
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective total synthesis of (−)-myrifabral A and B†
Tyler J.
Fulton
 a,
Anthony Y.
Chen
a,
Michael D.
Bartberger
b and
Brian M.
Stoltz
*a
a,
Anthony Y.
Chen
a,
Michael D.
Bartberger
b and
Brian M.
Stoltz
*a
aWarren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
b1200 Pharma LLC, 844 East Green Street, Suite 204, Pasadena, CA 91101, USA. E-mail: michael.bartberger@1200pharma.com
First published on 21st April 2020
Abstract
A catalytic enantioselective approach to the Myrioneuron alkaloids (−)-myrifabral A and (−)-myrifabral B is described. The synthesis was enabled by a palladium-catalyzed enantioselective allylic alkylation, that generates the C(10) all-carbon quaternary center. A key N-acyl iminium ion cyclization forged the cyclohexane fused tricyclic core, while vinyl boronate cross metathesis and oxidation afforded the lactol ring of (−)-myrifabral A. Adaptation of previously reported conditions allowed for the conversion of (−)-myrifabral A to (−)-myrifabral B.
Introduction
The Myrioneuron alkaloids are a small, yet growing family of structurally diverse polycyclic (tri-, tetra-, penta-, hexa-, and decacyclic) alkaloids believed to share a common biosynthetic origin from lysine (Fig. 1).1 The first Myrioneuron alkaloids from Myrioneuron nutans were reported in 2002, with altogether 10 structures reported to date.2 Since 2013, many new alkaloids have been isolated from Myrioneuron faberi,3Myrioneuron tonkinesis,4 and Myrioneuron effusum.5 In addition to their interesting structural features, a number of these alkaloids possess a range of biological activities such as antimalarial properties, KB cell cytotoxicity, antimicrobial, and hepatitis C virus (HCV) replication inhibition.1–5 Despite possessing promising biological properties and synthetically attractive motifs, relatively few of these alkaloids have been prepared by total synthesis efforts.2d,e,f,6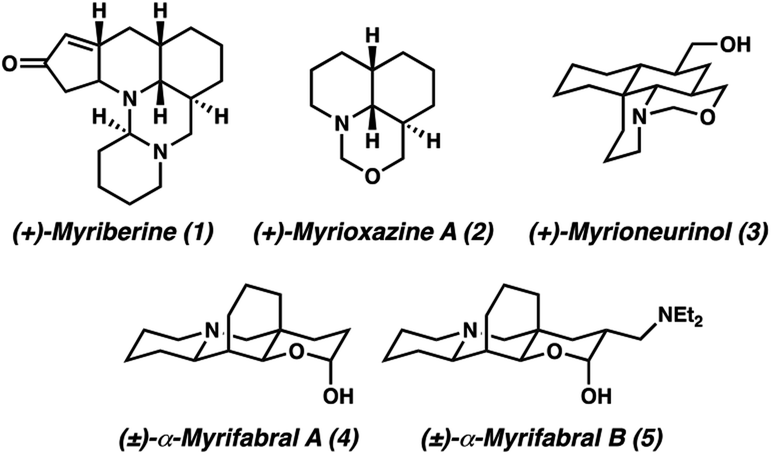 | ||
| Fig. 1 Representative Myrioneuron alkaloids. | ||
We became interested in (±)-myrifabral A (4) and (±)-myrifabral B (5) in particular due to their unique cyclohexane fused octahydroquinolizine skeletons which contain four contiguous stereogenic centers, including an all-carbon quaternary center embedded in the cyclohexane fusion. Interestingly, both 4 and 5 are isolated as racemic mixtures of α- and β-hydroxy epimers. Even as racemates, these clusters display promising HCV replication inhibition (EC50 = 4.7 μM for (±)-α,β-OH-4 and 2.2 μM (±)-α,β-OH-5, respectively) with significantly reduced liver cell cytotoxicity compared to commercial pharmaceutical HCV drug telaprevir (EC50 = 0.09 μM).3c She et al. recently reported a rapid total synthesis of (±)-α,β-OH-4 and (±)-α,β-OH-5, however, no asymmetric approaches have been disclosed to date.6c To enable further studies of these alkaloids in each enantiomeric series, we report herein a short, catalytic enantioselective synthesis of (−)-α,β-OH-4 and (−)-α,β-OH-5.
