for both RedAms and are notably different or even enantio-complementary in some instances (as seen in the case of ketoester 1–2, Fig. S3 in the ESI†). Remarkably, both the enantiopreference and extent of selectivity appeared to be both ketone- and amine-dependent, an observation which, given the sequence similarity between both enzymes (94% sequence ID), stresses the complexity of the mechanisms that govern the stereoselectivity of these reactions.Amination using ammonia
Following our preliminary observation,11 the initial assessment of the activity of NfRedAm and NfisRedAm with a range of substrates confirmed that these enzymes appeared to display superior activity when using ammonia (f) as a donor compared to previously studied RedAms such as AtRedAm and AdRedAm. For instance, with cyclohexanone (3), when using 5 equivalents of ammonia (f), 47% conversion to cyclohexylamine (4f) was observed in the AdRedAm-catalysed process, whereas 90% and 81% conversions were obtained for NfRedAm and NfisRedAm, respectively. Similarly, 14% conversion was obtained for AdRedAm in the reductive amination of 2-hexanone (4) and ammonia (f), whilst a 72% conversion was observed for NfRedAm under the same reaction conditions. This distinctive activity was further explored by screening these RedAms across a panel of structurally diverse ketones 3–20 (Fig. 2) using 1 M ammonia (100 equivalents) to mimic the optimal conditions in the AmDH-catalysed processes.15 Considering the possibility that the cofactor regeneration system could also be affected under such challenging reaction conditions, different NADPH regeneration strategies (glucose dehydrogenase from Bacillus subtilis, CDX-901 glucose dehydrogenase from Codexis and a variant of the phosphite dehydrogenase from Pseudomonas stutzeri (PtDH)28) and different ammonium sources [NH4Cl, (NH4)2SO4 and NH4OH] were evaluated (Fig. S5–S7 in the ESI†). For our reactions, GDH (CDX-901)/Glucose as NADPH regeneration system and 1 M NH4Cl in 100 mM Tris–HCl pH 9 were found to be the best conditions.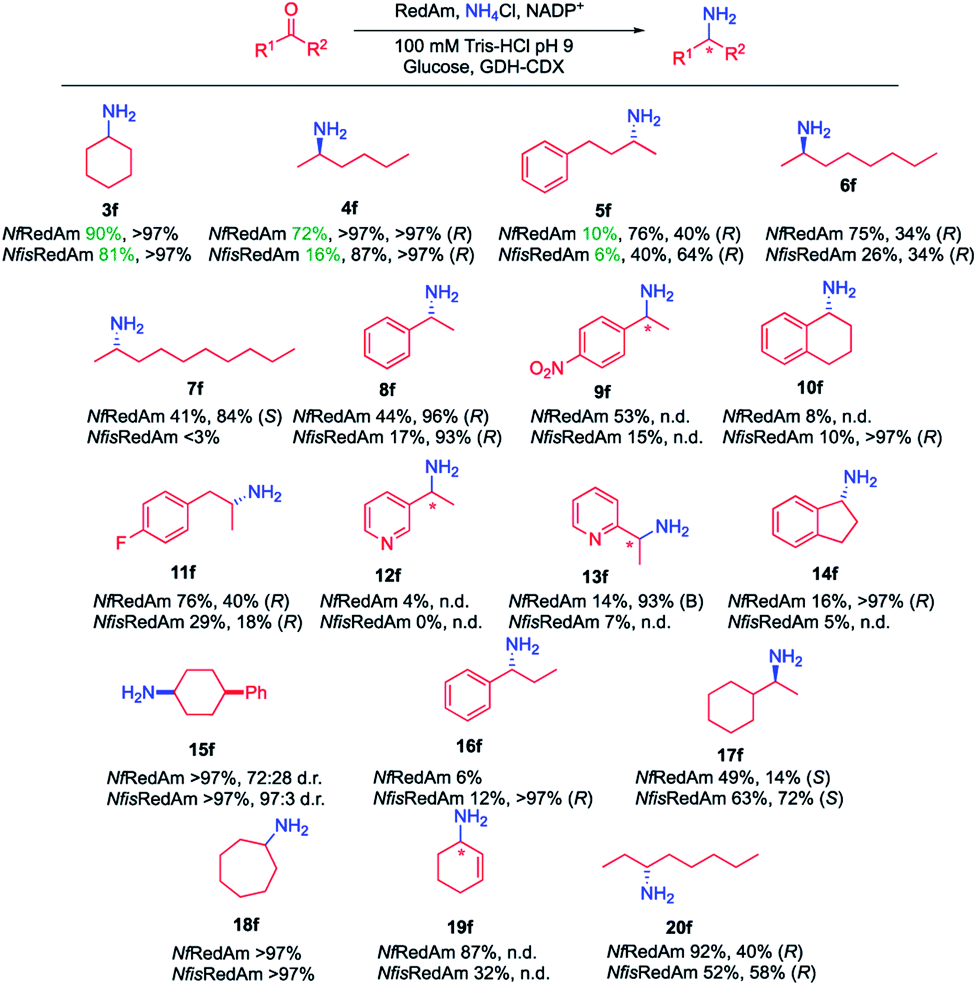 | ||
