
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox deracemization of β,γ-alkynyl α-amino esters†
Lu
Zhang‡
a,
Rongxiu
Zhu‡
 b,
Aili
Feng
b,
Changyin
Zhao
b,
Lei
Chen
a,
Guidong
Feng
a and
Lei
Liu
*ab
b,
Aili
Feng
b,
Changyin
Zhao
b,
Lei
Chen
a,
Guidong
Feng
a and
Lei
Liu
*ab
aSchool of Pharmaceutical Sciences, Shandong University, Jinan 250012, China. E-mail: leiliu@sdu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
First published on 21st April 2020
Abstract
The first non-enzymatic redox deracemization method using molecular oxygen as the terminal oxidant has been described. The one-pot deracemization of β,γ-alkynyl α-amino esters consisted of a copper-catalyzed aerobic oxidation and chiral phosphoric acid-catalyzed asymmetric transfer hydrogenation with excellent functional group compatibility. By using benzothiazoline as the reducing reagent, an exclusive chemoselectivity at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond over the C
N bond over the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond was achieved, allowing for efficient deracemization of a series of α-amino esters bearing diverse α-alkynyl substituent patterns. The origins of chemo- and enantio-selectivities were elucidated by experimental and computational mechanistic investigation. The generality of the strategy is further demonstrated by efficient deracemization of β,γ-alkenyl α-amino esters.
C bond was achieved, allowing for efficient deracemization of a series of α-amino esters bearing diverse α-alkynyl substituent patterns. The origins of chemo- and enantio-selectivities were elucidated by experimental and computational mechanistic investigation. The generality of the strategy is further demonstrated by efficient deracemization of β,γ-alkenyl α-amino esters.
Introduction
Deracemization, wherein a racemic mixture of a given molecule is completely transformed into a single enantiomer of exactly the same species in theoretically 100% yield, has emerged as an attractive and topologically straightforward alternative to conventional enantioselective synthesis or kinetic resolution strategies.1–3 Deracemization is a thermodynamically disfavored process and is commonly achieved by destroying and recreating stereocenters through oxidation and reduction chemistry, at least one of which should involve asymmetric manipulation. Despite the conceptual simplicity and potential practical benefits, pure chemically catalytic redox deracemization has remained underdeveloped4,5 and suffers from narrow substrate scope including secondary alcohols,6 cyclic amines,7,8 and cyclic ethers.9 Moreover, existing approaches typically require stoichiometric oxidants such as oxopiperidinium salt, NBS, and DDQ, which generate undesired waste.6,7,9 The use of molecular oxygen as an ideal oxidant in light of economic and ecological factors with H2O as the only byproduct has never been demonstrated for this process.10 Therefore, developing a deracemization method using molecular oxygen as the terminal oxidant to access enantioenriched molecules that are otherwise difficult to access is highly desirable.Optically active non-natural amino acids are fundamental subunits for a range of biologically significant molecules.11,12 In particular, optically pure β,γ-alkynyl α-amino acids represent a special class of these compounds,13 not only because the rich chemistry of alkynes allows for further synthetic elaboration on the β,γ-positions, but also because α-alkynyl moieties can change the biological properties of certain natural amino acids for potential therapeutic utility.14 But surprisingly, asymmetric synthesis of β,γ-alkynyl α-amino acids and their derivatives has remained challenging (Scheme 1). The groups of Chan and Rueping demonstrated enantioselective addition of terminal alkynes to α-imino esters (Scheme 1A).15 Albeit innovative, each method suffered from a narrow alkyne scope and was merely suitable for either aliphatic or electron-rich aryl alkynes with moderate to good ee (48–92%). Asymmetric hydrogenation of β,γ-alkynyl α-imino esters represents the other potentially efficient strategy.16 However, chemo- and enantio-selective reduction of the imine moiety with the alkyne intact proved to be challenging (Scheme 1B). The You group revealed chiral phosphoric acid (CPA) catalyzed asymmetric transfer hydrogenation with a Hantzsch ester, giving β,γ-alkenyl α-amino esters through an initial partial reduction of the CC bond followed by CN bond reduction.16a The Zhou group demonstrated that the alkyne group can be conserved using 5,6-dihydrophenanthridine as the hydrogen source, but the expected β,γ-alkynyl α-amino ester was only isolated in 26% yield with 36% ee.16b The hydride transfer mode (1,4 or 1,2) and hydrogen transfer ability have been demonstrated to generate a significant influence on the reactivity of target reactions.17 Moreover, benzothiazolines have exhibited superior reactivity and enantioselectivity to other hydride donors in several CPA catalyzed transfer hydrogenation reactions by carefully tuning the substituent at the C2 position.18 We envisioned that using benzothiazolines as the hydride donor might address the chemo- and enantio-selectivity issues encountered in Scheme 1B. Herein, we report a one-pot redox deracemization of β,γ-alkynyl α-amino acid derivatives (Scheme 1C). The strategically different approach consisted of an aerobic oxidation and CPA-catalyzed asymmetric transfer hydrogenation with benzothiazoline, allowing for asymmetric access to a series of non-natural α-amino esters bearing diverse α-alkynyl substituent patterns in high efficiency with excellent chemo- and enantio-selectivity.
