
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegio- and diastereoselective reactions of chiral secondary alkylcopper reagents with propargylic phosphates: preparation of chiral allenes†
Juri
Skotnitzki
,
Alexander
Kremsmair
,
Daniel
Keefer‡
 ,
Franziska
Schüppel
,
Brieuc
Le Cacher de Bonneville
,
Regina
de Vivie-Riedle
and
Paul
Knochel
*
,
Franziska
Schüppel
,
Brieuc
Le Cacher de Bonneville
,
Regina
de Vivie-Riedle
and
Paul
Knochel
*
Department of Chemistry, Ludwig-Maximilians-Universitaet, Butenandtstrasse 5-13, 81377 München, Germany. E-mail: paul.knochel@cup.uni-muenchen.de
First published on 14th May 2020
Abstract
The diastereoselective SN2′-substitution of secondary alkylcopper reagents with propargylic phosphates enables the preparation of stereodefined alkylallenes. By using enantiomerically enriched alkylcopper reagents and enantioenriched propargylic phosphates as electrophiles anti-SN2′-substitutions were performend leading to α-chiral allenes in good yields with excellent regioselectivity and retention of configuration. DFT-calculations were performed to rationalize the structure of these alkylcopper reagents in various solvents, emphasizing their configurational stability in THF.
Introduction
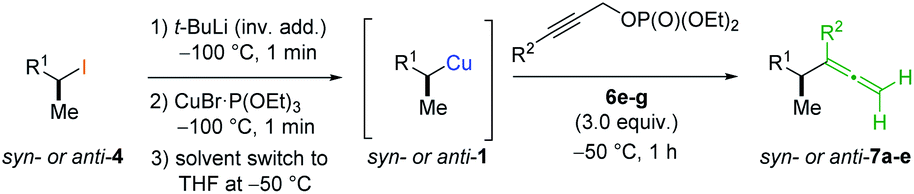
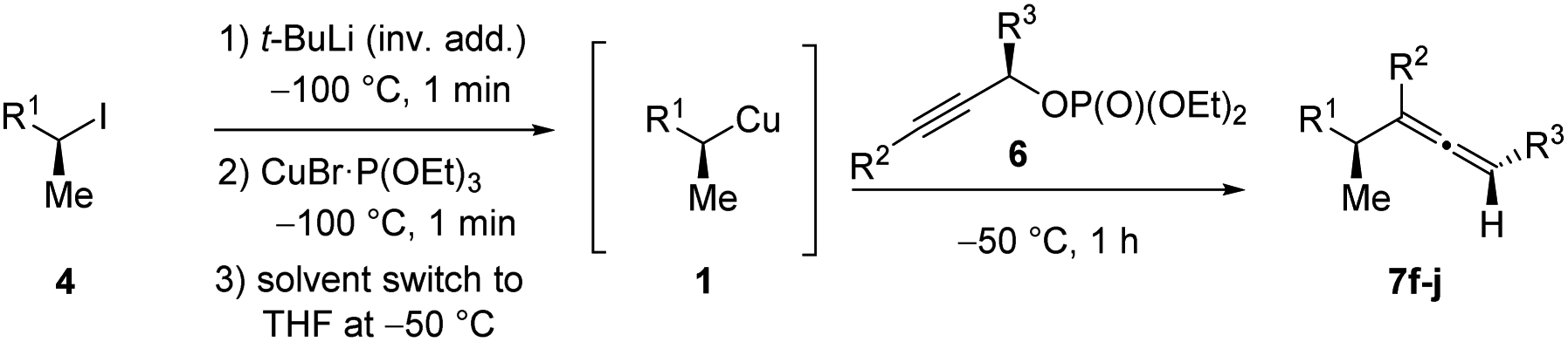
Allenes are common intermediates in organic synthesis and found in natural products.1 They are typically prepared by the substitution reaction of propargylic electrophiles with nucleophiles, such as organocopper reagents.2 Thereby, these propargylic reagents bear a good leaving group, such as acetates, ethers, epoxides, phosphates or halides.2–4 Axially chiral allenes are generally prepared from enantioenriched propargylic substrates3 or by the use of chiral ligands.4 The chirality transfer from the chiral propargylic substrate to the allene depends on the nature of the electrophile and nucleophile as well as on the solvent and temperature.1e However, the enantioselective preparation of axially chiral allenes bearing a stereocenter in α-position (“α-chiral allenes”) is rather difficult and only a few examples have been reported.5 Thereby, the stereochemistry of the α-position results from an asymmetric synthesis using chiral ligands.Recently, we reported a zinc-mediated anti-SN2′-substitution reaction of alkylcopper reagents of type 1 with allylic substrates (2) leading to chiral alkenes of type 3 with excellent regioselectivity and high