| Fig. 2 RedAm-catalysed reductive amination using ammonia to access chiral primary amines. Conditions: 10 mM ketone in 100 mM pH 9 Tris–HCl buffer containing 10 mM (green) or 1 M (black) NH4Cl, 1 mM NADP+, 40 mM glucose and 0.5 mg mL−1 CDX-GDH. Conversions determined after 24 h based on the areas of substrate and product by GC-FID. | ||
As previously observed for the reductive amination with primary amines,10,11 the best results were obtained for cyclic and aliphatic ketones. At 1 M NH4Cl, full conversions were found for cyclic ketones such as 3, 15 and 18. Cyclohexen-2-one (19) was also accepted affording cyclohex-2-en-1-amine (19f) as a sole product in up to 87% conversions. The corresponding saturated product cyclohexylamine (3f) was not detected in any case. For aliphatic substrates, moderate to excellent conversions were afforded although a drop in activity was observed with longer substrates. Remarkably, a switch in stereoselectivity was found in this series as well. (R)-2-Aminohexane (4f) can be obtained in full conversion and >97% ee. A 75% conversion was observed in the reductive amination of 6 to afford (R)-2-aminooctane (6f) in 34% ee and a switch to the S-product was observed for 2-decanone (7), obtaining (S)-2-aminodecane (7f) in 41% conversion and 84% ee. Other aliphatic substrates such as cyclohexyl methyl ketone (17) and 3-octanone (20) can also be aminated in this manner in moderate to good conversions (49–92%). For 17, a switch in the general stereoselectivity was again observed, obtaining (S)-17f in 14% and 72% ee for NfRedAm and NfisRedAm respectively. Aromatic ketones could also be aminated under these conditions. As previously described,29NfRedAm catalyzed the reductive amination of acetophenone (8) to amine 12f with 96% ee, 4-nitroacetophenone (9), 4-fluoro-acetophenone (11) or 4-phenyl-2-butanone (5) gave moderate to good conversions (44–76%) yielding the corresponding (R)-amine products 5f, 8f, 9f and 11f in moderate to excellent enantiomeric excess (40–96%). Other aromatic substrates such as 1-tetralone (10), 3-acetyl pyridine (12), 2-acetyl pyridine (13), 1-indanone (14) and propiophenone (16), low to poor conversions were obtained (up to 16%) although in high enantiomeric excess (>93%). In no cases were the alcohol products observed.
