
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA visible-light activated [2 + 2] cycloaddition reaction enables pinpointing carbon–carbon double bonds in lipids†
Guifang
Feng
 ,
Yanhong
Hao
,
Liang
Wu
and
Suming
Chen
*
,
Yanhong
Hao
,
Liang
Wu
and
Suming
Chen
*
Institute for Advanced Studies, Wuhan University, Wuhan, Hubei 430072, China. E-mail: sm.chen@whu.edu.cn
First published on 22nd June 2020
Abstract
The precise location of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in bioactive molecules is critical for a deep understanding of the relationship between their structures and biological roles. However, the traditional ultraviolet light-based approaches exhibited great limitations. Here, we discovered a new type of visible-light activated [2 + 2] cycloaddition of carbonyl with CC bonds. We found that carbonyl in anthraquinone showed great reactivities towards CC bonds in lipids to form oxetanes under the irradiation of visible-light. Combined with tandem mass spectrometry, this site-specific dissociation of oxetane enabled precisely locating the CC bonds in various kinds of monounsaturated and polyunsaturated lipids. The proof-of-concept applicability of this new type of [2 + 2] photocycloaddition was validated in the global identification of unsaturated lipids in a complex human serum sample. 86 monounsaturated and polyunsaturated lipids were identified with definitive positions of CC bonds, including phospholipids and fatty acids even with up to 6 CC bonds. This study provides new insights into both the photocycloaddition reactions and the structural lipidomics.
C bonds in bioactive molecules is critical for a deep understanding of the relationship between their structures and biological roles. However, the traditional ultraviolet light-based approaches exhibited great limitations. Here, we discovered a new type of visible-light activated [2 + 2] cycloaddition of carbonyl with CC bonds. We found that carbonyl in anthraquinone showed great reactivities towards CC bonds in lipids to form oxetanes under the irradiation of visible-light. Combined with tandem mass spectrometry, this site-specific dissociation of oxetane enabled precisely locating the CC bonds in various kinds of monounsaturated and polyunsaturated lipids. The proof-of-concept applicability of this new type of [2 + 2] photocycloaddition was validated in the global identification of unsaturated lipids in a complex human serum sample. 86 monounsaturated and polyunsaturated lipids were identified with definitive positions of CC bonds, including phospholipids and fatty acids even with up to 6 CC bonds. This study provides new insights into both the photocycloaddition reactions and the structural lipidomics.
Introduction
Carbon–carbon double bonds (CC) exist in a variety of bioactive molecules and play important roles in their structures and biological functions.1,2 The presence of CC locks the atoms around it into a fixed position. Thus, this rigid construction has a significant influence on the maintenance of specific shapes of biomolecules. Changes in the positions of CC bonds would form isomers, which might have dramatically different functions.3,4 Lipids are a group of biomolecules that play a multitude of crucial roles in the body, such as the structural elements of biological membranes, energy storage and signal transduction in cellular response pathways.5–10 Unsaturated lipids are a subclass of lipids containing either a single or multiple CC bonds in the fatty acyl chains. The CC bonds in positional isomers of unsaturated lipids endow them with different structures and functions in living organisms.2,11–13 For instance, lipids with cis-CC in the fatty acyl chain were found to modulate plasma membrane domain registration/anti-registration.14 Significant correlations between the CC positional isomer and the onset/progression of breast cancer were also observed.15 Therefore, the precise identification of CC isomers in lipid analysis would benefit the deep understanding of and reveal the underlying functional mechanisms of unsaturated lipids in physiological and pathological processes.16,17
The sensitivity and structural elucidation capabilities of mass spectrometry (MS) make it a preferred analytical technique for biomolecules.5,6,18–23 Some special methods for dissociation at the CC bonds in MS were developed to locate their positions.24–28 Meanwhile, selective chemical derivatizations or reactions of CC bonds prior to MS analysis also emerged, as well as the electrochemical epoxidation in nano-electrospray ionization MS developed very recently.18,29–34 Although these developed methods are promising in terms of identification of CC bond positions in specific lipid mixtures, their capability in comprehensive identification of unsaturated lipids has rarely been validated.35,36 Among these methods, photochemical reactions provide a feasible and clean approach for CC bond derivatization, such as the ozonolysis30 and Paternò–Büchi (PB) reactions.20 Nevertheless, the utilization of ultraviolet (UV) light in these reactions causes unexpected side reactions and potential health impairment.17,36–38 The mild visible-light activated reactions have great potential to overcome these limitations.39,40 Very recently, Ouyang et al. reported visible-light-driven photocycloaddition between benzophenone and CC bonds in unsaturated lipids, which provided new insight into utilizing noncovalent complexes for the synthesis of oxetanes.40 But no such reaction has been demonstrated for the comprehensive location of CC bonds in structural lipidomics.
In this study, we developed a new visible-light activated [2 + 2] cycloaddition (VACA) reaction coupled with tandem MS analysis towards the precise location of CC bonds. We found that carbonyl in anthraquinone (AQ) could be reacted with CC bonds in unsaturated lipids under the excitation of visible-light to form the unique oxetane ring. The subsequent breaking of this oxetane structure at the initial CC bond position in collision induced dissociation enabled pinpointing the CC bonds in lipids. The formation and dissociation of oxetane isomer products were confirmed by high resolution (HR) MS/MS. This is a reaction to realize [2 + 2] photocycloaddition of a quinone substrate with unsaturated lipids leading to the formation of oxetanes under visible-light. Based on this distinct VACA reaction, we have established an integrated solution for comprehensively locating the CC bonds in unsaturated lipids from a complex biological sample, and the results suggested the applicability of this approach in structural lipidomics.
Results and discussion
Validation of the VACA reaction with fatty acids
The photoexcited carbonyl compounds could react with CC to give the oxetane derived from a [2 + 2] cycloaddition reaction. However, almost all these traditional photocycloadditions need UV light to activate the carbonyl groups.41,42 To extend the wavelength of activation to the visible light region, we investigated various types of substrates and discovered that AQ possesses a large conjugated aromatic ring system and quinone structure43 exhibited high reactivity to the CC bonds in lipids induced by visible-light irradiation.