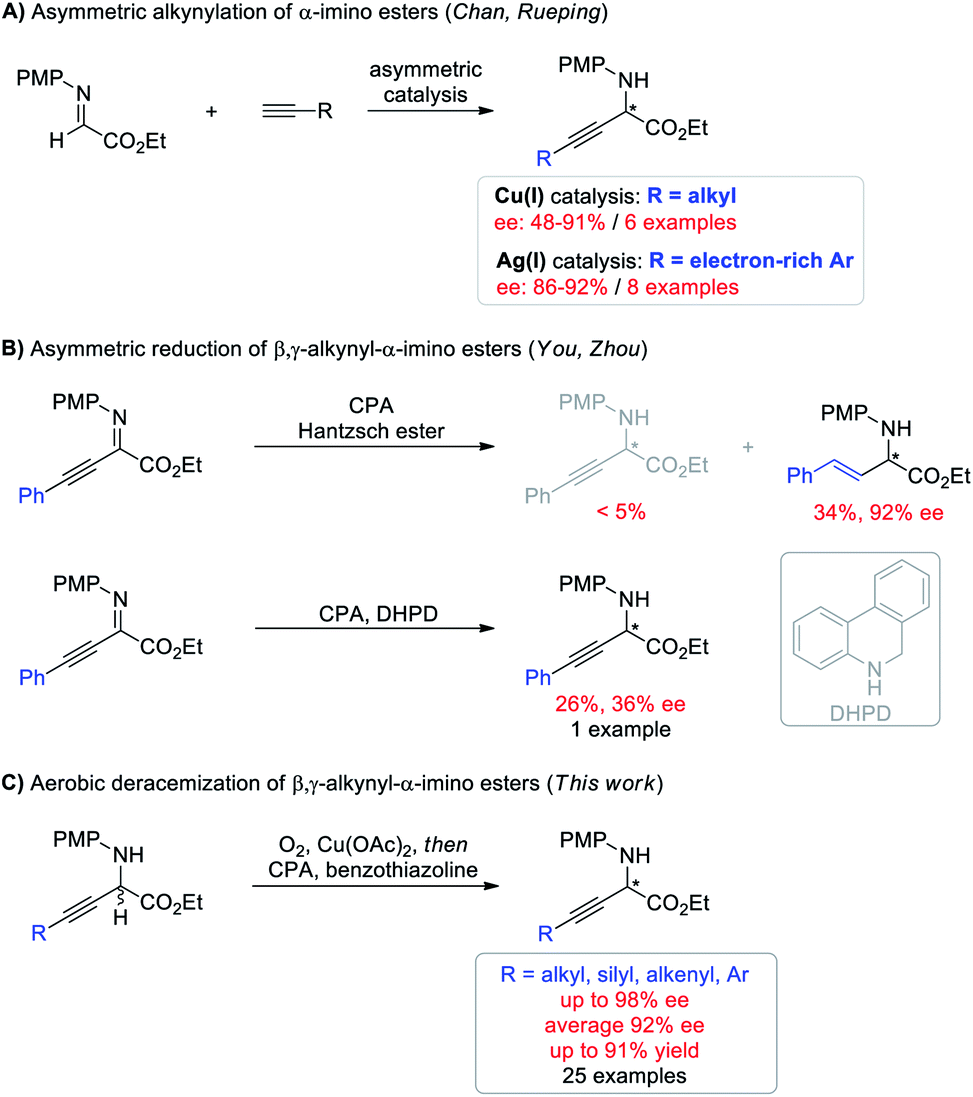 | ||
| Scheme 1 Overview of enantioselective access to β,γ-alkynyl α-amino acid derivatives. | ||
Results and discussion
Initially, redox deracemization of aliphatic alkynyl substituted α-amino ester rac-1a was selected as a model reaction using molecular oxygen as the terminal oxidant and the combination of CPA 2 and benzothiazoline 3 as the asymmetric transfer hydrogenation system (Table 1).19 The oxidation was performed prior to the addition of 2 to avoid reagent quenching. No oxidation was observed in the absence of any additive (entry 1, Table 1). A careful examination of metal salt additives revealed that reaction with 10 mol% of Cu(OAc)2 provided a clean and efficient aerobic oxidation at rt, and the expected (S)-1a was recovered in 85% yield with 6% ee in the presence of CPA 2a and benzothiazoline 3a (entries 2–7, Table 1). Notably, the reaction exhibited excellent chemoselectivity with the alkynyl moiety highly conserved. The solvent was found to be crucial to the enantioselectivity, and when asymmetric reduction was performed in a mixture of hexane and CH2Cl2, a remarkably improved ee of 73% was observed (entries 5 and 8, Table 1). The asymmetric transfer hydrogenation system was extensively investigated, and the combination of 2a and 3d afforded the highest level of enantiofacial discrimination (entries 8–14, Table 1). Further optimization of the solvent choice identified a mixture of decane and mesitylene to be optimal for the asymmetric reduction process (entries 15 and 16, Table 1). The reaction proved to be sensitive to moisture, as demonstrated by an obvious loss of enantioselectivity in the absence of molecular sieves (entries 16 and 17, Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | 2 | 3 | Recoveryb (%) | eec (%) |
a Reaction conditions: rac-1a (0.1 mmol) and additive (10 mol%) in CH2Cl2 (2 mL) under 1 atm molecular oxygen at rt for 3 h, followed by 2 (5 mol%), 3 (0.12 mmol), and 3 Å molecular sieves (40 mg) at rt for 4 h.
b Yield of the isolated product.
c Determined by chiral HPLC analysis.
d Asymmetric reduction in a mixture of hexane and CH2Cl2 (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
e Asymmetric reduction in a mixture of decane and CH2Cl2 (v/v = 1:1).
f Asymmetric reduction in a mixture of decane and mesitylene (v/v = 1:1).
g Reaction without 3 Å MS. PMP = 4-methoxyphenyl, n.o. = no oxidation, n.d. = not determined. :1).
e Asymmetric reduction in a mixture of decane and CH2Cl2 (v/v = 1:1).
f Asymmetric reduction in a mixture of decane and mesitylene (v/v = 1:1).
g Reaction without 3 Å MS. PMP = 4-methoxyphenyl, n.o. = no oxidation, n.d. = not determined.
