
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiofunctional Janus particles promote phagocytosis of tumor cells by macrophages†
Ya-Ru
Zhang
a,
Jia-Qi
Luo
b,
Jia-Xian
Li
a,
Qiu-Yue
Huang
a,
Xiao-Xiao
Shi
a,
Yong-Cong
Huang
b,
Kam W.
Leong
bf,
Wei-ling
He
*d and
Jin-Zhi
Du
 *ace
*ace
aGuangzhou First People's Hospital, Institutes for Life Sciences, School of Medicine, South China University of Technology, Guangzhou, 510006, China. E-mail: djzhi@scut.edu.cn
bSchool of Biomedical Sciences and Engineering, Guangzhou International Campus, South China University of Technology, Guangzhou, 510006, China
cNational Engineering Research Center for Tissue Restoration and Reconstruction, Key Laboratory of Biomedical Materials and Engineering of the Ministry of Education, Key Laboratory of Biomedical Engineering of Guangdong Province, Innovation Center for Tissue Restoration and Reconstruction, South China University of Technology, Guangzhou 510006, China
dDepartment of Gastrointestinal Surgery, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong 510080, China. E-mail: hewling@mail.sysu.edu.cn
eGuangzhou Regenerative Medicine and Health Guangdong Laboratory, Guangzhou 510005, China
fDepartment of Biomedical Engineering, Columbia University, New York, NY 10027, USA
First published on 5th May 2020
Abstract
Herein, a versatile strategy for the construction of biofunctional Janus particles (JPs) through the combination of Pickering emulsion and copper-free click chemistry is developed for the study of particle-mediated cell–cell interactions. A variety of biomolecules including bovine serum albumin (BSA), ferritin, transferrin (Tf), and anti-signal regulatory protein alpha antibodies (aSIRPα), etc., can be incorporated into the Janus platform in a spatially defined manner. JPs consisting of Tf and aSIRPα (Tf–SPA1–aSIRPα JPs) demonstrate a significantly improved binding affinity to either macrophages or tumor cells compared to their uniformly modified counterparts. More importantly, Tf–SPA1–aSIRPα JPs mediate more efficient phagocytosis of tumor cells by macrophages as revealed by real-time high-content confocal microscopy. This study demonstrates the potential advantages of JPs in mediating cell–cell interactions and may contribute to the emerging cancer immunotherapy.
Introduction
The fate and biological performance of synthetic particles are greatly influenced by their nano–bio interfaces.1 Many studies have shown that particle properties such as the size, shape, surface chemistry, and mechanical stiffness, have profound effects on particle–cell interactions.2,3 For example, Chan and co-workers have explored the use of DNA to construct nanostructures with shape-shifting properties so that they can alter folic acid presentation for the optimization of their biological functions.4 However, the cell response to nanoparticles is often not determined by single ligand-receptor recognition. In many cases, collective signals of different events initiate the biological process. For instance, T cell activation requires two simultaneous signals: T cell receptor-specific signals and costimulatory signals.5 In this context, different ligands are needed to be incorporated into the design of synthetic particles to obtain desirable particle–cell responses. Schneck's group has pioneered the design and development of synthetic particles as artificial antigen presenting cells (aAPCs) for in vitro activation of T cells.6,7 As a step forward, Chiang et al. incorporated anti-CD3, anti-CD28 and anti-PD-L1 antibodies onto IO@FuDex nanoparticles for simultaneous T-cell activation and checkpoint inhibition for more efficient cancer immunotherapy.8 In spite of these advances, the incorporation of different ligands onto the same particle is challenging and may cause unexpected mutual interference to compromise their biological functions.9Janus particles (JPs) that could spatially display ligands of different functions in one single entity may uniquely address this challenge. Developed for various biomedical applications including micro/nanoactuation,10–12 biosensing,13–15 drug delivery,16,17 and theranostics,18 JPs possess many promising features that are not offered by homogeneous particles. For example, Leong and co-workers developed a gene delivery system based on Janus nanorods that can simultaneously bind plasmid DNA and targeting ligands in separate parts, which avoided potential interference of DNA to the targeting ligands.19 The same nanorod could bridge the interactions of dendritic cells and T cells.20 The Yu group found that JPs entered cells differently from homogeneous ones, and further unraveled that JPs enabled the activation of T cells more efficiently than particles uniformly coated with the same number of ligands.21,22 All the studies demonstrate the potential advantages of JPs in manipulating particle–cell interactions. However, these studies only dealt with the interaction of JPs with a single type of cell.