retention of configuration (see Scheme 1(b and c)).6,7 These organocopper reagents were prepared from the corresponding alkyl iodide 4via I/Li-exchange reaction leading to alkyllithium reagent 5. Subsequent transmetalation with CuBr·P(OEt)3 afforded alkylcopper reagent 1.8 The regio-selectivity (SN2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SN2 ratio) of the substitution reactions highly depended on the choice of allylic electrophile 2 and the used organometallic species. The reaction of alkylcopper reagents 1 with allylic bromides 2a exclusively led to the SN2-product 3a (γ:α < 1:99; see Scheme 1(a)). The addition of zinc chloride and the use of chiral allylic phosphates 2b as electrophiles exclusively led to the SN2′-products 3b (γ:α > 99:1; (b)).6 Furthermore, we reported anti-SN2′-substitutions of secondary alkylcopper-zinc reagents with allylic epoxides 2c leading to chiral allylic alcohols of type 3c (γ:α > 95:5; (c)).7 This method was used in the total synthesis of the natural product (3S,6R,7S)-zingiberenol.7
:SN2 ratio) of the substitution reactions highly depended on the choice of allylic electrophile 2 and the used organometallic species. The reaction of alkylcopper reagents 1 with allylic bromides 2a exclusively led to the SN2-product 3a (γ:α < 1:99; see Scheme 1(a)). The addition of zinc chloride and the use of chiral allylic phosphates 2b as electrophiles exclusively led to the SN2′-products 3b (γ:α > 99:1; (b)).6 Furthermore, we reported anti-SN2′-substitutions of secondary alkylcopper-zinc reagents with allylic epoxides 2c leading to chiral allylic alcohols of type 3c (γ:α > 95:5; (c)).7 This method was used in the total synthesis of the natural product (3S,6R,7S)-zingiberenol.7
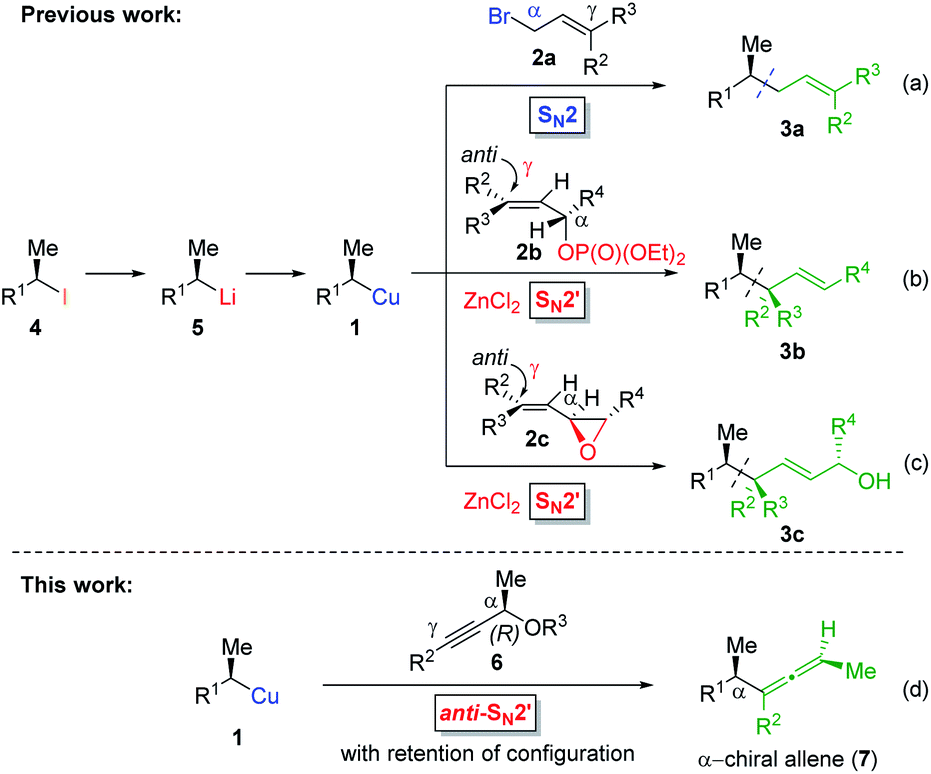 | ||
| Scheme 1 Stereoretentive preparation of chiral secondary alkylcopper reagents 1: (a–c): subsequent SN2- and zinc-mediated anti-SN2′-substitution reactions with allylic substrates. (d): Anti-SN2′-substitution with chiral propargylic phosphates leading to axially chiral allenes. | ||
Herein, we wish to report the anti-SN2′-substitution of secondary alkylcopper reagents 1 with chiral propargylic phosphates 6 leading to α-chiral allenes of type 7 with retention of the configuration (see Scheme 1(d)). Remarkably, this overall anti-SN2′-substitution reaction proceeded directly with the alkylcopper reagent 1 with transfer of chirality from the propargylic substrate 6 to the allene 7.