|
|||||
| 1 | — | — | — | n.o. | n.d. |
| 2 | CuBr2 | 2a | 3a | <5 | n.d. |
| 3 | CuCl2 | 2a | 3a | 17 | 3 |
| 4 | CuF2 | 2a | 3a | 63 | 5 |
| 5 | Cu(OAc)2 | 2a | 3a | 85 | 6 |
| 6 | Cu(OTf)2 | 2a | 3a | <5 | n.d. |
| 7 | CuOAc | 2a | 3a | 72 | 5 |
| 8d | Cu(OAc)2 | 2a | 3a | 86 | 73 |
| 9d | Cu(OAc)2 | 2a | 3b | 73 | 70 |
| 10d | Cu(OAc)2 | 2a | 3c | 71 | 47 |
| 11d | Cu(OAc)2 | 2a | 3d | 83 | 81 |
| 12d | Cu(OAc)2 | 2b | 3d | 76 | 77 |
| 13d | Cu(OAc)2 | 2c | 3d | <5 | n.d. |
| 14d | Cu(OAc)2 | 2d | 3d | 70 | 53 |
| 15e | Cu(OAc)2 | 2a | 3d | 79 | 85 |
| 16f | Cu(OAc)2 | 2a | 3d | 86 | 94 |
| 17f,g | Cu(OAc)2 | 2a | 3d | 79 | 55 |

|
|||||
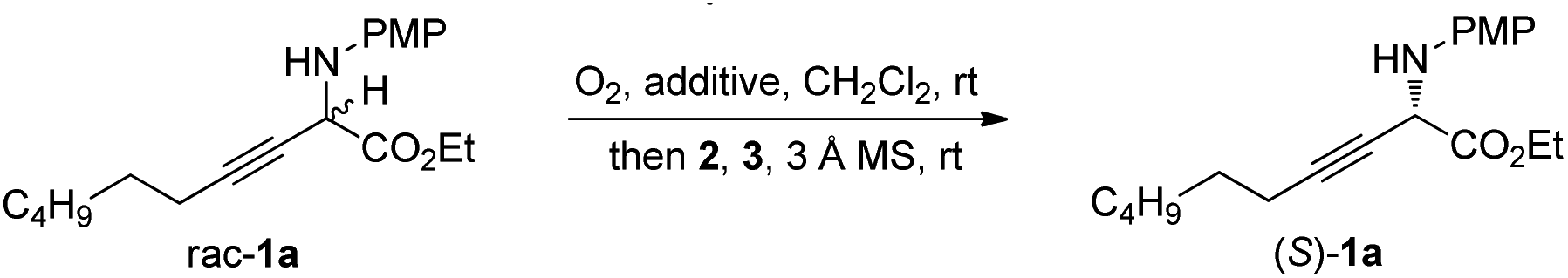
The scope of the one-pot redox deracemization of β,γ-alkynyl α-amino esters was explored (Scheme 2). The reaction was found to be fairly general for substrates bearing a wide range of aliphatic alkynes with varied chain lengths and sizes, affording corresponding optically pure 1a–1d in good yields with excellent ee (94–98% ee).20 The deracemization method exhibited excellent functional group compatibility with commonly encountered ones, including cyclopropane (1e), aryl (1f), benzyl ether (1g), silyl ether (1h), acetate (1i), and acetal (1j), well tolerated as additional functional handles for structurally diverse non-natural amino acid synthesis. Alkenyl substituted alkyne 1k was similarly effectively deracemized. Substrates containing electronically varied aryl and heteroaryl acetylenes were also suitable candidates for the protocol, as demonstrated by the efficient generation of enantiopure 1l–1p with high enantiocontrol. Silyl-substituted alkynes were further identified to be competent components for the process, furnishing respective TMS 1q and TBS 1r with 92% and 93% ee.
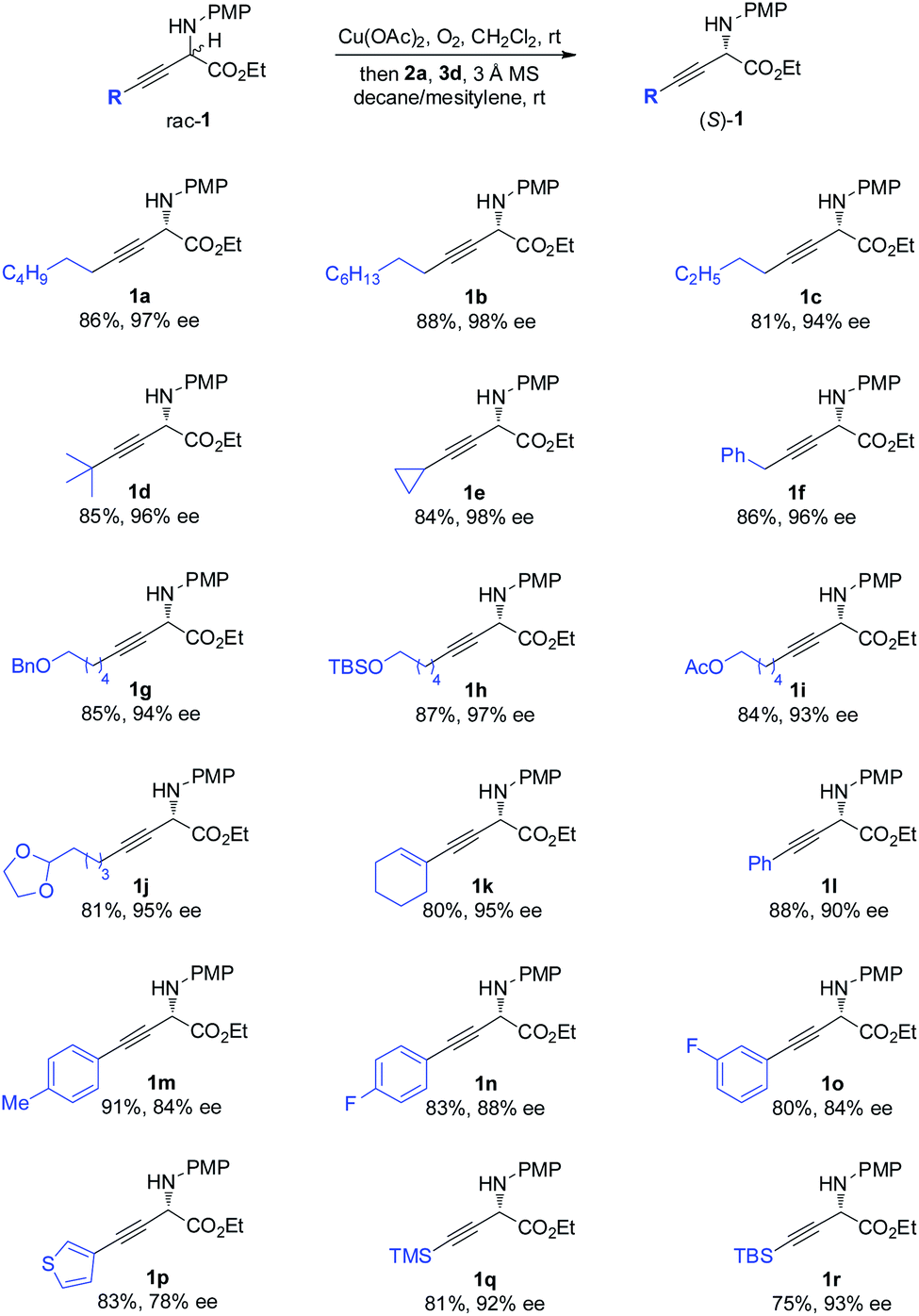 | ||
| Scheme 2 Scope of the α-alkynyl component of non-natural amino esters. | ||
The scope of the amino component was then investigated (Scheme 3). Besides the electron-rich PMP moiety, other electronically varied aryl groups (4a–4c) were also well tolerated. The method was found to be insensitive to the variant on the ester moiety, as demonstrated by effective deracemization of 4d–4g.