Many biological processes involve two or more biological entities in nature. For example, APCs such as macrophages are needed to phagocytize tumor cells for efficient antigen presentation.23 Ligand- and antibody-modified nanoparticles with homogeneous surface presentation have been developed for controlling the interaction of macrophages and tumor cells for effective cancer immunotherapy.9,24 However, the study of JP-mediated cell–cell interactions between different types of cells has been largely overlooked. In this study, we demonstrated a versatile strategy for the construction of biofunctional JPs through the combination of Pickering emulsion and copper-free click chemistry (Scheme S1†), and investigated how the JPs affect cell–cell interactions between macrophages and B16F10 tumor cells.
Results and discussion
To construct JPs with copper-free click chemistry, silica particles (SPs) functionalized with azide groups were prepared as reported.25 Three different azide-modified SPs (SPAs) with the sizes of 150 ± 8 nm, 450 ± 10 nm, and 2.30 ± 0.02 μm were obtained (Fig. S1A†), which were denoted as SPA1, SPA2, and SPA3, respectively. Transmission electron microscopy (TEM) showed that the particles have relatively monodisperse size distributions (Fig. 1A and S1C, D†). The immobilization of azide groups was evidenced by the peak at 2100 cm−1 in the FTIR spectrum (Fig. 1B), and such a change also slightly increased the particle zeta potential from −42 to −26 mV (Fig. S1B†). Then, the Pickering emulsion method26 using water as the aqueous phase and molten wax as the oil phase was employed to prepare SPA colloidosomes and precursors of the JPs. Scanning electron microscopy (SEM) images showed that all SPA1, SPA2, and SPA3 formed monolayer distribution on the surface of the colloidosomes (Fig. 1C and S1E, F†). | ||
| Fig. 1 Preparation and characterization of the biofunctional JPs. (A) TEM image of SPA1. (B) FTIR spectra of SP and SPA. (C) SEM micrograph of a wax colloidosome stabilized by SPA1. (D) Representative TEM images of PEG–SPA1–ferritin JNPs showing the distribution of silicon (green) and iron elements (red). (E) Fluorescence image of FITC–SPA3 JMPs. (F) Flow cytometry analysis of FITC–SPA3–RB JMPs. | ||
Subsequent modifications were performed on the as-made colloidosomes to prepare JPs. To confirm the Janus structure, TEM and fluorescence imaging were employed. For TEM observation, Janus nanoparticles (JNPs) made from SPA1 were functionalized with poly(ethylene glycol) (PEG) and ferritin in opposite faces (denoted as PEG–SPA1–ferritin JNPs). As indicated in Fig. 1D, ferritin patches (∼10 nm) could be seen on one side of PEG–SPA1–ferritin JNPs, which was also confirmed by the enrichment of Fe element in the energy-dispersive X-ray spectroscopy (EDS) mapping mode. For fluorescence imaging, Janus microparticles (JMPs) made from SPA3 were labeled with fluorescein isothiocyanate (FITC) on one hemisphere (denoted as FITC–SPA3 JMPs), and the green fluorescence mainly distributed on one side of the particles (Fig. 1E). We further prepared FITC and rhodamine B (RB)-labeled BSA and attached them to the opposite sides of SPA3 to generate FITC–SPA3–RB JMPs. Fluorescence microscopy observation demonstrated that the green and red fluorescence were mainly distributed in a spatially segregated manner over the surface of the particles, while more even distribution of the two fluorescence was observed in uniform FITC–SPA3–RB UMPs (Fig. S2†). Flow cytometry analysis quantitatively demonstrated that about 84.6% of the particles were simultaneously stained with two different dyes (Fig. 1F). To demonstrate the versatility of the approach, we constructed a series of JPs with different surface properties, as summarized in Table S1.†
Next, JNPs modified with transferrin (Tf) and BSA on opposite sides (denoted as Tf–SPA1–BSA JNPs) were prepared to examine their targeting ability towards B16F10 cells since this cell line overexpresses transferrin receptors (TfRs).27 For fluorescence imaging, FITC-labeled Tf–SPA1–BSA JNPs were used. As shown in Fig. S3,† confocal microscopy demonstrated that FITC-labeled Tf–SPA1–BSA JNPs showed significantly enhanced green fluorescence on the membrane or inside B16F10 cells in comparison with nontargeted FITC-labeled SPA1 particles. However, if the cells were pretreated with free Tf, the cellular uptake of Tf–SPA1–BSA JNPs was obviously reduced due to the competitive binding of free Tf to TfRs. Quantitative analysis with flow cytometry was in good agreement with confocal microscopy imaging (Fig. 2A and B).