Results and discussion
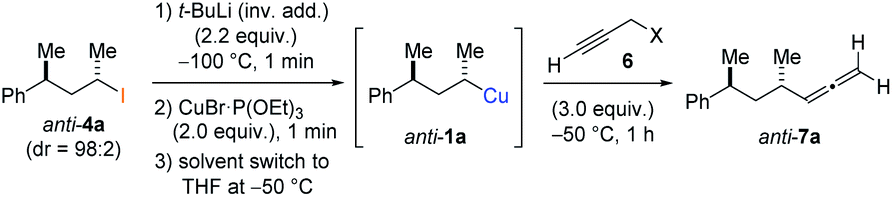
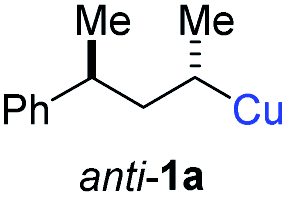
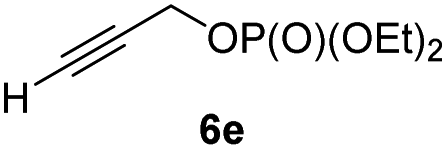
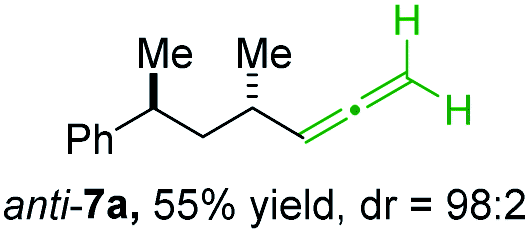
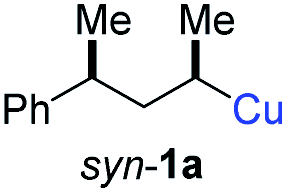
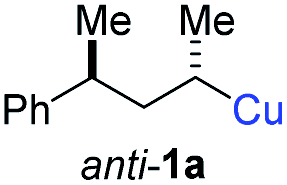
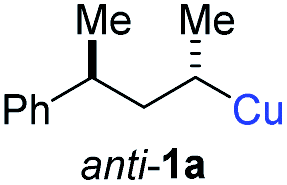
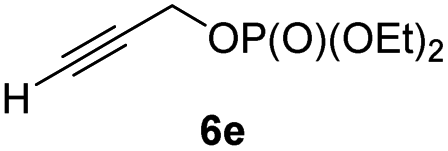
In preliminary experiments, we examined the leaving group of the propargylic electrophile for achieving the desired SN2′-reaction. Thus, we prepared the secondary alkyllithium reagent anti-5avia I/Li-exchange of the corresponding alkyl iodide anti-4a at −100 °C in pentane/diethyl ether-mixture (3:2) using t-BuLi (2.2 equiv.) followed by subsequent treatment with CuBr·P(OEt)3 (2.0 equiv.) leading to alkylcopper reagent anti-1a (see Table 1). This alkylcopper reagent was configurationally stable in THF up to −50 °C and thus, we performed a solvent switch at this temperature.6 Subsequent addition of the propargylic bromide9a (6a, 3.0 equiv.) furnished only traces of the desired allene anti-7a (see Table 1; entry 1) after stirring for 1 h at −50 °C. The use of propargylic acetate (6b)9b showed a similar result (entry 2). Switching to pentafluorobenzoate (6c)9c or diphenylphosphate (6d)9d as leaving groups afforded anti-7a in good yields, but with moderate stereoretention (48–50% yield, dr up to 93:7; entries 3 and 4). However, using the propargylic diethyl phosphate 6e9e as electrophile significantly increased the stereoretention of the secondary alkylcopper center (anti-7a, 59% yield, dr = 98:2). The same reaction afforded anti-7a in only 40% yield and dr = 92:8 when no solvent switch was performed, demonstrating the necessity of THF as solvent.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Br
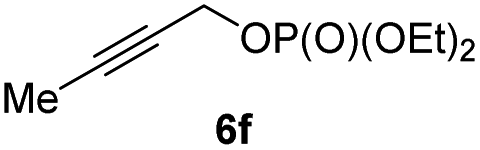
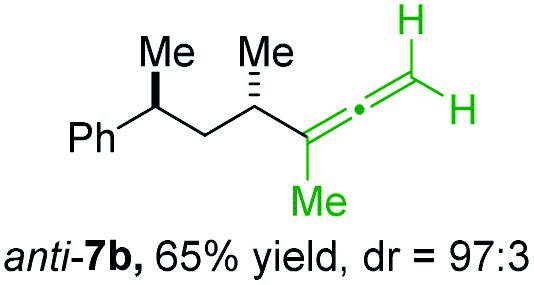
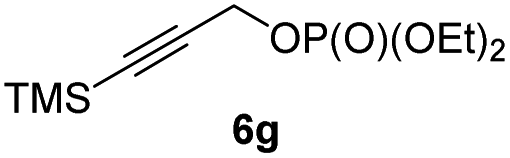
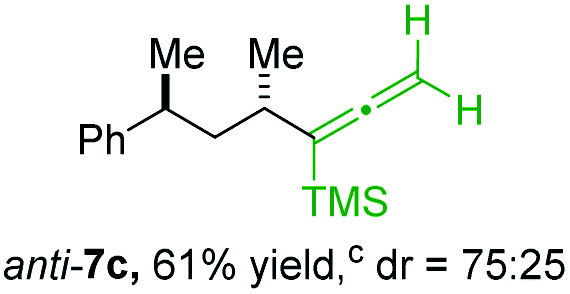
BrWith these results in hand, we performed stereoselective reactions with various diastereomerically pure alkyl iodides syn- or anti-4a–d and propargylic phosphates 6e–g leading to allenes 7a–e in 42–65% yield and with dr higher than 95:5 (see Table 2).10,11 In most cases, a high retention of configuration was observed. However, using the TMS-substituted propargylic phosphate 6g as electrophile led to allene anti-7c in 61% yield with moderate diastereoselectivity (dr = 75:25; entry 4). The reaction of anti-1a with the propargylic phosphate bearing a terminal methyl-group 6f led to the methyl-substituted allene anti-7b in 65% yield and dr = 97:3 (see Table 2; entry 3). Furthermore, the 1,2-substituted secondary alkylcopper reagents anti- and syn-1b reacted with 6e to the corresponding allenes anti-7d (58% yield, dr = 98:2; entry 5) and syn-7d (42% yield, dr = 6:94; entry 6). The OTBS-substituted allenes anti-7e (50% yield, dr = 95:5; entry 7) and syn-7e (44% yield, dr = 4:96; entry 8) were prepared with high retention of configuration as well.
|
|
|||
|---|---|---|---|
| Entry | Alkylcopper | Electrophile 6 | Product of type 7a,b |
|
a The diastereoselectivity (dr; anti:syn ratio) was determined by 1H- or 13C-NMR analysis.
b The SN2′ to SN2 ratio was higher than 99:1.
c The yield was determined by GC-analysis using dodecane as internal standard.