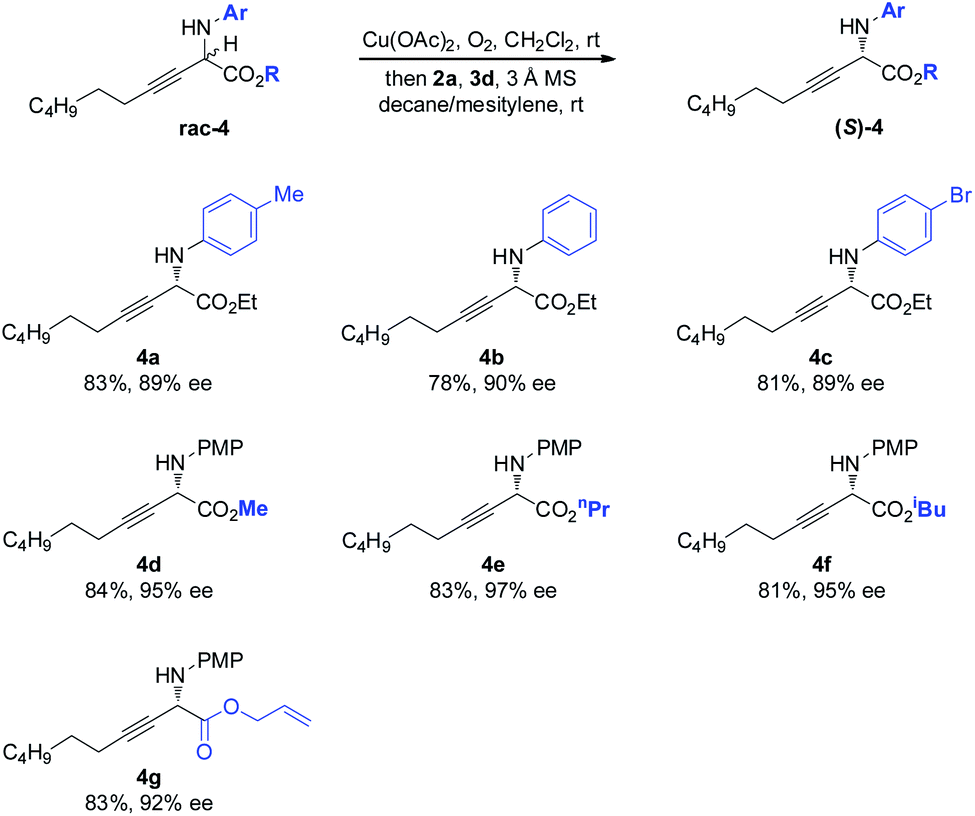 | ||
| Scheme 3 Scope of the amino and ester components of non-natural amino esters. | ||
The gram-scale aerobic deracemization proceeded smoothly without obvious loss of efficiency and enantioselectivity, and (S)-1a was obtained in 77% yield with 94% ee (Scheme 4a). The redox deracemization technology was also applicable to β,γ-alkenyl α-amino esters, as illustrated by the formation of enantiopure (S)-5 in 82% yield with 91% ee (Scheme 4b).21
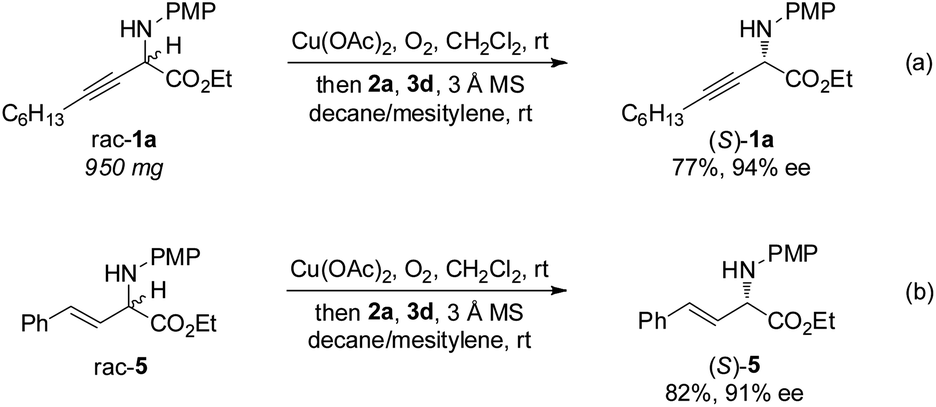 | ||
| Scheme 4 Gram-scale study and redox deracemization of β,γ-alkenyl α-amino ester. | ||
Control experiments were conducted to gain further insights into the reaction mechanism (Scheme 5). In the presence of a catalytic amount of Cu(OAc)2 under a molecular oxygen atmosphere, α-amino ester rac-1l was transformed to an intermediate detected by TLC analysis, which was identified as α-imino ester 6 (Scheme 5a). Subjecting 6 to standard conditions in the absence of oxidation elements furnished the desired (S)-1l with a comparable ee to that obtained by the deracemization process, indicating the intermediacy of α-imino ester 6 (Scheme 5b). Unlike the studies in Scheme 1B, CPA-catalyzed asymmetric transfer hydrogenation of 6 with benzothiazoline as the hydride donor exhibited exclusive chemoselectivity at the CN bond, and reduction of the CC bond was not observed. Diphenyl phosphate 7 catalyzed reduction of 6 with benzothiazoline 3d proceeded smoothly, providing rac-1l in 90% yield, suggesting that the excellent chemoselectivity should not derive from the chiral elements on the CPA catalyst (Scheme 5c). Similarly, the chemoselectivity of reduction with a Hantzsch ester at the CC bond over the CN bond in Scheme 1B should also not originate from the chiral environment on CPA, as demonstrated by diphenyl phosphate catalyzed reduction of 6 with Hantzsch ester 8 affording rac-5 in 35% yield (Scheme 5d).