The reactivities of AQ with the CC bonds in lipids (Fig. S1 and S2 in the ESI†) and the possibility of producing decent diagnostic fragment ions in tandem MS were studied systematically. These reactions were conducted in a micro volume autosampler vial insert, and a visible-light LED laser (405 nm, 150 mW) was placed on top of the glass vial to irradiate the reaction solution in methanol. The reaction solution could be easily used for further ultra-high performance liquid chromatography (UHPLC)-MS or direct-injection MS analysis. Fatty acids (FA) 20:1 (11Z) were first utilized to validate the reaction with AQ. As shown in Fig. 1a, AQ and unsaturated lipids were excited by visible-light and then formed oxetane between one of the carbonyl groups in quinones and the CC bond in lipids.41 Two isomeric oxetanes which were defined as P1 and P2 in this paper could be generated depending on the relative positions of carbonyl and the CC bond. When energy such as heat is applied to the VACA reaction products, a retro reaction could be carried out through two possible pathways.41 Besides the pathway returning to the original reactants, another pathway was the cleavage of the C–C bond at the initial CC bond position and C–O bond at the initial carbonyl group. This pathway would produce the fragments P1a, P1b, P2a and P2b, which could indicate the CC bond position in lipids (Fig. 1a). We anticipated that the same products generated from the VACA reaction and fragments could be detected by MS and tandem MS. In the HR mass spectra of the reaction solution of AQ and FA 20:1 (11Z) shown in Fig. 1b and c, the appearance of peaks with the mass increase of 208 Da compared with the mass of the original substrate in both negative (m/z 517.3230, Fig. 1b) and positive ion (m/z 541.3277, Fig. 1c) modes clearly indicated the formation of VACA products. The conversion of FA was about 50% within 1 min (Fig. S3†). We chose the reaction time of 5 min in the following studies. Although the isomer products P1/P2 couldn't be differentiated by the direct MS scan method, the LC separated peaks revealed the presence of two isomers with an area ratio of about 1.4:1 (P1/P2) (inset in Fig. 1b). This ratio implied the slightly different probability of these two opposite orientations between CO and CC bonds in the VACA reaction. Interestingly, only one isomer could be clearly observed in positive ion mode after LC separation (inset in Fig. 1c). The MS/MS spectrum at 7.77 min detected with [P1a + Na]+ at m/z 399.2 indicated that this isomer is product P1 (Fig. S4†). This difference of peak intensities might be caused by the low ionization efficiency of P2 in the positive ion mode of electrospray ionization. If we increase the injected amount of the products in LC-MS, the product P2 can be observed (Fig. S5†). Similar results were observed in the reaction of AQ with FA 20:1 (11E) (Fig. S6†). In order to further confirm the structure of these VACA reaction products, a CID process was conducted for the products P1 and P2. Only the FA anion (m/z 309.2794) and AQ radical anion (m/z 208.0529) (Fig. 1d) were detected via CID for reaction products P1 and P2 in the negative ion mode, while more characteristic fragments such as P1a (m/z 399.1936), P2a (m/z 223.1310), and even P2b (m/z 341.1877) could be observed in the positive ion mode for products [M + Na]+ (Fig. 1e). The neutral loss of fragment P1b (consisting of an aldehyde) was not detected (Fig. 1f), but its formula and degree of unsaturation could be a direct indication of CC bond positions. These characteristic fragments provided not only unambiguous evidence of the formation of VACA reaction products (Fig. 1f) but also diagnostic ions for the identification of the position of CC bonds. The ion pair P1a and P2a has a mass difference of 176 Da (C14H8) and could be simply used as “diagnostic ions” for the presence of CC bonds.
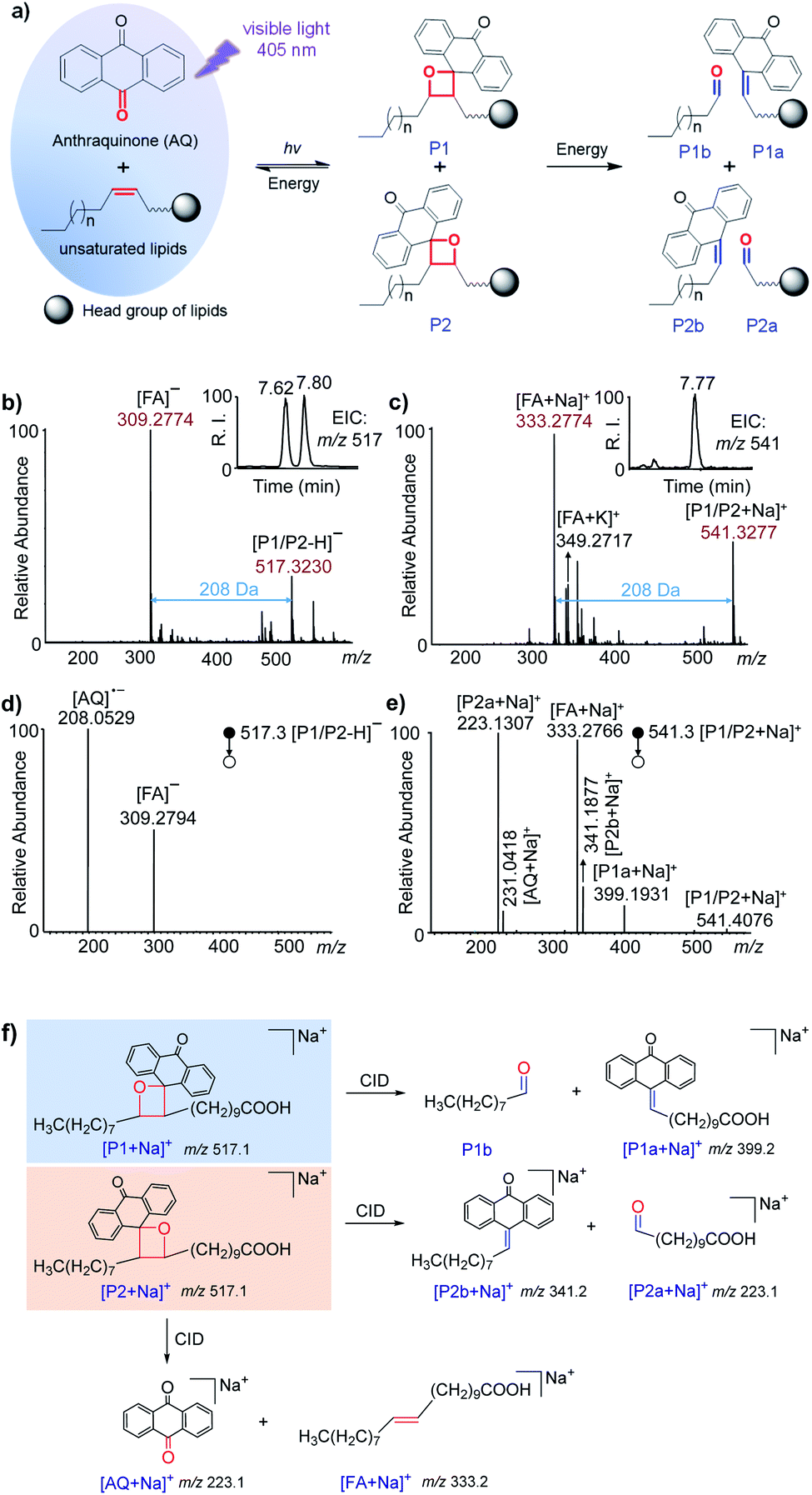 | ||
| Fig. 1 Visible-light activated [2 + 2] cycloaddition (VACA) reaction of anthraquinone (AQ) to unsaturated lipids. (a) Schematic illustration of the reaction between AQ and unsaturated lipids, and the dissociation pathways of reaction products; (b and c) MS spectra of the VACA reaction solution of AQ and FA 20:1 (11E) in (b) negative and (c) positive ion modes. Insets show the retention times of the VACA reaction products in LC-MS in (b) negative and (c) positive modes, respectively; (d and e) MS/MS spectra of the VACA reaction products in (d) negative (m/z 517.1, [M − H]−) and (e) positive (m/z 541.4 , [M + Na]+) ion modes; (f) proposed dissociation pathways and fragments of the VACA reaction product isomers. | ||
