
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpeciation of Be2+ in acidic liquid ammonia and formation of tetra- and octanuclear beryllium amido clusters†
Matthias
Müller
a,
Antti J.
Karttunen
b and
Magnus R.
Buchner
 *a
*a
aAnorganische Chemie, Nachwuchsgruppe Berylliumchemie, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35032 Marburg, Germany. E-mail: magnus.buchner@chemie.uni-marburg.de; Fax: +49 6421 2825669; Tel: +49 6421 2825668
bDepartment of Chemistry and Materials Science, Aalto University, 00076 Aalto, Finland
First published on 5th May 2020
Abstract
The hexa-μ2-amido-tetraammine-tetraberyllium compounds [Be4(NH2)6(NH3)4]X2 (X = Cl, Br, I, CN, SCN, N3) have been prepared from beryllium metal and NH4X or [Be(NH3)4]X2 in liquid ammonia at ambient temperature. The obtained compounds feature an adamantyl shaped complex cation, which was examined via X-ray diffraction, IR, Raman and NMR spectroscopy. The speciation in solution was studied via15N labeling experiments supplemented with quantum chemistry. Hereby, the intermediates [Be2(NH2)(NH3)6]3+ and [Be3(NH2)3(NH3)6]3+ were identified. Reactivity studies provided bis(N-acetimidoylacetamidinato-N,N′)-beryllium(II), when [Be4(NH2)6(NH3)4]2+ was treated with acetonitrile. While the unprecedented octa-nuclear complex cation [Be8O(NH2)12(C5H5N)4]2+ was received from pyridine. This cluster proves that the [Be4O]6+ core can be stabilized without bidentate O-donor ligands.
1 Introduction
Over the last decades, beryllium chemistry has been considerably encumbered by concerns over its severe toxicity.1,2 This is due to the development of berylliosis, a severe lung disease, in some people who have inhaled minute amounts of beryllium containing dust.3 While the physiological processes liable for the development of beryllium related diseases are relatively well understood,4 this can not be said for the underlying molecular mechanisms.3 This is mainly caused by the lack of simple beryllium compounds with low-molecular, bio-relevant ligands, like amino acids.5 The obstacle for the synthesis of these model systems is mainly due to solubility problems of either the ligands or the beryllium precursor. In general, bio-molecules are well soluble in aqueous media at physiological pH values. However, in this pH range the dominating beryllium species is Be(OH)2, which is practically insoluble, under these conditions.5 If more acidic or basic solutions are used, aqueous beryllium chemistry is dominated by aqua and hydroxide complexes and the huge excess of water present, often prevents the coordination of the investigated ligands.5 Therefore, another, water related solvent system is needed, which exhibits hydrogen bonding as well as autoprotolysis, but coordinates significantly weaker to the Be2+ ion.Liquid ammonia is quite similar to water concerning its solvent properties6 and is able to dissolve organic molecules like amino acids as well as beryllium salts readily.7 Therefore, it is a promising solvent to simulate the aqueous conditions in vivo, without the solubility issues in H2O. Analogous to this, Be(NH2)2 represents a suitable model for Be(OH)2, which is the most likely beryllium species to be taken up by an organism.8 Additionally, Be(NH2)2 is a potential starting material to gain access to homoleptic beryllium compounds with bio-relevant ligands, because it should cleave off NH3 when treated with protic compounds. This would enable the study of the toxicity of beryllium from a coordination chemist's point of view, without the generation of non-volatile byproducts. This is of particular importance, since beryllium chemistry is commonly restricted to the use of the beryllium halides as starting materials. While they are readily prepared from the elements and relatively easy to handle,9 they come with several drawbacks. The presence of halide ions often leads to the formation of halide salt as byproducts, which can hardly be removed if the beryllium complex is insoluble in non-coordinating or aprotic solvents. Though, this is the common case if bio-relevant ligand systems are investigated.
Complex cations are postulated to be the species liable for the formation of beryllium associated diseases.10–12 Yet here another problem with the use of beryllium halides emerges, since the halide anions can hardly be cleaved completely.13,14 This obstacle is most prominent for BeF2. Here, the Be–F bond is so strong that there is no compound known, that could abstract the fluoride from the Be2+ cation. Even an excess of H2O or NH3 is just enough to recrystallize BeF2 or form simple solvent adducts.15,16 This is less pronounced in the other halides, but still present. There are plenty of adducts of BeCl2 and even an excess of ligand does not always lead to displacement of the halides.17,18 Thus, routes to related beryllium precursors with more weakly coordinating counter-ions are desirable.