In devising our strategy, we targeted (−)-myrifabral A (4), which can be directly converted to (−)-myrifabral B (5), as reported by She (Scheme 1).6c Retrosynthetically, we envisioned (−)-myrifabral A (4) could be simplified to tricyclic lactam (6). Importantly, the versatile ketone, allyl, and lactam functional handles in tricyclic lactam 6 provide ample opportunity for future diversification of the natural product scaffold for medicinal chemistry efforts and potential derivative synthesis. We envisioned the critical C(6)–C(7) bond could arise by means of a diastereoselective N-acyl iminium ion cyclization of enantioenriched ketone 7.7 Finally, the C(10) all-carbon quaternary center could be forged in an enantioselective manner by means of asymmetric allylic alkylation of glutarimide 8. In turn, glutarimide 8 could be prepared from β-ketoester 9.
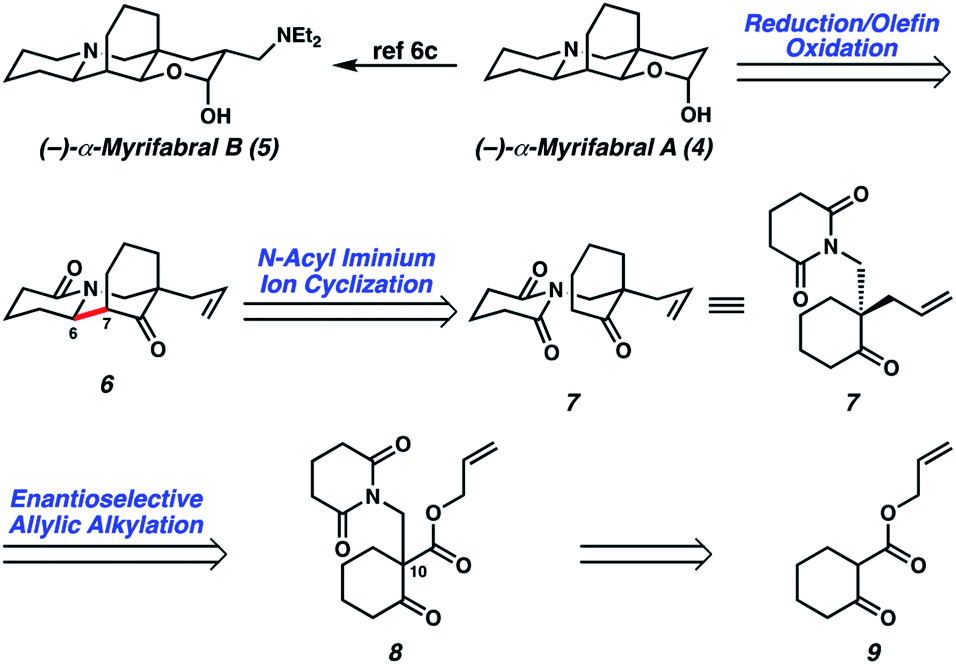 | ||
| Scheme 1 Retrosynthetic analysis of (−)-myrifabral A. | ||
Results and discussion
Our synthetic efforts commenced with the preparation of glutarimide 8 (Scheme 2). Alkylation of β-ketoester 9 (ref. 8) with sulfonylmethyl carbamate 10 in the presence of Cs2CO3 proceeded smoothly on a 30.0 g scale to afford β-aminoketone 11 in 95% yield.9 Elaboration of the Boc-protected amine to glutarimide 8 with standard protocols10 was low yielding and required three separate reactions. This inspired us to develop a more practical, one-pot procedure to enable the installation of the glutarimide moiety (Table 1). Initially, trifluoroacetic acid mediated conditions were explored, with the most promising results observed in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF/TFA at refluxing temperature (entry 1) and 3:1 1,4-dioxane/TFA (entry 2). Further investigation revealed that stoichiometric boric acid could mediate the transformation in excellent yield in xylenes at 140 °C (entry 3). While aryl boronic acids11 and boric acid12 have been demonstrated as effective catalysts for amidation of carboxylic acids with amines, we were surprised to observe concomitant Boc removal, amidation, and glutarimide cyclization. Control experiments revealed the reaction only proceeds with the complete set of reagents (entries 4–6). Furthermore, Boc protected β-aminoketone 11 was stable to xylenes at reflux for 120 h (entry 7). Additional examination revealed catalytic boric acid (10 mol%) with 2 equivalents of glutaric anhydride (12) performed equally well as our best conditions (entry 8). Replacing glutaric anhydride (12) with glutaric acid (13) as a cheaper alternative did not affect the reaction time or yield (entry 9). Pleasingly, the reaction time could be reduced by utilizing catalytic PhB(OH)2 as a soluble boronic acid (entry 10), although this benefit was tempered on larger scales (entry 11). Optimal results were obtained by utilizing the electron deficient catalyst 4-CF3PhB(OH)2 (entry 12).
