
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Demonstration of the utility of DOS-derived fragment libraries for rapid hit derivatisation in a multidirectional fashion†
Sarah L.
Kidd‡
 a,
Elaine
Fowler‡
a,
Till
Reinhardt
a,
Thomas
Compton
a,
Natalia
Mateu
a,
Hector
Newman
bc,
Dom
Bellini
b,
Romain
Talon
cd,
Joseph
McLoughlin
e,
Tobias
Krojer
f,
Anthony
Aimon
cd,
Anthony
Bradley
c,
Michael
Fairhead
d,
Paul
Brear
e,
Laura
Díaz-Sáez
df,
Katherine
McAuley
c,
Hannah F.
Sore
a,
Andrew
Madin
g,
Daniel H.
O'Donovan
h,
Kilian V. M.
Huber
df,
Marko
Hyvönen
e,
Frank
von Delft
cdi,
Christopher G.
Dowson
b and
David R.
Spring
*a
a,
Elaine
Fowler‡
a,
Till
Reinhardt
a,
Thomas
Compton
a,
Natalia
Mateu
a,
Hector
Newman
bc,
Dom
Bellini
b,
Romain
Talon
cd,
Joseph
McLoughlin
e,
Tobias
Krojer
f,
Anthony
Aimon
cd,
Anthony
Bradley
c,
Michael
Fairhead
d,
Paul
Brear
e,
Laura
Díaz-Sáez
df,
Katherine
McAuley
c,
Hannah F.
Sore
a,
Andrew
Madin
g,
Daniel H.
O'Donovan
h,
Kilian V. M.
Huber
df,
Marko
Hyvönen
e,
Frank
von Delft
cdi,
Christopher G.
Dowson
b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bSchool of Life Sciences, University of Warwick, Coventry, UK
cDiamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot OX11 0QX, UK
dStructural Genomics Consortium (SGC), University of Oxford, Oxford, OX3 7DQ, UK
eDepartment of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 1GA, UK
fTarget Discovery Institute, Nuffield Department of Medicine, University of Oxford, Oxford, UK
gHit Discovery, Discovery Sciences, R&D, AstraZeneca, Cambridge, UK
hOncology R&D, AstraZeneca, Cambridge, CB4 0WG, UK
iDepartment of Biochemistry, University of Johannesburg, Auckland Park 2006, South Africa
First published on 14th May 2020
Abstract
Organic synthesis underpins the evolution of weak fragment hits into potent lead compounds. Deficiencies within current screening collections often result in the requirement of significant synthetic investment to enable multidirectional fragment growth, limiting the efficiency of the hit evolution process. Diversity-oriented synthesis (DOS)-derived fragment libraries are constructed in an efficient and modular fashion and thus are well-suited to address this challenge. To demonstrate the effective nature of such libraries within fragment-based drug discovery, we herein describe the screening of a 40-member DOS library against three functionally distinct biological targets using X-Ray crystallography. Firstly, we demonstrate the importance for diversity in aiding hit identification with four fragment binders resulting from these efforts. Moreover, we also exemplify the ability to readily access a library of analogues from cheap commercially available materials, which ultimately enabled the exploration of a minimum of four synthetic vectors from each molecule. In total, 10–14 analogues of each hit were rapidly accessed in three to six synthetic steps. Thus, we showcase how DOS-derived fragment libraries enable efficient hit derivatisation and can be utilised to remove the synthetic limitations encountered in early stage fragment-based drug discovery.
Introduction
In the twenty years since its conception, fragment-based drug discovery (FBDD) has evolved into a mainstream approach to develop bioactive compounds. Three drugs originating from this technique have now been approved, whilst over 30 FBDD-derived clinical candidates remain under evaluation highlighting the effectiveness of this strategy.1,2 The fundamental challenge of developing potent molecules from the small, weakly bound initial hits that are identified by this method, however, should not be underestimated. Hits must be strategically optimised through fragment growing,3 linking4 or merging,5 often guided by structural information. In early development, this can be achieved using commercial compounds via an SAR-by catalogue approach,6,7 however, with less trivial fragments and as the research evolves, this rapidly becomes challenging. In this context, organic synthesis is a vital component that can contribute to the viability of a given early-stage drug discovery project.Since the emergence of this strategy, physicochemical constraints have been used to assemble collections of molecules to screen based upon the properties of successful hits from early campaigns, now termed the Rule of Three (Ro3).8 Indeed, several commercial libraries adhering to these criteria are now readily available from many vendors. However, in recent years, in addition to the Ro3 compliance, synthetic accessibility and the ability to derivatise fragment molecules have been noted as important but arguably less-well represented features.9,10 As a result, calls from leaders within the field have focused on the necessity for the development of novel fragments featuring multidirectional exit vectors with synthetic tractability, including demonstration of available growth vectors.10 Thus, within the community there has been a sustained effort to design novel fragment libraries featuring 3-dimensional (3-D) elements11–15 (such as high fraction of sp3 carbons) and polar functionality,16 both of which enable facile fragment elaboration. Moreover, despite the debate within the literature on the relevance of 3-D fragments,17,18 recent examples have validated the utility of enriching screening libraries with these motifs.19–21
Diversity-oriented synthesis (DOS) is a strategy by which libraries of structurally diverse compounds are constructed in a rapid and synthetically efficient manner through the employment of divergent synthetic manipulations.22–25 Whilst traditionally efforts in this field were focussed on larger molecules, in recent years the application of this methodology toward the synthesis of novel 3-D fragments has emerged.26 Herein, we demonstrate the relevance and utility of such libraries within FBDD. Firstly, we validate the importance for diversity in enabling identification of hits against several targets. In this case, fragment binders for three distinct proteins from different protein families were found from our recently published small but shape diverse 40-member library (Fig. 1).27 This included novel hits for challenging protein targets with no previously reported small molecule binders. Secondly, we highlight how molecules of this origin allow for analogues to be accessed in a synthetically efficient manner, including complex quaternary centre-containing compounds, in three to six steps from cheap (<£3 per gram) and readily available starting materials. Finally, we exemplify how the inherently modular chemistry can enable fragment elaboration from a variety of vectors, with derivatives of each hit exploring a minimum of four different positions.