|
|||
| 1 |
|
|
|
| 2 |
|
6e |

|
| 3 |
|
|
|
| 4 |
|
|
|
| 5 |
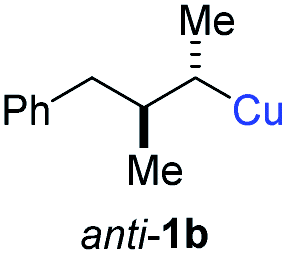
|
6e |
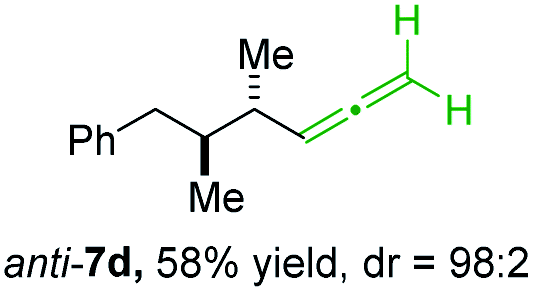
|
| 6 |
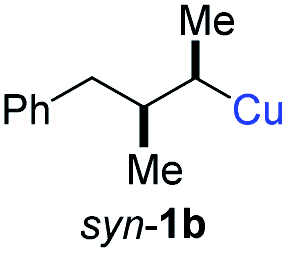
|
6e |
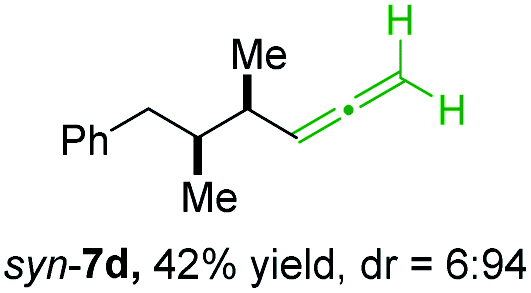
|
| 7 |
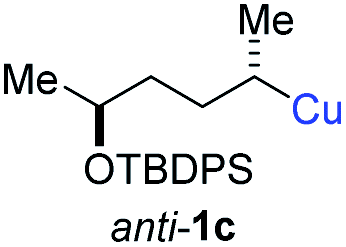
|
6e |
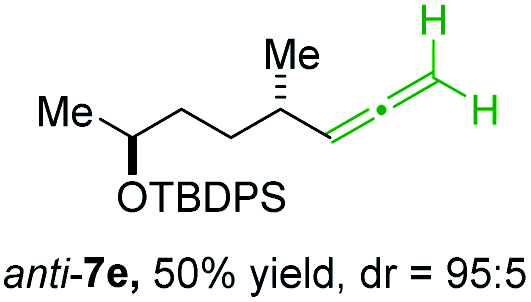
|
| 8 |
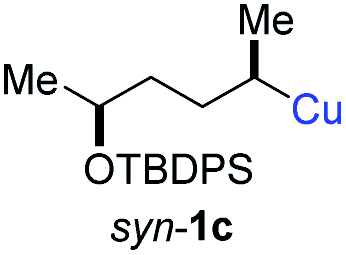
|
6e |

|
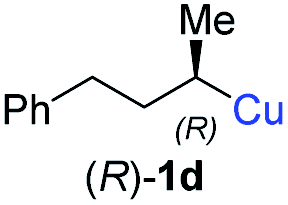
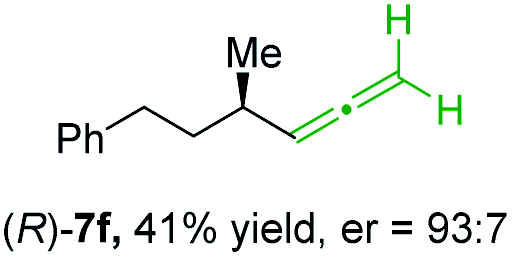
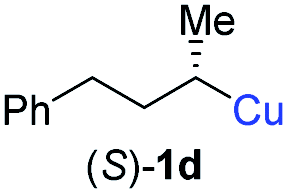
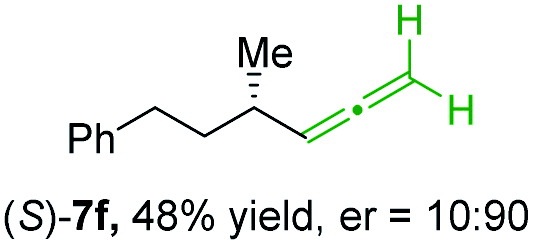
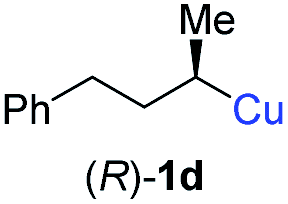
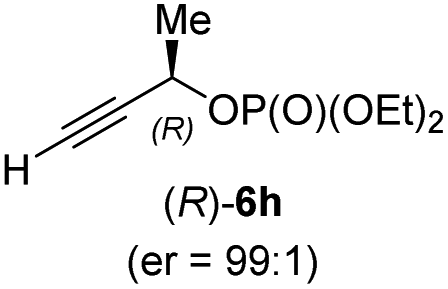
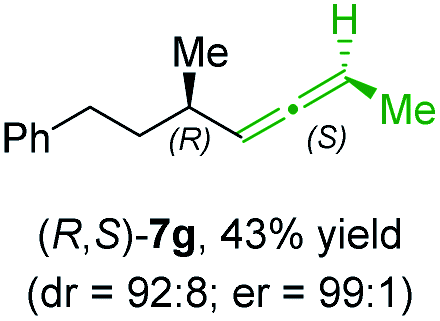
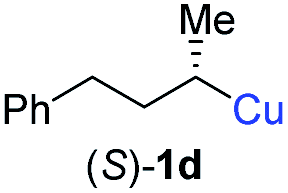
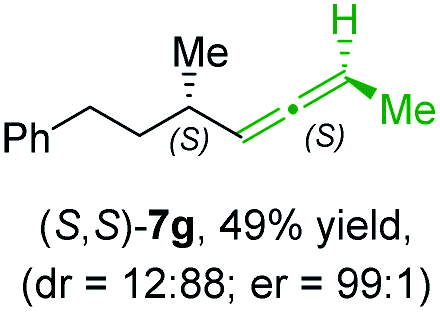
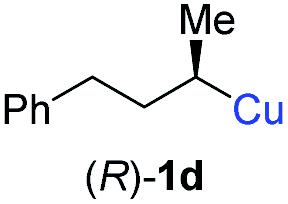
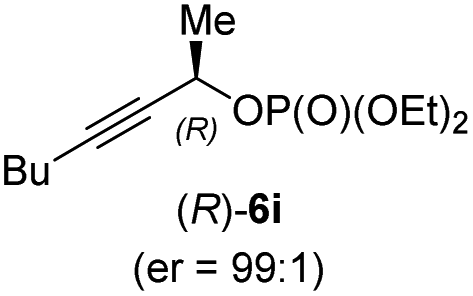
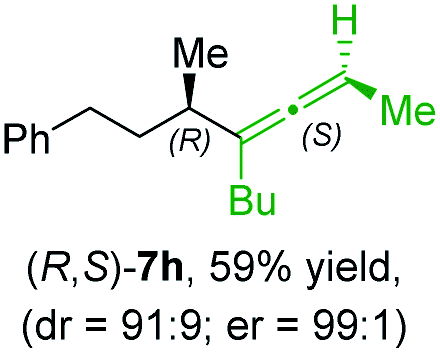
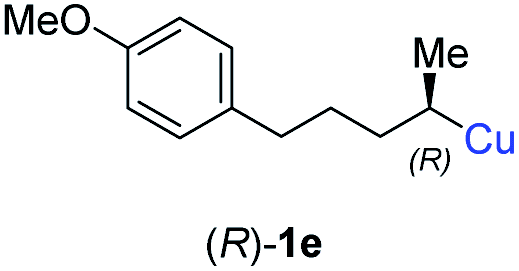
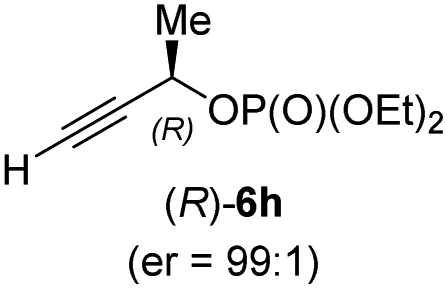
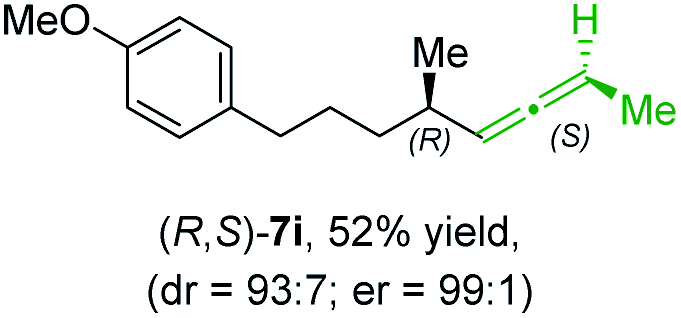
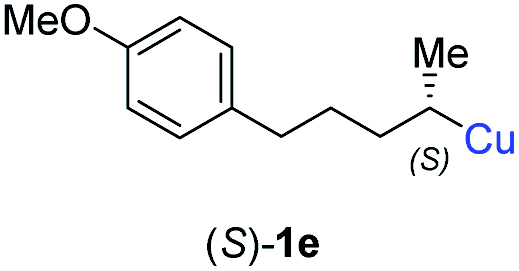
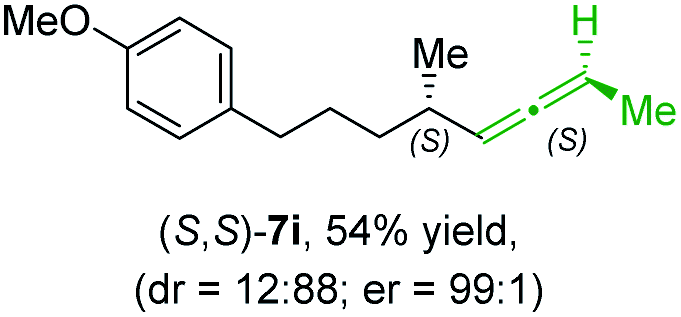
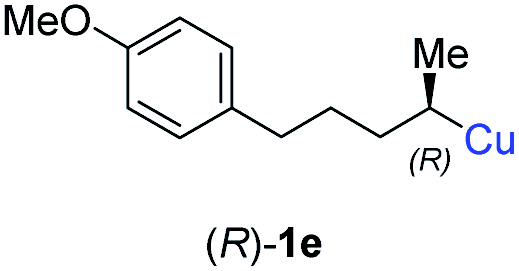
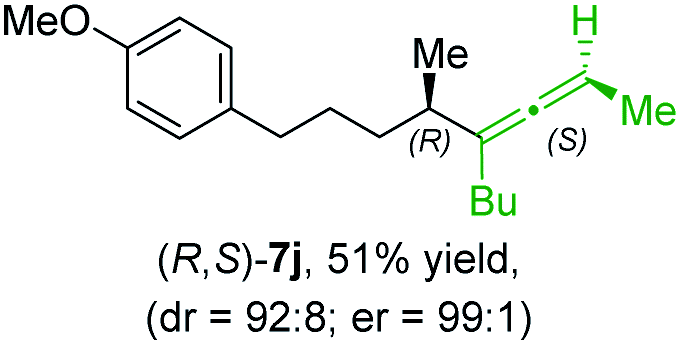
In addition, this anti-selective substitution was extended to optically enriched alkylcopper reagents 1d–e (see Table 3). Thus, the reaction of the secondary alkylcopper reagent (R)-1d with propargylic phosphate 6e furnished (R)-7f in 41% yield and er = 93:7 (see Table 3; entry 1). Analogously, the corresponding (S)-enantiomer (S)-7f was prepared in 48% yield and er = 10:90 (entry 2). To our delight, chiral alkylcopper reagents reacted also with higher substituted chiral propargylic phosphates 6h–i leading to axially chiral allenes bearing a stereocenter in the α-position (see Table 3; entries 3–8). Thus, the reaction of the alkylcopper (R)-1d with enantioenriched propargylic phosphate (R)-6h, prepared from the corresponding 3-butyn-2-ol,12 led to the α-chiral disubstituted allene (R,S)-7g13 in 43% yield with high anti-SN2′-substitution ratio (dr = 92:8; er = 99:1, entry 3). Similarly, the allene (S,S)-7g was prepared from organocopper (S)-1d and the chiral phosphate (R)-6h in 49% yield (dr = 12:88; er = 99:1;14 entry 4). Moreover, (R)-oct-3-yn-2-yl diethyl-phosphate (R)-6i was prepared according to literature from the corresponding optically enriched propargylic alcohol.3e,6,14 Subsequent reaction of alkylcopper (R)-1d with phosphate (R)-6i furnished the α-chiral trisubstituted allene (R,S)-7h in 59% yield (dr = 91:9, er = 99:1; entry 5). It was also possible to convert the methoxy-substituted secondary alkyl iodide (R)- and (S)-4e to the corresponding alkylcopper reagents (R)- and (S)-1e and after reaction with (R)-6h the α-chiral disubstituted allenes (R,S)-7i (52% yield, dr = 93:7, er = 99:1; entry 6) and (S,S)-7i (54% yield, dr = 12:88, er = 99:1; entry 7) were obtained. Furthermore, the reaction of (R)-1e with (R)-6i led to the trisubstituted allene (R,S)-7j in 51% yield