Structure of NfRedAm
The structure of NfRedAm was determined in two forms: an apo-form, at a resolution of 2.25 Å, in space group P3221 with one molecule in the asymmetric unit (asu), and a complex with the NADP+ cofactor, in space group P1, at a lower resolution of 2.77 Å, with eight molecules in the asu forming four dimers.‡ However, size-exclusion chromatography-multiangle light scattering (SEC-MALS) analysis confirmed that NfRedAm exists as a dimer in solution (Fig. S3 in the ESI†). The structure was solved by molecular replacement using the structure of AtRedAm (PDB 6EOD, 55% sequence identity)11 as a model. Data collection and refinement statistics can be found in Table S2 in the ESI.† The structure of the dimeric form of the enzyme is shown in Fig. 3A. The structure conforms to the canonical model determined for IRED-family enzymes, featuring two monomers, each composed of an N-terminal Rossmann domain, connected to a C-terminal helical bundle by a long alpha helix. The dimer is formed through reciprocal domain sharing, with the active sites formed at the interface between the N-terminal domain of one subunit, and the C-terminal domain of the other. Analysis using the DALI server31 shows that the monomer superimposed with its closest homologues – the RedAms from Aspergillus oryzae (PDB 5G6R; 58% sequence identity) and Aspergillus terreus (5OJL; 57%) with rmsd values of 1.3 and 1.8 Å over 289 and 284 C-alpha atoms respectively.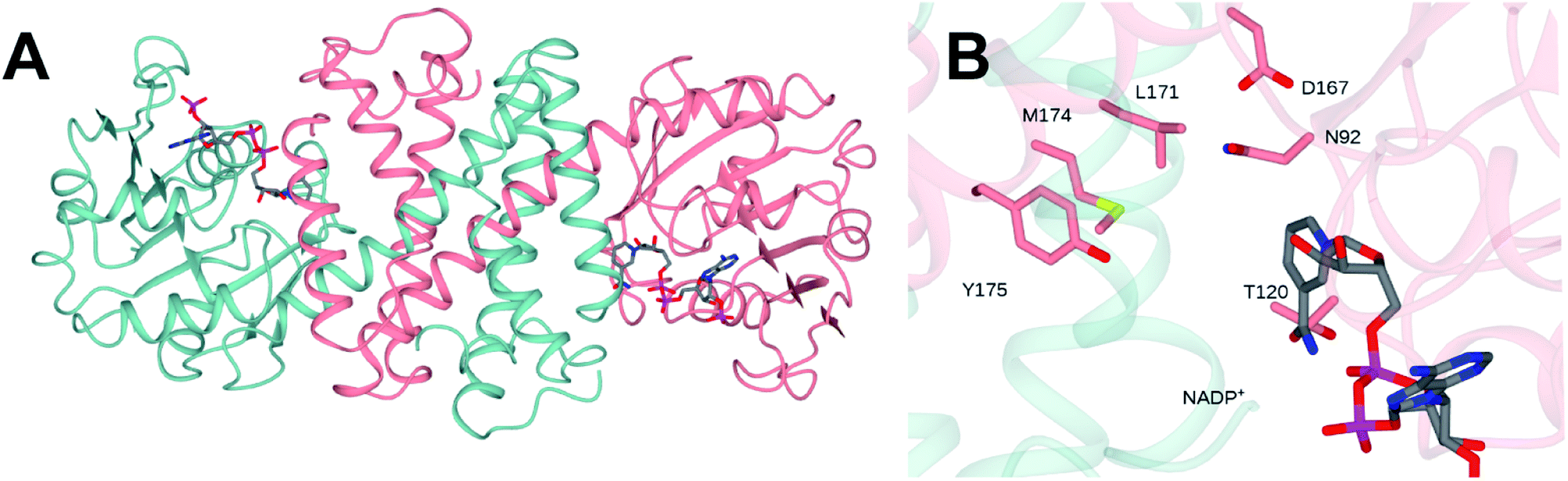 | ||
| Fig. 3 (A) Structure of dimer of NfRedAm in ribbon format with subunits shown in blue and pink. (B) Detail of active site of NfRedAm with NADP+ shown in cylinder format with carbon atoms in grey. | ||
The structure of the active site is shown in Fig. 3B. The majority of active site residues are conserved between NfRedAm and AspRedAm, including D167 (NfRedAm numbering) and Y175, thought to be involved in the activation of small amine nucleophiles and in securing the carbonyl group of the ketone substrate respectively. T120 in NfRedAm replaces V121 in AspRedAm behind the NADP+ cofactor as viewed in Fig. 3B. With many active site residues conserved, the reasons for the improved reduction of primary imines was not clear. In order to further investigate the comparative biophysical characteristics of the RedAm enzymes we determined the melting temperature of AspRedAm, NfRedAm, NfisRedAm using circular dichroism (Fig. 4).
