
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ortho C–H arylation of arenes at room temperature using visible light ruthenium C–H activation†
Arunachalam
Sagadevan
 ,
Anastasios
Charitou
,
Fen
Wang
,
Maria
Ivanova
,
Martin
Vuagnat
and
Michael F.
Greaney
*
,
Anastasios
Charitou
,
Fen
Wang
,
Maria
Ivanova
,
Martin
Vuagnat
and
Michael F.
Greaney
*
School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michael.greaney@manchester.ac.uk
First published on 7th April 2020
Abstract
A ruthenium-catalyzed ortho C–H arylation process is described using visible light. Using the readily available catalyst [RuCl2(p-cymene)]2, visible light irradiation was found to enable arylation of 2-aryl-pyridines at room temperature for a range of aryl bromides and iodides.
Introduction
The integration of visible light photoredox catalysis with transition metal C–H activation catalysis creates new pathways for bond formation, that frequently operate under mild conditions. The two catalysis regimes can be meshed via two separate catalyst entities, with two corresponding catalysis cycles;1 or alternatively, a single dual-function catalyst system can be used.2 We are interested in this latter approach to exploit the facility of some Ru catalysts to absorb visible light, such that their native C–H activation function is enhanced in terms of improved rates, substrate scope, and environmental impact. We, along with the Ackermann group, recently demonstrated this concept for the Ru-catalysed meta-alkylation reaction (Scheme 1).3 The tert-butylation of 2-phenylpyridine 1, typically carried out under thermal conditions (ca. 100 °C), could proceed at room temperature under blue-light irradiation using the widely employed catalyst [RuCl2(p-cymene)]2 to give the alkylated product 3 in good yield.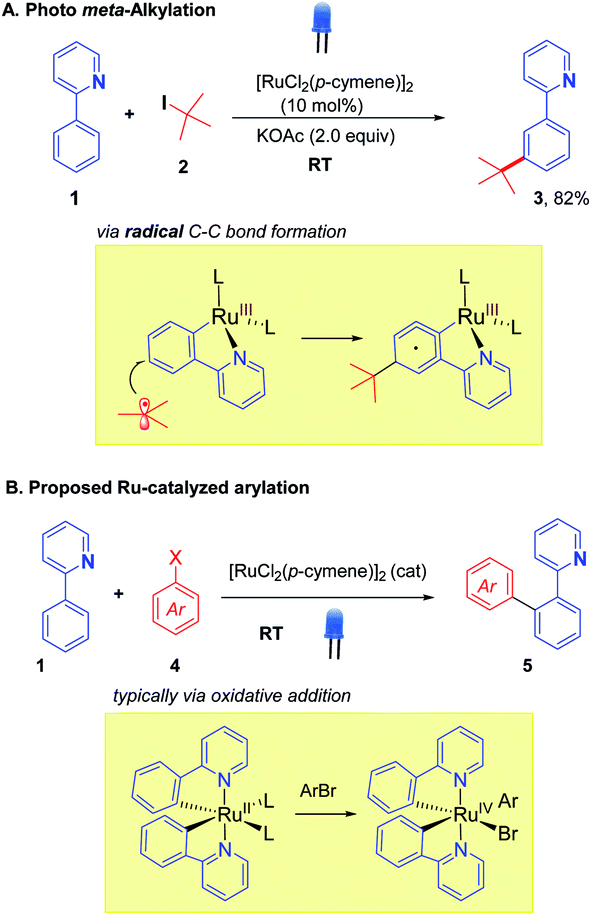 | ||
| Scheme 1 Ruthenium photocatalysis for meta-alkylation (A) and proposed ortho-arylation (B). | ||
We were interested in exploring this concept of photo-Ru C–H activation in the arylation regime. Ru-catalysed ortho-arylation is a powerful approach to C–C bond formation that has seen extensive development in recent years.4 Proceeding via Ru(II)/Ru(IV) catalytic cycles, the process has excellent scope for aryl halides (including aryl chlorides), is very tolerant of water and air, and the cost of Ru compares favorably with the far more expensive alternatives of Pd and Rh that are frequently used for ortho C–H arylation. High reaction temperatures are standard, however, when using [RuCl2(p-cymene)]2 as catalyst.5,6 A photo Ru arylation reaction could activate alternative mechanistic paths and offer the possibility of room temperature reaction.7 The two Ru-catalyzed processes, meta-alkylation and ortho-arylation, operate through very different frameworks in the thermal regime, with the former thought to involve discrete 2° and 3° carbon-centered radicals adding to Ru(III) metallacycles (Scheme 1A),8 and the latter involving more typical oxidative additions of aryl halides to a Ru(II) center.9 The role of photoexcitation on a putative arylation process was thus interesting to examine in light of this dichotomy; as the photoreductive formation of highly reactive aryl radicals in analogy to Ru-meta alkylation chemistry was unlikely to be a significant factor.