 | ||
| Scheme 5 Control experiments. | ||
Due to the different hydride transfer modes (1,4 vs. 1,2) and hydrogen transfer abilities, benzothiazolines have exhibited superior reactivity and enantioselectivity to widely used Hantzsch esters in several CPA catalyzed transfer hydrogenation reactions.17,18 However, to our knowledge, different chemoselectivities have never been observed for these two types of hydride donors for the same substrate. To elucidate the origin of different chemoselectivities for benzothiazoline and the Hantzsch ester, density functional theory (DFT) calculations have been performed on the transition structures for respective transfer hydrogenation of the CN bond (TS1 and TS3) and CC bond (TS2 and TS4) at the M06/6-311+G(d,p)//B3LYP/6-31G(d,p) level with the polarizable continuum model (PCM) (Fig. 1, see the ESI† for details).22,23 For the reduction with benzothiazoline, TS1 is 6.3 kcal mol−1 more favorable than TS2 in terms of free energy. In the case of the Hantzsch ester as the hydride donor, TS3 for CN bond reduction is 2.7 kcal mol−1 higher in energy than TS4 for the CC bond. The results are consistent with the experimentally observed chemoselectivities.
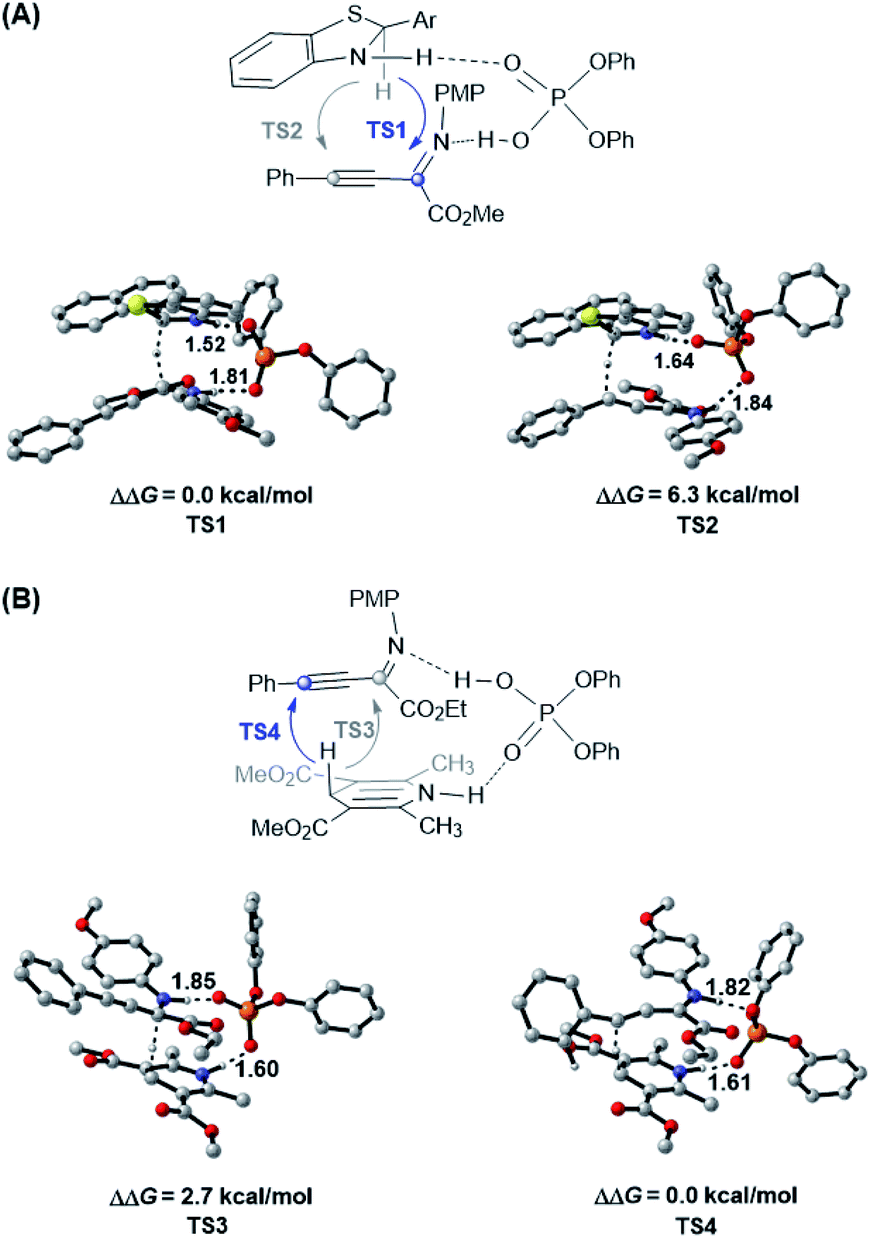 | ||
| Fig. 1 Optimized structures and the relative Gibbs free energies of chemoselectivity-determining transition states at the PCM-M06/6-311+G(d,p)//B3LYP/6-31G(d,p) level in toluene. (A) TS1 and TS2 for benzothiazoline-mediated transfer hydrogenation. (B) TS3 and TS4 for Hantzsch ester-mediated transfer hydrogenation. Key hydrogen-bonding interactions are marked with bond lengths in Å. Noncritical hydrogen atoms are omitted for clarity. | ||
To further understand this intriguing chemoselectivity, we conducted distortion/interaction analysis for the aforementioned four transition state structures at the PCM-M06/6-311+G(d,p)//B3LYP/6-31G(d,p) level in toluene (Table 2).24 We compared the energies of distortion of the CPA catalyst (ΔEcatdist), hydride donor (ΔEhyddist), and substrate (ΔEsubdist) components, as well as the interaction energies among these three parts. Despite the small difference in catalyst and substrate distortion, drastically different hydride donor distortion energies were found, favoring reduction of the CC bond for both reductants. On the other hand, an obvious interaction energy difference was observed, favoring reduction of the CN