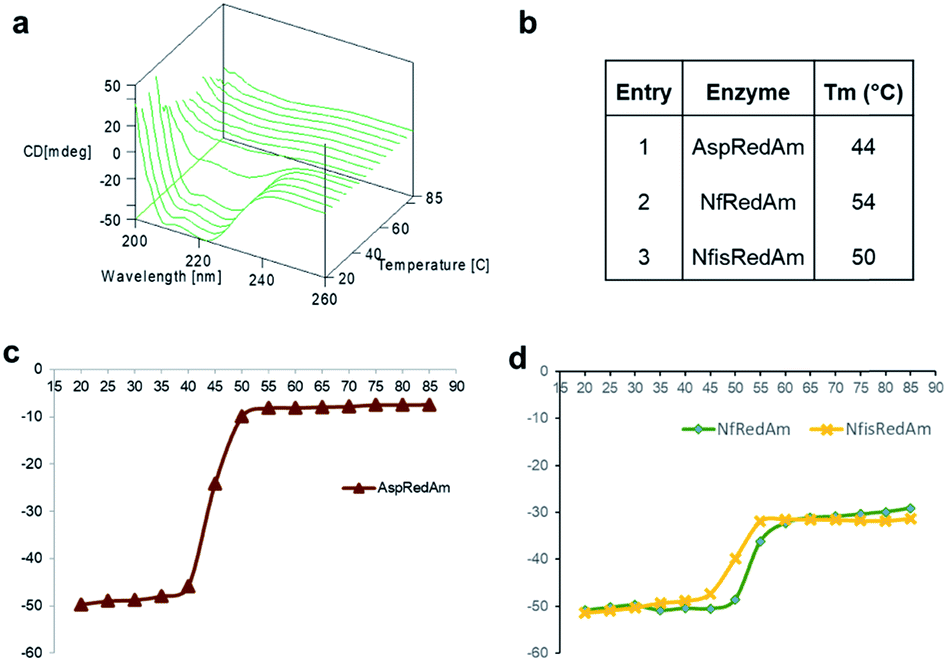 | ||
| Fig. 4 Thermal denaturation studies as a measure of thermodynamic stability. (a) CD spectra of NfRedAm as the temperature is increased from 20 to 85 °C. (b) Melting temperatures of AspRedAm, NfRedAm and NfisRedAm. CD220 plotted against temperature to calculate the melting temperatures of (c) AspRedAm and (d) NfRedAm and NfisRedAm. | ||
The unfolding of the enzymes was studied by recording CD222 at different temperatures (20–85 °C) as a measure of thermodynamic stability (Fig. 4). Importantly, the melting temperatures (Tm) of the homologues were found to be 6–10 °C higher than AspRedAm, which demonstrated the lowest Tm value amongst these fungal RedAms.
We also evaluated the thermostability of NfRedAm compared to AspRedAm by measuring the residual activity of both enzymes after incubation at 50 °C at different times (Fig. 5). In this case, the reductive amination of cyclohexanone (3) and allylamine (b) at equimolar concentrations and 30 °C was selected as a control reaction as both enzymes display high activity for this transformation. After an incubation time of only 20 min, a 50% decrease in activity was observed for AspRedAm whilst a slight drop was found for NfRedAm. Remarkably, NfRedAm does not seem to be significantly affected by the thermal treatment and retains most of its reductive amination activity after 1 h at 50 °C.