Results and discussion
Photoarylation of 2-arylazines with aryl halides
We began by examining the archetypal arylation system of 2-phenylpyridine 1a with bromoanisole 4a (Table 1). Using [RuCl2(p-cymene)]2 as catalyst in the presence of KOAc, under blue LED irradiation, we were pleased to observe successful arylation at room temperature to give a good 70% combined conversion to mono and di-ortho-arylated products 5a and 5aa. As is typical for Ru ortho-arylations of 2-phenyl pyridine, the monoarylated product is a superior substrate than the starting material, giving the diarylated material as the major product.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additives | Solvent | Yieldb [%] |
| a Reaction conditions: 1a (0.3 mmol), 4a (0.6 mmol), [Ru] catalyst (10 mol%), additive (10 mol%), base 2.0 equiv., solvent (1 mL). The mixture was irradiated with blue LEDs (40–90 W power) for 24 h under N2 atmosphere. b Yields in brackets refer to combined 1H NMR yield using 1,3,5-trimethylbenzene (mesitylene) as the internal standard, unbracketed yields are isolated. THF = tetrahydrofuran, 2-MeTHF = 2-methyl tetrahydrofuran, DME = 1,2-dimethoxyethane, DMF = dimethyl formamide, DCM = dichloromethane. | ||||
| 1 | [RuCl2(p-cymene)]2 | MesCO2H | 1,4-Dioxane | (70) |
| 2 | [RuCl2(p-cymene)]2 | MesCO2H | DME | 60 |
| 3 | [RuCl2(p-cymene)]2 | MesCO2H | THF | 65 |
| 4 | [RuCl2(p-cymene)]2 | MesCO2H | 2-MeTHF | (72) |
| 5 | [RuCl2(p-cymene)]2 | N-Ac-L-isoleu | 2-MeTHF | (74) |
| 6 | [RuCl2(p-cymene)]2 | N-Ac-L-isoleu | 2-MeTHF | (78) |
| 7 | [RuCl2(p-cymene)]2 | N-Ac-L-isoleu/H2O | 2-MeTHF | (80) |
| 8 | [RuCl 2 (p-cymene)] 2 | None/H 2 O | 2-MeTHF | (80) |
| 9 | [RuCl2(p-cymene)]2 | N-Ac-L-isoleu | MeOH, MeCN, DCM, or DMF | Trace |
| 10 | RuCl3·H2O | None | 2-MeTHF | 0 |
| 11 | Ru(PPh3)4Cl2 | N-Ac-L-isoleu/H2O | 2-MeTHF | 0 |
| 12 | Ru(bpy)3Cl2 | None | 2-MeTHF | 0 |