:1 DMF/TFA at refluxing temperature (entry 1) and 3:1 1,4-dioxane/TFA (entry 2). Further investigation revealed that stoichiometric boric acid could mediate the transformation in excellent yield in xylenes at 140 °C (entry 3). While aryl boronic acids11 and boric acid12 have been demonstrated as effective catalysts for amidation of carboxylic acids with amines, we were surprised to observe concomitant Boc removal, amidation, and glutarimide cyclization. Control experiments revealed the reaction only proceeds with the complete set of reagents (entries 4–6). Furthermore, Boc protected β-aminoketone 11 was stable to xylenes at reflux for 120 h (entry 7). Additional examination revealed catalytic boric acid (10 mol%) with 2 equivalents of glutaric anhydride (12) performed equally well as our best conditions (entry 8). Replacing glutaric anhydride (12) with glutaric acid (13) as a cheaper alternative did not affect the reaction time or yield (entry 9). Pleasingly, the reaction time could be reduced by utilizing catalytic PhB(OH)2 as a soluble boronic acid (entry 10), although this benefit was tempered on larger scales (entry 11). Optimal results were obtained by utilizing the electron deficient catalyst 4-CF3PhB(OH)2 (entry 12).
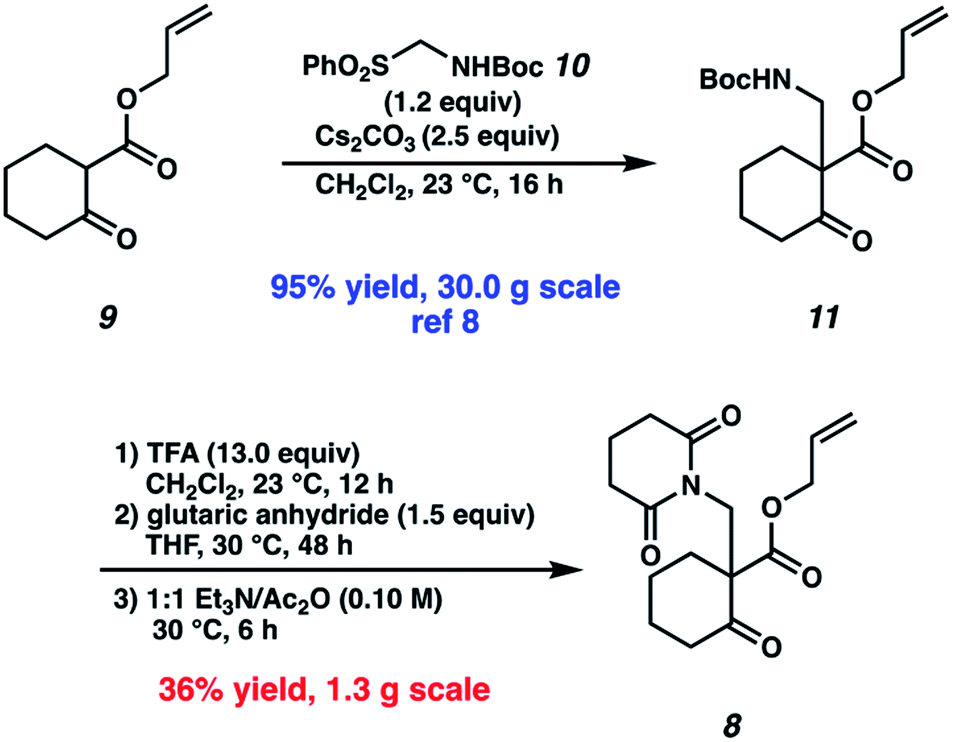 | ||
| Scheme 2 Initial synthesis of glutarimide 8. | ||
|
|
||
|---|---|---|
| Entry | Conditionsa | % yieldb |
| a Reaction performed on a 0.16 mmol scale unless otherwise stated. b Isolated yield; ranges reflect yields obtained from 3–4 reactions. c Reaction performed on a 0.80 mmol scale. | ||
| 1 | 3:1 DMF/TFA (0.10 M), 12 (4 equiv.), reflux, 48 h |
60 |
| 2 | 3:1 1,4-dioxane/TFA (0.10 M), 12 (4 equiv.), reflux, 48 h |
70 |