 | ||
| Fig. 2 Targeting of Tf–SPA1–BSA JNPs towards B16F10 tumor cells in vitro. Representative flow cytometry histograms (A) and corresponding mean fluorescence intensity (B) of B16F10 cells incubated with FITC-labeled SPA1 and FITC-labeled Tf–SPA1–BSA JNPs in the absence or presence of free Tf. (C) Saturation binding of FITC-labeled Tf–SPA1–BSA JNPs and Tf–SPA1–BSA UNPs to B16F10 cells. (D) Enhancement in the binding affinity of the two nanoparticles in comparison to free Tf. Data are presented as mean ± SD (n = 3). | ||
Next, we studied the binding affinity of Tf–SPA1–BSA JNPs to B16F10 cells in comparison with their uniformly modified counterparts (denoted as Tf–SPA1–BSA UNPs), in which the same amount of BSA and Tf was attached (Table S3†). Scatchard analysis was used to estimate the dissociation constant (KD) of Tf–SPA1–BSA JNPs, Tf–SPA1–BSA UNPs, and free Tf.29 As calculated, the KD values of Tf–SPA1–BSA UNPs and Tf–SPA1–BSA JNPs were 0.70 ± 0.05 nM and 0.20 ± 0.06 nM, respectively, representing approximately 110-fold and 400-fold enhancement in binding affinity in comparison with free Tf (KD = 80.38 ± 1.20 nM) (Fig. 2C, D and S4†). Such an enhancement demonstrates the multivalent effect of nanoparticle-based systems due to the increased local surface ligand density in nanoparticle-ligand conjugates.28,29 More importantly, the KD value of Tf–SPA1–BSA JNPs was also lower than that of Tf–SPA1–BSA UNPs, indicating that JNPs have higher binding affinity than uniformly modified nanoparticles. This may lay the foundation for possible enhanced particle–cell interactions.
The above studies demonstrated the construction of a series of JPs with different biofunctionalities. More interestingly, we have discovered that JPs showed a higher binding affinity to tumor cells than UNPs under the same conditions. However, particle–cell interaction studies were conducted with a single cell type. Many important biological processes usually involve different types of cells. For example, in cancer immunotherapy, macrophages or dendritic cells are needed to phagocytose tumor cells for antigen processing. Here, we wonder whether such JPs would be effective in mediating tumor cell–macrophage interaction. Tumor cells overexpress CD47 on membranes, which binds to SIRPα on macrophages to inhibit phagocytosis.30 Blockade of the CD47–SIRPα axis has been shown to increase tumor cell phagocytosis.31,32 Thus, in this study, we chosen Tf and aSIRPα to prepare Tf–SPA1–aSIRPα JNPs for the study of particle-mediated interaction between tumor cells and macrophages. On top of this, we would like to validate whether the Tf–SPA1–aSIRPα JNPs could bind to TfRs on tumor cells and SIRPα on macrophages separately. eFluor 670-labeled B16F10 cells or anti-F4/80 labeled bone marrow-derived macrophages (BMDMs) were incubated with Tf–SPA1–aSIRPα JNPs and subjected to flow cytometry analysis (Fig. S5† and 3A, B). As can be seen, both functionalized NPs showed stronger cell binding to either B16F10 tumor cells or BMDMs than each free ligand. Moreover, significantly enhanced cell binding of Tf–SPA1–aSIRPα JNPs in comparison to Tf–SPA1–aSIRPα UNPs was observed in either tumor cells (99.4% vs. 56.7%, p < 0.001) or macrophages (99.5% vs. 41.0%, p < 0.001), indicating the advantage of Janus structures in mediating cell–particle interactions.