| 13 | [RuCl2(p-cymene)]2 (5 mol%) | N-Ac-L-isoleu/H2O | 2-MeTHF | (72) |
| 14 | No catalyst | N-Ac-L-isoleu | 2-MeTHF | 0 |
| 15 | [RuCl2(p-cymene)]2 dark | N-Ac-L-isoleu/H2O | 2-MeTHF | 0 |
| 16 | [RuCl2(p-cymene)]2 with air | N-Ac-L-isoleu/H2O | 2-MeTHF | 0 |
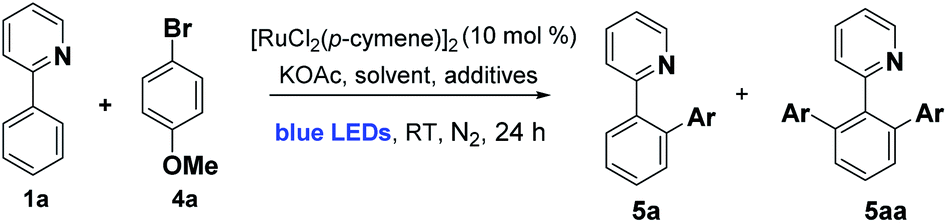
The reaction worked well in ethereal solvents (entries 1–4), and a screen of carboxylate and amino acid additives did not yield substantial improvements, although the acetylisoleucine derivative gave slightly improved yields and was retained for substrate screening (entry 7). Common solvents such as MeOH, DCM, MeCN, or DMF were not effective (entry 8), nor were the simple Ru salts shown in entries (10–12). Control experiments established that both light and catalyst were essential for reaction at room temp (entries 13–15), and as with our previous Ru meta systems,10 the reaction was found to be air sensitive and require inert atmospheres to proceed.
With these conditions in hand, we established the scope of the reaction with respect to the C–H component using p-bromoanisole as the arylating agent. Alkyl, alkoxy, carboxy, and phenyl substitution was tolerated in the 4-position of the arene, favoring the disubstituted product (5aa–5ff). Substitution in the 3-position with MeO, Me, Cl, and fused ring of the naphthyl group acted as a steric control element, suppressing the second ortho ruthenation step and yielding mono-arylated products exclusively (5g–5j). Some alterations to the directing group were possible, with alkylated pyridines being effective in the reaction, along with bicyclic quinoline and isoquinoline directing groups (5kk–5o) and the pyrazine 5pp. Finally, changing the arene C–H to the 5-membered heteroarene thiophene C–H was partially successful in the photoarylation, affording the novel pyridyl thiophene 5q but in diminished yield (Scheme 2).
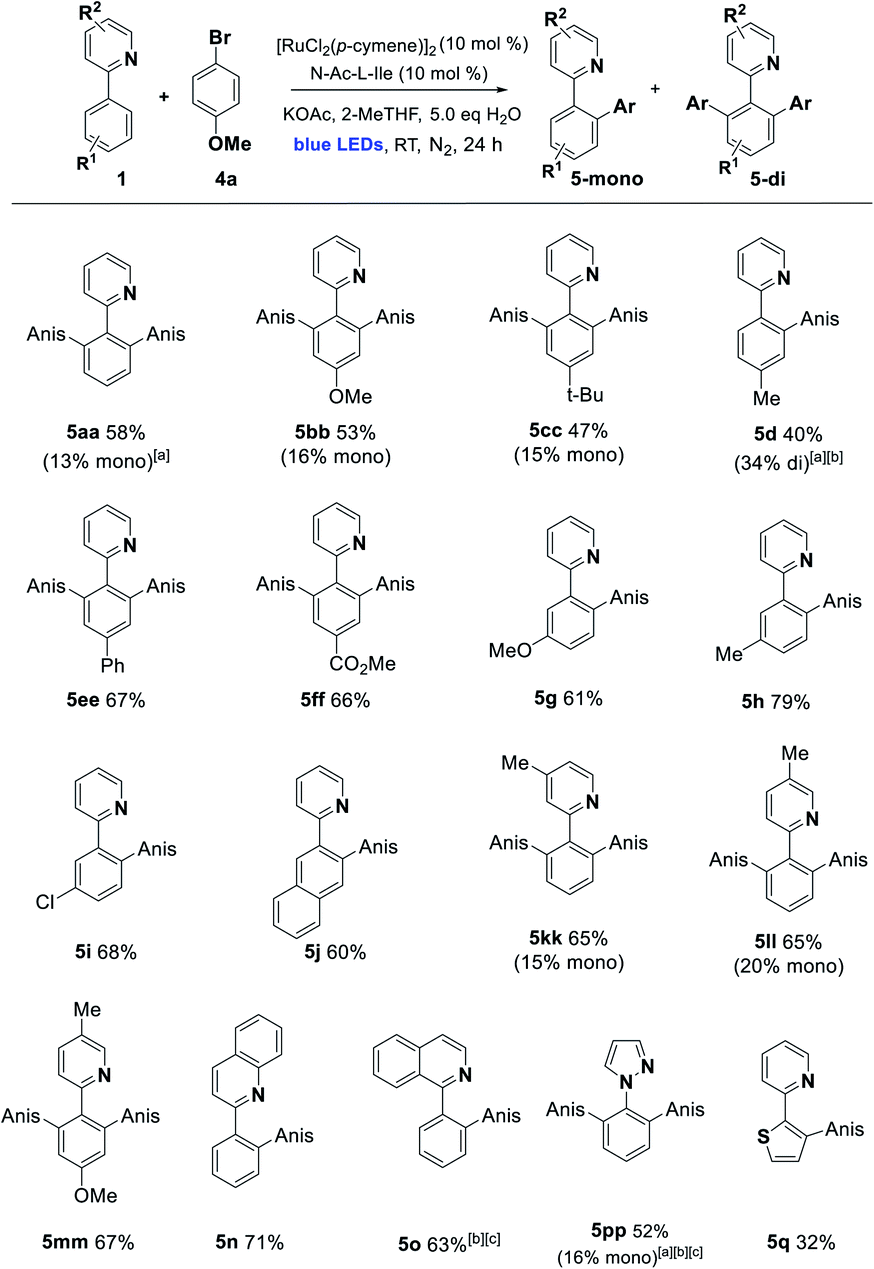 | ||