| 3 | B(OH)3 (3.0 equiv.), 12 (4 equiv.), xylenes, Dean–Stark, reflux, 36 h | 90–95 |
| 4 | B(OH)3 (3.0 equiv.) xylenes, Dean–Stark, reflux, 72 h | >95% 11 |
| 5 | 12 (4 equiv.), xylenes, Dean–Stark, reflux, 72 h | >95% 11 |
| 6 | 13 (4 equiv.), xylenes, Dean–Stark, reflux, 72 h | >95% 11 |
| 7 | Xylenes, Dean–Stark, reflux, 120 h | >95% 11 |
| 8 | B(OH)3 (10 mol%), 12 (2 equiv.), xylenes, Dean–Stark, reflux, 36 h | 90–95 |
| 9 | B(OH)3 (10 mol%), 13 (2 equiv.), xylenes, Dean–Stark, reflux, 36 h | 90–95 |
| 10 | PhB(OH)2 (10 mol%), 13 (2 equiv.), xylenes, Dean–Stark, reflux, 24 h | 90–95 |
| 11 | PhB(OH)2 (10 mol%), 13 (2 equiv.), xylenes, Dean–Stark, reflux, 36 hc | 90–95 |
| 12 | 4-CF3PhB(OH)2 (10 mol%), 13 (2 equiv.), xylenes, Dean–Stark, reflux, 24 hc | 90–95 |
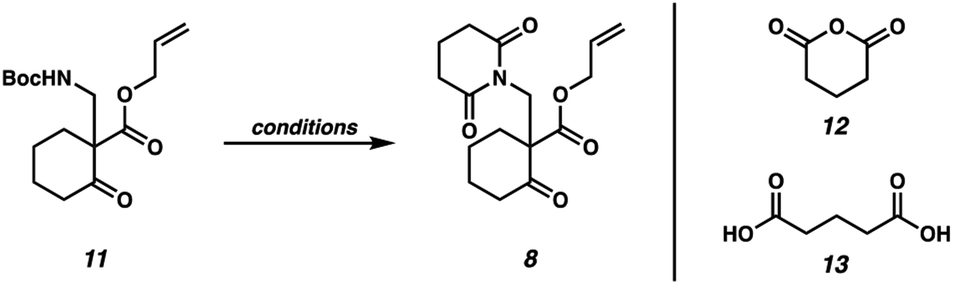
These optimized conditions for elaboration of Boc protected β-aminoketone 11 to glutarimide 8 performed well on a 15.0 g scale, enabling us to press forward in our synthetic campaign (Scheme 3). Palladium-catalyzed decarboxylative asymmetric allylic alkylation of glutarimide 8 established the C(10) all-carbon quaternary center, affording ketone 7 in 94% yield and 88% ee. The absolute configuration of the all-carbon quaternary center was established as (S) via experimental and computational vibrational circular dichroism (VCD) and optical rotation analyses.13 To affect the key N-acyl iminium ion cyclization, the ketone was first protected as ethyl vinyl ether 14. A one-pot protocol was then employed to affect mono-reduction of the glutarimide with LiEt3BH followed by BF3·OEt2 mediated N-acyl iminium ion cyclization of intermediate N-acyl hemiaminal 15, furnishing tricyclic lactam 6 as a single diastereomer in 89% yield.