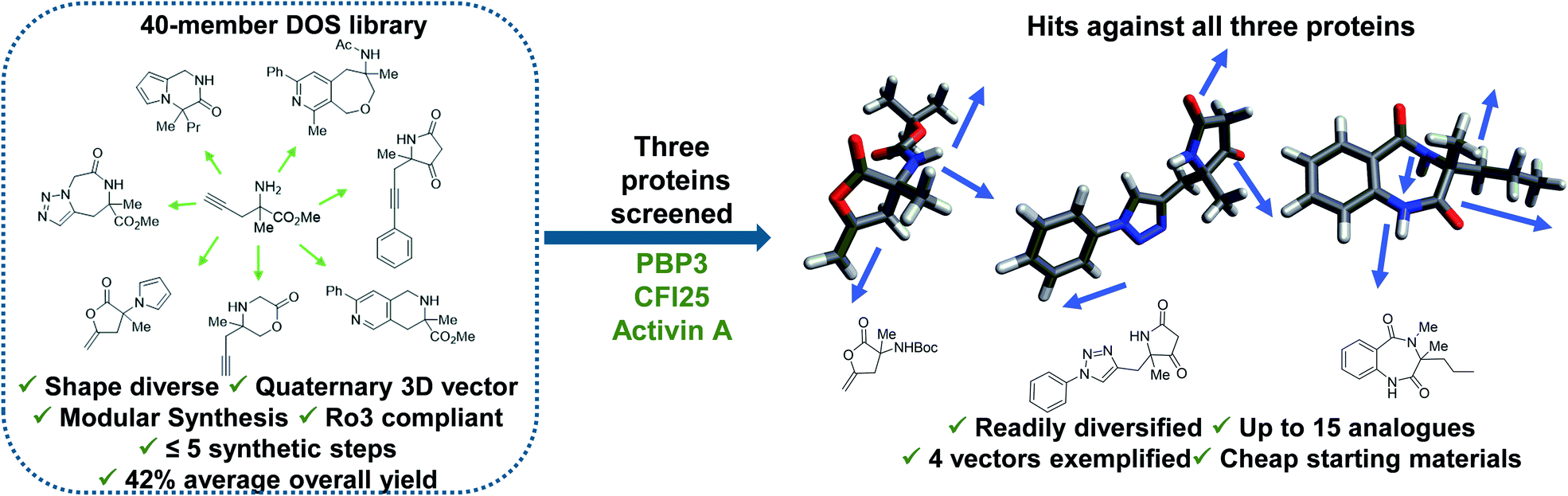 | ||
| Fig. 1 Demonstration of the utility of DOS libraries in X-ray based fragment screening and the ability to enable rapid analogue generation along multiple 3-D vectors around initial hits in a facile manner. See ref. 27 for chemistry towards the library. | ||
Results
With advances in foundational technologies such as third-generation synchrotrons and high-throughput technologies,9,28–30 X-ray crystallography methods have since become one of the most well-used techniques for hit finding within the field of FBDD.31 Thus, this method was selected as the primary screening technique conducted through a collaboration with the XChem platform.32 The DOS library was screened in a racemic fashion to provide both enantiomers and was used in a 500 mM§ format in d6-DMSO.Penicillin binding protein 3
The first protein screened with our DOS library was penicillin binding protein 3 (PBP3). The PBP family is responsible for the synthesis and cross-linking of peptidoglycan, the major component in the bacterial cell wall. The cell wall plays a pivotal role in controlling the shape and integrity of the cell and inhibition of the PBPs leads to cell lysis due to turgor pressure.33,34 The penicillin-binding domain contains a catalytic serine residue, which is vital for its function and a useful target for inhibition.35 Due to their essential role in cell division and elongation, PBPs are attractive targets for antibiotics with many β-lactam antibiotics developed for this purpose.36 However, the efficacy and wide-spread use of β-lactams has driven the alarming growth of bacterial resistance. Novel scaffolds capable of inhibiting the PBP family are greatly needed to overcome resistance mechanisms and restore activity against common infections.37 Due to this imminent need for new antibiotic leads, our DOS library was screened against P. aeruginosa PBP3 using the XChem platform.This initial screen resulted in the serendipitous discovery of 1 as a covalent binder of PBP3 (PDB: 6Y6Z, Fig. 2), which strikingly was the first binder identified amongst approximately 1300 previous fragment soaks (see Table S1† for further details). The core enol lactone scaffold was found to react with Ser294, the catalytic residue found within the conserved SXXK motif of the β-lactam binding pocket. Upon incubation of the compound with the crystals, resulting electron density maps suggested a linear bound compound, which was hypothesised to result from lactone ring-opening followed by enol tautomerization to afford the linear ketone derivative. In addition to the covalent bond, hydrogen bonding interactions were observed with neighbouring residues Asn351, Ser349 and Thr487.
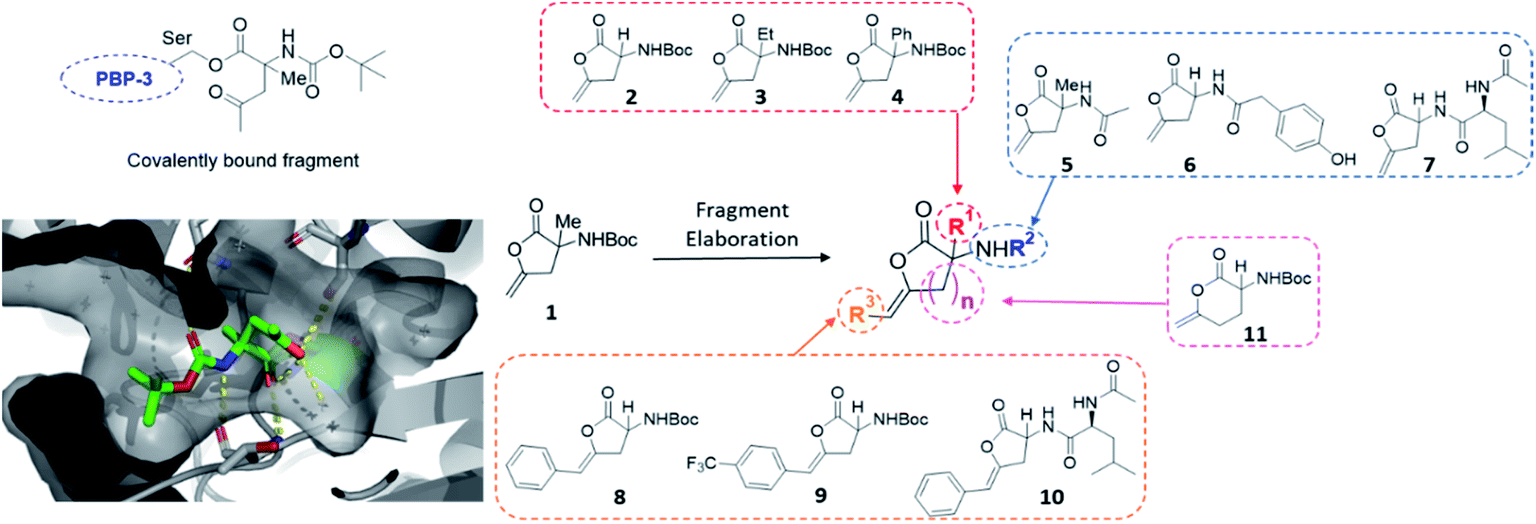 | ||
| Fig. 2 Initial hit compound 1 bound to PBP3, highlighted vectors suitable for diversification and the analogues synthesised to validate the initial hit. | ||
Considering these findings, it was proposed that the identified hit could be rapidly diversified through four primary vectors to comprehensively probe the PBP3 binding pocket. It was envisaged that the quaternary centre could be substituted at R1 with different alkyl (2, 3) and aryl groups (4), amide couplings could be used to functionalise R2 (5–7), the terminal alkene could be substituted at R3 (8, 9, 10) and the lactone ring size could be altered (11). In this manner, exploring different ring sizes would allow for core scaffold modification, which is often difficult to incorporate into early fragment development. Importantly, substituents chosen for elaboration at R2 were designed with key β-lactam inhibitors in mind.