In general the solution behaviour of (pseudo) main group metals in liquid ammonia is little investigated, with exception of the first row. This chemistry is dominated by mononuclear complex cations and anions.19–23 Recently, we reported on the chemistry of beryllium metal in either acidic or basic liquid ammonia. Thereby, we obtained the tetraammineberyllium salts [Be(NH3)4]X2 (X = Cl, Br, I, CN, SCN, N3), when the metal is exposed to NH4X.7 On the other hand, [Be(NH2)4]2−, [Be(NH2)3]− and Be(NH2)2 can be isolated when metallic beryllium is reacted with the alkali metals in ammonia.24 However, in order to utilize the ammonia solvent system as an adequate model for physiological, aqueous conditions, more insights into the pH-dependent speciation in liquid NH3 are mandatory. Accordingly, we explored the boundaries of the oxidation of beryllium metal in acidic ammonia solutions and the subsequent formation of beryllium amido compounds.
2 Results and discussion
When an excess of beryllium was reacted with ammonium salts in liquid ammonia at ambient temperature we observed a vivid evolution of dihydrogen, analogous to the synthesis of [Be(NH3)4]X2.7 Different to the [Be(NH3)4]2+ salts, where clear, colorless solutions were obtained,7 the reactions stop after several days with significant amounts of beryllium left in the reaction tubes. During the course of the reactions between beryllium and NH4X (X = Cl, Br, I, CN, SCN, N3), whereas NH4CN and NH4N3 were generated in situ through the use of TMS-CN and TMS-N3 respectively, cuboctahedral single crystals grew on top of the beryllium chips. The same observation was made, when BeCl2, BeBr2 or BeI2 were reacted with beryllium metal in liquid NH3. The single crystals were isolated at −78 °C and analyzed via X-ray diffraction.The hexa-μ2-amido-tetraammin-tetraberyllium(II) cation ([Be4(NH2)6(NH3)4]2+, 1) was obtained from all these reactions (Scheme 1). Within cation 1 four beryllium atoms are μ2-linked via six amido ligands to form an adamantyl-shaped complex cation in which the tetrahedral coordination around the beryllium cations is completed by four ammine ligands in total, as depicted in Fig. 1.
 | ||
| Scheme 1 Synthesis of 1a–f through oxidation of beryllium in the presence of [Be(NH3)4]X2 in liquid NH3 at ambient temperature. X = Cl (a), Br (b), I (c), CN (d), SCN (e), N3 (f). | ||
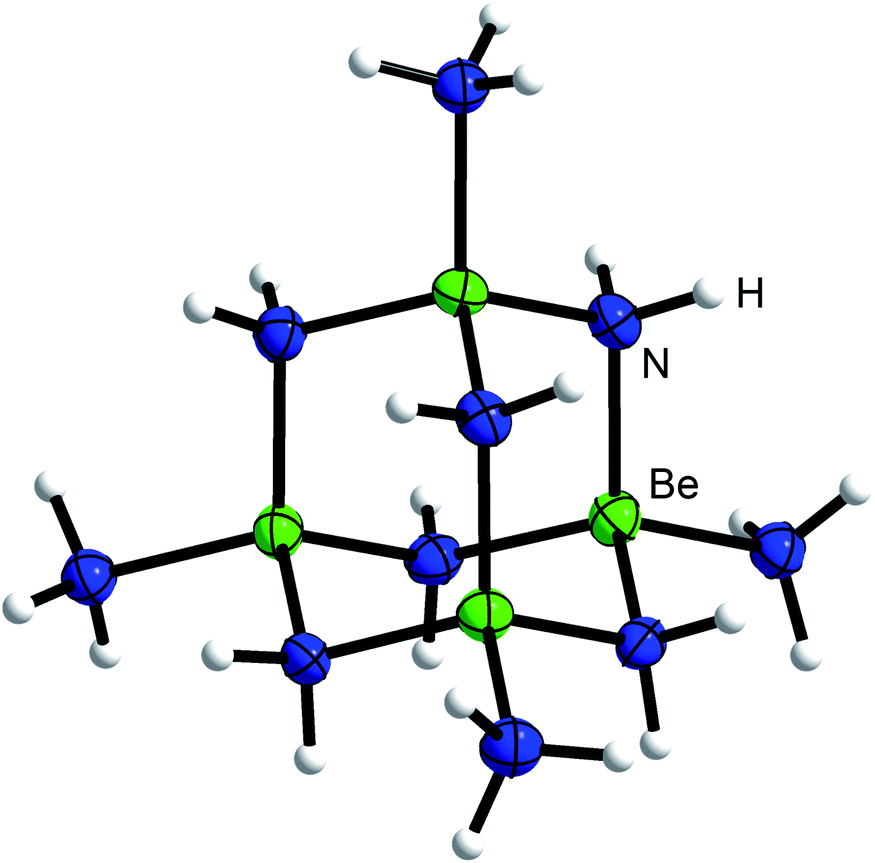 | ||
| Fig. 1 Molecular structure of the [Be4(NH2)6(NH3)4]2+ cation (1) of 1a. Ellipsoids are depicted at 70% probability at 100 K. Hydrogen atoms are depicted with arbitrary radii. | ||
The structural motif of cation 1 is known from the aqueous chemistry of beryllium in the form of the decahydroxido-tetraberyllium-dianion [Be4(OH)10]2− (ref. 25) as well as from the solid state structure of Be(NH2)2,26 in which adamantyl-shaped [Be4(NH2)6]2+ fragments are linked via μ2-[NH2]− anions to each other similar to β-BeCl2.27 Therefore, cation 1 is not an unusual coordination polyhedron for ionic beryllium compounds. The Be–NH2 atomic distances in the cations 1 range from 1.703(16) to 1.73(1) Å. This is in the range of the Be–NH3 atomic distances (1.707(3)–1.744(4) Å) reported for the [Be(NH3)4]2+ cation.7,28 Compared to those, the corresponding Be–NH3 atomic distances in 1 (1.78(2)–1.807(2) Å) are significantly longer. We assume that these distinct elongated atomic distances are due to a strong interaction between Be2+ and μ2-[NH2]−. Fig. 2 illustrates the Be–N atomic distances towards NH3 and μ2-[NH2]− in the cations 1 depending on the counter-ion. Here it becomes evident that despite numerous hydrogen bonds among the ions, no significant influence on the Be–N atomic distances is discernible.