Investigating the versatility of this VACA reaction with different unsaturated lipids
To investigate the generality of this VACA reaction and demonstrate its capability in CC bond position location, different types of unsaturated lipids were taken for further study. First, the VACA reaction of AQ with polyunsaturated linoleic acid 18:2 (9Z, 12Z) was examined. The LC-MS results evidenced the formation of oxetane products (Fig. S7a,†m/z 511.3, [P1/P2 + Na]+). As expected, the VACA reaction product isomers at m/z 511.3 generated two pairs of characteristic ions via CID considering two CC bonds (Fig. 2a). The mass difference of every diagnostic ion pair P1a/P2a (m/z 411.2 and 235.1, m/z 371.2 and 195.0) generated from products P1/P2 was exactly 176 Da (Fig. 1a), which indicated the VACA reaction of AQ with CC bonds. The generic chemical formula of the fragment P1b (consisting of an aldehyde) is CnH2nO when there is no CC bond inside, and its mass could be calculated by subtracting the mass of the P1a ion from the mass of the P1 ion, namely 511.3 − 411.2 = 100.1 Da (C6H12O). Then this CC bond could be readily deduced to be at Δ12 counting from the carbon at the carboxyl group in the C18 chain. Another CC position could be deduced to be at Δ9 from the mass of the other P1b fragment (511.3–371.2 = 140.1 Da, C9H16O). The 40 Da mass difference of fragments P1a (Δ9 vs. Δ12, m/z 371.2 and 411.2) and P2a (Δ9 vs. Δ12, m/z 195.0 and 235.1) at different CC positions also indicated the three-carbon shift (C3H4) of the CC position. Given that one carbon space between CC bonds in lipids is typical, this 40 Da mass difference could be considered to be another signature for CC bond identification in polyunsaturated lipids. In addition, the VACA reaction of AQ with TG 18:1 (9Z)/18:1 (9Z)/18:1 (9Z) was also carried out and the products were detected (Fig. S7b†). The diagnostic ion pair P1a and P2a was observed at m/z 973.9 and 797.7, respectively, when CID was applied to the sodium adducts of P1/P2. Phospholipids (PLs) are a large family of lipids containing phosphorus, a polar head, and nonpolar tails. They are essential lipids of biological membranes, so we next examined this VACA reaction with PLs. The isomer products of lysophosphatidylethanolamine (LPE) 18:1 (9Z) with AQ could be well separated by LC (Fig. S7c†). The MS/MS spectra at retention times (RTs) of 2.12 (Fig. 2c) and 2.65 (Fig. 2d) min indicated the generation of isomers P1 and P2, respectively. The diagnostic ion pair P1a and P2a at m/z 568.2 and 392.1 was produced from the sodiated P1/P2 ions because of the cleavage of the formed oxetane ring at Δ9. However, only one of the diagnostic ions, P1a, could be detected in the MS/MS spectra of the protonated products (Fig. S9†). For the VACA reaction of PC 16:0/18:1 (9Z), two peaks of protonated isomer products were observed in the LC-MS spectrum (Fig. S7d†), as well as the characteristic ion pair of P1a and P2a at m/z 826.5 and 650.4, respectively. It should be noted that this VACA reaction of AQ with another steric isomer PC 18:1 (9Z)/16:0 showed similar reactivity and produced the same fragments of P1a (m/z 826.5) and P2a (m/z 650.4) via CID, respectively (Fig. S10a†). This result implied that this VACA reaction was not suitable for the recognition of sn-steric isomers of unsaturated lipids. However, the potential of this reaction for locating CC bond positions would enable a deep understanding of structural lipidomics. The generality of this VACA reaction for positioning the CC bonds in unsaturated lipids was also validated with more phospholipids, such as phosphatidic acid (PA) 16:0/18:1 (9Z), PE 18:1 (9Z)/18:1 (9Z), phosphatidylglycerol (PG) 18:0/18:1 (9Z), phosphatidylserine (PS) 16:0/18:1 (9Z), and ceramide (Cer) d18:1/16:0 (Fig. S10b–f†). Based on the above results, we found that the nature of the adduct ion has an important impact on forming double bond diagnostic ions. For most of the lipids, the sodium adducts of their derivatized products could form decent diagnostic ion pairs for double bond identification by CID, whereas the protonated product of PC shows better characteristic diagnostic ions than the sodiated one. Thus, the proton adducts for PC and sodium adducts for other lipids were used for the identification of double bonds in unsaturated lipids in the following experiments.