| Scheme 2 Photoarylation of 2-arylazines with p-bromoanisole (Anis = 4-BrC6H4). Unless otherwise noted, reaction conditions were as follows: 1 (1.0 eq., 0.3 mmol), 4a (2.0 eq., 0.6 mmol), [Ru] catalyst (10 mol%), additive (10 mol%), base (2.0 eq.), solvent (1 mL), H2O (5 eq.). The mixture was irradiated with blue LEDs (40–90 W power) for 24 h under N2 (1 atm). The yields refer to isolated yields after purification by column chromatography on silica gel (major product illustrated). aReaction performed using 0.250 mmol of 1 (1.0 eq.) and 0.375 mmol of 4a (1.5 eq.). bReaction performed without the addition of N-Ac-L-Ile. cReaction run for 40 h. | ||
We then turned our attention to the aryl halide coupling partner, and were pleased to find broad substrate scope across a variety of aryl bromides and iodides (Scheme 3). We used 3-methyl-2-phenylpyridine (1b) as the substrate in the majority of cases, to simplify the reaction pathway for monoarylation. We observed good efficiencies for 4-alkyl and moderate efficiencies for 4-phenyl aryl halides (6a–6cc), along with the 2-thienyliodide substrate (6dd). Halogens were well tolerated (6e–6gg), and electron poor (4-keto and 4-carboxy ester groups) along with electron rich (4-methoxy) aryl halides reacted smoothly in each case. Ortho-substituted aryl halides were not generally effective in this protocol, although 1-bromo-2-methoxybenzene did react successfully to give 6k in 50% yield. This ortho substitution pattern is known to favour an oxidative dimerization pathway for aryl halide substrates.11
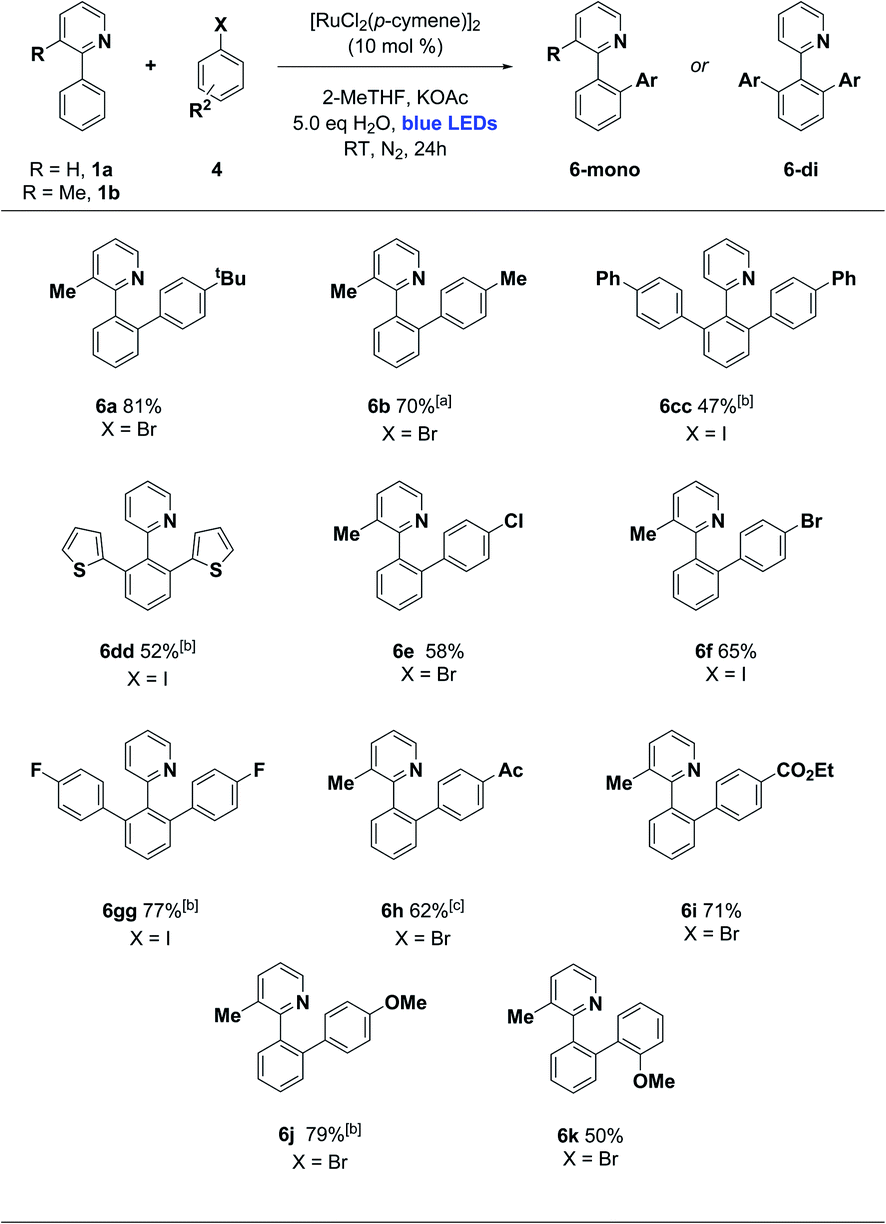 | ||
| Scheme 3 Photoarylation of 2-arylpyridines with various aryl halides. Unless otherwise noted, reaction conditions were as follows: 1 (1.0 eq., 0.25–0.30 mmol), 4 (1.5 eq., 0.375–0.45 mmol), [Ru] catalyst (10 mol%), base (2.0 eq.), solvent (1 mL), H2O (5 eq.). The mixture was irradiated with blue LEDs (40–90 W power) for 24 h in N2 (1 atm). The yields refer to isolated yields after purification by column chromatography on silica gel. aReaction run for 72 h. bReaction performed using 0.3 mmol of 1 (1.0 eq.) and 0.6 mmol of 4 (2.0 eq.) in the presence of N-Ac-L-Ile (10 mol%). cReaction run for 40 h. | ||