 | ||
| Fig. 3 Tf–SPA1–aSIRPα JNPs facilitate phagocytosis in vitro. Relative binding ability of different treatments to B16F10 cells (A) and BMDMs (B). B16F10 cells were labelled with eFluor 670 and BMDMs were labelled with APC anti-F4/80. Data are presented as mean ± SD (n = 3). ***p < 0.001; ****p < 0.0001. (C) Schematic representation of phagocytosis assay (upper) and representative flow cytometric analysis images of phagocytosis assays treated with different formulations (lower). (D) Relative quantification of phagocytosis of tumor cells by BMDMs. Data are presented as mean ± SD (n = 3). **p < 0.01. (E) Representative confocal images of phagocytosis assays. Scale bar: 20 μm. In (C), (D) and (E), B16F10 cells were labelled with CFSE (green) and BMDMs were labelled with eFluor 670 (red). | ||
On this basis, we further investigated how the nanoparticles mediated the interaction between the macrophages and tumor cells. eFluor 670-labeled BMDMs and CFSE-labeled B16F10 cells were co-cultured with Tf–SPA1–aSIRPα JNPs and other formulations under the same conditions. Flow cytometry analysis (Fig. 3C and D) indicated that Tf–SPA1–aSIRPα UNPs were able to facilitate phagocytosis of B16F10 cells by BMDMs compared with a physical mixture of free Tf, free aSIRPα, and SPA1 (denoted as Tf + aSIRPα + SPA1) to some extent. The Tf–SPA1–aSIRPα JNPs could mediate an even higher phagocytosis than Tf–SPA1–aSIRPα UNPs (19.4% vs. 25.3%, p < 0.01), presumably due to their increased binding affinity to both tumor cells and BMDMs. Moreover, we also employed confocal microscopy to visually observe the nanoparticle-mediated B16F10–BMDM interaction (Fig. 3E), and confirmed that more B16F10 cells could be phagocytosed by BMDMs after co-culture with Tf–SPA1–SIRPα JNPs.
To observe the phagocytosis of B16F10 cells by BMDMs, 2.30 μm Tf–SPA3–aSIRPα JMPs were utilized and appeared as blue in pseudo-color, while BMDMs and B16F10 cells showed red and green fluorescence, respectively. As shown in the confocal images (Fig. 4A), we could see that more blue particles bound to BMDMs and B16F10 cells after 2 h co-incubation with Tf–SPA3–aSIRPα JMPs than other groups. With the incubation time extending to 4 h, B16F10 cells became phagocytosed by BMDMs for Tf–SPA3–aSIRPα JMP treatment, which indicates that Tf–SPA3–aSIRPα JMPs might mediate a stronger cell–cell interaction due to their enhanced binding affinity to both cells. To further observe the details, time-course tracking of phagocytosis was performed on a high-content analysis system. In Fig. 4B, we could clearly see the blue Tf–SPA3–aSIRPα JMPs attached to B16F10 cells and BMDMs, with B16F10 cells coming into contact with BMDM gradually and culminating with the phagocytosis of the tumor cells and JMPs together.
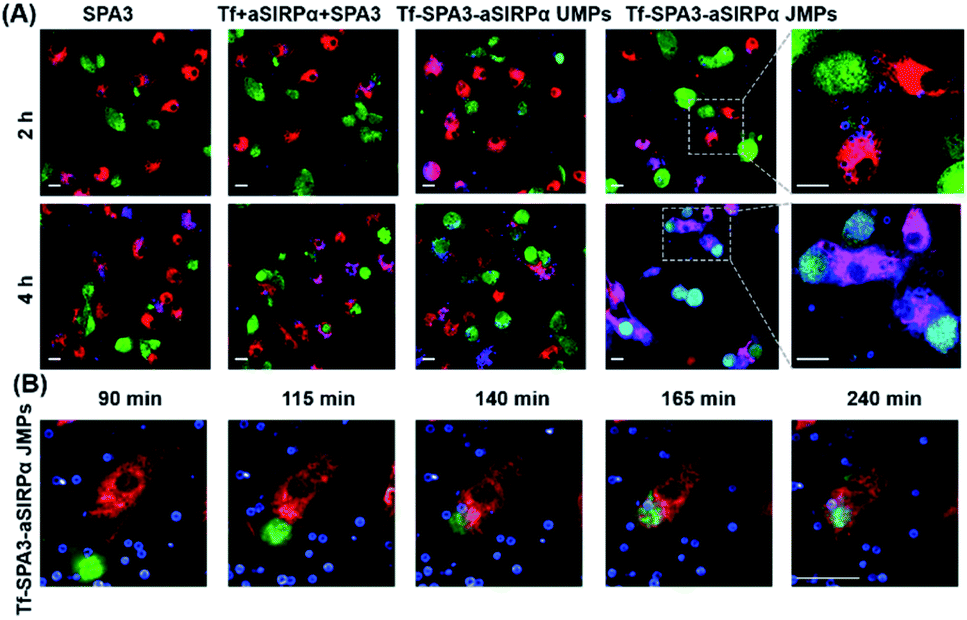 | ||