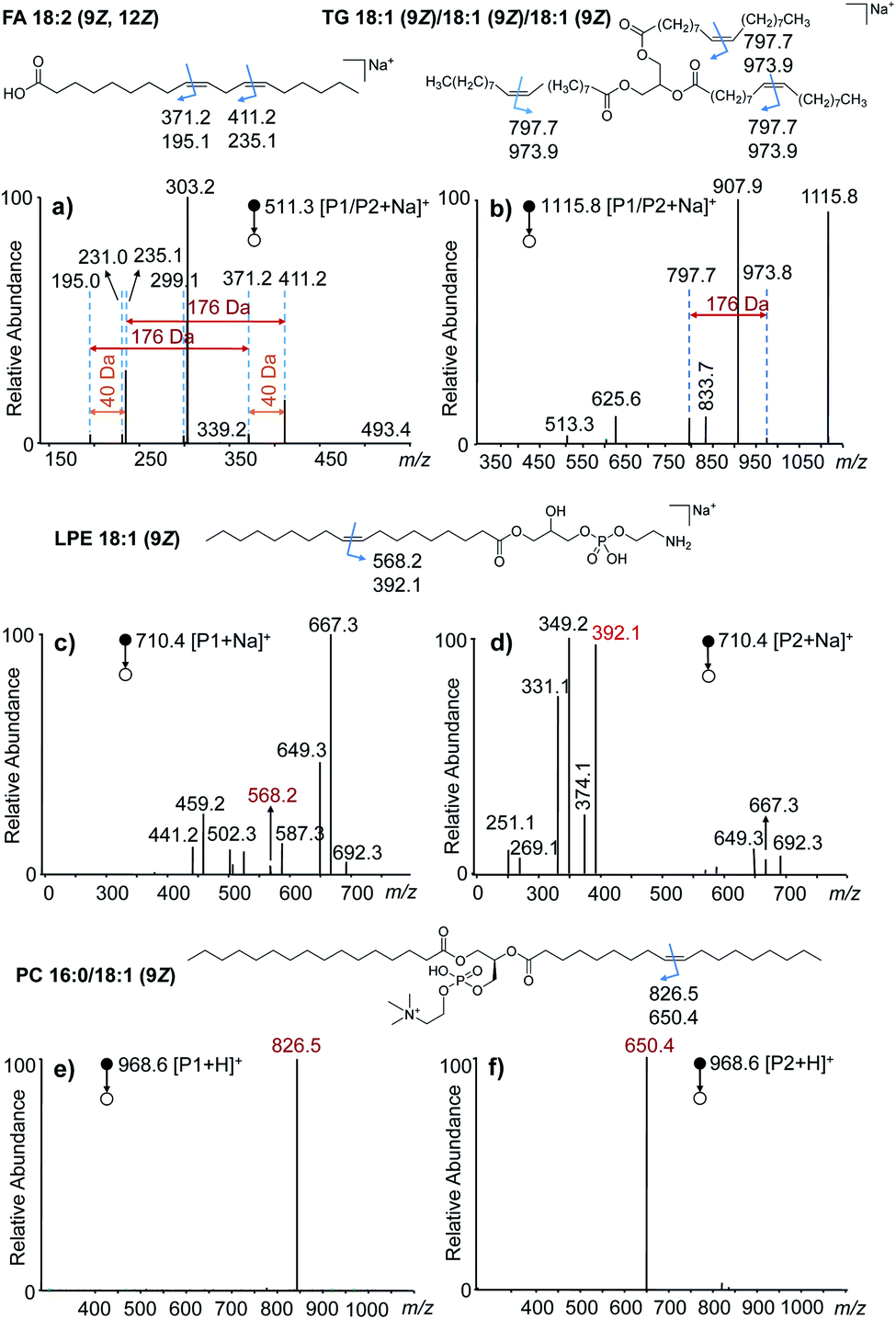 | ||
| Fig. 2 VACA reaction of anthraquinone with different unsaturated lipids. (a–f) The MS/MS spectra of the VACA product ions produced from the reaction of anthraquinone with (a) FA 18:2 (9Z, 12Z), (b) TG 18:1 (9Z)/18:1 (9Z)/18:1 (9Z), (c and d) LPE 18:1 (9Z), and (e and f) PC 16:0/18:1 (9Z). The chemical structures at the top of each MS/MS spectrum shows the positions of CC bonds and the m/z values of the possible diagnostic fragment ions of VACA reaction products via CID. The fragment ions in panels (c) and (d) were mainly obtained by the fragmentation of the ethanolamine head group of LPE. The detailed dissociation pathways are shown in Fig. S8 in the ESI.† | ||
Quantitative analysis of lipid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C location isomers
C location isomers
To further examine the quantitative capability of this method, isomers of FAs and PCs with different CC locations were quantitatively investigated. Fig. 3a and c show the results of the absolute quantification of FA 18:1 (9E), 18:1 (11Z), and PC 18:1 (9Z). For the analysis of FAs, FA 18:1 (6Z) was chosen as an internal standard (IS) at a constant concentration, while the concentration of FA 18:1 (9E) and 18:1 (11Z) was varied from 0.75 to 12.5 μM. The peak areas for each pair of CC diagnostic ions (P1a/P2a), i.e., m/z 195 and 371 from the 9E isomer or m/z 223 and 391 from the 11Z isomer, and m/z 153 and 379 from the 6Z internal standard, were summed, and the peak area ratios (A9E/A6Z or A11Z/A6Z) were plotted against the varied concentrations (Fig. 3a). Good linearities (R2 = 0.9956 for the 9E isomer and R2 = 0.9983 for the 11Z isomer) were obtained with reasonable accuracy (relative error < 10%, relative standard deviation (RSD) < 7%, n = 6). A limit of identification of 0.5 μM was obtained for each FA 18:1 isomer. Likewise, a good linearity (R2 = 0.9922) was obtained for PC 18:1 (9Z)/18:1 (9Z) from 1 nM to 1 μM with PC 18:1 (6Z)/18:1 (6Z) as the internal standard (Fig. 3c). The limit of identification was as low as 0.1 nM.
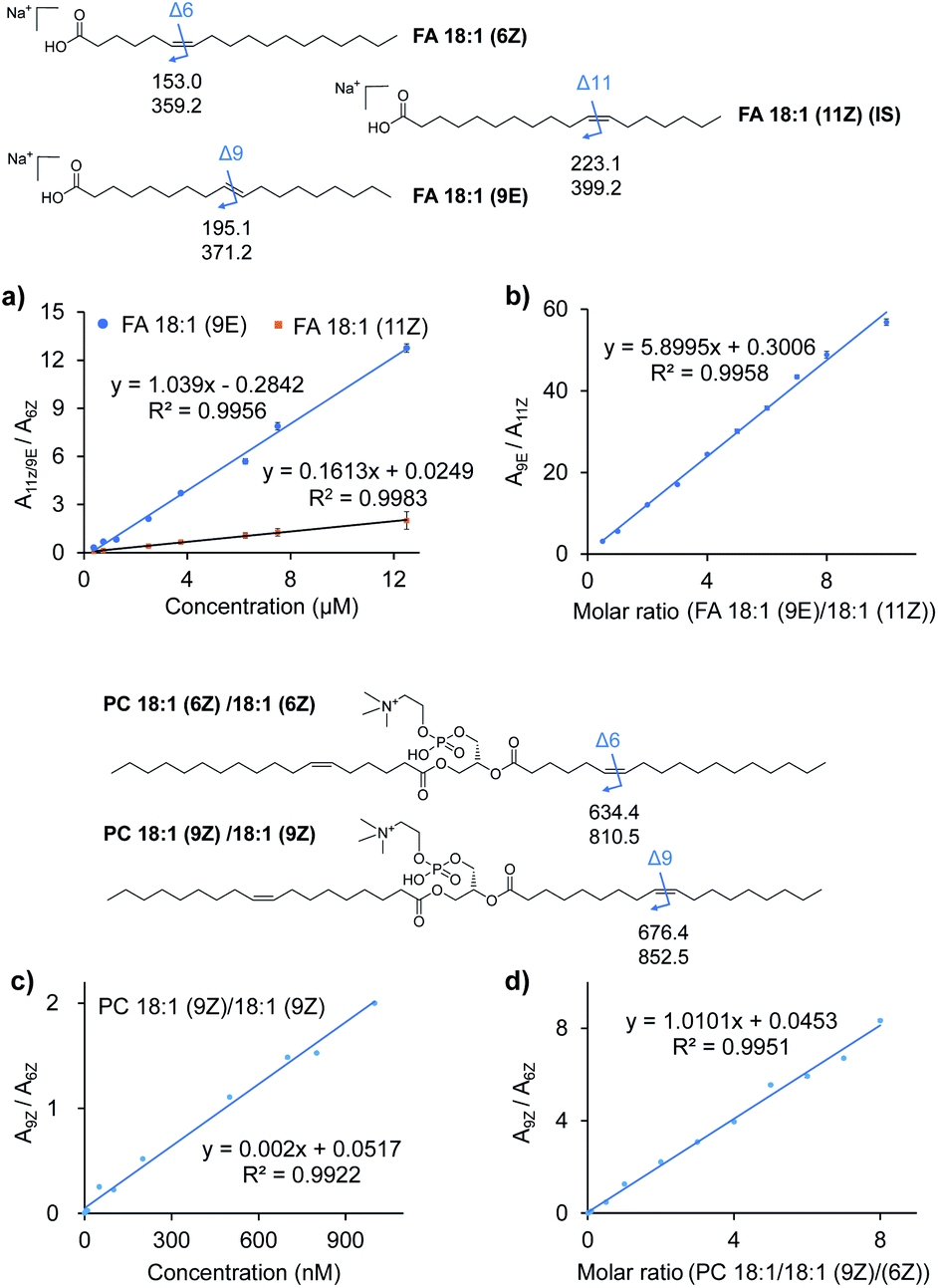 | ||
| Fig. 3 Quantitative analysis of lipid CC location isomers. (a) Calibration curves for quantitation of FA 18:1 (9E) and FA 18:1 (11Z) using FA 18:1 (6Z) as the internal standard. (b) Linear relationship established between the peak area ratio (A9E/A11Z) of the diagnostic ions and molar ratio (C9E/C11Z) of the two FA 18:1 CC location isomers. (c) Calibration curves for quantitation of PC 18:1 (9Z)/18:1 (9Z) using PC 18:1 (6Z)/18:1 (6Z) as the internal standard. (d) Linear relationship established between the peak area ratio (A9Z/A6Z) of the diagnostic ions and molar ratio (C9Z/C6Z) of the two PC 18:1/18:1 CC location isomers. The chemical structures of the lipids show the positions of CC bonds and the m/z values of the possible diagnostic fragment ions of VACA reaction products via CID. | ||
Quantification of the ratio of the CC isomers was performed with a series of mixtures of FA 18:1 isomers and PC 36:2 isomers. As shown in Fig. 3b for the FA 18:1 isomers, the ratios of the total summed peak areas from each pair of CC diagnostic ions (P1a/P2a), i.e., m/z 195 and 371 from the 9E isomer and m/z 223 and 391 from the 11Z isomer, were found to be directly proportional to the molar ratios of these two isomers with the total concentration kept constant (50 μM). A good linearity (R2 = 0.9958) was obtained with molar ratios from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:2. A similar relative quantitation of CC isomers was also conducted for the mixture of PC 18:1 (6Z)/18:1 (6Z) and PC 18:1 (9Z)/18:1 (9Z) at a constant concentration of 1 μM (Fig. 3d). The ratios of peak areas of the diagnostic ions (m/z 852 and 674 from the 9Z isomer and m/z 810 and 634 from the 6Z isomer) from each isomer and their molar ratios showed a good linear relationship (R2 = 0.9983) in a wide dynamic range (from 0.1:1 to 8:1). These results demonstrated the good quantitative capability of the proposed VACA reaction-based method for lipid CC isomers.