bond for both reductants. In the benzothiazoline-mediated reaction, the interaction energy is responsible for the observed selectivity of CN bond reduction, as evidenced by the stronger hydrogen bonding interaction in TS1 than in TS2 (1.52 Å vs. 1.64 Å and 1.81 Å vs. 1.84 Å). In the Hantzsch ester-involved reaction, the distortion energy is responsible for the observed selectivity of CC bond reduction, probably due to the reacting hydride lying in a distal position with reference to the hydrogen-binding N–H moiety.
| TS | ΔE‡ | ΔEcatdist | ΔEhyddist | ΔEsubdist | ΔEdistb | ΔEintc |
|---|---|---|---|---|---|---|
| a Calculated at the PCM-M06/6-311+G(d,p)//B3LYP/6-31G(d,p) level in toluene. All energies are given in kcal mol−1. b ΔEdist = ΔEcatdist + ΔEhyddist + ΔEsubdist. c ΔEint = ΔE‡ − ΔEdist. | ||||||
| TS1 | 9.1 | 2.1 | 25.7 | 15.1 | 42.9 | −33.8 |
| TS2 | 15.4 | 1.9 | 14.6 | 18.9 | 35.4 | −20.0 |
| TS3 | 11.8 | 1.5 | 21.4 | 14.6 | 37.5 | −25.7 |
| TS4 | 9.1 | 1.0 | 10.0 | 16.7 | 27.7 | −18.6 |
The enantioselective transfer hydrogenation of β,γ-alkynyl-α-imino ester with CPA 2a and benzothiazoline 3d was next investigated by DFT calculations at the PCM-M06/6-311+G(d,p)//B3LYP/6-31G(d,p) level in n-decane (Fig. 2). After exploring the possible transition structures for the process (see the ESI† for details), the most stable diastereomeric TSSR giving the major enantiomer and TSRS giving the minor enantiomer were compared. TSSR is 4.8 kcal mol−1 more favorable than TSRS in terms of free energy, which is in agreement with the experimentally observed enantioselectivity. A closer inspection of the two transition structures reveals that TSRS suffers from steric repulsion between the PMP group of the α-imino ester and the bulky triisopropylphenyl moiety of CPA when the si-face of the substrate is attacked by 3d, while such steric repulsion could be avoided when the attack occurs from the re-face. The hydrogen bonding interactions of CPA with the respective substrate and 3d are the second factor that differentiates the diastereomeric transition states. There are stronger NH⋯O interactions in TSSR than in TSRS, evidenced by shorter NH⋯O hydrogen bonding distances in TSSR than in TSRS (1.57 and 1.82 Å in TSSRvs. 1.70 and 1.95 Å in TSRS). Therefore, our calculations suggest that the steric effect and hydrogen bonding interactions differentiate the enantiomeric reduction process.