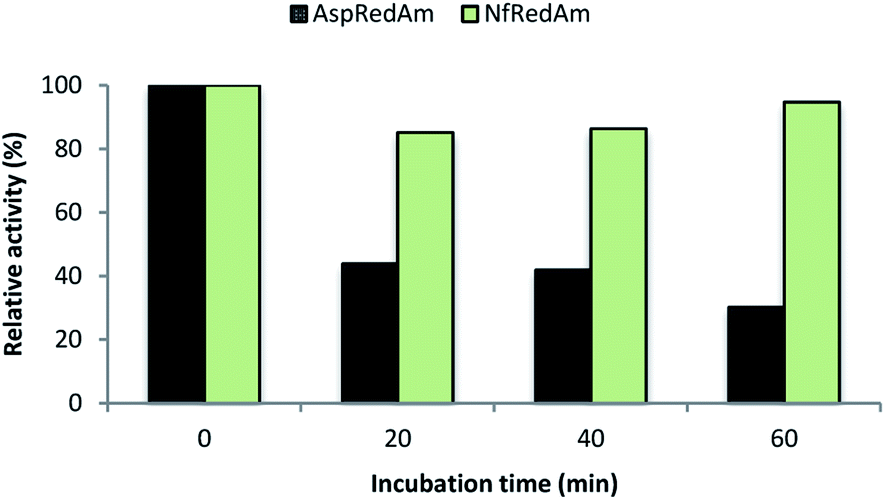 | ||
| Fig. 5 Relative activity of AspRedAm (black) and NfRedAm (green) in the equimolar bio-reductive amination of 3 and b at 30 °C after incubation at 50 °C at different times. 100% activity corresponds to 1.03 ± 0.03 U mg−1 for AspRedAm and 0.77 ± 0.11 U mg−1 for NfRedAm. | ||
Synthetic potential for large-scale processes
Given the potential of RedAms for large-scale processes,32 we used 4-phenyl cyclohexanone (15) and ammonia (f) as model reaction components to test higher substrate loadings (Fig. S8 and S9 in the ESI†). In this case, turnover numbers (TN) up to 1400 were obtained at 40 mM concentration (7 g L−1) although this system is restricted by the limited solubility of the starting material and further reaction engineering would be necessary to reach higher productivities. With conversion at 97%, a space time yield (STY) of 0.28 g L−1 h−1 was obtained, and a biocatalyst productivity of 6.8 gprod genz−1.In the reductive amination of 2-hexanone (4), TN up to 3250 were obtained at 80 mM substrate concentration (8 g L−1). This represented a low STY of only 0.34 g L−1 h−1, but with biocatalyst loading at only 0.5 mg mL−1 it delivered a reasonable productivity of 16 gprod genz−1. However, TN decreased at higher concentrations and additional DMSO loadings to increase the solubility of the starting material led to lower productivities. The use of continuous flow was investigated to try to improve the process conditions for these enzymes.33 More specifically, immobilisation of NfRedAm was investigated as the immobilised preparations are often stabilised by the solid supports, and can be applied in continuous packed-bed reactors.34 We applied EziG, a porous silica-based affinity resin, as we had demonstrated previously that it could stabilise AspRedAm.35–37NfRedAm was immobilised and tested under several conditions with 2-hexanone (4) (Section S6 in the ESI†). The optimal conditions permitted the continuous production of (R)-2-aminohexane ((R)-4f) for up to 12 h. The optimised conditions (see ESI Fig. S12, entry 6†) delivered a STY of 8.1 g L−1 h−1, which is much higher than the equivalent batch reaction. The biocatalyst productivity was 4.86 gprod genz−1, with TN up to 1400. Isolation issues must be overcome to allow this to be applied on a larger scale, however this initial proof-of-concept study demonstrates the versatility of these biocatalysts for process intensification (see ESI† for full details of flow experiments). We also tested these enzymes for reductive amination with primary amines. At 100 mM 3 and 2 equivalents of methylamine, TN of 2900 and 3400 were obtained for NfRedAm and NfisRedAm, respectively. At higher substrate concentrations, NfRedAm displayed up to 14000 TN in the equimolar reductive amination of 3 and b. In the reaction of 5 (100 mM) and cyclopropylamine (400 mM), TN around 2000 were obtained whilst remarkably, for 4, full conversions were observed. We reduced the amine concentration to 200 mM (2 eq.) observing no decrease in conversion. 4 was also tested at 200 mM (400 mM c) observing TN up to 6800 with NfRedAm. Encouraged by these results, we also looked at other combinations beyond equimolar ratios of both partners (Fig. S10 in the ESI†). In particular, we focused on acetophenone (8) and 1-(4-fluorophenyl)propan-2-one (11), given that secondary amines containing these scaffolds are valuable intermediates in different industries, and a–c as amine partners. For 8, cyclopropylamine (c) was found to be the best amine partner with conversions up to 56% with NfisRedAm. For a and b as amine donors, NfRedAm performed better although moderate conversions were achieved (21 and 25%, respectively). For 11, b was found to be the best nucleophilic partner, observing full conversion to the corresponding chiral secondary amine 11b for both NfRedAm and NfisRedAm. In all cases, excellent enantiomeric ratios were detected (from 91 to >97%). These results constitute a promising starting point and show the synthetic versatility and potential of these enzymes for industrial biocatalysis.