| Fig. 4 Tf–SPA3–aSIRPα JMPs promote the interaction and subsequent phagocytosis of B16F10 cells by BMDMs. (A) Representative confocal images of phagocytosis assays treated with different formulations for 2 or 4 h, respectively. (B) Time-dependent of phagocytosis treated with Tf–SPA3–aSirpα JMPs. In (A) and (B), B16F10 cells were labelled with CFSE (green), BMDMs were labelled with eFluor 670 (red) and particles were labelled with RB (blue). Scale bar: 20 μm. | ||
Conclusion
In summary, we developed a versatile strategy for the construction of JPs that can be spatially selective for the immobilization of different biomolecules on a single particle. Using Tf as a targeting ligand, we discovered that the dissociation constants of the JPs were significantly lower than those of particles coated uniformly with the same number of ligands, demonstrating that the Janus structure leads to an enhanced binding affinity to tumor cells with Tf-receptor overexpression. More importantly, by manipulating the spatial arrangement of Tf and aSIRPα on the particles, we demonstrated that the biofunctional JPs enable simultaneous targeting of macrophages and tumor cells, and thus facilitate the subsequent phagocytosis of tumor cells. Time-course tracking imaging indicated that the JPs could mediate the phagocytosis process. This study supports the continued development of Janus structures with spatially defined surface functionality for unique biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0205600), National Natural Science Foundation of China (31771091, 51922043, and 81871994), Guangdong Natural Science Funds for Distinguished Young Scholar (2017A030306018 and 2019B151502063), Guangdong Provincial Programs (2017ZT07S054 and 2017GC010304), Outstanding Scholar Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110102001), Guangzhou Science and Technology Planning Program (201902020018), and Fundamental Research Funds for Central Universities.Notes and references
- M. T. Zhu, G. J. Nie, H. Meng, T. Xia, A. Nel and Y. L. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- T. J. Merkel, S. W. Jones, K. P. Herlihy, F. R. Kersey, A. R. Shields, M. Napier, J. C. Luft, H. Wu, W. C. Zamboni and A. Z. Wang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 586–591 CrossRef CAS PubMed.
- J. Cui, Y. Ju, K. Liang, H. Ejima, S. Lörcher, K. T. Gause, J. J. Richardson and F. Caruso, Soft Matter, 2014, 10, 2656–2663 RSC.
- S. Ohta, D. Glancy and W. C. W. Chan, Science, 2016, 351, 841–845 CrossRef CAS PubMed.
- L. J. Eggermont, L. E. Paulis, J. Tel and C. G. Figdor, Trends Biotechnol., 2014, 32, 456–465 CrossRef CAS PubMed.
- A. K. Kosmides, K. Necochea, J. W. Hickey and J. P. Schneck, Nano Lett., 2018, 18, 1916–1924 CrossRef CAS PubMed.
- J. W. Hickey, F. P. Vicente, G. P. Howard, H.-Q. Mao and J. P. Schneck, Nano Lett., 2017, 17, 7045–7054 CrossRef CAS PubMed.
- C. S. Chiang, Y. J. Lin, R. Lee, Y. H. Lai, H. W. Cheng, C. H. Hsieh, W. C. Shyu and S. Y. Chen, Nat. Nanotechnol., 2018, 13, 746–753 CrossRef CAS PubMed.
- H. Yuan, W. Jiang, C. A. von Roemeling, Y. Qie, X. Liu, Y. Chen, Y. Wang, R. E. Wharen, K. Yun and G. Bu, Nat. Nanotechnol., 2017, 12, 763–769 CrossRef CAS PubMed.