The synthetic strategy used to access the proposed analogues was based upon the original chemistry used to prepare the library and utilised a common amino ester substrate in a divergent process (Scheme 1). All four vectors were accessed in five synthetic steps. Firstly, to diversify the quaternary centre, the R1 group in the commercially available ketoesters of type 12 could be varied. All examples shown here were purchased from Sigma Aldrich for under £3 per gram. To begin, 12a–d were condensed with p-anisidine to generate the p-methoxyphenyl (PMP)-imine, which was subjected to a Barbier-type coupling to install the alkyne handle, giving protected amines 13a–d. The PMP-group was subsequently removed from 13a–d using cerium ammonium nitrate (CAN), giving amines 15a–d. Alternatively, to access the tertiary carbon centre a simple substitution then deprotection of commercially available imine 14 allowed for easy access of amine 15e in high yields. In this case, an alkyne featuring an extra methylene linker was employed to enable downstream formation of the larger six-membered ring derivative of 1. From these amine intermediates, a diverse range of analogues were subsequently rapidly accessed using a simple toolkit of reliable chemistries. HATU-mediated amide couplings were exploited to connect a variety of motifs (R2) to the amine using substrates 16a–d. Elaboration of this vector proved to be highly efficient since the final scaffold could be accessed in just three steps from the common amine intermediate, and hence many groups were explored with little synthetic effort. Following amide formation, the ester groups within 17a–h were readily hydrolysed using LiOH, yielding acids 18a–f. These final precursors could be cyclised using Cu(I)Br to form the unsaturated lactone scaffolds 2–7, 10 and 11. Alternatively, a procedure inspired by a reported one-pot Pd-catalysed cyclisation-coupling reaction38,39 was used to vary the R3 alkene substitution, enabling exploration of the final vector and providing access to 8–10. This late-stage diversification provided extremely efficient access to a variety of novel analogues from cheap, commercially available aryl iodides.
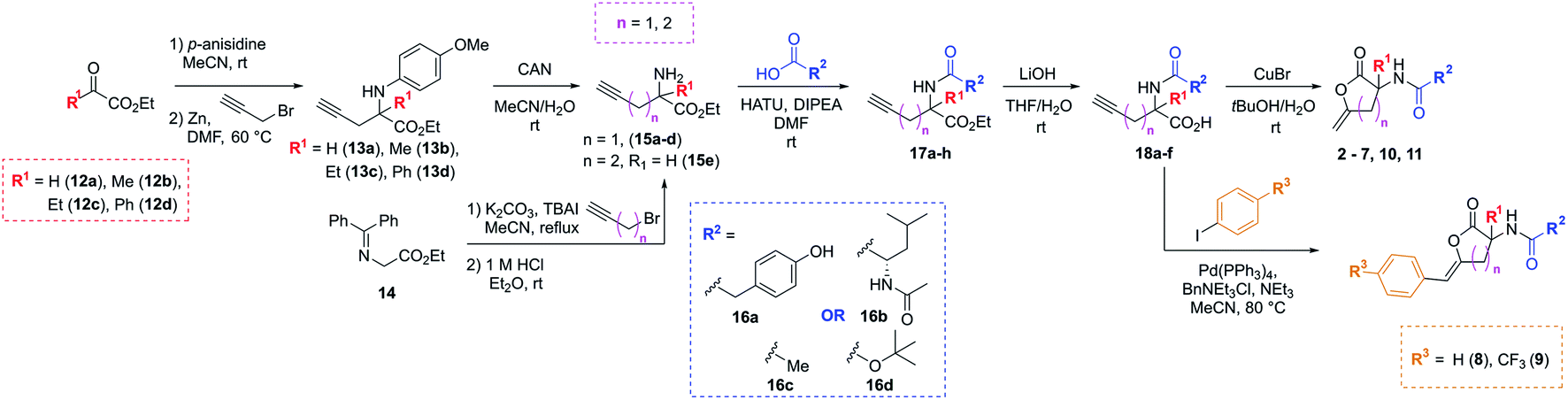 | ||
| Scheme 1 Synthetic route to analogues of the initial PBP3 hit 1 in five steps through key amine of type 15. | ||
Due to the inherent design features of our fragment library diversification of this fragment hit was simple, rapid and used robust chemistry. The 10 elaborated analogues were then screened using X-ray crystallography to validate the initial hit and observe the effects of vector derivatisation on the PBP3 binding preference. All compounds except for 8–11 were identified as PBP3 binders using this method. This preliminary data proved to be extremely useful in validating this hit with all analogues covalently binding to Ser294 in a similar fashion to that of 1, whilst the specificity for the 5-membered lactone and terminal alkene could be inferred.
We found that fragment elaboration from the amine vector (R2) was well-tolerated, including a variety of functionalities and sizes (5–7). Interestingly, the phenol group of 6 was found to project into a hydrophobic cleft within the pocket, appearing to make π–π interactions with proximal aromatic residues Tyr407 and Tyr409 (PDB: 6Y6U, Fig. 3, orange sticks), whilst maintaining previously observed hydrogen bonding interactions. These additional π–π interactions could prove extremely useful for further medicinal chemistry efforts in the downstream fragment evolution process.
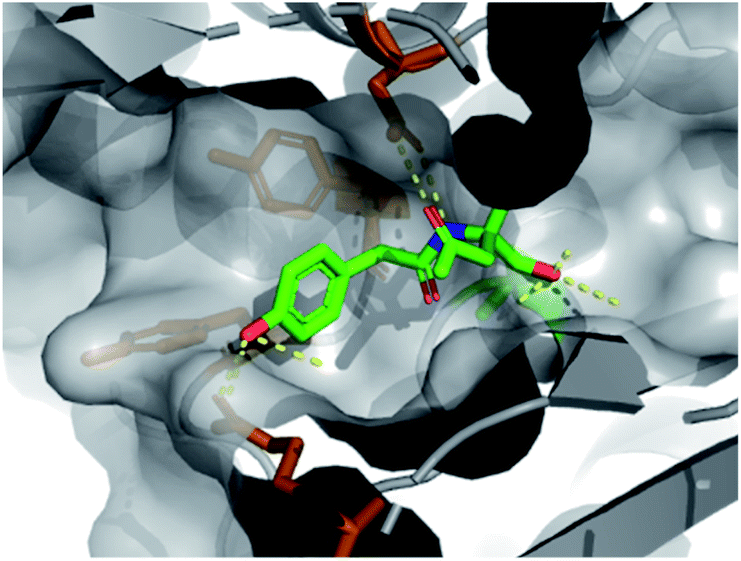 | ||
| Fig. 3 X-ray structure of 6 bound to PBP3 with conserved H-bonds highlighted. | ||
CFI25
To further demonstrate the utility of our library a second target CFI25 (cleavage factor 25 kDa), an essential sub-unit of the pre-mRNA cleavage factor Im, was screened. This heterotetramic complex comprises two units of CFI25 with two further units of either CFI59 or CFI68.40,41 Numerous studies have shown CFI25 to play a key role in determining the size of the 3′ untranslated region of mRNA, due to its involvement in the alternative polyadenylation (APA).42 This important mechanism is involved in gene regulation, ultimately contributing to the generation of different mRNA isoforms.43 Crucially, several studies have implicated CFI25 in oncology44 and neuropsychiatric disease45 settings, yet to date no small molecule modulators of this target are known to enable the further elucidation of its function. Thus, this served as an interesting target to explore with our novel DOS fragment library.Upon analysis of the resulting PanDDA event maps,46 two X-ray hits were identified (PDBs: 5R4P and 5R4Q, Fig. 4 and S1†) in a putative allosteric site away from the known mRNA substrate channel.41,47 Importantly, as a result of the diverse nature of the DOS library these hits related to distinctly different chemotypes, highlighting the potential of this collection to deliver hits of varied molecular architecture.