Conclusions
The activity of two enzymes from the fungal reductive aminase (RedAm) clade, NfRedAm and NfisRedAm, has been characterised. While they possess a similar substrate scope to previously described RedAms in the reductive amination with primary amines, their ability to enable the reductive amination of ketones using ammonia was found to be much higher. This distinctive activity was further explored showing a broad substrate scope, which highlights the versatility of these enzymes to serve as scaffolds for evolution towards fit-to-purpose biocatalysts in industrial processes. These RedAms also display a greater thermostability compared to other enzymes within the same family. Finally, an initial assessment of the industrial potential of NfRedAm and NfisRedAm was carried out by performing reactions at high substrate loadings of selected substrates obtaining TN up to 14000, as well as reactions at higher amine-to-ketone ratios to expand their synthetic operational landscape. Reactions in continuous flow were also performed, optimising the system for the production of (R)-2-aminohexane with a STY of 8.1 g L−1 h−1, which show the extraordinary capabilities and synthetic potential of these biocatalysts for large-scale use.
Experimental
Gene expression and protein purification
The plasmids containing the genes for target enzymes were used to transform E. coli BL21(DE3) competent cells for gene expression. Pre-cultures were grown in LB-medium (5 mL) containing 30 μg mL−1 kanamycin for 18 h at 37 °C with shaking at 180 rpm. 0.6 L volume cultures were inoculated with the pre-culture (6 mL) and incubated at 37 °C, with shaking at 180 rpm until an OD600 of 0.6–0.8 was reached. Gene expression was induced by addition of IPTG (1 mM) and shaking was continued overnight at 20 °C with shaking at 180 rpm. The cells were then harvested by centrifugation at 5000g for 20 min and resuspended in 50 mM Tris–HCl buffer pH 7.5, containing 300 mM NaCl. Cells were disrupted by ultrasonication for 3 × 5 min, with 30 s on and 30 s off cycles, and the suspension was centrifuged at 50000g for 30 min to yield a clear lysate. The N-terminal His6-tagged proteins were purified using immobilised-metal affinity chromatography (IMAC) using Ni-NTA column.
RedAm-catalysed bio-reductive amination
A 500 μL reaction mixture contained 20 mM D-glucose, 0.5 mg mL−1 GDH (Codexis, CDX-901), 0.5 mM NADP+, 1 mg mL−1 purified RedAm, 10 mM ketone, in 1 M NH4Cl pH 9 NaPi buffer adjusted and 2% v/v DMSO. Reactions were incubated at 30 °C with 250 rpm shaking for 24 h, after which they were quenched by the addition of 30 μL of 10 M NaOH and extracted twice with 500 μL tert-butyl methyl ether. The organic fractions were combined and dried over anhydrous MgSO4 and analysed by HPLC or GC-FID on a chiral stationary phase.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
J. M.-S. and M. S. were funded by grant BB/M006832/1 from the UK Biotechnology and Biological Sciences Research Council. N. J. T. is grateful to the ERC for the award of an Advanced Grant (Grant number 742987). S. C. C. would like to acknowledge the EPSRC UK Catalysis Hub for funding. We also thank Dr James Galman from the Manchester Institute of Biotechnology for the plasmid containing the phosphite dehydrogenase variant from Pseudomonas stutzeri (PtDH).Notes and references
- C. Wang and J. Xiao, in Stereoselective Formation of Amines, ed. C. Wang and J. Xiao, Springer-Verlag Berlin Heidelberg, 2014, pp. 261–282 Search PubMed.
- V. F. Batista, J. Galman, D. C. Gouveia Alves Pinto, A. M. S. Silva and N. J. Turner, ACS Catal., 2018, 8, 11889–11907 CrossRef CAS.
- I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS.
- M. Sharma, J. Mangas-Sanchez, N. J. Turner and G. Grogan, Adv. Synth. Catal., 2017, 359, 2011–2025 CrossRef CAS PubMed.
- G. Grogan, Curr. Opin. Chem. Biol., 2018, 43, 15–22 CrossRef CAS PubMed.
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Rev., 2011, 111, 3998–4035 CrossRef CAS PubMed.
- K. Mitsukura, M. Suzuki, K. Tada, T. Yoshida and T. Nagasawa, Org. Biomol. Chem., 2010, 8, 4533–4535 RSC.