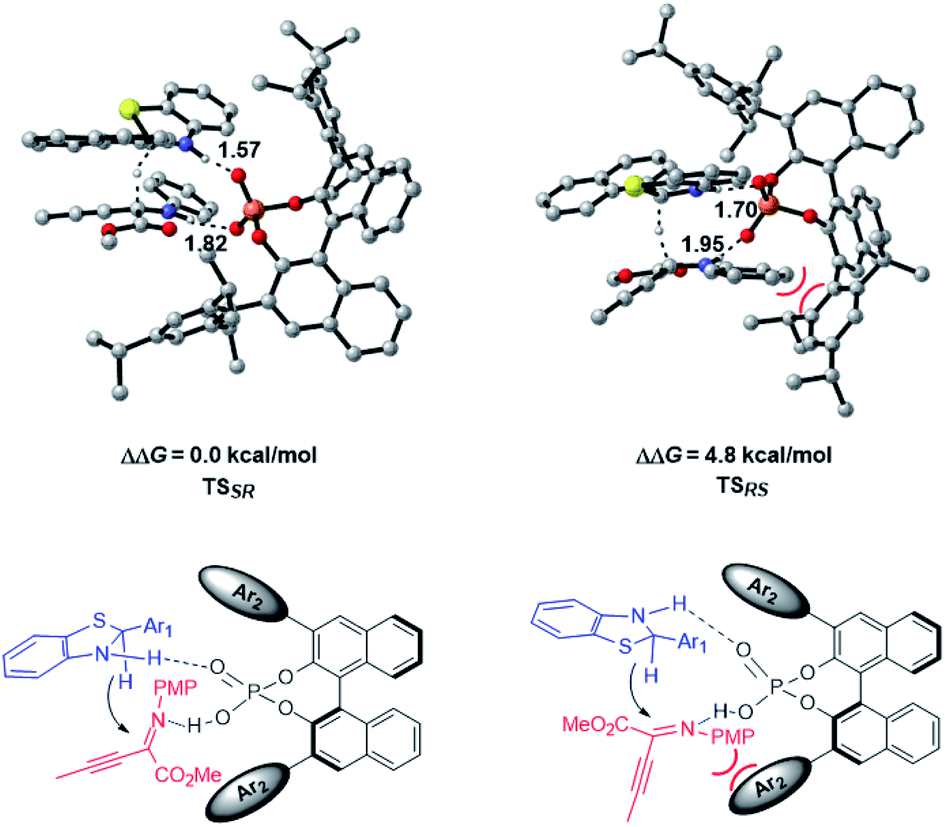 | ||
| Fig. 2 Lower-lying transition states and their relative Gibbs free energies for CPA-catalyzed transfer hydrogenation of β,γ-alkynyl-α-imino ester at the PCM-M06/6-311+G(d,p)//B3LYP/6-31G(d,p) level in n-decane. Key hydrogen-bonding interactions are marked with bond lengths in Å. | ||
Conclusions
In summary, the first non-enzymatic redox deracemization method using molecular oxygen as the terminal oxidant has been revealed. The one-pot deracemization of β,γ-alkynyl α-amino acid derivatives consisted of a copper-catalyzed aerobic oxidative process and CPA-catalyzed asymmetric transfer hydrogenation with excellent functional group compatibility. By using benzothiazoline as the reductant, an exclusive chemoselectivity at the CN bond over the CC bond was achieved, allowing for efficient deracemization of a series of α-amino esters bearing diverse α-alkynyl substituent patterns. The generality of the strategy is further demonstrated by efficient deracemization of β,γ-alkenyl α-amino esters. Mechanistic exploration by combined experiments and computations elucidated the origins of chemo- and enantio-selectivities. We envision that the sustainable method outlined herein will have potential applications in increasingly significant proteomics and peptide based drug discovery research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Science Foundation of China (21722204 and 21971148), Fok Ying Tung Education Foundation (151035), and Youth Interdiscipline Innovative Research Group of Shandong University (2020QNQT009).Notes and references
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, 1999, vol. I–III Search PubMed.
- (a) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5 CrossRef CAS; (b) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed; (c) H. Pellissier, Adv. Synth. Catal., 2011, 353, 1613 CrossRef CAS; (d) F. F. Huerta, A. B. E. Minidis and J.-E. Bäckvall, Chem. Soc. Rev., 2001, 30, 321 RSC.
- Deracemization used in this text is the definition in the strict sense, and the only difference between the product and the starting material is that the latter is enantioenriched. Relevant reviews, see: (a) K. Faber, Chem.–Eur. J., 2001, 7, 5004 CrossRef CAS PubMed; (b) C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789 CrossRef CAS; (c) N. J. Turner, Curr. Opin. Chem. Biol., 2010, 14, 115 CrossRef CAS PubMed; (d) M. Rachwalski, N. Vermue and F. P. J. T. Rutjes, Chem. Soc. Rev., 2013, 42, 9268 RSC; (e) R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129 CrossRef CAS.
- Current deracemization studies rely heavily on enzymatic catalysis with alcohol and amine substrates, see: (a) C. V. Voss, C. C. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 741 CrossRef CAS PubMed; (b) C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969 CrossRef CAS PubMed; (c) C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224 CrossRef CAS PubMed; (d) D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863 CrossRef CAS PubMed; (e) K. Yasukawa, S. Nakano and Y. Asano, Angew. Chem., Int. Ed., 2014, 53, 4428 CrossRef CAS PubMed.
- An innovative example of photochemical deracemization of allenes enabled by chiral sensitizer showing different energy transfer efficiencies for two enantiomers, see: (a) A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240 CrossRef PubMed; (b) A. Tröster, A. Bauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 3538 CrossRef PubMed.
- (a) Y. Shimada, Y. Miyake, H. Matsuzawa and Y. Nishibayashi, Chem.–Asian J., 2007, 2, 393 CrossRef CAS PubMed; (b) G. R. A. Adair and J. M. J. Williams, Chem. Commun., 2007, 2608 RSC; (c) P. Qu, M. Kuepfert, S. Jockusch and M. Weck, ACS Catal., 2019, 9, 2701 CrossRef CAS.
- (a) A. D. Lackner, A. V. Samant and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14090 CrossRef CAS PubMed; (b) Y. Ji, L. Shi, M.-W. Chen, G.-S. Feng and Y.-G. Zhou, J. Am. Chem. Soc., 2015, 137, 10496 CrossRef CAS PubMed.
- An innovative example of photochemical deracemization of α-substituted 2-imidazolidinones through a sequence of electron, proton, and hydrogen-atom transfer steps, see: N. Y. Shin, J. M. Ryss, X. Zhang, S. J. Miller and R. R. Knowles, Science, 2019, 366, 364 CrossRef CAS PubMed.
- (a) M. Wan, S. Sun, Y. Li and L. Liu, Angew. Chem., Int. Ed., 2017, 56, 5116 CrossRef CAS PubMed; (b) R. Lu, Y. Li, J. Zhao, J. Li, S. Wang and L. Liu, Chem. Commun., 2018, 54, 4445 RSC.
- (a) N. Gulzar, B. Schweitzer-Chaput and M. Klussmann, Catal. Sci. Technol., 2014, 4, 2778 RSC; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC.