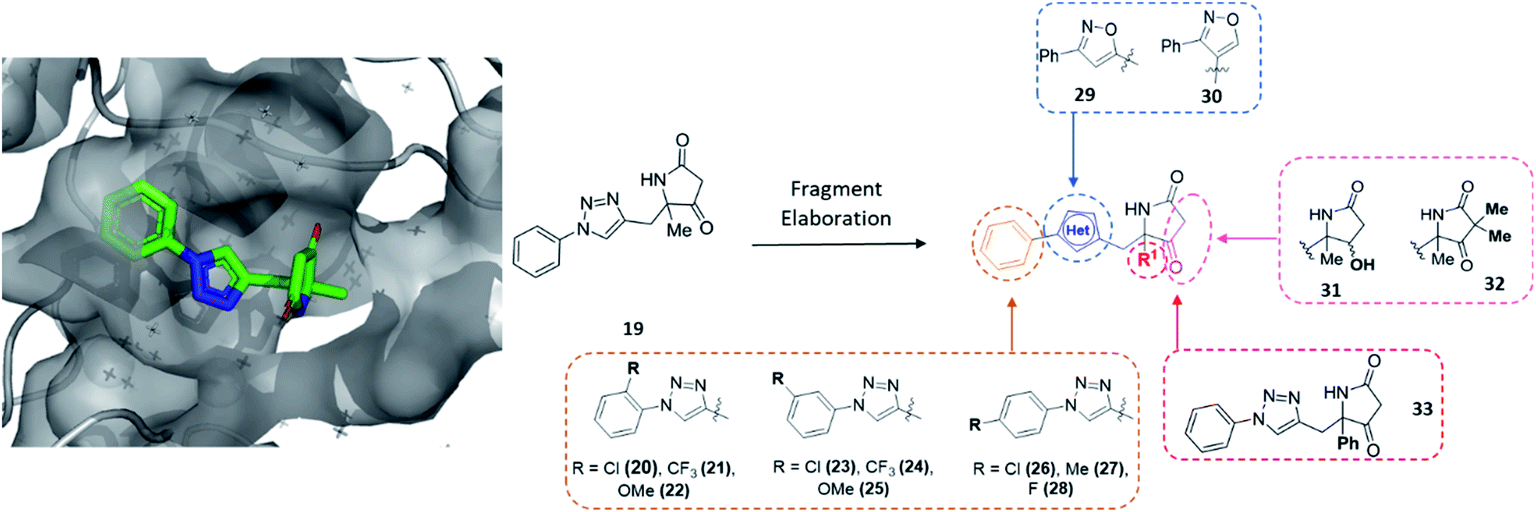 | ||
| Fig. 4 Initial hit compound 19 bound to CFI25, highlighted vectors suitable for diversification and the analogues synthesised to validate the initial hit. | ||
To exemplify the ability of modular DOS methodologies to enable rapid construction of varied analogues, hit 19 was further investigated. The amenability of the DOS chemistry toward multidirectional vector growth could be demonstrated via derivatisation to almost every functionality within 19 (Fig. 4). Specifically, in line with the structural data, it was thought that these investigations could include modification of the benzene ring through substitution (20–28), variation in the bridging heterocycle (29, 30), derivatisation of the pyrrolidinone heterocycle via α-alkylation or ketone modification (31, 32) and finally modification of the quaternary substituent R1 (33).
Importantly, in an analogous fashion to the explorations around 1 all derivatives were directly formed from the same quaternary amine intermediates of type 15 (Scheme 2). Firstly, to access analogues bearing substituents (R2) on the benzene ring amine 15a was acylated to give amide 34a. Next, Cu-mediated click chemistry was performed on 34a using a variety commercially available substituted azides 35a–j. In all cases, the resultant triazole products 36a–j were obtained in good yields. Next, precursors 36a–j were taken forward for cyclisation to afford the desired pyrrolidinone analogues 20–28via subjection to Dieckmann condensation conditions followed by thermal decarboxylation, which proceeded with good yields.
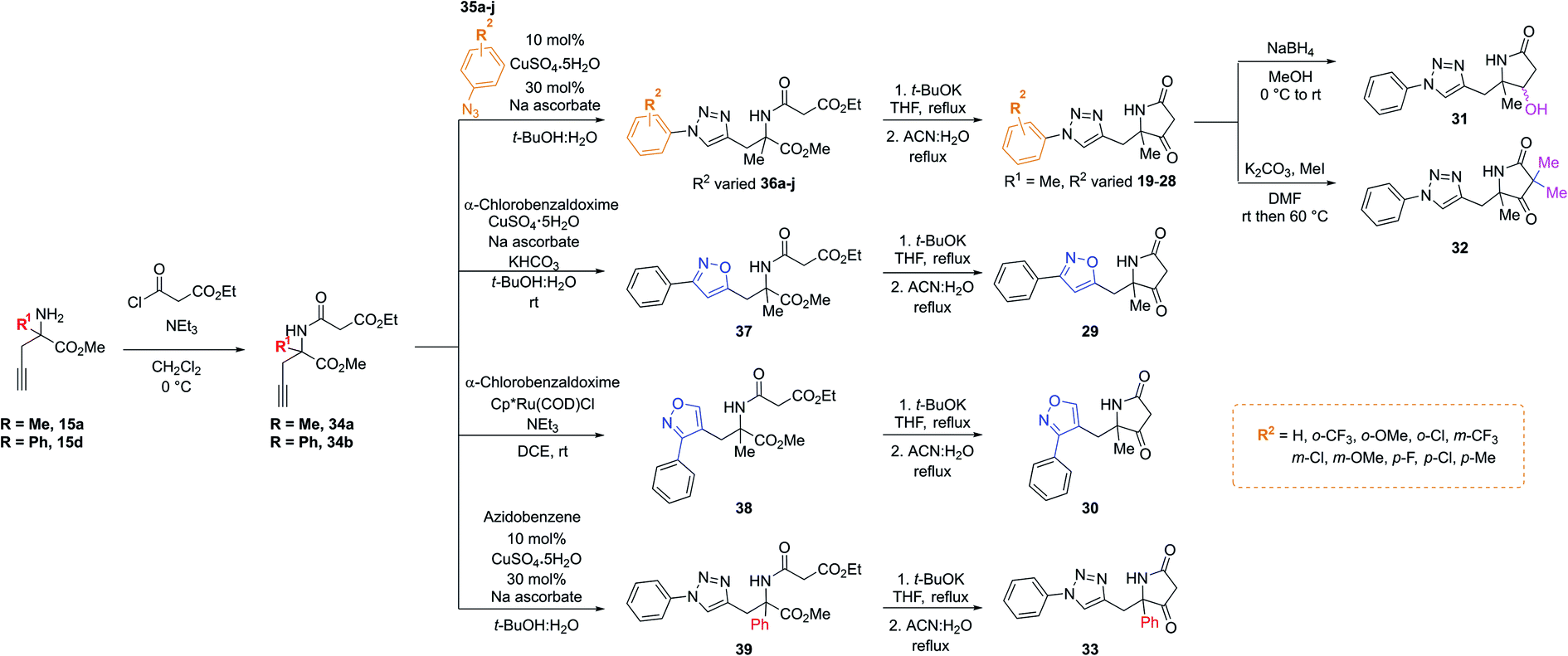 | ||
| Scheme 2 A divergent scheme was harnessed to access analogues 20–33 starting from the key quaternary amine intermediate of type 15. | ||
Next, using 34a in the same click reaction but exchanging the azide component to α-chlorobenzaldoxime48 furnished the 1,4-substituted isoxazole intermediate 37, which could again be cyclised by employing the same conditions to afford 29. Alternatively, the 1,5-isoxazole variant could be obtained using the same strategy but using a Ru-based catalyst,49 affording 38. Once more, Dieckmann condensation followed by decarboxylation yielded 30. Finally, derivatives containing pyrrolidinone modifications were accessed through a late-stage modification strategy from 19 through either ketone reduction to give 31, as a diastereomeric mixture, or α-deprotonation followed methylation to give 32. In a similar fashion, modifying the R1 position could be achieved using the phenyl quaternary amine 15d, which was subjected to the above sequence to give 34b followed by 39, and cyclised to give 33.