- K. Mitsukura, M. Suzuki, S. Shinoda, T. Kuramoto, T. Yoshida and T. Nagasawa, Biosci., Biotechnol., Biochem., 2011, 75, 1778–1782 CrossRef CAS PubMed.
- K. Mitsukura, T. Kuramoto, T. Yoshida, N. Kimoto, H. Yamamoto and T. Nagasawa, Appl. Microbiol. Biotechnol., 2013, 97, 8079–8086 CrossRef CAS PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- M. Sharma, J. Mangas-Sanchez, S. P. France, G. A. Aleku, S. L. Montgomery, J. I. Ramsden, N. J. Turner and G. Grogan, ACS Catal., 2018, 8, 11534–11541 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8, 254 CrossRef.
- M. J. Abrahamson, E. Vázquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969–3972 CrossRef CAS PubMed.
- T. Knaus, W. Böhmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- V. Tseliou, T. Knaus, M. F. Masman, M. L. Corrado and F. G. Mutti, Nat. Commun., 2019, 10, 3717 CrossRef PubMed.
- G. Rehn, P. Adlercreutz and C. Grey, J. Biotechnol., 2014, 179, 50–55 CrossRef CAS PubMed.
- M. Höhne, S. Kühl, K. Robins and U. T. Bornscheuer, ChemBioChem, 2008, 9, 363–365 CrossRef PubMed.
- J. L. Galman, I. Slabu, N. J. Weise, C. Iglesias, F. Parmeggiani, R. C. Lloyd and N. J. Turner, Green Chem., 2017, 19, 361–366 RSC.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- M. D. Truppo, J. David Rozzell and N. J. Turner, Org. Process Res. Dev., 2010, 14, 234–237 CrossRef CAS.
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119–1122 CrossRef CAS.
- F. F. Chen, G. W. Zheng, L. Liu, H. Li, Q. Chen, F. L. Li, C. X. Li and J. H. Xu, ACS Catal., 2018, 8, 2622–2628 CrossRef CAS.
- A. Pushpanath, E. Siirola, A. Bornadel, D. Woodlock and U. Schell, ACS Catal., 2017, 7, 3204–3209 CrossRef CAS.
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 CrossRef CAS.
- O. Mayol, S. David, E. Darii, A. Debard, A. Mariage, V. Pellouin, J. L. Petit, M. Salanoubat, V. De Berardinis, A. Zaparucha and C. Vergne-Vaxelaire, Catal. Sci. Technol., 2016, 6, 7421–7428 RSC.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J. L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Catal., 2019, 2, 324–333 CrossRef CAS.
- D. Wetzl, M. Gand, A. Ross, H. Müller, P. Matzel, S. P. Hanlon, M. Müller, B. Wirz, M. Höhne and H. Iding, ChemCatChem, 2016, 8, 2023–2026 CrossRef CAS.
- G. Grogan, D. Gonzalez-Martinez, A. Cuetos, M. Sharma, M. Garcia-Ramos, I. Lavandera and V. Gotor-Fernandez, ChemCatChem, 2020 DOI:10.1002/cctc.201901999.
- G. D. Roiban, M. Kern, Z. Liu, J. Hyslop, P. L. Tey, M. S. Levine, L. S. Jordan, K. K. Brown, T. Hadi, L. A. F. Ihnken and M. J. B. Brown, ChemCatChem, 2017, 9, 4475–4479 CrossRef CAS.
- L. Holm and L. M. Laakso, Nucleic Acids Res., 2016, 44, W351–W355 CrossRef CAS PubMed.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G.-D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- J. Britton, G. A. Weiss and J. Britton, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- K. Engelmark Cassimjee, M. Kadow, Y. Wikmark, M. Svedendahl Humble, M. L. Rothstein, D. M. Rothstein and J. E. Bäckvall, Chem. Commun., 2014, 50, 9134–9137 RSC.
- K. Engelmark Cassimjee and H.-J. Federsel, in Biocatalysis: An Industrial Perspective, The Royal Society of Chemistry, 2018, pp. 345–362 Search PubMed.
- M. P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02253e |
| ‡ The coordinates for NfRedAm-apo and NfRedAm-NADP+ complex were deposited in the PDB with accession codes 6SKX and 6SLE respectively. |
| This journal is © The Royal Society of Chemistry 2020 |