and good diastereoselectivity (dr = 92:8, er = 99:1; entry 8). Unfortunately, the preparation of tertiary propargylic phosphates was unsuccessful although the subsequent preparation of axially chiral tetrasubstituted allenes would be of high interest for organic synthesis.
|
|
|||
|---|---|---|---|
| Entry | Alkylcopper of type 1 | Propargylic phosphate 6 | Product of type 7a,b,c |
|
a The diastereoselectivity (dr; anti:syn ratio) was determined by 1H- or 13C-NMR analysis.
b The SN2′ to SN2 ratio was higher than 99:1.
c The enantiomeric ratio (er) was determined by chiral GC-analysis.
|
|||
| 1 |
|
|
|
| 2 |
|
6e |
|
| 3 |
|
|
|
| 4 |
|
(R)-6h (er = 99:1) |
|
| 5 |
|
|
|
| 6 |
|
|
|
| 7 |
|
(R)-6h (er = 99:1) |
|
| 8 |
|
(R)-6i (er = 99:1) |
|
To get a better understanding of the regioselectivity, we have prepared the racemic phosphate 6j, which contains a propargylic moiety (see Scheme 2).15 The nucleophilic organocopper reagent rac-1d can undergo a substitution either in the α-position (SN2-substitution of the phosphate), the γ-position (SN2′-attack on the propargylic site) or γ′-position (SN2′-attack on the allylic site). Interestingly, the reaction of 1d with 6j afforded the allene 7k, the SN2-product 7l and the alkene 7m in 58% yield16 with a ratio of 2.6:1.0:6.4 = γ:α:γ′. This selectivity could be explained by steric hindrance of the α-position and favoured direct SN2′-substitution of the allylic phosphate (γ′-position) compared to the propargylic moiety (γ-position).