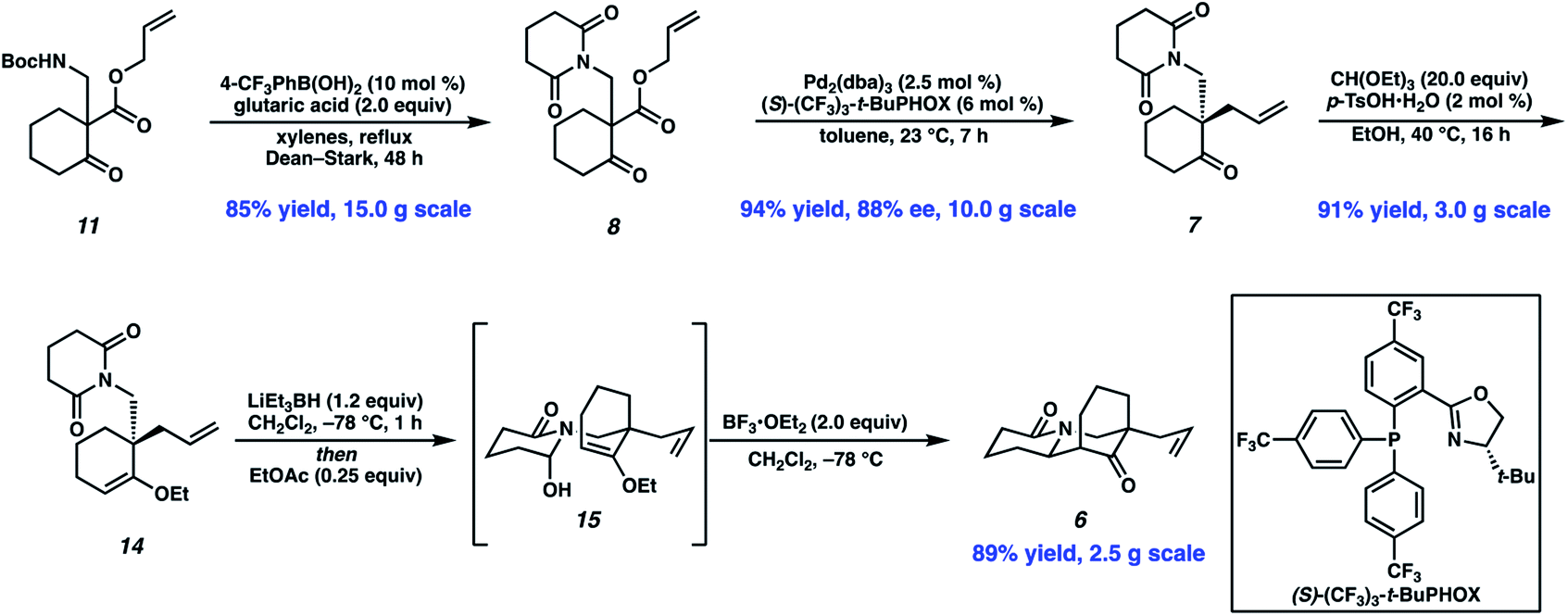 | ||
| Scheme 3 Enantioselective synthesis of tricyclic lactam 6. | ||
Completion of the synthesis required reduction of the lactam and ketone and elaboration of the terminal olefin to an aldehyde (Scheme 4). Toward that end, a one-pot procedure was developed wherein the ketone was first reduced by L-selectride with exceptional diastereoselection (>19:1). Following the reduction of the ketone, addition of LiAlH4 and heating to reflux afforded full reduction of the lactam to the corresponding tertiary amine. A modified Fieser work up then provided desired amino alcohol 16 in 97% yield. Initially, we found the terminal olefin recalcitrant to both direct and two-step oxidations to the aldehyde due to challenges with olefin isomerization and undesired or poor reactivity. To our delight, olefin cross metathesis using Hoveyda–Grubbs II catalyst of amino alcohol 16 with vinyl boronic acid pinacol ester (17) as an aldehyde surrogate smoothly afforded metathesis product 18. Elaboration of the vinyl boronate was then affected by deprotection and oxidation of the boronic acid, with in situ lactolization providing (−)-myrifabral A in 50% yield. X-ray crystallography allowed for the determination of the absolute stereochemistry of the (−)-myrifabral A enantiomeric series. Adaptation of She's conditions for the synthesis of (±)-myrifabral B then provided access to (−)-myrifabral B in 70% yield (Scheme 5).6c Interestingly, both (−)-myrifabral B, and (−)-myrifabral A are isolated as oils, whereas racemates of these compounds are isolated as solids.3c,6c Spectroscopic data obtained were in excellent agreement with the natural compound (see ESI†).