:1 to 1:2. A similar relative quantitation of CC isomers was also conducted for the mixture of PC 18:1 (6Z)/18:1 (6Z) and PC 18:1 (9Z)/18:1 (9Z) at a constant concentration of 1 μM (Fig. 3d). The ratios of peak areas of the diagnostic ions (m/z 852 and 674 from the 9Z isomer and m/z 810 and 634 from the 6Z isomer) from each isomer and their molar ratios showed a good linear relationship (R2 = 0.9983) in a wide dynamic range (from 0.1:1 to 8:1). These results demonstrated the good quantitative capability of the proposed VACA reaction-based method for lipid CC isomers.
Application of this VACA reaction in structural lipidomics
To validate the applicability of the developed visible-light activated [2 + 2] cycloaddition (VACA) reaction in structural lipidomics, a human serum sample was used for the proof-of-concept test. Three steps were used for identification of the position of CC bonds as shown in Fig. 4a. First, the lipids in the serum sample were extracted and analyzed by LC-MS/MS (see the details in the Experimental section). Based on the retention time (RT) and exact mass, the class of lipids could be identified preliminarily. The numbers of the isomers for lipids with the same molecular mass were judged relying on the number of peaks after LC separation. Then, the MS/MS spectra were used for the identification of lipids from every UHPLC peak (Fig. S11†). The glycerol phospholipids and glycerides were identified by matching the MS/MS data with those in LipidBlast,44 while the FA was identified by matching the extract mass and formula from a developed in-house library. The structures of every lipid were confirmed preliminarily with the molecular formula, subclass, fatty acyls, and number of CC bonds. Finally, the VACA reaction products with AQ were verified by extracting the precursor ions with a mass increase of 208 Da (C14H8O2). The diagnostic ion pair P1a and P2a could be easily distinguished in the MS/MS spectra due to their characteristic mass difference of 176 Da. As mentioned above, the formula of the neutral loss fragment P1b (CxHyO) could be simulated by using its exact mass, which is calculated by subtracting the mass of the P1a ion from the mass of the P1 ion. Then the CC bond positions can be readily deduced one by one with de novo analysis.
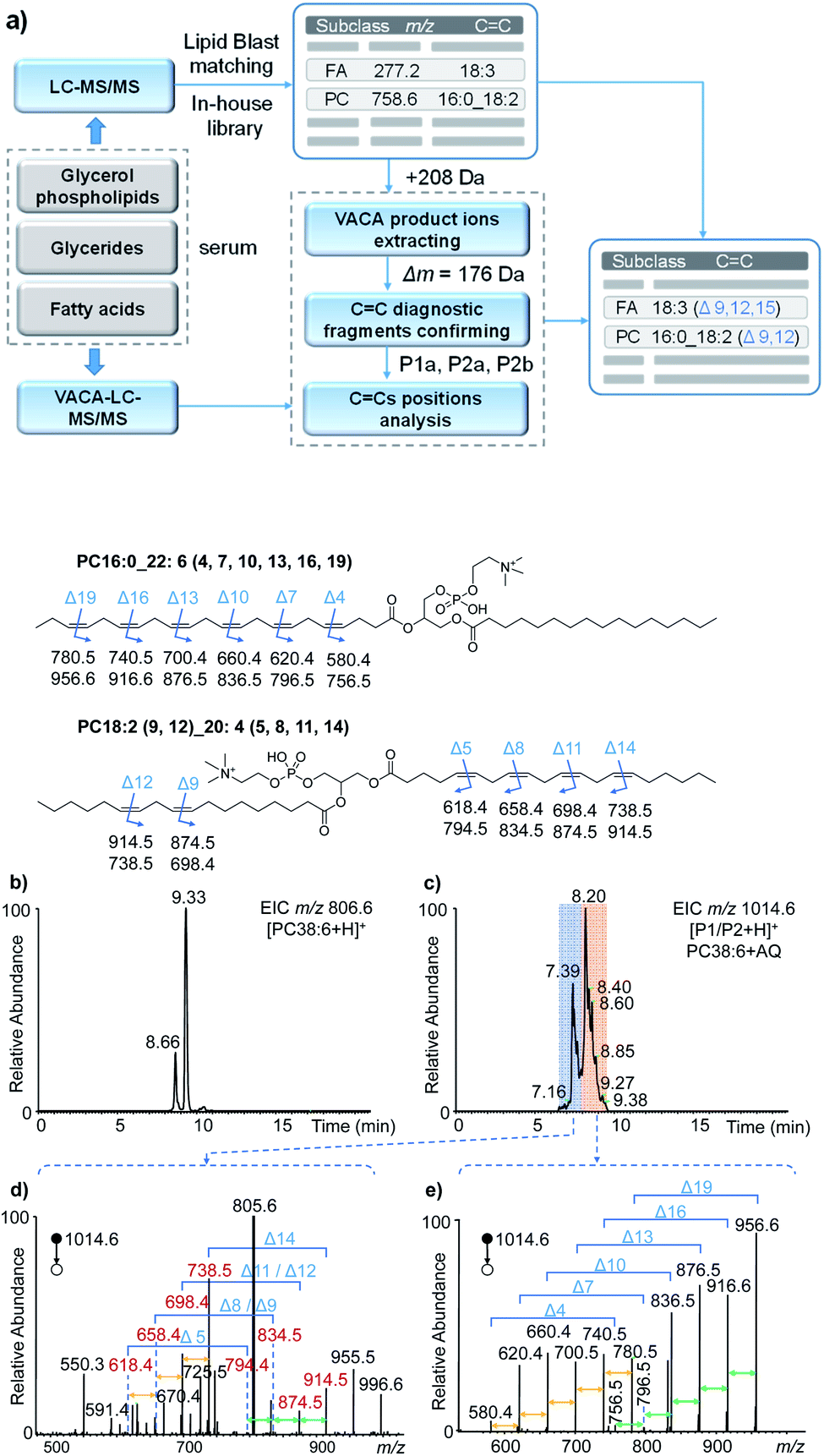 | ||
| Fig. 4 The analysis of unsaturated lipids in human serum. (a) Workflow for the identification of the positions of CC bonds via the VACA reaction and LC-MS/MS. (b and c) The extracted ion chromatograms of PC 38:6 (b) before and (c) after the VACA reaction. (d and e) The averaged MS/MS spectra of VACA products of PC 38:6 at m/z 1014.6 at (d) 7.9–9.3 min and (e) 6.2–7.9 min. The fragment ions at m/z 805.6 in panel (d) denote [PC 38:6]+, m/z 955.5 denotes [P1/P2–C3H9N + H]+, and m/z 996.6 denotes [P1/P2–H2O + H]+. The chemical structures show the positions of CC bonds in lipids and the m/z values of the possible diagnostic fragment ions. | ||
With the workflow we developed in this study, 213 lipids in serum were identified (Tables S1–S8 in the ESI†) and 86 of them were further identified at the CC location level (Table S2†). These position-identified unsaturated lipids included PC (45), PE (5), TG (21), sphingomyelin (SM, 1), and FA (12). It is worth mentioning that the VACA reaction has very good compatibility with polyunsaturated lipids with different degrees of unsaturation. The CC bond positions in phospholipids and fatty acids possessing even six CC bonds could be located. Taking PC 38:6 (m/z 806.6, [M + H]+) as an example, the extracted ion chromatogram (EIC) at m/z 806.6 of the serum extract before the VACA reaction gave two peaks at RTs of 8.66 and 9.33 min (Fig. 4b), which meant the existence of two isomers. The MS/MS spectra of these two peaks showed PC-like fragmentation pathways, namely the neutral losses of the head group and fatty acyls (Fig. S12†). The compositions