- (a) G. C. Barrett, Chemistry and Biochemistry of the Amino Acids, Chapman & Hall, London, 1985 CrossRef; (b) R. M. Williams, Synthesis of Optically Active α-Amino Acids, Pergamon, Oxford, 1989 Search PubMed; (c) R. O. Duthaler, Tetrahedron, 1994, 50, 1539 CrossRef CAS; (d) L. S. Hegedus, Acc. Chem. Res., 1995, 28, 299 CrossRef CAS; (e) J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang and P. G. Schultz, Science, 2002, 301, 964 CrossRef PubMed; (f) L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2004, 44, 34 CrossRef PubMed; (g) T. A. Cropp and P. G. Schultz, Trends Genet., 2004, 20, 625 CrossRef CAS PubMed.
- (a) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS PubMed; (b) A. E. Taggi, A. M. Hafez and T. Lectka, Acc. Chem. Res., 2003, 36, 10 CrossRef CAS PubMed; (c) B. Lygo and B. I. Angrews, Acc. Chem. Res., 2004, 37, 518 CrossRef CAS PubMed; (d) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed.
- (a) S. A. Abdulganeeva and K. B. Erzhanov, Russ. Chem. Rev., 1991, 60, 676 CrossRef; (b) R. M. Williams, D. J. Aldous and S. C. Aldous, J. Org. Chem., 1990, 55, 4657 CrossRef CAS; (c) A. L. Castelhano, S. Horne, G. J. Taylor, R. Billedeau and A. Krantz, Tetrahedron, 1988, 44, 5451 CrossRef CAS.
- (a) R. R. Rando, Methods in Enzymology, Academic Press, New York, San Francisco, London, 1977 Search PubMed; (b) R. R. Rando, Nature, 1974, 250, 586 CrossRef CAS PubMed; (c) N. Relyea and R. R. Rando, Biochem. Biophys. Res. Commun., 1975, 67, 392 CrossRef CAS PubMed.
- (a) J.-X. Ji, J. Wu and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11196 CrossRef CAS PubMed; (b) M. Rueping, A. P. Antonchick and C. Brinkmann, Angew. Chem., Int. Ed., 2007, 46, 6903 CrossRef CAS PubMed.
- (a) Q. Kang, Z.-A. Zhao and S.-L. You, Org. Lett., 2008, 10, 2031 CrossRef CAS PubMed; (b) M.-W. Chen, B. Wu, Z.-P. Chen, L. Shi and Y.-G. Zhou, Org. Lett., 2016, 18, 4650 CrossRef CAS PubMed.
- Selected studies on hydride donor abilities of the hydride source, see: (a) D. Richter and H. Mayr, Angew. Chem., Int. Ed., 2009, 48, 1958 CrossRef CAS PubMed; (b) X.-Q. Zhu, Y. Liu, B.-J. Zhao and J.-P. Cheng, J. Org. Chem., 2001, 66, 370 CrossRef CAS PubMed; (c) X.-Q. Zhu, H.-R. Li, Q. Li, T. Ai, J.-Y. Lu, Y. Yang and J.-P. Cheng, Chem.–Eur. J., 2003, 9, 871 CrossRef CAS PubMed; (d) X.-Q. Zhu, Q.-Y. Liu, Q. Chen and L.-R. Mei, J. Org. Chem., 2010, 75, 789 CrossRef CAS PubMed; (e) Z.-P. Chen, M.-W. Chen, R.-N. Guo and Y.-G. Zhou, Org. Lett., 2014, 16, 1406 CrossRef CAS PubMed.
- (a) C. Zhu, K. Saito, M. Yamanaka and T. Akiyama, Acc. Chem. Res., 2015, 48, 388 CrossRef CAS PubMed; (b) C. Zhu and J. R. Falck, ChemCatChem, 2011, 3, 1850 CrossRef CAS; (c) C. Zhu and T. Akiyama, Org. Lett., 2009, 11, 4180 CrossRef CAS PubMed; (d) A. Henseler, M. Kato, K. Mori and T. Akiyama, Angew. Chem., Int. Ed., 2011, 50, 8180 CrossRef CAS PubMed; (e) K. Horiguchi, E. Yamamoto, K. Saito, M. Yamanaka and T. Akiyama, Chem.–Eur. J., 2016, 22, 8078 CrossRef CAS PubMed; (f) K.-H. Kim, T. Akiyama and C.-H. Cheon, Chem.–Asian J., 2016, 11, 274 CrossRef CAS PubMed; (g) W. Wen, Y. Zeng, L.-Y. Peng, L.-N. Fu and Q.-X. Guo, Org. Lett., 2015, 17, 3922 CrossRef CAS PubMed.
- (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) M. Terada, Synthesis, 2010, 42, 1929 CrossRef; (c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (d) I. Čorić and B. List, Nature, 2012, 483, 315 CrossRef PubMed; (e) C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC; (f) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
- The absolute configuration was assigned by comparing the optical rotation and HPLC analysis according to ref. 15.
- The absolute configuration was assigned by comparing the optical rotation and HPLC analysis according to ref. 16a.
- (a) L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741 CrossRef PubMed; (b) Y. Shibata and M. Yamanaka, J. Org. Chem., 2018, 78, 3731 CrossRef PubMed; (c) K. Saito, K. Horiguchi, Y. Shibata, M. Yamanaka and T. Akiyama, Chem.–Eur. J., 2014, 20, 7616 CrossRef CAS PubMed.
- The oxidized β,γ-alkynyl α-imino esters preferred an E geometry assigned by X-ray analysis, see: M. Hatano, K. Yamashita, M. Mizuno, O. Ito and K. Ishihara, Angew. Chem., Int. Ed., 2015, 54, 2707 CrossRef CAS PubMed.
- (a) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646 CrossRef CAS PubMed; (b) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187 CrossRef CAS PubMed; (c) W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118 RSC; (d) F. M. Bickelhaupt, J. Comput. Chem., 1999, 20, 114 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00944j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |