DOI:
10.1039/D0SC01153C
(Edge Article)
Chem. Sci., 2020,
11, 13137-13142
A single amino acid Gly-tag enables metal-free protein purification†
Received
26th February 2020
, Accepted 26th October 2020
First published on 26th October 2020
Abstract
Analytically pure proteins are indispensable for diverse applications, including therapeutics. Here, we report a methodology where a single amino acid, glycine, enables metal-free protein purification. This robust platform is enabled by a Gly-tag resin for site-specific capture, enrichment, and release through chemically triggered C–C bond dissociation by resonance-assisted electron density polarization.
Introduction
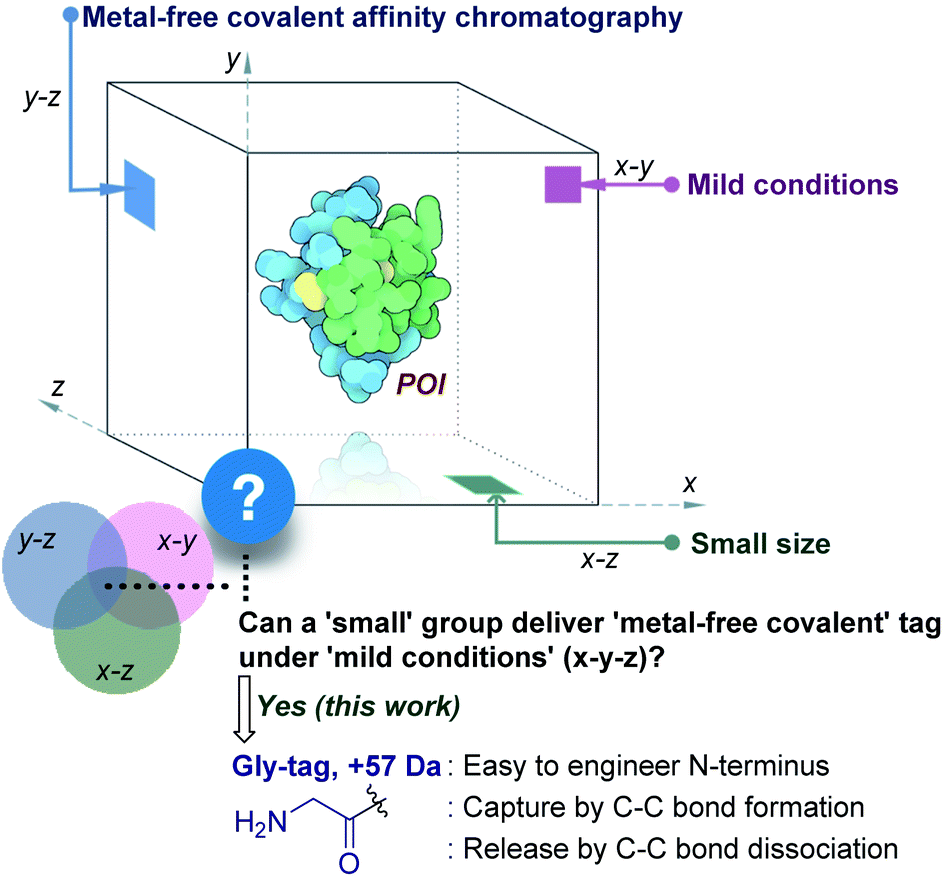
Living systems are intricate, and a broad set of proteins drive their complex machinery. Elucidating the biological role of these individual proteins requires investigation of their physical, chemical, and structural properties. In turn, it necessitates the production of a pure functional protein of interest (POI). Classic natural sources took a back seat with time,1 while recombinant protein expression caters to the growing academic and industrial demands.2 The latter approach requires the isolation of the POI from the cell extract. The fishing out of a specific protein among thousand others with similar features is highly demanding. It needs a methodology to synchronize diverse aspects of chemical reactivity and selectivity. Besides, the restriction to operate under mild physiological conditions amplifies the complexity.
The initial efforts involved the development of affinity tags that can show unique capture and release attributes. In this perspective, immobilized metal-affinity chromatography is one of the most prominent techniques.3 Here, a sequence of His residues (His tag) installed in a protein provides preferential binding to a metal complex (x–z plane, Fig. 1). However, the non-specific binding to other residues in the proteins and leaching of metals are unavoidable. The quest for metal-free techniques and specific non-covalent interactions led to the development of peptide and protein-based fusion tags that operate under mild conditions (x–y plane, Fig. 1). The specificity in these cases requires a large recognition motif, either as a part of the protein or as the capture ligand on a resin.4,5 Nevertheless, the loss of the protein is unavoidable due to the participation of multiple dynamic interactions that provide a gradient of binding energy. In contrast, the covalent immobilization of the POI at a specific site through a well-defined bond could offer a discrete switch ON mechanism for its capture. In this perspective, recent advances in the field of site-selective protein bioconjugation are encouraging.6–17 However, a chemical technology for the release of the POI under physiological conditions poses the next monumental challenge. This has been the prime reason behind the lack of methods for covalent affinity chromatography (y–z plane, Fig. 1). The problem is addressed indirectly by coding an additional enzyme-cleavable fragment, as in the case of HaloTag.18 Here, a protein with the tag can be installed on resin and allows stringent washing with minimal loss of the POI. Although, the tag serves as a necessary evil due to its size (∼34 kDa), making its removal an essential step. TEV protease releases the POI leaving behind the HaloTag on the resin. Consequently, the resin cannot be recycled, and the separation of the protease from the POI requires an additional step.
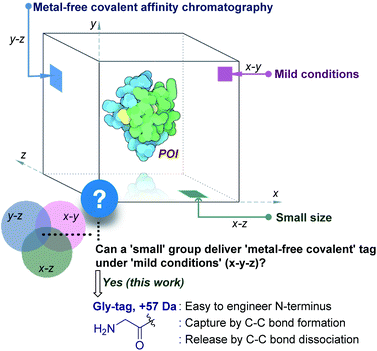 |
| | Fig. 1 Methods for purification of a protein under physiological conditions and the unmet technological demands. | |
This necessitates a method that can enable covalent affinity chromatography under mild conditions with a small tag (x–y–z, Fig. 1), ideally a single residue. This technological demand requires a methodology that can offer residue specificity and result in the single-site labeling of a protein in a complex mixture. Subsequently, it would need the translation of chemistry to the solid phase for covalent immobilization of the POI without compromising its selectivity. Finally, the development of an on-demand chemically triggered release of the POI under mild conditions would be critical for a successful metal-free protein purification technology.
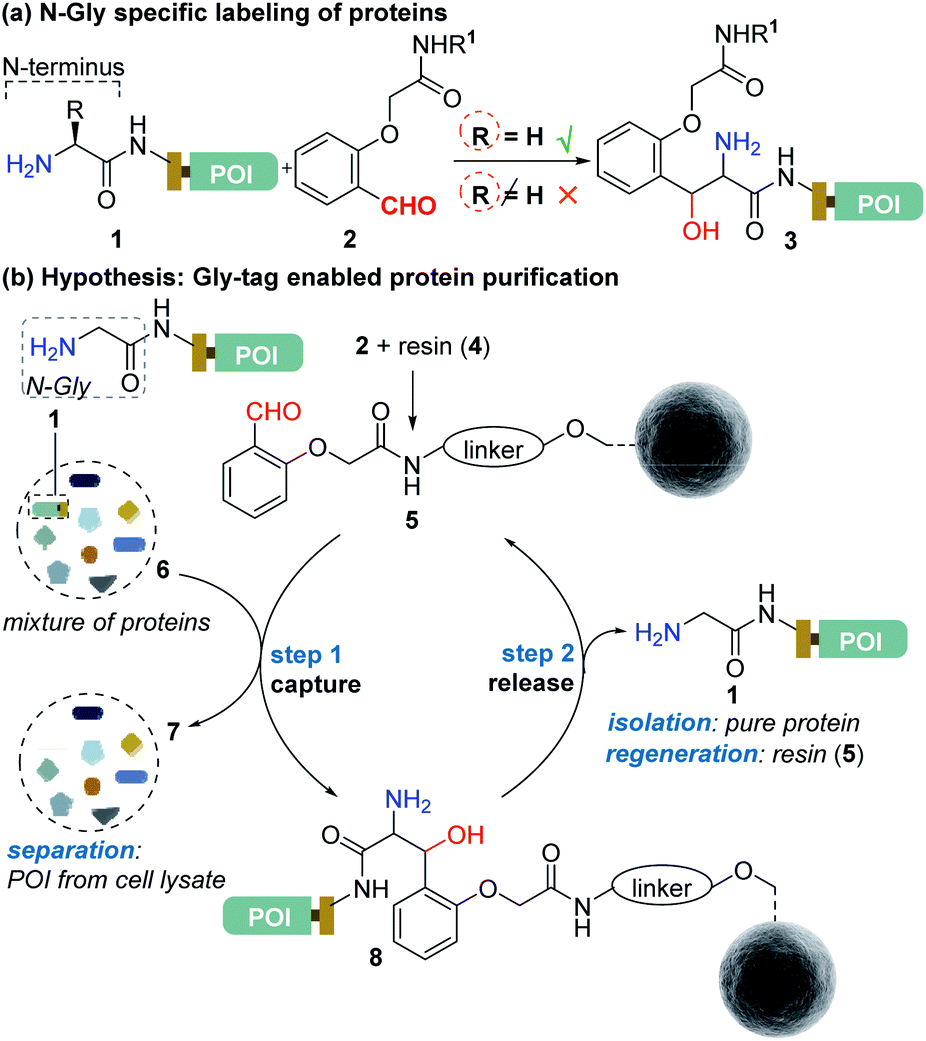
Protein bioconjugation mostly relies on carbon–heteroatom bond formation.19,20 Such bonds are easy to maneuver as they avoid the high energy required for C–C bond formation or dissociation. Recently, we developed the residue-specific labeling of N-Gly in proteins (Fig. 2a).11 It is noteworthy that the installation of a single Gly residue at the N-terminus of a protein during its expression is convenient.21 Here, we demonstrate that a single residue (Gly) at the N-terminus can facilitate site-specific immobilization of proteins with a stable C–C bond (step 1, aldol reaction, Fig. 2b). Importantly, we have successfully developed a methodology that enables chemically triggered release of the protein (step 2, retro-aldol reaction, Fig. 2b). The orthogonal nature of the C–C bond formation, its dissociation, and efficiency under the mild operating conditions are remarkable. The functionalized Gly-tag purification resin developed for this technology is robust and regenerated for multiple uses. The combination of all the attributes enables a single Gly residue at the N-terminus of a protein (Gly-tag) for metal-free covalent affinity purification.
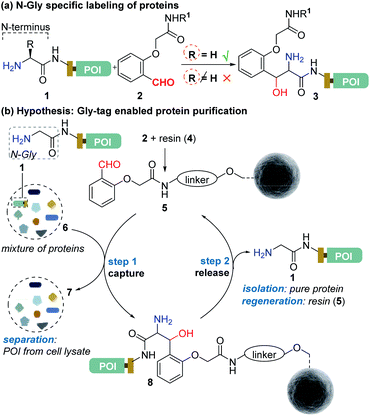 |
| | Fig. 2 (a) Selective labeling of N-terminus Gly containing proteins.11 (b) Hypothesis for Gly-tag enabled protein purification includes successful realization of protein capture (step 1) through engineered Sepharose resin 5 and its release (step 2) under mild operating conditions. | |
Results and discussion
Immobilization of the Gly-capture reagent on NHS Sepharose resin
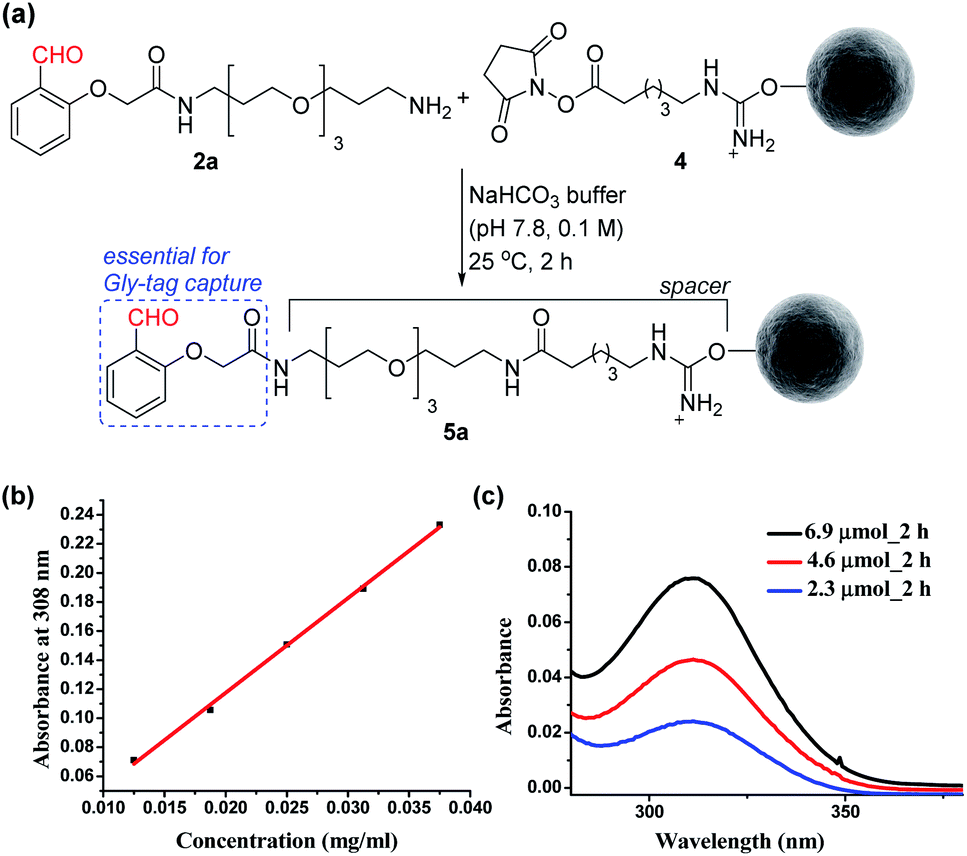
At first, we synthesized the aldehyde with proximal hydrogen bond acceptors (2a, Fig. 3a) in four steps (Fig. S1 and S2†). We tethered this Gly-tag capture element to a PEG diamine and placed a nucleophilic amine functionality at the other end of the reagent (2a). The PEG linker regulates the surface availability of the reagent upon its immobilization.22 On the other hand, the amine functionality provides the nucleophilic handle to conjugate it with the electrophilic NHS ester functionalized resin (4). Next, we quantified the extent of immobilization of the Gly-capture reagent on the Sepharose resin by UV analysis (Fig. 3a). Initially, we monitored the concentration of NHS at 260 nm (λmax). However, the reagent 2a also contributes to the absorption at this wavelength. To avoid ambiguity, we selected the additional absorbance of the unbound reagent 2a at 308 nm to monitor the extent of immobilization. Initially, we derived the standard calibration curve from 2a at different concentrations of the reagent (Fig. 3b and S4†). Subsequently, we determined the molar extinction coefficient of the reagent (6.518 M−1 cm−1) 2a. Withthis information in hand, we were able to monitor the synthesis of the Gly-tag purification resin through the concentration of the unbound reagent. Interestingly, just two equivalent of reagent 2a (4.6 μmol with 2.3 μmol of resin 4) was enough to result in 20 μmol ml−1 loading. It is consistent with the expected loading capacity of commercially available NHS ester functionalized Sepharose resin 4 (16–23 μmol ml−1) (Fig. 3c). The UV spectrum of the washed fraction allowed the estimation of the adsorbed reagent. Also, the higher stoichiometry of the reagent did not result in noticeable improvement of the loading (Fig. S4†).
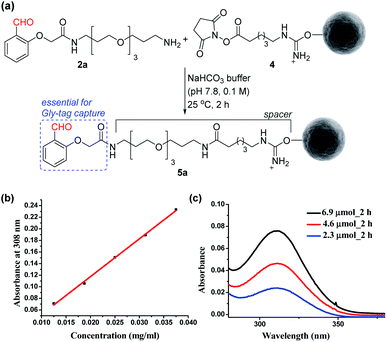 |
| | Fig. 3 (a) Synthesis of the Gly-tag purification resin. (b) Determination of the molar extinction coefficient of the reagent 2a. (c) Estimation of unbound reagent 2a by UV absorption after reaction with resin 4 (2.3 μmol). | |
Immobilization of N-terminus Gly proteins on modified resin
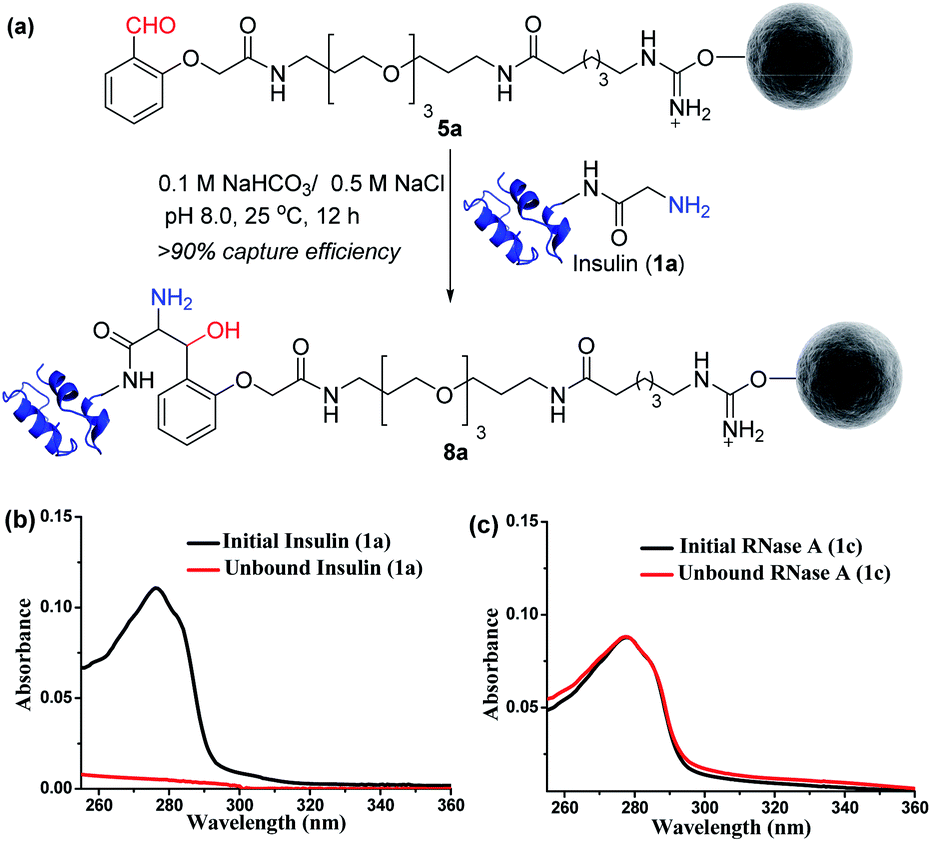
Next, we selected a therapeutic protein (insulin, 1a) for examining the method for immobilization of N-terminus Gly containing proteins on Gly-tag purification resin (5a). Insulin (1a) has two chains where Nα–NH2 of chain B is Phe, and that of chain A is a Gly residue. We anticipated that the N-terminus Gly would form a stable aminoalcohol product 8a with the resin 5a, thereby immobilizing the insulin (Fig. 4a). An aldol reaction would result in the C–C bond formation between an enolate and aldehyde. We analyzed the insulin-capture using the absorbance of unbound insulin (at 280 nm). When 20 nmol of insulin was vortexed with 400 μl of the resin 5a, we observed the complete disappearance of the insulin absorption peak for the supernatant, indicating excellent binding (>90% efficiency, Fig. 4b). Subsequently, the resin was thoroughly washed to remove any adsorbed protein. ESI-MS and UV-Vis analysis confirmed that no protein remained in the solution. Also, we were able to rinse the resin with several portions of buffer without releasing the bound protein. The robust immobilization through C–C bond formation facilitates ordered single-site immobilization and opens a gateway for developing other biochemical tools and biomaterials.23–26 Alongside, we selected myoglobin (1b, N-Gly) and SUMO1 (1d, N-Gly) to re-validate the efficiency of the Gly-tag purification resin (Fig. S5 and S8†). Interestingly, the method translated well for their immobilization. In the control experiment, RNase A (1c, N-terminus Lys) did not result in any reaction with 5a (Fig. 4c). This confirmed the selectivity of the Gly-tag purification resin towards the Gly at the N-terminus of the protein.
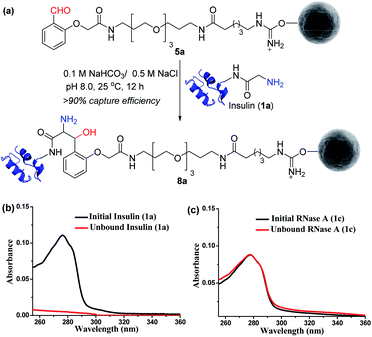 |
| | Fig. 4 (a) Capture by immobilization of N-Gly containing insulin on Gly-tag purification resin 5a. (b) UV spectra for the quantification of insulin immobilized on resin 5a (for myoglobin and SUMO1, see Fig. S5 and S8†). (c) Control experiment: UV spectra suggesting no capture of RNase A 1c (lacks N-Gly). | |
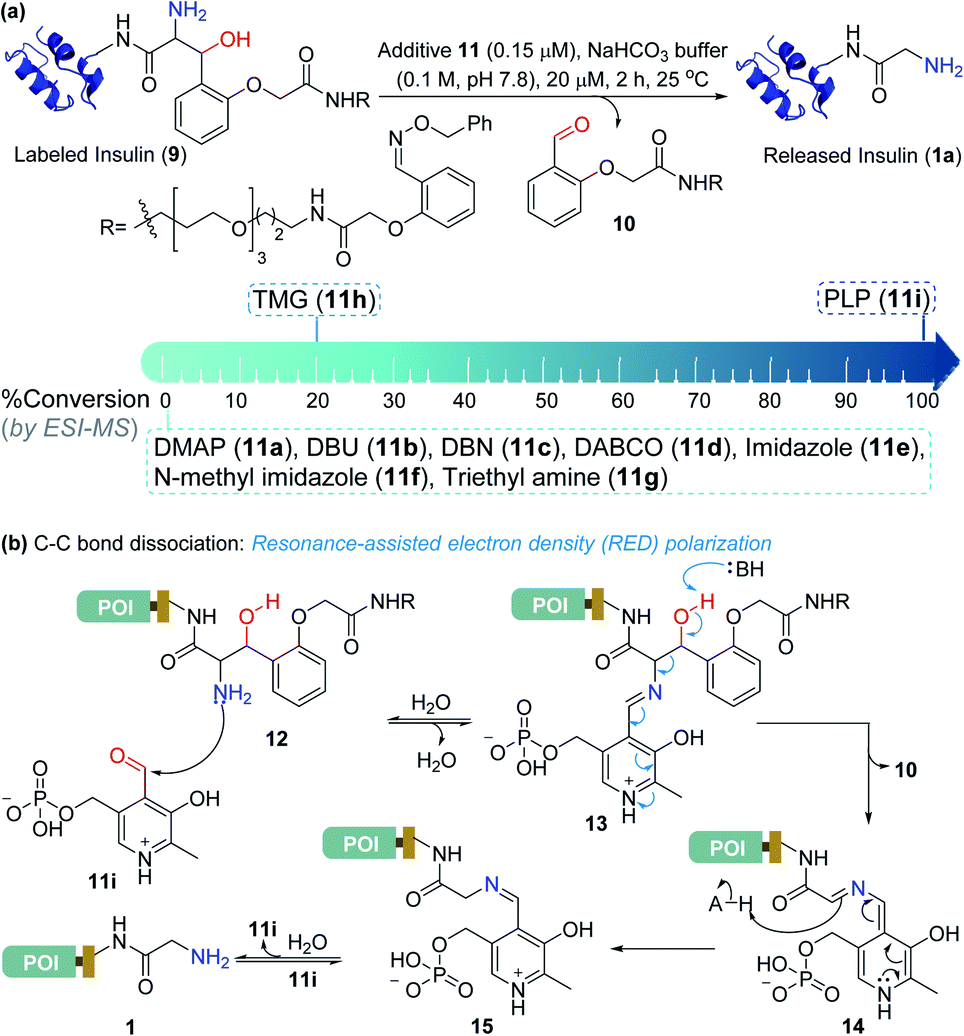
Screening of additives for C–C bond dissociation in the aminoalcohol
At this stage, we were poised to tackle the most critical challenge. The final element of the technology required a methodology for aminoalcohol C–C bond dissociation under physiological conditions. Initially, we thought that base-mediated deprotonation of hydroxyl could trigger a retro-aldol reaction. To examine the theory, we employed various organic bases (11a–11g, Fig. 5a) and mixed them with the labeled insulin (9). However, all these additives were unable to initiate the release. With a stronger organic base, TMG (11h, Fig. 5a), we were able to observe a retro-aldol reaction (20% conversion, see Fig. S7†). However, it was too slow indicating that direct employment of a base would not serve the purpose. We reasoned that lowering the pKa of hydroxyl might be critical for performing the reaction under mild conditions. We hypothesized that transforming the amine (12, Fig. 5b) to imine (13) combined with an appropriate resonance-assisted electron density (RED) polarization might address the requirement. We postulated that pyridoxal 5′-phosphate (PLP, 11i) could provide the extended conjugation and activate the aminoalcohol 12 (Fig. 5b). Hence, the formation of imine (13) could enable the abstraction of the hydroxyl proton. The feasibility would depend on the additional pull effect provided by the PLP (11i). To test this conjecture, we decided to monitor the reaction of the aminoalcohol product with PLP (11i) in the solution phase. To generate the aminoalcohol product (12), we modified the N-terminus of native insulin with a bis-aldehyde (S8) (Fig. S6†).11 We were delighted to note that PLP (11i, 50 equiv., 0.1 M NaHCO3 buffer, pH 7.8) promoted the retro-aldol reaction with 98% conversion within 2 h (Fig. 5a and Table S1;† for myoglobin and SUMO1, see Fig. S8†). Importantly, we do not observe the potential side-reaction of N-aldehyde introduction under our reaction conditions.27 Further, we revalidated the absence of the aldehyde by mixing the reaction mixture with benzyloxyamine. The remarkable chemoselectivity of our release methodology is noteworthy. Also, it is absolutely essential for the success of Gly-tag technology.
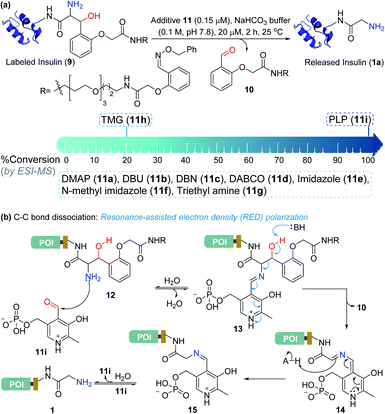 |
| | Fig. 5 Release under mild conditions. (a) Screening of reagents for C–C bond dissociation in the aminoalcohol. (b) Potential role of resonance-assisted electron-density (RED) polarization in C–C bond dissociation and release. | |
Purification of the protein of interest from a mixture of proteins and cell lysate
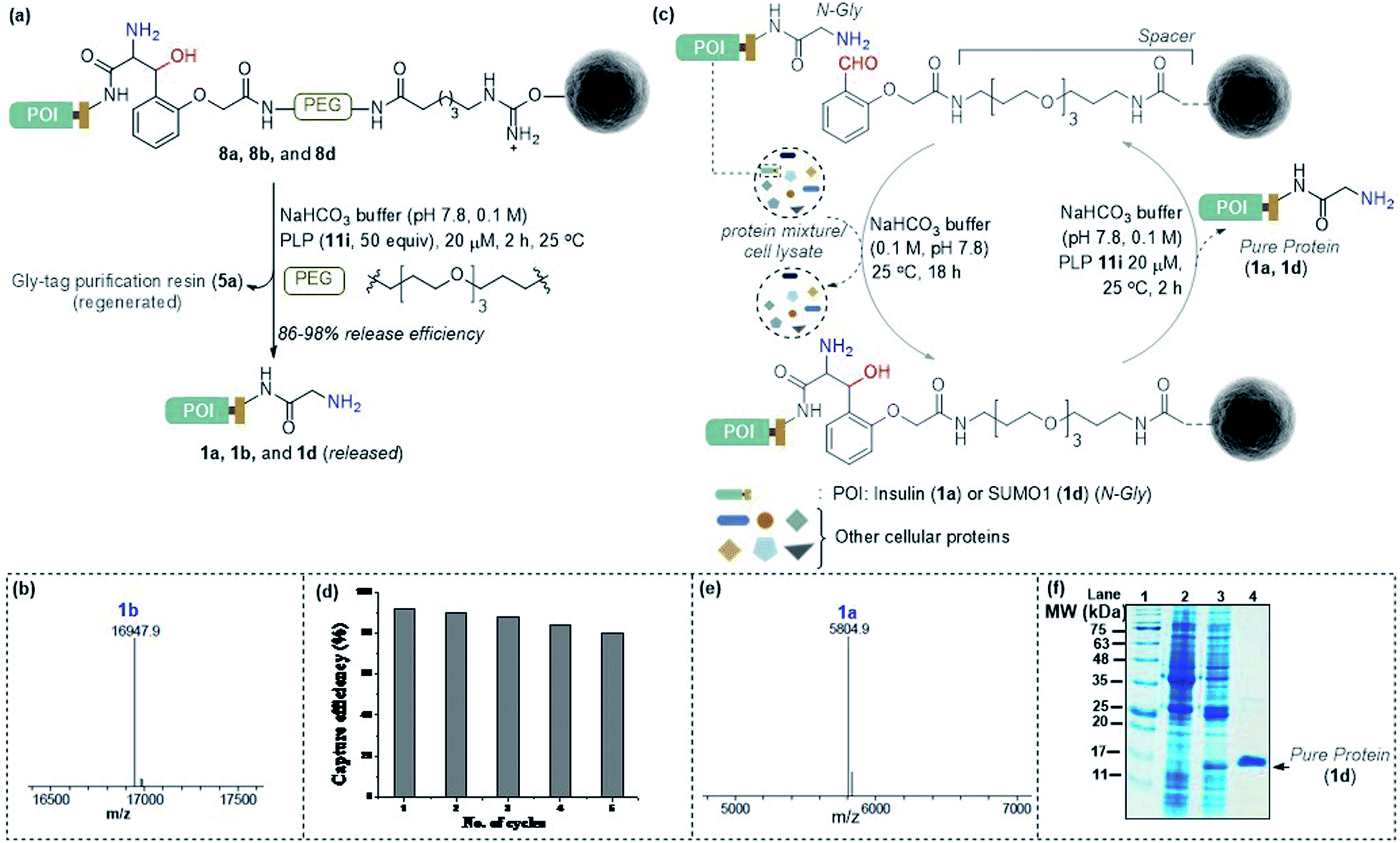
Subsequently, we investigated the retro-aldol reaction with immobilized proteins (Fig. 6a). The PLP (1 μmol in 0.1 M NaHCO3 buffer, pH 7.8) mediated C–C bond dissociation results in 86–98% release of insulin and myoglobin within 2 h (Fig. 6b and S8†). We removed the PLP 11i by centrifugal spin concentration and isolated the pure insulin (1a) with a conserved structure (for CD, see Fig. S9†). Similarly, immobilized myoglobin (8b) also yielded pure protein 1b upon PLP assisted release (Fig. 6b and S8†). To investigate the efficiency of the methodology in selective capture and release of the N-Gly protein, we selected a mixture of proteins having diverse residues at the N-terminus. The protein mixture comprised insulin 1a, aprotinin 1e, ubiquitin 1f, cytochrome C 1g, lysozyme C 1h, β-lactoglobulin 1i, and chymotrypsinogen A 1j (Fig. 6c). To our delight, we were able to purify insulin 1a from the mixture of proteins while conserving the efficiency (Fig. 6e and S10†). Also, the protocol allows excellent recovery and recyclability (five times) of the Gly-tag purification resin without substantial loss of activity (Fig. 6d; for recyclability with cell lysate, see Fig. S10c†). Finally, we checked if we could purify a protein of interest from the cell lysate (Fig. 6c). We overexpressed the protein modifier, SUMO1 protein (1d), in the E. coli BL21 (DE3) strain, with the recognition sequence for preScission protease. The post-proteolytic digestion resulted in a Gly residue at the N-terminus. Remarkably, the protein is captured and released selectively to yield pure SUMO1 (1d, Fig. 6f, 50% recovery yield, and 30-fold enrichment by SDS-PAGE). The protocol also extends conveniently to the purification of insulin from the cell lysate (Fig. S10c†). Moreover, the activity of purified SUMO1 and insulin remains unperturbed (Fig. S11 and S12†).
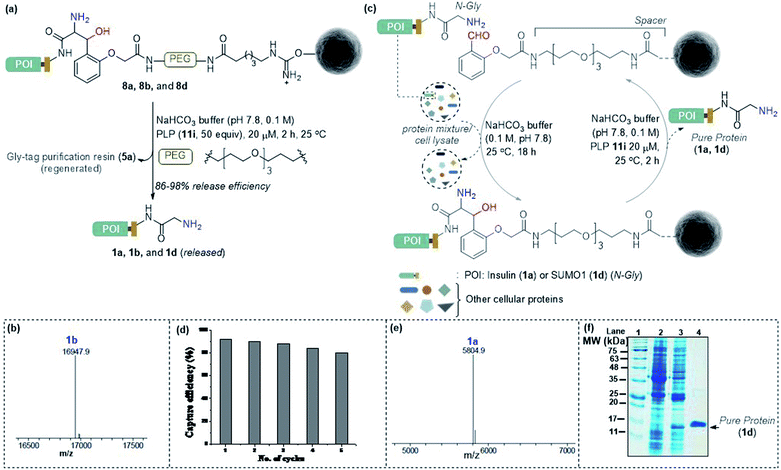 |
| | Fig. 6 (a) Release of immobilized proteins by a retro-aldol reaction. (b) ESI-MS spectrum of released myoglobin 1b. (c) Purification of the protein of interest from a mixture of proteins or cell lysate. For capture and release of insulin, see Fig. S8.† (d) Recycling efficiency of the Gly-tag purification resin with insulin. (e) ESI-MS spectrum of insulin purified from the mixture of proteins. (f) SDS-PAGE of purified SUMO1 from cell lysate; lanes 1, 2,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3, and 4 represent the ladder, GST-SUMO1, GST-clipped SUMO1 in cell lysate, and purified SUMO1, respectively. 3, and 4 represent the ladder, GST-SUMO1, GST-clipped SUMO1 in cell lysate, and purified SUMO1, respectively. | |
Conclusions
In summary, we developed a chemical technology that establishes a single amino acid (N-Gly) as the covalent affinity tag. We built the initial platform based on our residue-specific N-Gly labeling strategy. It enabled us to develop a Gly-capture reagent that delivered high efficiency even in the solid phase. The resulting functionalized Gly-tag purification resin facilitated selective capture of the protein of interest (POI) while leaving the other proteins in solution. Importantly, we were successful in developing a methodology for C–C bond dissociation under mild conditions. This on-demand reversibility proved critical for the operation of the overall technology and enabled the release of the POI. Besides, the recovery and recycling of the Gly-tag purification resin allowed us to reduce the cost of operation. The technology facilitates the separation and isolation of N-Gly tagged proteins from a mixture of proteins and cell lysates.
Experimental
Procedure for N-Gly protein purification
N-Hydroxy succinimidyl Sepharose beads 4 (400 μl, resin loading: 23 μmol ml−1) were taken in a 5 ml fritted polypropylene chromatography column with end tip closures. Sodium bicarbonate buffer (0.1 M, pH 7.8, 3 × 1 ml) was used to wash the beads and they were re-suspended (sodium bicarbonate buffer, 360 μl, 0.1 M, pH 7.8). To this solution, 2a (13.8 μM) in DMSO (40 μl) from a freshly prepared stock solution was added and vortexed at 25 °C. The progress of the immobilization of the reagent on Sepharose resin was monitored using the UV-absorbance of the supernatant. Subsequently, the supernatant was removed and the beads were washed with aqueous buffer (0.1 M NaHCO3/0.5 M NaCl pH 8.0, 3 × 1 ml; 0.1 M acetate/0.5 M NaCl pH 4.0, 3 × 1 ml) and H2O (3 × 1 ml) to remove the adsorbed reagent from the resin (stored at 4 °C). The Sepharose beads 5a were washed with the aqueous buffer (0.1 M NaHCO3/0.5 M NaCl pH 8.0, 3 × 1 ml) and re-suspended (aqueous buffer, 375 μl, 0.1 M NaHCO3/0.5 M NaCl pH 8.0). To this solution, the native protein (20 nmol) dissolved in sodium bicarbonate buffer (25 μl, 0.1 M, pH 7.8) was added and vortexed at 25 °C. Binding was ensured using UV-Vis analysis. The beads were washed thoroughly with aqueous buffer (0.1 M NaHCO3/0.5 M NaCl pH 8.0, 3 × 1 ml), 1 N KCl (3 × 1 ml) and H2O (3 × 1 ml) to remove any non-specifically bound protein from the resin. This was confirmed by analyzing the final wash fraction using LC-MS. For eluting out the bound protein, pyridoxal 5′-phosphate 11i (50 equiv.) in 0.1 M NaHCO3 buffer (pH 7.8) was added to the resin and vortexed for 2 h at 25 °C. Hydroxyl amine was added before a final round of spin concentration. The eluted protein was analysed by using ESI-MS and SDS-PAGE.