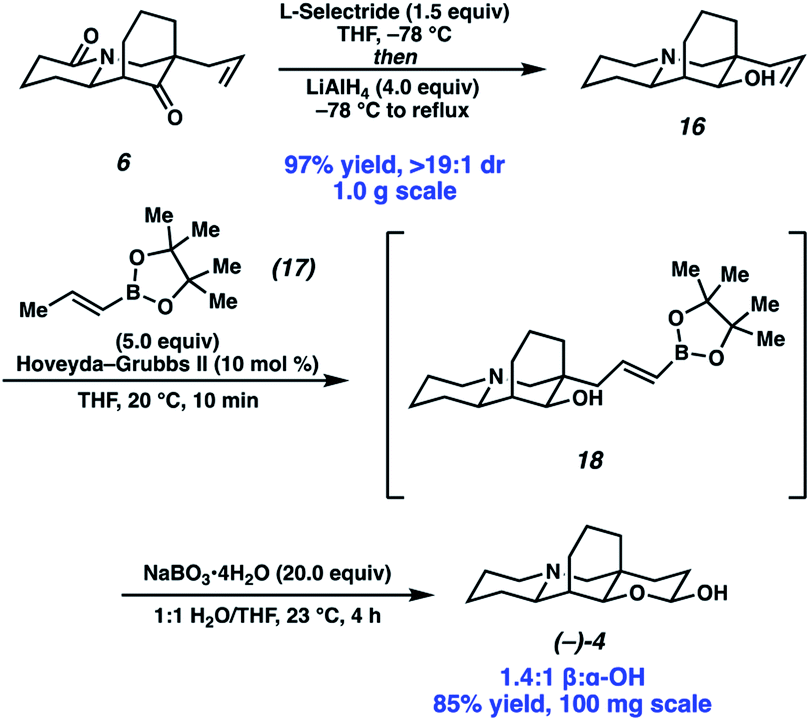 | ||
| Scheme 4 End game for (−)-myrifabral A. | ||
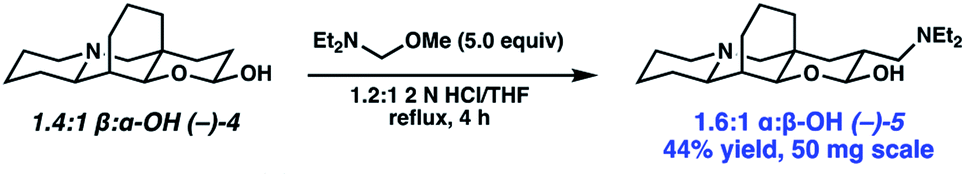 | ||
| Scheme 5 Synthesis of (−)-myrifabral B. | ||
Conclusions
We have described the first enantioselective total synthesis of (−)-myrifabral A and B. Critical to the success of this strategy was the development of a direct and high yielding one-pot conversion of a Boc-protected amine (11) to the glutarimide (8). Palladium catalyzed decarboxylative asymmetric allylic alkylation provided the C(10) all-carbon quaternary center in 88% ee, setting the stage for ketone protection, glutarimide reduction, and an exquisitely diastereoselective N-acyl iminium ion cyclization. Following ketone and lactam reduction, cross metathesis with vinyl boronic acid pinacol boronate and subsequent boronic acid deprotection and oxidation afforded (−)-myrifabral A. Utilizing previously reported conditions, (−)-myrifabral A was converted to (−)-myrifabral B. This marks the first catalyst-controlled asymmetric synthesis of myrifabral A and B, enabling future biological study of individual enantiomeric series.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NIH-NIGMS (R01GM080269) and Caltech for financial support. Dr Scott Virgil (Caltech) is thanked for instrumentation and SFC assistance. We thank Dr David Vander Velde (Caltech) for NMR expertise, and Dr Mona Shahgholi (Caltech) and Naseem Torian (Caltech) for mass spectrometry assistance.Notes and references
- For a review see: E. Gravel and E. Poupon, Biosynthesis and biomimetic synthesis of alkaloids isolated from plants oft he Nitraria and Myrioneuron genera: an unusual lysine-based metabolism, Nat. Prod. Rep., 2010, 27, 32–56 RSC.
- (a) V. C. Pham, A. Jossang, T. Sévenet, V. H. Nguyen and B. Bodo, Absolute configuration of Myrobotinol, New Fused-Hexacyclic Alkaloid Skeleton from Myrionueron nutans, J. Org. Chem., 2007, 72, 9826–9829 CrossRef CAS PubMed; (b) V. C. Pham, A. Jossang, T. Sévenet, V. H. Nguyen and B. Bodo, Myrioneurinol: a novel alkaloid skeleton from Myrioneuron nutans, Tetrahedron, 2007, 63, 11244–11249 CrossRef CAS; (c) V. C. Pham, A. Jossang, T. Sévenet, V. H. Nguyen and B. Bodo, Novel Alkaloids from Myrioneuron nutans, Eur. J. Org. Chem., 2009, 74, 1412–1416 CrossRef; (d) V. C. Pham, A. Jossang, A. Chiaroni, T. Sévenet and B. Bodo, Asymmetric synthesis of myrioxazines A and B, novel alkaloids of Myrioneuron nutans, Tetrahedron Lett., 2002, 43, 7565–7568 CrossRef CAS; (e) V. C. Pham, A. Jossang, A. Chiaroni, T. Sévenet, V. H. Nguyen and B. Bodo, Solution and Crystal Conformations of Myrionine, a new 8β-Alkyl-cis-decahydroquinoline of Myrioneuron nutans, Org. Lett., 2007, 9, 3531–3534 CrossRef CAS PubMed; (f) V. C. Pham, A. Jossang, P. Grellier, T. Sévenet, V. H. Nguyen and B. Bodo, Structure and Total Synthesis of (–)-Myrionidine and (–)-Schoberine, Antimalarial Alkaloids from Myrioneuron nutans, J. Org. Chem., 2008, 73, 7565–7573 CrossRef CAS PubMed.