of the fatty acyl chains could be calculated from the exact mass of each lost fatty acyl chain. By matching the LC-MS/MS results with the LipidBlast database (Fig. S13 and S14†), the possible structure at 9.33 min is PC 16:0_22:6 and another one at 8.66 min is PC 18:2_20:4 without the definite information of CC bond positions. To further analyze the locations of double bonds, the VACA reaction of AQ and lipid extract was conducted and the resultant solution was subjected to LC-MS analysis (Fig. 4c). The EIC of m/z 1014.6 (+208 Da) exhibited two clusters of peaks around 7.39 and 8.20 min, and the corresponding MS/MS spectra are shown in Fig. 4d and e. As shown in Fig. 4e, all the six diagnostic ion pairs (Δm = 176 Da) could be observed in the MS/MS spectrum of VACA products (RT 7.9–9.3 min), which indicated six CC bonds in this PC. That the mass difference of 40 Da was also detected between every adjacent ion pair suggested the existence of one carbon between adjacent CC bonds. In addition, the formulae of the smallest diagnostic ion P2a at m/z 580.4 (C28H55NO9P) showed no CC bonds in the other fatty acyl chain. Thus, PC 16:0_22:6 should be the right structure. The 58 Da mass difference between m/z 956.6 [P1a + H]+ and 1014.6 [P1/P2 + H]+ meant that the formula of the neutral loss aldehyde was C3H6O. This formula indicated terminal CC located between the third and fourth carbons counting from the end of the fatty acyl chain. Likewise, locations of five other CC bonds could be obtained. Therefore, the above evidence indicated that the detailed structure for one of the PC 38:6 isomers should be PC 16:0_22:6 (4, 7, 10, 13, 16, 19).
The MS/MS spectrum of another isomer PC 18:2_20:4 around 8.2 min displayed four diagnostic ion pairs (Fig. 4d), namely m/z 914.5 and 738.5, m/z 874.5 and 698.4, m/z 834.5 and 658.4, and m/z 794.4 and 618.4. Although some of them showed low intensities in this averaged MS/MS spectrum (6.2–7.9 min), the EICs of these ions further confirmed the presence of these peaks (Fig. S15a†). Based on these diagnostic ions, the formulae of the neutral aldehyde fragments P1b were calculated to be C6H12O (100 Da, no CC bond), C9H16O (140 Da containing one CC bond), C12H20O (180 Da, two CC bonds), and C15H24O (220 Da, three CC bonds), respectively, which meant the locations of CC bonds were Δ5, Δ8, Δ11 and Δ14 for the fatty acyl chain 20:4. For the other fatty acyl chain 18:2, only the diagnostic ions at Δ9 and Δ12 were detected (Fig. 4d, m/z 914.5 and 738.5; m/z 874.5 and 698.4) and extracted (Fig. S15b†). The peaks for other diagnostic ions simulated from other possible CC locations (Fig. S14†) were not observed in the extracted ion chromatograms (Fig. S15b†). Hence, we speculated that the structure of another isomer of PC 38:6 should be PC 18:2 (9, 12)_20:4 (5, 8, 11, 14). The successful identification of FA 22:6 (4, 7, 10, 13, 16, 19) and TG 18:0_18:1 (9)_20:4 (5, 8, 11, 14) was also described in Fig. S16–S18 in the ESI.† These facts undoubtedly demonstrated the capability of the newly developed visible-light activated [2 + 2] cycloaddition reaction for CC bond positioning in lipids with complex structures.
Conclusions
In summary, we discovered a novel visible-light activated [2 + 2] cycloaddition reaction of anthraquinone and unsaturated lipids. This VACA reaction exhibited great capability for locating the CC bonds of different kinds of lipids in complex biological samples. By using visible light, this reaction could alleviate the problems of side reactions and potential health impairment in the structural analysis of lipids. Other analytical advantages include the simple experimental setup for the reaction, non-requirement for MS instrument modification, easy-to-interpret mass spectra, and good compatibility with LC-MS/MS analysis. This visible-light driven cycloaddition reaction provides an attractive new paradigm for CC bond location in structural lipidomics.