Mechanistic studies
The exclusive formation of ortho-arylated products, allied with the high reduction potential of aryl halides (ca. −2.5 eV)12 would likely preclude any discrete aryl radical generation through SET from a Ru(II) catalytic species in the reaction. This was supported with radical quenching experiments, where the reaction proceeded to reasonable conversion in the presence of both BHT and 1,1-diphenylethene (DHP), although TEMPO was observed to completely inhibit the reaction forming an insoluble black suspension. A competition experiment between the electron-rich bromoanisole 4a and electron-poor bromo-benzoate afforded a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of products in favour of the benzoate 6i, in line with what is commonly observed in high-temperature Ru ortho-arylation through an oxidative addition mechanism.13
:1 ratio of products in favour of the benzoate 6i, in line with what is commonly observed in high-temperature Ru ortho-arylation through an oxidative addition mechanism.13
Recent investigations from Larrosa and co-workers into the mechanism of Ru(II)-ortho arylation have identified the inhibitory role of the cymene ligand in [RuCl2(p-cymene)]2 catalysis.6,14 While this ligand affords an air-stable, easy to use catalyst, it retards activity in catalytic cycles, and must de-complex to enable the formation of the active bis-cycloruthenated complex C (Scheme 4E). We analysed the free cymene formed in the reaction under room temperature blue light irradiation against an ambient light control. Decomplexation was observed in both cases, but with a clear rate increase under blue