- (a) S.-D. Huang, Y. Zhang, M.-M. Cao, Y.-T. Di, G.-H. Tang, Z.-G. Peng, J.-D. Jiang, H.-P. He and X.-J. Hao, Myriberine A, a New Alkaloid with an Unprecedented Heteropentacyclic Skeleton from Myrioneuron faberi, Org. Lett., 2013, 15, 590–593 CrossRef CAS PubMed; (b) M.-M. Cao, S.-D. Huang, Y.-T. Di, C.-M. Yuan, G.-Y. Zuo, Y.-C. Gu, Y. Zhang and X.-J. Hao, Myrifabine, the First Dimeric Myrioneuron Alkaloid from Myrioneuron faberi, Org. Lett., 2014, 16, 528–531 CrossRef CAS; (c) M.-M. Cao, Y. Zhang, X.-H. Li, Z.-G. Peng, J.-D. Jiang, Y.-C. Gu, Y.-T. Di, X.-N. Li, D.-Z. Chen, C.-F. Xia, H.-P. He, S.-L. Li and X.-J. Hao, Cyclohexane-Fused Octahydroquinolizine Alkaloids from Myrioneuron faberi with Activity against Hepatitis C Virus, J. Org. Chem., 2014, 79, 7945–7950 CrossRef CAS PubMed; (d) M.-M. Cao, Y. Zhang, S.-D. Huang, Y.-T. Di, Z.-G. Peng, J.-D. Jiang, C.-M. Yuan, D.-Z. Chen, S.-L. Li, H.-P. He and X.-J. Hao, Alkaloids with Different Carbon Units from Myrioneuron faberi, J. Nat. Prod., 2015, 78, 2609–2616 CrossRef CAS PubMed; (e) M.-M. Cao, Y. Zhang, Z.-G. Peng, J.-D. Jiang, Y.-J. Gao and X.-J. Hao, Schoberine B, an alkaloid with an unprecedented straight C5 side chain, and myriberine B from Myrioneuron faberi, RSC Adv., 2016, 6, 10180–10184 RSC; (f) M.-M. Cao, Y. Zhang, S.-D. Huang, Z.-G. Peng, J.-D. Jiang and X.-J. Hao, Three new alkaloids from Myrioneuron faberi, Tetrahedron Lett., 2016, 57, 4021–4023 CrossRef CAS; (g) M.-M. Cao, J.-H. Zhang, Y. Zhang, Z.-G. Peng, J.-D. Jiang and X.-J. Hao, New findings of cyclohexane-fused octahydroquinolizine alkaloids from Myrioneuron faberi, Tetrahedron Lett., 2016, 57, 5632–5635 CrossRef CAS.
- X.-H. Li, Y. Zhang, J.-H. Zhang, X.-N. Li, M.-M. Cao, Y.-T. Di, Z.-G. Peng, J.-D. Jiang and X.-J. Hao, Myritonines A–C, Alkaloids from Myrioneuron tonkinesis Based on a Novel Hexacyclic Skeleton, J. Nat. Prod., 2016, 79, 1203–1207 CrossRef CAS PubMed.
- Z.-H. Zhang, J.-J. Guo, Y.-X. Yuan, Y.-H. Fu, Y. C. Gu, Y. Zhang, D.-Z. Chen, S.-L. Li, Y.-T. Di and X.-J. Hao, Four new tetracyclic alkaloids with cis-decahydroquinoline motif from Myrioneuron effusum, Fitoterapia, 2016, 112, 217–221 CrossRef PubMed.
- (a) A. J. Nocket and S. M. Weinreb, Total Synthesis of the Tetracyclic Antimalarial Alkaloid (±)-Myrioneurinol, Angew. Chem., Int. Ed., 2014, 53, 14162–14165 CrossRef CAS PubMed; (b) A. J. Nocket, Y. Feng and S. M. Weinreb, Construction of the Myrioneuron Alkaloids: A Total Synthesis of (±)-Myrioneurinol, J. Org. Chem., 2015, 80, 1116–1129 CrossRef CAS PubMed; (c) D. Song, Z. Wang, R. Mei, W. Zhang, D. Ma, D. Xu, X. Xie and X. She, Short and Scalable Total Synthesis of Myrioneuron Alkaloids (±)-α,β-Myrifabral A and B, Org. Lett., 2016, 18, 669–671 CrossRef CAS PubMed.
- A. Plas, F. Marchand, A. Eschalier, Y. Troin and P. Chalard, Stereoselective Synthesis and In Vivo Evaluation of the Analgesic Activity of Polysubstituted Bispidines, Eur. J. Org. Chem., 2012, 6070–6079 CrossRef CAS.
- J. T. Mohr, M. R. Krout and B. M. Stoltz, Preparation of (S)-2-allyl-2-methylcyclohexanaone, Org. Synth., 2009, 86, 194–211 CrossRef CAS.
- Y. Numajiri, B. P. Pritchett, K. Chiyoda and B. M. Stoltz, Enantioselective Synthesis of α-Quaternary Mannich Adducts by Palladium-Catalyzed Allylic Alkylation: Total Synthesis of (+)-Sibirinine, J. Am. Chem. Soc., 2015, 137, 1040–1043 CrossRef CAS PubMed.
- (a) Y. Kawanaka, K. Kobayashi, S. Kusuda, T. Tatsumi, M. Murota, T. Nishiyama, K. Hisaichi, A. Fujii, K. Hirai, M. Naka, M. Komeno, Y. Odagaki, H. Nakai and M. Toda, Design and Synthesis of Orally Bioavailable Inhibitors of Inducible Nitric Oxide Synthase. Identification of 2-Azabicyclo[4.1.0]heptan-3-imines, Bioorg. Med. Chem., 2003, 11, 1723–1743 CrossRef CAS; (b) I. S. del Villar, A. Gradillas and J. Pérez-Castells, Synthesis of 2-Azabicyclo[4.1.0]heptanes through Stereoselective Cyclopropanation Reactions, Eur. J. Org. Chem., 2010, 5850–5862 CrossRef.
- (a) K. Ishihara, S. Ohara and H. Yamamoto, 3,4,5-Trifluorobenzeneboronic Acid as an Extremely Active Amidation Catalyst, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed; (b) K. Ishihara, S. Kondo and H. Yamamoto, 3,5-Bis(perfluorodecyl)phenylboronic Acid as an Easily Recyclable Direct Amide Condensation Catalyst, Synlett, 2001, 9, 1371–1374 CrossRef; (c) K. Ishihara, S. Ohara and H. Yamamoto, (3,4,5-trifluorophenyl)boronic Acid-catalyzed Amide Formation from Carboxylic Acids and Amines: N-benzyl-4-phenylbutyramide, Org. Synth. Coll. Vol., 2004, 10, 80–86 Search PubMed; (d) R. Al-Zoubi, O. Marion and D. Hall, Direct and Waste-Free Amidations and Cycloadditions by Organocatalytic Activation of Carboxylic Acids at Room Temperature, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS PubMed; (e) D. Hall, N. Gernigon, R. Al-Zoubi and P. D. Thornton, Boronic acid catalysts and methods of use thereof for activation and transformation of carboxylic acids, WO2012109749A1, 2012; (f) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, Direct Amidation of Carboxylic Acids Catalyzed by ortho-Iodo Arylboronic Acids: Catalyst Optimization, Scope, and Preliminary Mechanistic Study Supporting a Peculiar Halogen Acceleration Effect, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed.
- P. Tang and H. Krause, Boric Acid Catalyzed Amide Formation from Carboxylic Acids and Amines: N-benzyl-4-phenylbutyramide, Org. Synth., 2005, 81, 262–272 CrossRef CAS.
- See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and IR spectra, SFC traces, X-ray crystallographic data. See DOI: 10.1039/d0sc01141j |
| This journal is © The Royal Society of Chemistry 2020 |