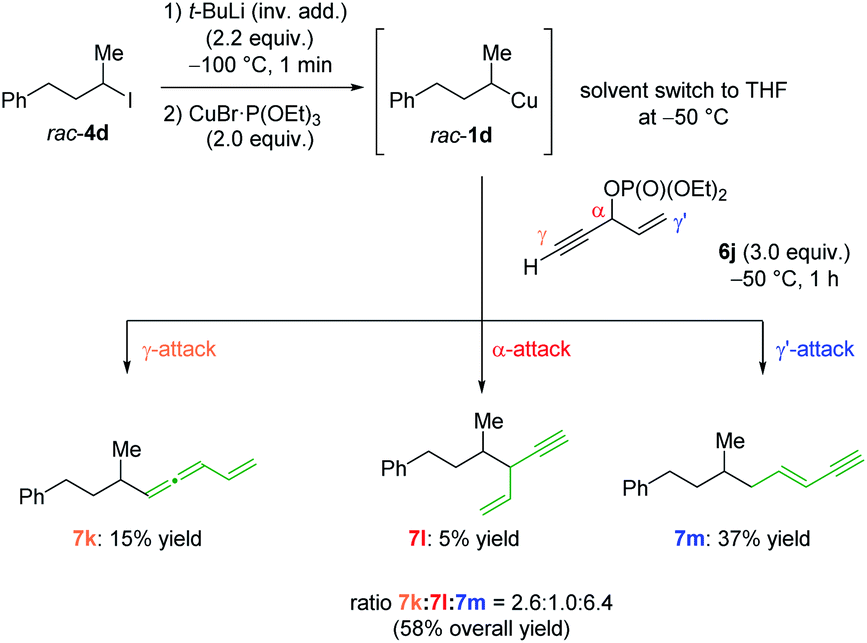 | ||
| Scheme 2 Regioselective addition of secondary alkylcopper reagent 1d to allylic and propargylic moiety containing phosphate 6f. | ||
Computational calculations
Furthermore, DFT-calculations17 were performed to rationalize the high configurational stability of these chiral secondary alkylcopper reagents. Solvation effects were accounted for by the Polarizable Continuum Model (PCM).18 First, we determined the structure of secondary alkylcopper reagent anti-1a in solution. Thus, we calculated the free energies of anti-1a with coordination to all possible ligands, namely triethyl phosphite (P(OEt)3; anti-8), tetrahydrofuran (THF; anti-9) and diethyl ether (Et2O; anti-10; see Scheme 3, (1–2)).19 Comparison of the free energies of anti-8 with the free energies of anti-9 showed that the coordination to P(OEt)3 is thermodynamically more stable (ΔG = +4.6 kcal mol−1; see Scheme 3, (1)). Similar results were obtained for the substitution of P(OEt)3 with Et2O (ΔG = +6.8 kcal mol−1, (2)) showing again the high affinity of phosphor to copper. These calculations emphasized that anti-8 is the thermodynamically most stable structure. The direct comparison of anti-9 and anti-10 shows that the THF coordinated structure 9 is 3.9 kcal mol−1 more stable compared to the Et2O coordinated structure 10. In addition, the bond energies and bond lengths of the carbon–copper bond for anti-8 (53.9 kcal mol−1, 198.5 pm), anti-9 (51.3 kcal mol−1, 195.9 pm) and anti-10 (50.6 kcal mol−1, 195.8 pm) were determined showing that the carbon–copper bond is most stable when the copper is coordinated to P(OEt)3. Comparison of the free energies of anti-8 and syn-8 showed that the anti-isomer is thermodynamically more stable (ΔG = +2.9 kcal mol−1; see Scheme 3). This result is in agreement with previous reported findings.20 | ||
| Scheme 3 Theoretical calculations for the structure determination of anti-1a and the epimerization of secondary alkylcopper reagent anti-8 to syn-8. | ||
Next, we investigated the epimerization of anti-8 to the corresponding syn-isomer syn-8via cleavage of the carbon–copper bond or a planar transition state ts-8 (see Scheme 3). The high carbon–copper bond energy of 54.0 kcal mol−1 as well as the transition state energy of 51.9 kcal mol−1 corroborate the high stability of anti-8 towards epimerization at −50 °C.21 However, the slight epimerization of the secondary alkylcopper reagents (1) may be due to polymolecular exchange reactions between these copper reagents.22
Conclusions
In conclusion, we have reported the enantioselective preparation of axially chiral allenes bearing a stereocontrolled α-chiral center via anti-SN2′-substitution reaction of chiral secondary alkylcopper reagents with enantioenriched propargylic phosphates with retention of configuration. DFT-calculations were performed to determine the structure of these alkylcopper reagents and rationalize the high configurational stability in THF. Further extensions are currently under investigation in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Excellence Cluster “e-conversion” and the Munich-Centre for Advanced Photonics (MAP) for financial support. We also thank Albemarle for the generous gift of chemicals. J. S. thanks the FCI Foundation for a fellowship. D. K. and J. S. acknowledge financial support by the Dr Klaus Roemer Foundation through their PhD thesis award.Notes and references
- (a) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, vol. 1 and 2 Search PubMed. For reviews see: (b) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed; (c) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed; (d) R. K. Neff and D. E. Frantz, ACS Catal., 2014, 4, 519–528 CrossRef CAS; (e) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210–1224 RSC.
- (a) P. Rona and P. Crabbe, J. Am. Chem. Soc., 1968, 90, 4733–4734 CrossRef CAS; (b) R. S. Brinkmeyer and T. L. Macdonald, J. Chem. Soc., Chem. Commun., 1978, 876–877 RSC; (c) A. C. Oehlschlager and E. Czyzewska, Tetrahedron Lett., 1983, 24, 5587–5590 CrossRef CAS; (d) A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, Tetrahedron Lett., 1989, 30, 2387–2390 CrossRef CAS; (e) A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, Tetrahedron, 1991, 47, 1677–1696 CrossRef CAS; (f) J. A. Marshall and K. G. Pinney, J. Org. Chem., 1993, 58, 7180–7184 CrossRef CAS; (g) J. P. Varghese, P. Knochel and I. Marek, Org. Lett., 2000, 2, 2849–2852 CrossRef CAS PubMed.