Labeling of the protein in the solution phase
In a 1.5 ml Eppendorf tube, protein 1a (3 nmol) was mixed with sodium bicarbonate buffer (120 μl, 0.1 M, pH 7.8). To this solution, S8 (1500 nmol) in DMSO (30 μl) from a freshly prepared stock solution was added and vortexed at 25 °C. The overall concentration of the protein and S8 was 20 μM and 10 mM respectively. After 24–48 h, the reaction mixture was diluted with acetonitrile:water (10:90, 3000 μl). Unreacted 2-(2-formylphenoxy)acetic acid and salts were removed by using an Amicon® Ultra-0.5 ml 3 kDa or 10 kDa MWCO centrifugal filter spin concentrator. The protein mixture was further washed with Millipore Grade I water (5 × 0.4 ml). The sample was analyzed by ESI-MS. The aqueous sample was concentrated by lyophilization before subjecting it to digestion, peptide mapping, and sequencing by MS-MS.
Author contributions
L. P. and U. V. B. contributed equally. V. R., L. P., and U. V. B. conceived the research. D. G. R., S. D. M., N. C. R., and S. R. A. designed and performed synthesis and bioconjugation experiments. M. G., S. T. K., and R. K. M. designed and performed the bioassays. All the authors wrote the manuscript.
Conflicts of interest
V. R. is the founder of Plabeltech Private Limited. A patent application has been filed on this work with V. R. as the inventor (patent application no. WO2017158612-A1, Sept. 21, 2017).
Acknowledgements
This work was supported by SERB, DBT, DAE, and IISER Bhopal. L. P., U. V. B., D. G. R., S. D. M., M. G., S. T. K., N. C. R., and S. R. A. are recipients of a research fellowship from CSIR, UGC, and IISER Bhopal. V. R. is a recipient of Swarnajayanti fellowship (DST/SJF/CSA-01/2018-19; SB/SJF/2019-20/01).
Notes and references
-
W. Nes, Protein Purification: Principles, High-Resolution Methods, and Applications, ed. J. C. Janson and L. Rydén, John Wiley & Sons, New York, 2nd edn, 1988 Search PubMed.
- S. Gräslund,
et al.
, Nat. Methods, 2008, 5, 135–146 CrossRef.
- J. Porath, J. Carlsson, I. Olsson and G. Belfrage, Nature, 1975, 258, 598–599 CrossRef CAS.
- D. Smith and K. Johnson, Gene, 1988, 67, 31–40 CrossRef CAS.
- M. Kimple, A. Brill and R. Pasker, Curr. Protoc. Protein Sci., 2013, 73, 9 Search PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS.
- D. G. Rawale, K. Thakur, S. R. Adusumalli and V. Rai, Eur. J. Org. Chem., 2019, 6749–6763 CrossRef CAS.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS.
- S. R. Adusumalli, D. G. Rawale, U. Singh, P. Tripathi, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS.
- L. Purushottam, S. R. Adusumalli, U. Singh, V. B. Unnikrishnan, D. G. Rawale, M. Gujrati, R. K. Mishra and V. Rai, Nat. Commun., 2019, 10, 2539 CrossRef.
- H. Ren, F. Xiao, K. Zhan, Y.-P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS.
- G. J. L. Bernardes, M. Steiner, I. Hartmann, D. Neri and G. Casi, Nat. Protoc., 2013, 8, 2079–2089 CrossRef CAS.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- X. Zheng, Z. Li, W. Gao, X. Meng, X. Li, L. Y. P. Luk, Y. Zhao, Y.-H. Tsai and C. Wu, J. Am. Chem. Soc., 2020, 142, 5097–5103 CrossRef CAS.
- N. C. Reddy, M. Kumar and R. Molla, Org. Biomol. Chem., 2020, 18, 4669–4691 RSC.
- C. G. England, H. Luo and W. Cai, Bioconjugate Chem., 2015, 26, 975–986 CrossRef CAS.
- T. A. Egorov, A. Svenson, L. Rydén and J. Carlsson, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3029–3033 CrossRef CAS.
- M. M. Zegota, T. Wang, C. Seidler, D. Y. Wah Ng, S. L. Kuan and T. Weil, Bioconjugate Chem., 2018, 29, 2665–2670 CrossRef CAS.
- D. S. Waugh, Protein Expression Purif., 2011, 80, 283–293 CrossRef CAS.
- R. Trzcinska, P. Suder, A. Bodzon-Kulakowska, M. Skalska, A. Kubacki, J. Marcinkowski, R. Pedrys, J. Silberring, A. Dworak and B. Trzebicka, Anal. Bioanal. Chem., 2013, 405, 9049–9059 CrossRef CAS.
- L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS.
- N. R. Mohamada, N. H. C. Marzukia, N. A. Buanga, F. Huyopb and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef.
- M. Shen, J. Rusling and C. K. Dixit, Methods, 2017, 116, 95–111 CrossRef CAS.
- J. A. Shadish and C. A. DeForest, Matter, 2020, 2, 50–77 CrossRef.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kah, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01153c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a