LED irradiation (Scheme 4C). Photo-decomplexation of η6-arenes such as cymene is well known, and has been used as a triggering mechanism for both chemical and biological activity in Ru(II) complexes.15 However, a light/dark experiment (Scheme 4D) demonstrated that continuous irradiation was necessary for complete conversion. This suggests that either reversible cymene re-complexation can inhibit the reaction, or there is an additional role for visible light in the catalytic cycle (e.g. photoexcitation of complexes C or D to facilitate either oxidative addition or reductive elimination, respectively). Stern–Volmer experiments with stoichiometric pre-catalyst A and aryl halides did not show clear evidence of photoluminescence quenching, but we cannot rule out an additional role for visible light in subsequent steps in the catalytic cycle at this time.
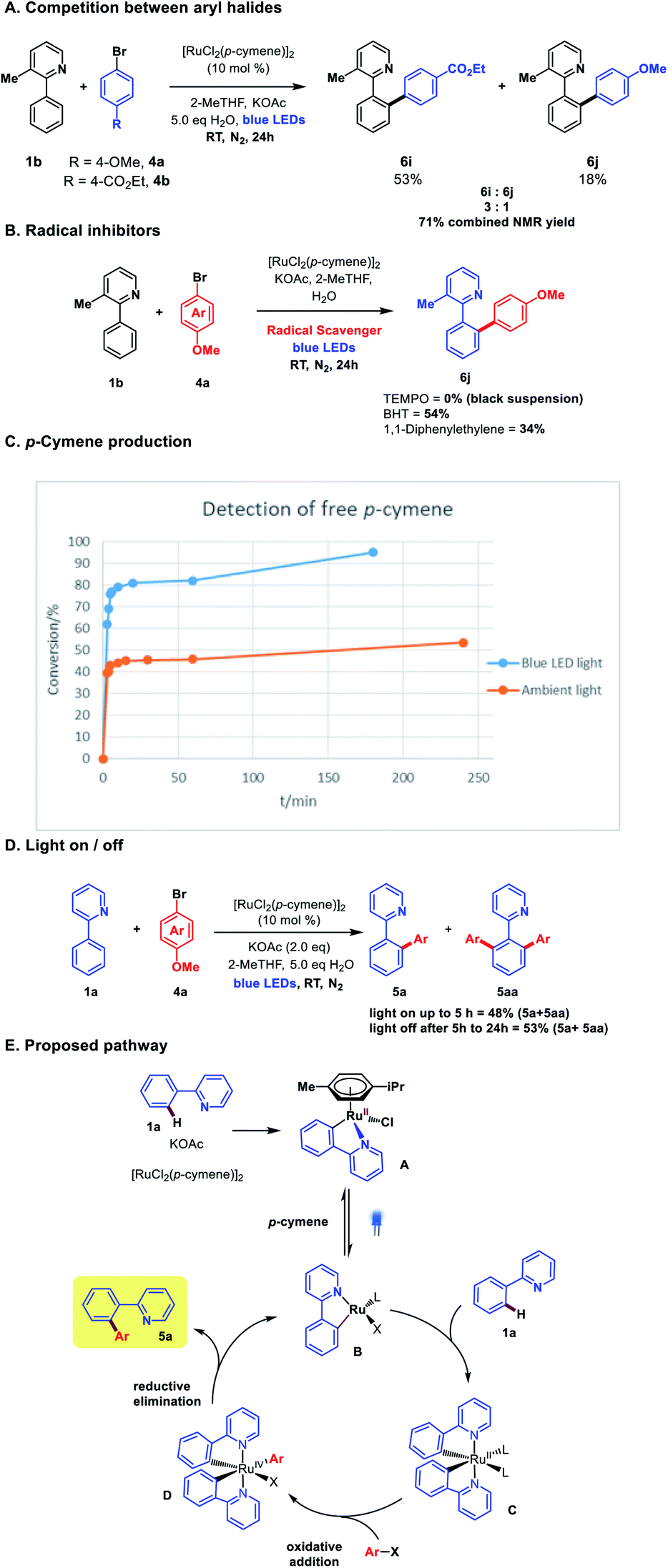 | ||
| Scheme 4 (A) Intermolecular competition experiment between aryl halides. (B) Reaction in the presence of radical inhibitors. (C) Detection of free p-cymene. Conversion refers to amount of p-cymene produced. (D) Light on/off experiment. (E) Proposed pathway. BHT = butylated hydoxytoluene. | ||
Conclusions
We have established a room temperature, ruthenium catalysed ortho-arylation reaction that proceeds at room temperature under the agency of visible light irradiation. The reaction encompasses a wide selection of aryl halides, producing C–H arylated products that are typically accessed at temperatures in excess of 100 °C. Initial observations point to a photo-decomplexation of the cymene ligand from the ruthenium pre-catalyst as playing a key role in the catalytic cycle. Future work will extend the photoarylation to new C–H substrate classes, and further delineate the role of visible light in the mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC (EP/P00850X/1), The Royal Society (Newton Fellowship to F. W.), and the EU (Marie Skłodowska-Curie Actions Individual Fellowship to M. I.). We thank Dr Louise Natrajan (University of Manchester) for helpful discussions.Notes and references