Experimental
Materials
Lipid standards including phosphatidylcholine (PC) 16:0/18:1 (9Z), PC 18:1 (9Z)/16:0, PC 18:1 (9Z)/18:1 (9Z), PC 18:1 (6Z)/18:1 (6Z), phosphatidylethanolamine (PE) 18:1 (9Z)/18:1 (9Z), lysophosphatidylethanolamine (LPE) 18:1 (9Z), phosphatidylglycerol (PG) 18:0/18:1 (9Z), phosphatidic acid (PA) 16:0/18:1 (9Z), phosphatidylserine (PS) 16:0/18:1 (9Z) and ceramide (Cer) d18:1/16:0 were purchased from Avanti Polar Lipids (AL, USA). Fatty acids including FA 18:1 (9Z), FA 18:1 (9E), FA 18:1 (6Z), FA 18:1 (11Z), FA 20:1 (11Z), FA 20:1 (11E), FA 18:2 (9Z, 12Z) and arachidonic acid FA 18:4 (5Z, 8Z, 11Z, 14Z), and triacylglycerol (TG) 18:1 (9Z)/18:1 (9Z)/18:1 (9Z) were purchased from Sigma-Aldrich. Other solvents were purchased from Innochem (Beijing, China) and meet or exceed the analytical grade standard. The structures of all pure lipids are shown in Fig. S1 and S2.† Anthraquinone was purchased from Rhawn (Shanghai, China).Sample preparation
The human serum was provided by a healthy volunteer and collected at Renmin Hospital of Wuhan University. The use of human samples was approved by the Renmin Hospital of Wuhan University Review Board (Wuhan, China).The glycerol phospholipid (GPL) extraction protocol is described below:45,46 100 μL human serum samples were added to 300 μL butanol/methanol (3:1, v/v) and vortexed for 1 min. Then 150 μL heptane/EtAc (3:1, v/v) was added twice to the tube and the solution was vortexed for 1 min, and then 300 μL of 50 mM LiCl was added to the test tube to induce phase separation. The mixture was centrifuged at 1800g for 10 min to collect the upper organic layer. This part was flushed with nitrogen, capped, and stored at −20 °C.
The protocol of fatty acid extraction from human serum is described below:44 300 μL DPBS solution was added to 200 μL serum, followed by addition of 600 μL methanol. The mixed solution was added to 1 M HCl to reach a final concentration of 25 mM. Then 1 mL isooctane was added before vortexing. The mixture was centrifuged at 3000g for 1 min to collect the upper layer. This part was dried under a nitrogen flow and stored at −20 °C. Then 150 μL of a 1 mM AQ methanol solution was used for re-dissolution of lipid extracts. Before the VACA reaction, they were centrifuged at 16000g for 10 min.
VACA reaction
A visible LED laser (λ = 405 nm, 150 mW, Fulei Technology Co., Ltd, Shenzhen, China) was used as the light source for the visible-light activated cycloaddition (VACA) reaction. 100 μL of a solution containing anthraquinone (AQ, 100 μM) and olefins (10 μM) was placed in a micro volume autosampler vial insert, and the visible-light laser was placed on top of the glass vial to irradiate the reaction solution in methanol. The distance between the lamp and the liquid surface was 2 cm. The solution mixture was irradiated for 5 min at room temperature and then subjected to mass spectrometric analysis.Quantitative analysis
FAs and PCs were chosen for absolute and relative quantitative analysis. For plotting the calibration curves, one IS was defined, namely FA 18:1 (6Z) (6.25 μM) for FA and PC 18:1 (6Z)/18:1 (6Z) (0.5 μM) for PC. The concentrations of FA 18:1 (9E) and 18:1 (11Z) were varied from 0.75 μM to 12.5 μM while that of PC 18:1 (9Z)/18:1 (9Z) was varied from 1 nM to 1 μM. The quantitation of the ratios of two CC bond isomers was performed with a constant total concentration (50 μM for FA and 1 μM for PC) while the molar ratios of the isomers varied (1:2 to 10:1 for FA and 0.1:1 to 8:1 for PC). The total volume of all the reaction solutions was kept at 300 μL and the concentration of AQ in each solution was 0.5 mM. The reaction was conducted with 5 min irradiation using a visible light LED laser (λ = 405 nm, 150 mW) for all samples, and then subjected to further liquid chromatographic and mass spectrometric analysis.
Liquid chromatographic and mass spectrometric analysis
MS analyses were performed on an LTQ and LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific, Germany). The main mass parameters were set up as below: sheath gas (N2): 40 arbitrary units; auxiliary gas (N2): 5 units; capillary temperature: 350 °C; spray voltage: 3.8 kV; collision energy: 40 V; the resolution of the full mass scan, 60000. The parameters of MS/MS scanning were set as below: the top 5 ions with high intensity were selected for fragmentation analysis, and the dynamic exclusion function was turned on for 10 s. The MS/MS scan was induced by collision dissociation (CID), with a resolution of 30000.
A Waters ACQUITY UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 mm) was used in an UltiMate 3000 UPLC system (DIONEX, Thermo Scientific, Germany) and maintained at 40 °C for separation of lipid species. The flow rate was set as 0.3 μL min−1. Gradient elution consisted of mobile phase A (ACN/H2O (60:40, v/v) mixed with 10 mM NH4OAC and 0.1% formic acid) and mobile phase B (IPA/ACN (90:10, v/v) mixed with 10 mM NH4OAC and 0.1% formic acid). The injection volume was 10 μL. The optimal chromatographic gradient program for biological samples and GPL standards was as follows: 20–30% B at 0–2 min, 30–52% B at 2–5 min, 52–55% B at 5–8 min, 55–58% B at 8–9 min, 58–63% B at 9–12 min, 63–67% B at 12–14 min, 67–75% B at 14–15 min, 75–80% B at 15–16 min, 80–100% B at 16–17 min, 100% B at 17–19 min. Gradient elution consisting of mobile phase A (water, 0.1% formic acid) and mobile phase B (ACN) was performed for other FAs. The optimal chromatographic gradient program for arachidonic acid is described below: 0–5 min, 50% B; 5–30 min, 0–100% B; 30–40 min, 100% B. That of other FAs is set as below: 0–3 min, 40% B; 3–5 min, 40–100% B; 5–9 min, 100% B. The quantitative analysis of FAs was conducted with a water–methanol solvent system (phase A, water; phase B, methanol), and the optimal chromatographic gradient program was set as below: 0–0.5 min, 70% B; 0.5–10 min, 70–100% B; 10–12 min, 100% B.
Data analysis
LipidBlast is a collection of in silico electrospray tandem mass spectral libraries for the identification of neutral and polar lipid species. It was developed by Fiehn Lab with known theoretical fragmentation and experimental fragmentation from tandem mass spectra (https://fiehnlab.ucdavis.edu/). The NIST MS Search program was used for easy identification of every lipid. LipidBlast can be used to annotate lipids from large MS/MS files with NIST MSPepSearch.Because FAs respond weakly in the positive ion mode, it was difficult to confirm FAs in the serum extract in the positive ion mode. We established a list that contains molecular formula and mass information for all possible FAs. The number of carbon atoms in these compounds ranged from 12 to 30. And the number of unsaturated bonds of these FAs ranged from 0 to 7. These compounds were extracted in the negative ion mode in the serum extract. Then the detectable FA was analyzed for the position of its CC bond in the positive ion mode. As described in the text, adduct [M + Na]+ of the VACA product was used for MS/MS analysis. By setting the number of C, H and O atoms, the theoretical molecular formula was simulated. Then the neutral loss molecules were calculated for the analysis of the position of CC bonds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the start-up funds of Wuhan University and the “Youth Thousand Talents Plan” of China.References
- B. Antonny, S. Vanni, H. Shindou and T. Ferreira, Trends Cell Biol., 2015, 25, 427–436 CrossRef CAS PubMed
.