In this example, the highly modular and divergent DOS strategy successfully enabled the rapid synthesis of 14 derivatives of hit 19. These analogues were subsequently screened for binding using a further round of X-ray crystallography. This data revealed of the nine substituted aromatic analogues (20–28), only the p-fluorine analogue 28 was tolerated within the crystals (PDB: 5R4T, Fig. 5A, green sticks). Here, it was found that the aromatic portion of the molecule bound in a similar fashion to initial hit 19, with the amide carbonyl interacting with Lys56 within the protein backbone. Similarly, the binding of 29 (PDB: 5R4U, Fig. 5A, cyan sticks) revealed that the alternative isoxazole bridging heterocycle was also tolerated, again in a similar binding pose to the original hit 19. Importantly, selectivity for the 1,4-regioisomer could be inferred from these results since no binding of the 1,5-isomer 30 was identified. In an analogous fashion, 32 also exhibited this binding mode (PDB: 5R4R, Fig. 5A, yellow sticks). Interestingly, in this case the gem-dimethyl substituents and quaternary centre were oriented toward different channels within the protein, suggesting these positions could be utilised as two alternative 3-D growth vectors from the molecule.
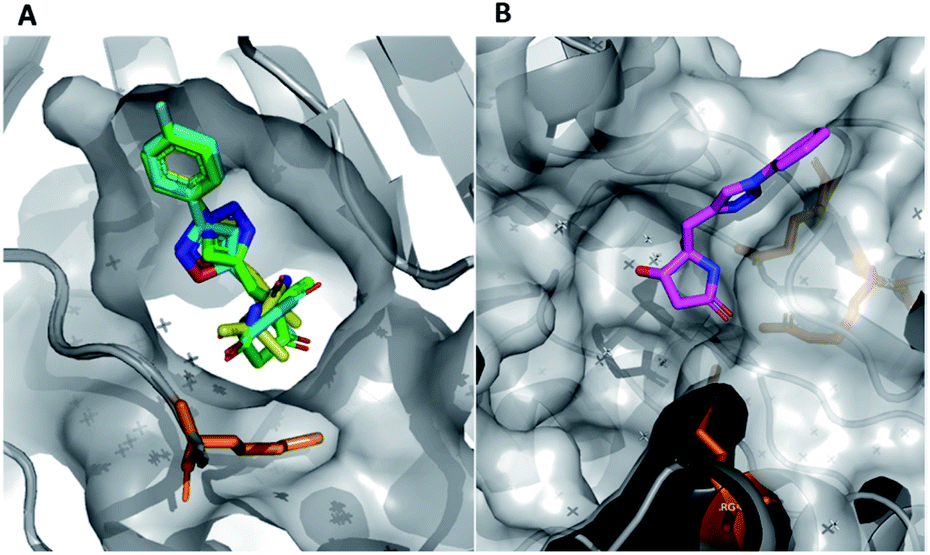 | ||
| Fig. 5 (A) Hits that bound in allosteric site, 28 = green sticks, 29 = cyan sticks, 32 = yellow sticks and (B) 31 was found to bind in the substrate channel. | ||
Conversely, soaking of 31 revealed this compound instead bound within the distal mRNA substrate channel with a putative polar interaction between the amide carbonyl and the key Arg63 residue known to mediate binding of the UUGUAU RNA motif (PDB: 5R4S, Fig. 5B, binding protein residues in orange).41,47 Additional interactions toward Arg150 and Gln157 further stabilised this binding. It is worth mentioning, as with all bound derivatives, the electron density for the aromatic region proved to be much more defined, whilst that of the quaternary heterocycle was more ambiguous to assign. Thus, whilst these interactions could be hypothesised, screening of the single enantiomer or diastereomer variants of all four binders would provide vital information about the true binding preference and spatial orientation of the heterocycle. Building on previous research in the field of DOS fragments for hit evolution,50 in this example we have demonstrated how this can be achieved in a multidirectional fashion through leveraging the inherent modularity, the quaternary motif and sp3 carbons to provide insights into the most effective strategy for fragment growth.
Activin A
The final protein to be screened against our DOS-fragment library was activin A. Activins are members of the transforming growth factor β (TGF-β) superfamily of growth factors, which play essential roles in homeostasis and development and have been studied for many years.51 Research has shown activins mediate an intramolecular signalling cascade via binding of the extracellular domains of transmembrane serine/threonine kinases known as type I or type II receptors, ultimately conducting the phosphorylation of Smad proteins involved in target gene expression.52–54 Importantly, in this context, binding of the type II receptors has been shown to be crucial for type I receptor binding and therefore vital to initiate the first step of this signalling pathway. Several studies have associated the role of activin A signalling with the regulation of embryogenesis, stem cell differentiation and wound healing, among other processes. Moreover, dysregulation of activin A signalling or expression has been linked to human diseases such as inflammatory conditions, cancer and fibrodysplasia ossificans progressiva.55–58 Nevertheless, despite the potential of this target, to the best of our knowledge no small molecule modulators of this protein exist to enable further investigations into the associated biology. Thus, an XChem screen was conducted leading to the identification of 40 as a binding partner for activin A (PDB: 6Y6N, Fig. 6). This data suggested a key hydrogen bonding interaction between the benzylic amide carbonyl and the Trp28 residue within the site. This pocket is in the predicted binding sites for the activin A type I receptor ACVR1B/ALK4, proving an interesting avenue to pursue.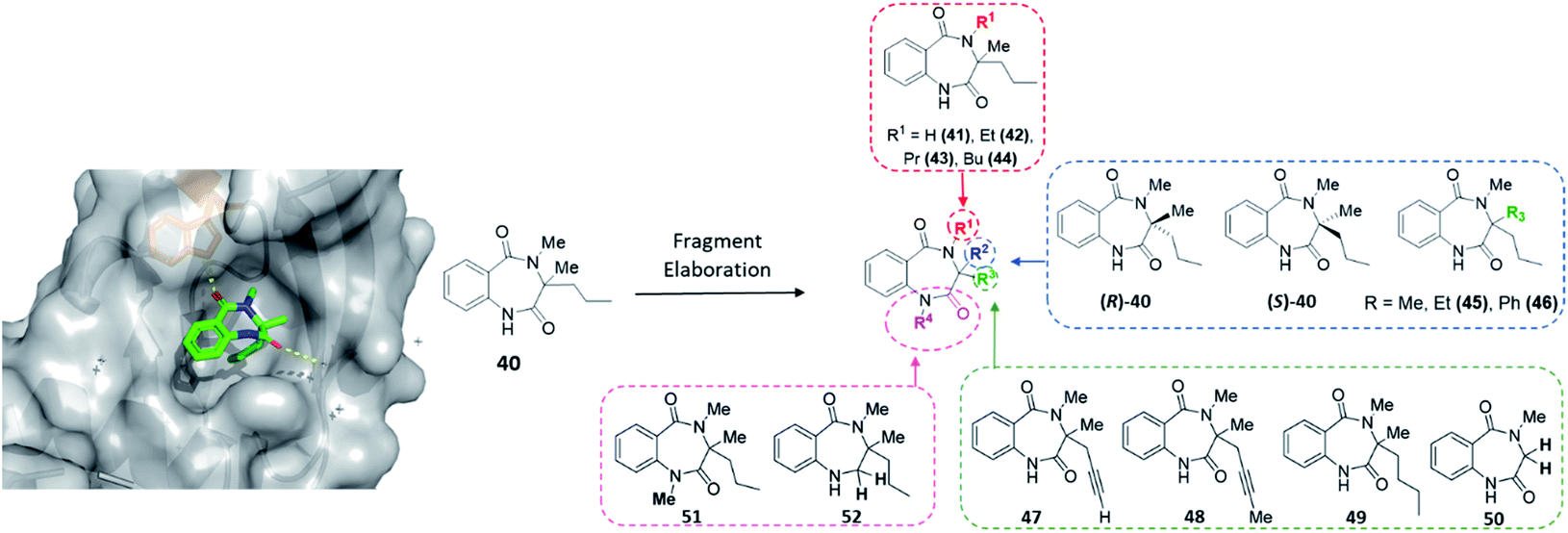 | ||
| Fig. 6 Initial hit compound 40 bound to activin A, highlighted vectors suitable for diversification and the analogues (41–52) synthesised to validate initial hit. | ||
With the crystal structure and modular DOS route in mind, several analogues were once more explored to showcase the chemistry. It was proposed that the benzodiazepine core of the molecule provided an opportunity for several points of derivatisation, such as simple N-alkylation (R1) to form 41–44, quaternary substituent modifications at R2 (45, 46), including enantiopure derivatives ((R)/(S)-40), alkyne chain modifications (R3, 47–49), removal of the quaternary substituents (50) and finally amide modifications (R4) e.g.51 and 52. Once more, this was proposed to commence via a divergent process (Scheme 3A), utilising the same key amine intermediates 15b–d.