- Reviews: (a) M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (c) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152 CrossRef CAS PubMed; (d) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227 RSC.
- Selected recent examples: (a) W. Ding, L.-Q. Lu, Q. Q. Zhou, Y. Wei, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 63 CrossRef CAS PubMed; (b) Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850 CrossRef CAS PubMed; (c) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681 CrossRef CAS PubMed; (d) J. M. Ahn, T. S. Ratani, K. I. Hannoun, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 12716 CrossRef CAS PubMed; (e) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef CAS PubMed; (f) A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896 CrossRef CAS PubMed; (g) H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed; (h) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320 CrossRef CAS PubMed; (i) J. Thongpaen, R. Manguin, V. Dorcet, T. Vives, C. Duhayon, M. Mauduit and O. Basle, Angew. Chem., Int. Ed., 2019, 58, 15244 CrossRef CAS PubMed; (j) S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 1522 CrossRef CAS; (k) I. Abdiaj, A. Fontana, M. Vitoria Gomez, A. de la Hoz and J. Alcazar, Angew. Chem., Int. Ed., 2018, 57, 8473 CrossRef CAS PubMed; (l) B. D. Ravetz, J. Y. Wang, K. E. Ruhl and T. Rovis, ACS Catal., 2019, 9, 200 CrossRef CAS; (m) R. Kancherla, K. Muralirajan, B. Maity, C. Zhu, P. E. Krach, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3412 CrossRef CAS PubMed.
- (a) A. Sagadevan and M. F. Greaney, Angew. Chem., Int. Ed., 2019, 58, 9826 CrossRef CAS PubMed; (b) P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 9820 CrossRef CAS PubMed.