- (a) I. Marek, P. Mangeney, A. Alexakis and J. F. Normant, Tetrahedron Lett., 1986, 27, 5499–5502 CrossRef CAS; (b) A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, J. Am. Chem. Soc., 1990, 112, 8042–8047 CrossRef CAS; (c) M. T. Crimmins and K. A. Emmitte, J. Am. Chem. Soc., 2001, 123, 1533–1534 CrossRef CAS PubMed; (d) M. Leclère and A. G. Fallis, Angew. Chem., Int. Ed., 2008, 47, 568–572 CrossRef PubMed; (e) H. Ohmiya, U. Yokobori, Y. Makida and M. Sawamura, Org. Lett., 2011, 13, 6312–6315 CrossRef CAS PubMed.
- (a) R. K. Dieter, N. Chen and V. K. Gore, J. Org. Chem., 2006, 71, 8755–8760 CrossRef CAS PubMed; (b) H. Li, D. Müller, L. Guénée and A. Alexakis, Org. Lett., 2012, 14, 5880–5883 CrossRef CAS PubMed; (c) D. Qian, L. Wu, Z. Lin and J. Sun, Nat. Commun., 2017, 8, 567 CrossRef PubMed.
- Extensive studies were done by S. Ma and others: (a) M. O. Frederick, R. P. Hsung, R. H. Lambeth, J. A. Mulder and M. R. Tracey, Org. Lett., 2003, 5, 2663–2666 CrossRef CAS PubMed; (b) X. Jiang, C. Fu and S. Ma, Chem.–Eur. J., 2008, 14, 9656–9664 CrossRef CAS PubMed; (c) Q. Li, C. Fu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 11783–11786 CrossRef CAS PubMed; (d) Q. Li, C. Fu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 6511–6514 CrossRef CAS PubMed; (e) J. Dai, X. Duan, J. Zhou, C. Fu and S. Ma, Chin. J. Chem., 2018, 36, 387–391 CrossRef CAS; (f) B. Wang, X. Wang, X. Yin, W. Yu, Y. Liao, J. Ye, M. Wang and J. Liao, Org. Lett., 2019, 21, 3913–3917 CrossRef CAS PubMed.
- (a) The reactivity and configurational stability is considerably higher in THF. For details, see: J. Skotnitzki, L. Spessert and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 1509–1514 CrossRef CAS PubMed; (b) For a recent review, see: J. Skotnitzki, A. Kremsmair and P. Knochel, Synthesis, 2020, 52, 189–196 CrossRef CAS.
- J. Skotnitzki, A. Kremsmair, B. Kicin, R. Saeb, V. Ruf and P. Knochel, Synthesis, 2020, 52, 873–881 CrossRef CAS.
- (a) V. Morozova, J. Skotnitzki, K. Moriya, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 5516–5519 CrossRef CAS PubMed; (b) J. Skotnitzki, V. Morozova and P. Knochel, Org. Lett., 2018, 20, 2365–2368 CrossRef CAS PubMed.
- (a) Propargyl bromide is commercially available as a solution in toluene; (b) Propargyl acetate is commercially available (Sigma-Aldrich); (c) N. N. Solodukhin, N. E. Borisova, A. V. Churakov and K. V. Zaitsev, J. Fluorine Chem., 2016, 187, 15–23 CrossRef CAS; (d) J. Eisenblaetter, M. Bruns, U. Fehrenbacher, L. Barner and C. Barner-Kowollik, Polym. Chem., 2013, 4, 2406–2413 RSC; (e) M. Hojo, R. Sakuragi, S. Okabe and A. Hosomi, Chem. Commun., 2001, 357–358 RSC . For details, see ESI.†.
- The use of a phenyl group in α-position was unsuccessful due to dimerisation of the corresponding benzyl–alkylcopper reagent. Furthermore, we prepared racemic alkyl iodides bearing a n-butyl and cyclohexyl substituent in α-position, which could be used successfully for the preparation of allenes. However, the preparation of the corresponding chiral alkyl alcohols is more challenging and under investigation in our laboratories.
- The addition of ZnCl2 to the alkylcopper reagent syn-1a as in ref. 6 and 7 led to the corresponding alkylcopper-zinc reagent. After addition of propargylic substrate 6e comparable regioselectivity was achieved leading to syn-7a, however in lower diastereomeric ratio and yield (dr = 91:9 and 40% yield).
- (R)-(+)-3-Butyn-2-ol is commercially available (TCI; er >99:1).
- The enantiomeric ratio was determined by chiral GC analysis or chiral HPLC analysis. For details, see ESI.†.
- The enantiomeric ratio was determined by chiral GC analysis. For details, see ref. 6.
- A. Czepa and T. Hofmann, J. Agric. Food Chem., 2004, 52, 4508–4514 CrossRef CAS PubMed.
- The yield was determined by GC-analysis using dodecane as internal standard.
- A detailed description of the theoretical methodology, along with optimized structures and energies of all investigated compounds can be found in the ESI.†.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef; (b) Continuum Solvation Models in Chemical Physics, John Wiley & Sons, Ltd, 2007, DOI:10.1002/9780470515235; (c) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- Coordination of more than one solvent molecule decreased the free energy. For details, see ESI.†.
- J. Skotnitzki, A. Kremsmair, D. Keefer, Y. Gong, R. de Vivie-Riedle and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 320–324 CrossRef CAS PubMed.
- We also performed DFT-calculations for the transition state energy with THF (ts-9) and diethyl ether (ts-10) as ligands. The energies are slightly higher (55.7 kcal mol−1 and 57.4 kcal mol−1). For details, see ESI.†.
- All attempts to investigate the bimolecular epimerization pathway were unsuccessful due to inconclusive results from the DFT calculations.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05982b |
| ‡ Present address: Department of Chemistry, University of California Irvine, California 92697, United States. |
| This journal is © The Royal Society of Chemistry 2020 |