 | ||
| Scheme 3 Two synthetic strategies to access analogues of 40, (A) via key amine 15, (B) from sarcosine methyl ester hydrochloride. | ||
Firstly, analogues bearing N-amide substituent variations at the R1 position were pursued. Starting from 15b the acylated intermediate 53a was accessed in good yield. To form 41 with R1 as hydrogen, 53a was subjected to nitro reduction using palladium catalysis and finally hydride-mediated cyclisation of the resultant amine toward the remaining ester functionality. Alternatively, alkylation of 53a with a variety of alkyl halides and catalytic TBAI afforded 54a–d. These acyclic precursors could then undergo the same synthetic sequence of reduction and cyclisation to give 42–44. Following this synthetic route, R2 was explored using amines 15c and 15d, bearing variation of the quaternary substituent. These were converted to 54e and 54f, followed by 45 and 46 as previously described.
Next, to showcase the ability to readily access enantiopure derivatives of both the key amine 15b and related analogues, asymmetric routes to both (R)- and (S)-15b were pursued (synthetic procedure, see ESI†). Following previously established and reported chemistry from within the group,27 these variants were also rapidly accessed from the same commercially available ketoester starting material 12b in just four steps. Following the previously described procedure, both enantiomers were converted to (R)-40 and (S)-40via (R)- or (S)-53a and -54a.
To modify the propyl chain (R3) of 40 and form 47–49, in this instance 15b was acylated with 2-azidobenzoyl chloride to form 55. Surprisingly, upon subjection of this to the standard methylation procedure, both 56a and 56b were formed as an inseparable mixture of products. At this stage, the crude material was telescoped into the zinc-mediated reduction step, followed by cyclisation to generate separable material. Indeed, both 47 and 48 were isolated. To further explore this vector in a divergent fashion, the alkyne moiety within 48 was reduced using palladium to give 49. Alternatively, to remove both substituents from the quaternary position (Scheme 3B), sarcosine methyl ester hydrochloride could be utilised, which was acylated to give 57 before reduction and cyclisation yielded 50. Finally, two late-stage diversifications of 40 were used to explore R4. This included further alkylation of the amide to give 51 and selective reduction of the unsubstituted amide to afford 52.
In total four vectors of the molecule were explored using the 14 analogues described. Once more, these compounds were rapidly accessed via short synthetic sequences from commercial materials, highlighting the utility of the chemistry described. In a subsequent round of crystallography, analogue 42 was found to bind in the same pocket as the original hit, with the key H-bond interaction toward the Trp28 residue conserved (PDB: 6Y6O, Fig. 7). This secondary binding data was useful to validate the original hit binding and show the ethyl variant to be tolerated, suggesting the substituted amide position to be viable growth vector for future synthetic efforts.
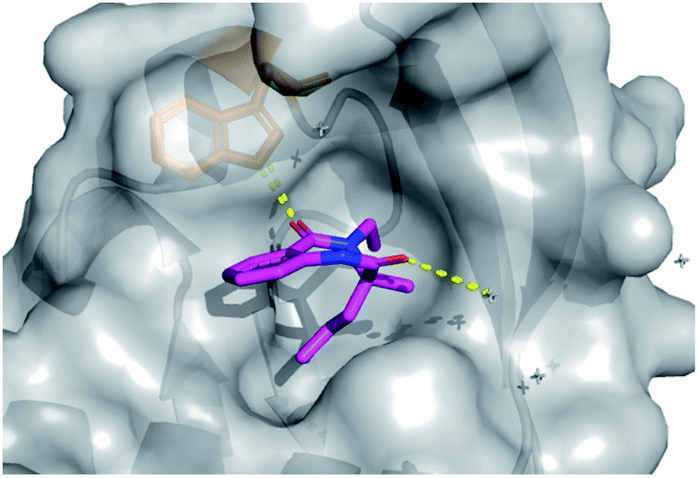 | ||
| Fig. 7 Follow-up analogue 42 bound to activin A with conserved H-bond to Trp28 highlighted. | ||
Discussion
Herein, we have exemplified the ability of fragment-focused DOS libraries to deliver diverse hits across numerous targets from distinct protein families, despite originating from the same amino ester building block. Screening of our DOS library containing 40 compounds gave several structurally distinct and tangible leads across all proteins considered. Importantly, PBP3, CFI25, and activin A are indicators for completely unrelated therapeutic areas, and as such these hits have the potential to serve as novel starting points for the development of inhibitors or chemical probes for a variety of biological purposes. The hit identified against PBP3 binds through a covalent mechanism, exemplifying the utility of our library towards the discovery of novel covalent ligands, in addition to reversible binders. Indeed, similar electrophilic fragments have been demonstrated to have enhanced utility in probe development due to their high duration of action and potency.59,60 We also report, to the best of our knowledge, the first small molecule binders of CFI25 and activin A.61 Furthermore, subsequent screens using the DOS library have also proven successful in delivering novel hits against additional antibiotic targets, with active discovery projects stemming from these results.For these three proteins, four hits were identified, three of which were then diversified to rapidly generate 10–14 analogues in just three to six steps. All ketoester starting materials described are commercially available, with costs under £3 per gram. Moreover, the reaction sequences used to access the key amine intermediates of type 15 were readily and reproducibly prepared on multi-gram scales. As a result, the timescales of downstream analogue formation could be further reduced since several analogues were accessed in a divergent manner from this material. It is worth mentioning, as some examples have highlighted, that removal of the quaternary substituent could also be utilised as a strategy to decrease the number of steps required to access analogues of this library. However, in all projects described this feature was retained to exemplify the ease of utilising this position as a growth vector. Indeed, derivatisation of an sp3 quaternary carbon centre could prove highly challenging for most fragment hits, yet due to the simple three-step procedure previously developed, we have demonstrated how analogues of library members with this feature could be prepared with no additional route design.