- Reviews: (a) P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721 CrossRef CAS; (b) L. Ackermann, Org. Process Res. Dev., 2015, 19, 260 CrossRef CAS; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed.
- Selected examples: (a) M. Drev, U. Grošelj, B. Ledinek, F. Perdih, J. Svete, B. Štefane and F. Požgan, Org. Lett., 2018, 20, 5268 CrossRef CAS PubMed; (b) A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576 CrossRef CAS PubMed; (c) B. Li, C. Darcel, T. Roisnel and P. H. Dixneuf, J. Organomet. Chem., 2015, 793, 200 CrossRef CAS; (d) Y.-Q. He and Y.-W. Zhong, Chem. Commun., 2015, 51, 3411 RSC; (e) M. Seki, ACS Catal., 2014, 4, 4047 CrossRef CAS; (f) L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299 CrossRef CAS PubMed; (g) S. Oi, K. Sakai and Y. Inoue, Org. Lett., 2005, 7, 4009 CrossRef CAS PubMed.
- For a recent mild Ru-catalyzed arylation using cyclometallated complexes see: M. Simonetti, D. M. Cannas, X. Just-Baringo, I. J. Vitorica-Yrezabal and I. Larrosa, Nat. Chem., 2018, 10, 724 CrossRef CAS PubMed.
- For a review of photoredox arylation, see: (a) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed. For a review of C–H activation under mild conditions, see: (b) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC.
- (a) A. J. Paterson, S. S. John-Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807 RSC; (b) J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894 CrossRef CAS PubMed; (c) F. Fumagalli, S. Warratz, S.-K. Zhang, T. Rogge, C. Zhu, A. C. Stückl and L. Ackermann, Chem.–Eur. J., 2018, 24, 3984 CrossRef CAS PubMed.
- C. Shan, L. Zhu, L.-B. Qu, R. Bai and Y. Lan, Chem. Soc. Rev., 2018, 47, 7552 RSC.
- (a) C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11677 CrossRef CAS PubMed; (b) H. L. Barlow, C. J. Teskey and M. F. Greaney, Org. Lett., 2017, 19, 6662 CrossRef CAS PubMed.
- T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 15640 CrossRef CAS PubMed.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat and J. M. Saveant, J. Am. Chem. Soc., 1979, 101, 3431 CrossRef CAS.
- L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032 CrossRef CAS PubMed.
- See also: (a) P. Marce, A. J. Paterson, M. F. Mahon and C. G. Frost, Catal. Sci. Technol., 2016, 6, 7068 RSC; (b) J. McIntyre, I. Mayoral-Soler, P. Salvador, A. Poater and D. J. Nelson, Catal. Sci. Technol., 2018, 8, 3174 RSC; (c) E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133(26), 10161 CrossRef PubMed.
- (a) E. E. Karslyan, D. S. Perekalin, P. V. Petrovskii, K. A. Lyssenko and A. R. Kudinov, Russ. Chem. Bull., 2008, 10, 2201 CrossRef; (b) E. E. Karslyan, D. S. Perekalin, P. V. Petrovskii, A. O. Borisova and A. R. Kudinov, Russ. Chem. Bull., 2009, 58, 585 CrossRef CAS; (c) F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098 CrossRef PubMed; (d) D. S. Perekalin, E. E. Karslyan, E. A. Trifonova, A. I. Konovalov, N. L. Loskutova, Y. V. Nelyubina and A. R. Kudinov, Eur. J. Inorg. Chem., 2013, 481–493 CrossRef CAS; (e) S. L. Saraf, T. J. Fish, A. D. Benninghoff, A. A. Buelt, R. C. Smith and L. M. Berreau, Organometallics, 2014, 33, 6341 CrossRef CAS; (f) P. Qin, S. K. Cope, H. Steger, K. M. Veccharelli, R. L. Holland, D. M. Hitt, C. E. Moore, K. K. Baldridge and J. M. O'Connor, Organometallics, 2017, 36, 3967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01289k |
| This journal is © The Royal Society of Chemistry 2020 |