- M. Xuan, J. Shao, C. Gao, W. Wang, L. Dai and Q. He, Angew. Chem., Int. Ed., 2018, 130, 12643–12647 CrossRef.
- S. Balasubramanian, D. Kagan, C. M. Jack Hu, S. Campuzano, M. J. Lobo-Castañon, N. Lim, D. Y. Kang, M. Zimmerman, L. Zhang and J. Wang, Angew. Chem., Int. Ed., 2011, 50, 4161–4164 CrossRef CAS PubMed.
- K. Lee, Y. Yi and Y. Yu, Angew. Chem., Int. Ed., 2016, 55, 7384–7387 CrossRef CAS PubMed.
- Y. Yi, L. Sanchez, Y. Gao and Y. Yu, Analyst, 2016, 141, 3526–3539 RSC.
- T. Yang, L. Wei, L. Jing, J. Liang, X. Zhang, M. Tang, M. J. Monteiro, Y. Chen, Y. Wang and S. Gu, Angew. Chem., Int. Ed., 2017, 56, 8459–8463 CrossRef CAS PubMed.
- R. Kadam, M. Zilli, M. Maas and K. Rezwan, Part. Part. Syst. Charact., 2018, 35, 1700332 CrossRef.
- G. Agrawal and R. Agrawal, ACS Appl. Nano Mater., 2019, 2, 1738–1757 CrossRef CAS.
- M. Zhang, L. Zhang, Y. Chen, L. Li, Z. Su and C. Wang, Chem. Sci., 2017, 8, 8067–8077 RSC.
- Y. Zhang, K. Huang, J. Lin and P. Huang, Biomater. Sci., 2019, 7, 1262–1275 RSC.
- A. K. Salem, P. C. Searson and K. W. Leong, Nat. Mater., 2003, 2, 668–671 CrossRef CAS PubMed.
- Y. J. Son, H. Kim, K. W. Leong and H. S. Yoo, ACS Nano, 2013, 7, 9771–9779 CrossRef CAS PubMed.
- Y. Gao and Y. Yu, J. Am. Chem. Soc., 2013, 135, 19091–19094 CrossRef CAS PubMed.
- K. Lee and Y. Yu, J. Mater. Chem. B, 2017, 5, 4410–4415 RSC.
- W. He, N. Kapate, C. W. Shields IV and S. Mitragotri, Adv. Drug Delivery Rev., 2019 DOI:10.1016/j.addr.2019.12.001.
- W. Nie, G. Wu, J. Zhang, L. L. Huang, J. Ding, A. Jiang, Y. Zhang, Y. Liu, J. Li, K. Pu and H. Xie, Angew. Chem., Int. Ed., 2020, 59, 2018–2022 CrossRef CAS PubMed.
- J. Chen, M. Liu, C. Chen, H. Gong and C. Gao, ACS Appl. Mater. Interfaces, 2011, 3, 3215–3223 CrossRef CAS PubMed.
- S. Jiang, M. J. Schultz, Q. Chen, J. S. Moore and S. Granick, Langmuir, 2008, 24, 10073–10077 CrossRef CAS PubMed.
- J. Y. Yhee, S. J. Lee, S. Lee, S. Song, H. S. Min, S.-W. Kang, S. Son, S. Y. Jeong, I. C. Kwon and S. H. Kim, Bioconjugate Chem., 2013, 24, 1850–1860 CrossRef CAS PubMed.
- C. H. J. Choi, C. A. Alabi, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1235–1240 CrossRef CAS PubMed.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145 CrossRef CAS PubMed.
- E. J. Lee, G. H. Nam, N. K. Lee, M. Kih, E. Koh, Y. K. Kim, Y. Hong, S. Kim, S. Y. Park and C. Jeong, Adv. Mater., 2018, 30, 1705581 CrossRef PubMed.
- Q. Chen, C. Wang, X. Zhang, G. Chen, Q. Hu, H. Li, J. Wang, D. Wen, Y. Zhang and Y. Lu, Nat. Nanotechnol., 2019, 14, 89–97 CrossRef CAS PubMed.
- F. Zhou, B. Feng, H. Yu, D. Wang, T. Wang, Y. Ma, S. Wang and Y. Li, Adv. Mater., 2019, 31, 1805888 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01146k |
| This journal is © The Royal Society of Chemistry 2020 |