The derivatives prepared explore at least four different fragment exit vectors utilising simple chemical transformations, offering significant incentives for library implementation in early FBDD programs. As discussed, one common hurdle within FBDD follow-up work remains the investigation of suitable points of hit modification to enable rapid and efficient exploration of a given binding pocket. Here, we have shown how novel libraries can be designed to alleviate this hurdle, allowing for facile initial exploratory chemistry, often where the molecules are low on the value-synthesis trajectory.62
In all cases, the preliminary X-ray data was used to deduce validation of each initial hit described, since at least one analogue of all three were additionally found to bind within the respective targets. In some cases, structural specificity could be inferred based upon the lack of density observed for some analogues. Thus, this initial scoping chemistry proved to be a valuable technique to also probe the binding pockets and derive potentially interesting vectors for further hit evolution during future project objectives.
In this work, we have demonstrated the advantages of using a DOS-derived library for FBDD lead validation and diversification. However, there are other factors which also limit hit progression in FBDD, including the difficulty in attaining additional biophysical characterisation required to generate structure–activity relationship data. The hits and follow-up compounds described in this work are currently under further investigation, with the overall aim to confirm binding in biophysical assays and ultimately produce potent lead compounds for each protein.
Conclusions
Herein, we have demonstrated that DOS-derived libraries are useful tools for the generation of novel hits across a variety of different biological targets. We identified four hits for PBP3, CFI25, and activin A, all of which are functionally diverse proteins with great relevance for developing novel therapeutics as well as biological function elucidation. This further strengthens the precedence for incorporation of 3-D, diverse fragments within screening collections to augment existing commercial compounds.We also evidence how the strategic design of novel libraries to incorporate modularity, whilst maintaining complexity, can result in alleviating chemistry as a limiting factor in early discovery projects. In this case, DOS methodology was exploited to facilitate rapid fragment elaboration, with up to 14 analogues of each hit readily accessed in short synthetic sequences despite the formation of challenging quaternary carbon centres. The additional advantage of these synthetic sequences is their use of cheap commercial materials, which reduces the requirement for lengthy and expensive initial explorative chemistry. The library described is currently available for screening via the XChem platform, where we hope it will be utilised by the scientific community to provide novel and more importantly tractable fragment hits for future development.
Author contributions
Research was conceived by S. L. K., E. F., N. M., H. F. S. and D. R. S. H. N., D. B., R. T., J. M., A. A., A. B., P. B., L. D. S., K. M. contributed toward data acquisition, analysis, and interpretation. Synthesis and characterisation carried out by S. L. K., E. F., T. R., T. C. with support from D. H. O. D. and A. M. Protein expression was carried out by M. F., D. B., H. N., and J. M. X-ray screening was conducted by S. L. K, D. B., H. N., R. T., J. M., with support from K. V. M. H., M. H., F. D., and C. G. D. All authors provided comments and advice on the manuscript during construction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EP/P020291/1), Royal Society (Wolfson Research Merit Award) and AstraZeneca. H. N. acknowledges a collaborative postgraduate award between the University of Warwick and Diamond Light Source (STU0212). C. G. D. acknowledges funding by the CHNUK grant (MR/S014934/1) from the MRC. J. M. was funded by BBSRC Doctoral Training Program studentship. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. We would like to acknowledge Diamond Light Source for time on i04-1/XChem under proposals lb18145, lb15649 and time on i03 and i24 under the proposals mx14303 and nt14493. We are grateful for access to and support of the X-ray crystallography facility at the Department of Biochemistry, University of Cambridge.References
- C. W. Murray, D. R. Newell and P. Angibaud, Medchemcomm, 2019, 10, 1509–1511 RSC.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- J. Schoepfer, W. Jahnke, G. Berellini, S. Buonamici, S. Cotesta, S. W. Cowan-Jacob, S. Dodd, P. Drueckes, D. Fabbro, T. Gabriel, J.-M. Groell, R. M. Grotzfeld, A. Q. Hassan, C. Henry, V. Iyer, D. Jones, F. Lombardo, A. Loo, P. W. Manley, X. Pellé, G. Rummel, B. Salem, M. Warmuth, A. A. Wylie, T. Zoller, A. L. Marzinzik and P. Furet, J. Med. Chem., 2018, 61, 8120–8135 CrossRef CAS PubMed.
- C. De Fusco, P. Brear, J. Iegre, K. H. Georgiou, H. F. Sore, M. Hyvönen and D. R. Spring, Bioorg. Med. Chem., 2017, 25, 3471–3482 CrossRef CAS PubMed.
- P. O. Nikiforov, S. Surade, M. Blaszczyk, V. Delorme, P. Brodin, A. R. Baulard, T. L. Blundell and C. Abell, Org. Biomol. Chem., 2016, 14, 2318–2326 RSC.
- N. Baurin, F. Aboul-Ela, X. Barril, B. Davis, M. Drysdale, B. Dymock, H. Finch, C. Fromont, C. Richardson, H. Simmonite and R. E. Hubbard, J. Chem. Inf. Comput. Sci., 2004, 44, 2157–2166 CrossRef CAS PubMed.
- B. Lamoree and R. E. Hubbard, Essays Biochem., 2017, 61, 453–461 CrossRef PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, J. Med. Chem., 2016, 59, 8189–8206 CrossRef PubMed.
- C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 55, 488–492 CrossRef CAS PubMed.
- D. G. Twigg, N. Kondo, S. L. Mitchell, W. R. J. D. Galloway, H. F. Sore, A. Madin and D. R. Spring, Angew. Chem., Int. Ed., 2016, 55, 12479–12483 CrossRef CAS PubMed.
- N. Palmer, T. M. Peakman, D. Norton and D. C. Rees, Org. Biomol. Chem., 2016, 14, 1599–1610 RSC.
- D. J. Foley, P. G. E. E. Craven, P. M. Collins, R. G. Doveston, A. Aimon, R. Talon, I. Churcher, F. von Delft, S. P. Marsden and A. Nelson, Chem.–Eur. J., 2017, 23, 15227–15232 CrossRef CAS PubMed.
- K. F. Morgan, I. A. Hollingsworth and J. A. Bull, Chem. Commun., 2014, 50, 5203–5205 RSC.
- H. Hassan, S. P. Marsden and A. Nelson, Bioorg. Med. Chem., 2018, 26, 3030–3033 CrossRef CAS PubMed.
- O. B. Cox, T. Krojer, P. Collins, O. Monteiro, R. Talon, A. Bradley, O. Fedorov, J. Amin, B. D. Marsden, J. Spencer, F. Von Delft and P. E. Brennan, Chem. Sci., 2016, 7, 2322–2330 RSC.
- M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS PubMed.
- A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert, S. Hoelder, A. Jordan, S. V. Ley, A. Merritt, D. Miller, M. E. Swarbrick and P. G. Wyatt, Drug Discovery Today, 2013, 18, 1221–1227 CrossRef PubMed.
- J. A. Johnson, C. A. Nicolaou, S. E. Kirberger, A. K. Pandey, H. Hu and W. C. K. Pomerantz, ACS Med. Chem. Lett., 2019, 10, 1648–1654 CrossRef CAS PubMed.
- U. Grädler, D. Schwarz, M. Blaesse, B. Leuthner, T. L. Johnson, F. Bernard, X. Jiang, A. Marx, M. Gilardone, H. Lemoine, D. Roche and C. Jorand-Lebrun, Bioorg. Med. Chem. Lett., 2019, 29, 126717 CrossRef PubMed.
- R. Zhang, P. J. McIntyre, P. M. Collins, D. J. Foley, C. Arter, F. von Delft, R. Bayliss, S. Warriner and A. Nelson, Chem.–Eur. J., 2019, 25, 6831–6839 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 1–13 Search PubMed.
- W. R. J. D. Galloway, A. Bender, M. Welch and D. R. Spring, Chem. Commun., 2009, 2446–2462 RSC.