- I. Cimen, Z. Yildirim, A. E. Dogan, A. D. Yildirim, O. Tufanli, U. I. Onat, N. UyenThao, S. M. Watkins, C. Weber and E. Erbay, Mol. Metab., 2019, 28, 58–72 CrossRef CAS PubMed
- K. Albracht-Schulte, N. S. Kalupahana, L. Ramalingam, S. Wang, S. M. Rahman, J. Robert-McComb and N. Moustaid-Moussa, J. Nutr. Biochem., 2018, 58, 1–16 CrossRef CAS PubMed
- X. Zhao, W. Zhang, D. Zhang, X. Liu, W. Cao, Q. Chen, Z. Ouyang and Y. Xia, Chem. Sci., 2019, 10, 10740–10748 RSC
- X. Han, Nat. Rev. Endocrinol., 2016, 12, 668–679 CrossRef CAS PubMed
- K. Yang and X. Han, Trends Biochem. Sci., 2016, 41, 954–969 CrossRef CAS PubMed
- A. Klupczynska, S. Plewa, M. Kasprzyk, W. Dyszkiewicz, Z. J. Kokot and J. Matysiak, Clin. Exp. Med., 2019, 19, 505–513 CrossRef CAS PubMed
- D. Wolrab, R. Jirasko, M. Chocholouskova, O. Peterka and M. Holcapek, TrAC, Trends Anal. Chem., 2019, 120, 115480 CrossRef
- M. R. Wenk, Cell, 2010, 143, 888–895 CrossRef CAS PubMed
- S. Chen, A. Datta-Chaudhuri, P. Deme, A. Dickens, R. Dastgheyb, P. Bhargava, H. Bi and N. J. Haughey, J. Circ. Biomarkers, 2019, 8, 1–12 Search PubMed
- J. Dong, X. Cai, L. Zhao, X. Xue, L. Zou, X. Zhang and X. Liang, Metabolomics, 2010, 6, 478–488 CrossRef CAS
- P. V. Subbaiah, D. Sircar, B. Aizezi and E. Mintzer, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 506–514 CrossRef CAS PubMed
- M. Flasiński, P. Wydro, M. Broniatowski, K. Hąc-Wydro and P. Fontaine, Langmuir, 2015, 31, 7364–7373 CrossRef PubMed
- S. Y. Zhang and X. B. Lin, J. Am. Chem. Soc., 2019, 141, 15884–15890 CrossRef CAS PubMed
- N. S. Kelley, N. E. Hubbard and K. L. Erickson, J. Nutr., 2007, 137, 2599–2607 CrossRef CAS PubMed
- T. Porta Siegel, K. Ekroos and S. R. Ellis, Angew. Chem., Int. Ed., 2019, 58, 6492–6501 CrossRef CAS PubMed
- S. E. Hancock, B. L. J. Poad, A. Batarseh, S. K. Abbott and T. W. Mitchell, Anal. Biochem., 2017, 524, 45–55 CrossRef CAS PubMed
- S. Tang, H. Cheng and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 209–214 CrossRef CAS PubMed
- J. Han, X. Huang, H. Liu, J. Wang, C. Xiong and Z. Nie, Chem. Sci., 2019, 10, 10958–10962 RSC
- X. Ma and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 2592–2596 CrossRef CAS PubMed
- D. Wu, J. Li, W. B. Struwe and C. V. Robinson, Chem. Sci., 2019, 10, 5146–5155 RSC
- S. M. Chen, Q. Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed., 2016, 55, 9345–9349 CrossRef CAS PubMed
- S. Chen, Q. Wan and A. K. Badu-Tawiah, J. Am. Chem. Soc., 2016, 138, 6356–6359 CrossRef CAS PubMed
- C. Cheng, E. Pittenauer and M. L. Gross, J. Am. Soc. Mass Spectrom., 1998, 9, 840–844 CrossRef CAS PubMed
- P. E. Williams, D. R. Klein, S. M. Greer and J. S. Brodbelt, J. Am. Chem. Soc., 2017, 139, 15681–15690 CrossRef CAS PubMed
- H. T. Pham, T. Ly, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Anal. Chem., 2012, 84, 7525–7532 CrossRef CAS PubMed
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, J. R. Nealon and S. J. Blanksby, Anal. Chem., 2008, 80, 303–311 CrossRef CAS PubMed
- H. Takahashi, Y. Shimabukuro, D. Asakawa, S. Yamauchi, S. Sekiya, S. Iwamoto, M. Wada and K. Tanaka, Anal. Chem., 2018, 90, 7230–7238 CrossRef CAS PubMed
- J. I. Zhang, W. A. Tao and R. G. Cooks, Anal. Chem., 2011, 83, 4738–4744 CrossRef CAS PubMed
- M. C. Thomas, T. W. Mitchell and S. J. Blanksby, J. Am. Chem. Soc., 2006, 128, 58–59 CrossRef CAS PubMed
- C. H. Lam and M. S. F. L. K. Jie, Chem. Phys. Lipids, 1976, 16, 181–194 CrossRef CAS
- Y. Kwon, S. Lee, D.-C. Oh and S. Kim, Angew. Chem., Int. Ed., 2011, 50, 8275–8278 CrossRef CAS PubMed
- G. W. Francis, Chem. Phys. Lipids, 1981, 29, 369–374 CrossRef CAS
- T.-H. Kuo, H.-H. Chung, H.-Y. Chang, C.-W. Lin, M.-Y. Wang, T.-L. Shen and C.-C. Hsu, Anal. Chem., 2019, 91, 11905–11915 CrossRef CAS PubMed
- W. Cao, S. Cheng, J. Yang, J. Feng, W. Zhang, Z. Li, Q. Chen, Y. Xia, Z. Ouyang and X. Ma, Nat. Commun., 2020, 11, 375 CrossRef CAS PubMed
- W. Zhang, D. Zhang, Q. Chen, J. Wu, Z. Ouyang and Y. Xia, Nat. Commun., 2019, 10, 79 CrossRef CAS PubMed
- X. Ma, L. Chong, R. Tian, R. Shi, T. Y. Hu, Z. Ouyang and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2573–2578 CrossRef CAS PubMed
- B. L. Poad, X. Zheng, T. W. Mitchell, R. D. Smith, E. S. Baker and S. J. Blanksby, Anal. Chem., 2017, 90, 1292–1300 CrossRef PubMed
- L. Marzo, S. K. Pagire, O. Reiser and B. Konig, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed
- H. F. Li, W. Cao, X. Ma, X. Xie, Y. Xia and Z. Ouyang, J. Am. Chem. Soc., 2020, 142, 3499–3505 CrossRef CAS PubMed
- M. D'Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 RSC
- M. Freneau and N. Hoffmann, J. Photochem. Photobiol., C, 2017, 33, 83–108 CrossRef CAS
-
K. T. Finley, in The chemistry of the quinonoid compounds, ed. S. Patai, John Wiley & Sons, 1974, pp. 465–530 Search PubMed
- T. Kind, K.-H. Liu, D. Y. Lee, B. DeFelice, J. K. Meissen and O. Fiehn, Nat. Methods, 2013, 10, 755–758 CrossRef CAS PubMed
- T. Xu, Z. Pi, F. Song, S. Liu and Z. Liu, Anal. Chim. Acta, 2018, 1028, 32–44 CrossRef CAS PubMed
- M. Cruz, M. Wang, J. Frisch-Daiello and X. Han, Lipids, 2016, 51, 887–896 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01149e |
| This journal is © The Royal Society of Chemistry 2020 |