- J. Kim, J. Jung, J. Koo, W. Cho, W. S. Lee, C. Kim, W. Park and S. B. Park, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799–6804 CrossRef CAS PubMed.
- N. Mateu, S. L. Kidd, L. Kalash, H. F. Sore, A. Madin, A. Bender and D. R. Spring, Chem.–Eur. J., 2018, 24, 13681–13687 CrossRef CAS PubMed.
- T. G. Davies and I. J. Tickle, in Fragment-Based Drug Discovery and X-Ray Crystallography, ed. M. Davies and Thomas G. Hyvönen, Springer, Berlin, Heidelberg, 2011, pp. 33–59 Search PubMed.
- X. Yin, A. Scalia, L. Leroy, C. M. Cuttitta, G. M. Polizzo, D. L. Ericson, C. G. Roessler, O. Campos, M. Y. Ma, R. Agarwal, R. Jackimowicz, M. Allaire, A. M. Orville, R. M. Sweet and A. S. Soares, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1177–1189 CrossRef CAS PubMed.
- D. Patel, J. D. Bauman and E. Arnold, Prog. Biophys. Mol. Biol., 2014, 116, 92–100 CrossRef CAS PubMed.
- D. Erlanson, Poll results: affiliation and fragment-finding methods, 2019, http://practicalfragments.blogspot.com/2019/12/poll-results-affiliation-and-fragment.html, accessed 10 January 2019 Search PubMed.
- P. J. McIntyre, P. M. Collins, L. Vrzal, K. Birchall, L. H. Arnold, C. Mpamhanga, P. J. Coombs, S. G. Burgess, M. W. Richards, A. Winter, V. Veverka, F. Von Delft, A. Merritt and R. Bayliss, ACS Chem. Biol., 2017, 12, 2906–2914 CrossRef CAS PubMed.
- E. Sauvage, A. Derouaux, C. Fraipont, M. Joris, R. Herman, M. Rocaboy, M. Schloesser, J. Dumas, F. Kerff, M. Nguyen-Distèche and P. Charlier, PLoS One, 2014, 9, e98042 CrossRef PubMed.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS PubMed.
- A. Zapun, C. Contreras-Martel and T. Vernet, FEMS Microbiol. Rev., 2008, 32, 361–385 CrossRef CAS PubMed.
- W. Chen, Y.-M. Zhang and C. Davies, Antimicrob. Agents Chemother., 2017, 61, e01651 CAS.
- R. J. Worthington and C. Melander, J. Org. Chem., 2013, 78, 4207–4213 CrossRef CAS PubMed.
- P. A. Jacobi, S. Lanz, I. Ghosh, S. H. Leung, F. Löwer and D. Pippin, Org. Lett., 2001, 3, 831–834 CrossRef CAS PubMed.
- W. G. O'Neal, W. P. Roberts, I. Ghosh and P. A. Jacobi, J. Org. Chem., 2005, 70, 7243–7251 CrossRef PubMed.
- S. Kim, J. Yamamoto, Y. Chen, M. Aida, T. Wada, H. Handa and Y. Yamaguchi, Genes Cells, 2010, 15, 1003–1013 CrossRef CAS PubMed.
- M. Coseno, G. Martin, C. Berger, G. Gilmartin, W. Keller and S. Doublié, Nucleic Acids Res., 2008, 36, 3474–3483 CrossRef CAS PubMed.
- B. Tian and J. L. Manley, Nat. Rev. Mol. Cell Biol., 2016, 18, 18–30 CrossRef PubMed.
- A. R. Gruber, G. Martin, W. Keller and M. Zavolan, RNA Biol., 2012, 9, 1405–1412 CrossRef CAS PubMed.
- C. P. Masamha, Z. Xia, J. Yang, T. R. Albrecht, M. Li, A. Bin Shyu, W. Li and E. J. Wagner, Nature, 2014, 510, 412–416 CrossRef CAS PubMed.
- V. A. Gennarino, C. E. Alcott, C. A. Chen, A. Chaudhury, M. A. Gillentine, J. A. Rosenfeld, S. Parikh, J. W. Wheless, E. R. Roeder, D. D. G. Horovitz, E. K. Roney, J. L. Smith, S. W. Cheung, W. Li, J. R. Neilson, C. P. Schaaf and H. Y. Zoghbi, Elife, 2015, 4, 1–13 CrossRef PubMed.
- N. M. Pearce, T. Krojer, A. R. Bradley, P. Collins, R. P. Nowak, R. Talon, B. D. Marsden, S. Kelm, J. Shi, C. M. Deane and F. Von Delft, Nat. Commun., 2017, 8, 15123 CrossRef PubMed.
- Q. Yang, G. M. Gilmartin and S. Doublié, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10062–10067 CrossRef CAS PubMed.
- D. V Vorobyeva, N. M. Karimova, I. L. Odinets, G.-V. Röschenthaler and S. N. Osipov, Org. Biomol. Chem., 2011, 9, 7335–7342 RSC.
- S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285–8287 CrossRef CAS PubMed.
- Y. Wang, J. Y. Wach, P. Sheehan, C. Zhong, C. Zhan, R. Harris, S. C. Almo, J. Bishop, S. J. Haggarty, A. Ramek, K. N. Berry, C. O'Herin, A. N. Koehler, A. W. Hung and D. W. Young, ACS Med. Chem. Lett., 2016, 7, 852–856 CrossRef CAS PubMed.
- X. Wang, G. Fischer and M. Hyvönen, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- L. Attisano, J. L. Wrana, E. Montalvo and J. Massagué, Mol. Cell. Biol., 1996, 16, 1066–1073 CrossRef CAS PubMed.
- J. L. Wrana, L. Attisano, R. Wieser, F. Ventura and J. Massagué, Nature, 1994, 370, 341–347 CrossRef CAS PubMed.
- S. Dennler, S. Itoh, D. Vivien, P. Ten Dijke, S. Phane Huet and J.-M. Gauthier, EMBO J., 1998, 17, 3091–3100 CrossRef CAS PubMed.
- M. Antsiferova and S. Werner, J. Cell Sci., 2012, 125, 3929–3937 CrossRef CAS PubMed.
- S. A. Pangas, C. J. Jorgez, M. Tran, J. Agno, X. Li, C. W. Brown, T. R. Kumar and M. M. Matzuk, Mol. Endocrinol., 2007, 21, 2458–2471 CrossRef CAS PubMed.
- Y. Togashi, A. Kogita, H. Sakamoto, H. Hayashi, M. Terashima, M. A. de Velasco, K. Sakai, Y. Fujita, S. Tomida, M. Kitano, K. Okuno, M. Kudo and K. Nishio, Cancer Lett., 2015, 356, 819–827 CrossRef CAS PubMed.
- S. J. Hatsell, V. Idone, D. M. A. Wolken, L. Huang, H. J. Kim, L. Wang, X. Wen, K. C. Nannuru, J. Jimenez, L. Xie, N. Das, G. Makhoul, R. Chernomorsky, D. D'Ambrosio, R. A. Corpina, C. J. Schoenherr, K. Feeley, P. B. Yu, G. D. Yancopoulos, A. J. Murphy and A. N. Economides, Sci. Transl. Med., 2015, 7, 303ra137 CrossRef PubMed.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- Four further hits were identified from 423 compounds during subsequent screening efforts carried out by our collaborators, see PDB 5R64, 5R65, 5R66 and 5R67.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01232g |
| ‡ These authors contributed equally to the work presented here. |
| § Three examples were screened at 250 mM concentrations due to solubility limitations. |
| This journal is © The Royal Society of Chemistry 2020 |