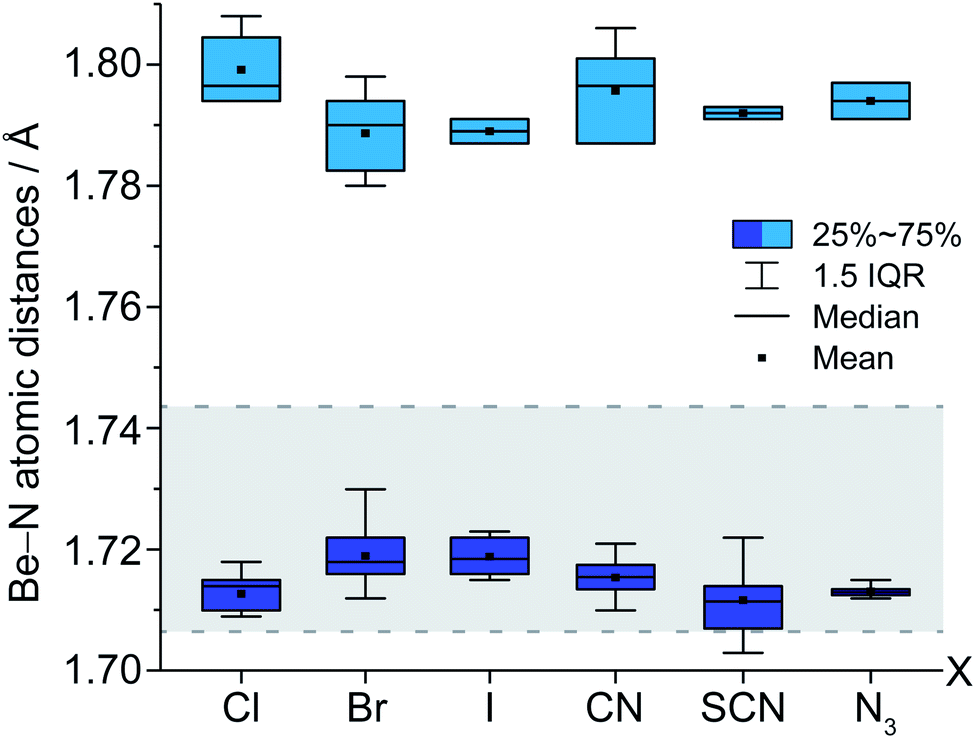 | ||
| Fig. 2 Box-plot of Be–NH3 atomic distances (light blue; 1.78(2)–1.807(2) Å) and Be–[NH2]− atomic distances (blue; 1.703(16)–1.73(1) Å) in compounds 1a–f. Be–NH3 atomic distances in [Be(NH3)4]2+ (gray area; 1.707(3)–1.744(4) Å).7,28 | ||
As similar as the cations are, they crystallize differently due to the variety of anions and crystal ammonia contents. Thus [Be4(NH2)6(NH3)4]Cl2·4NH3 (1a) and [Be4(NH2)6(NH3)4] Br2·4NH3 (1b) crystallize isostructurally in the trigonal space group P3 (143), while the iodide compounds were obtained as [Be4(NH2)6(NH3)4]I2·6NH3 (1c) in C2/c (15) or as [Be4(NH2)6(NH3)4]I2·9NH3 (1c′) in P3 (143), depending on the reaction temperature. The nona-ammoniate 1c′ was obtained when the reaction was performed at ambient temperature within days while the hexa-ammoniate 1c was formed at −40 °C in the course of several weeks. This proves that this reaction can also be carried out at lower temperatures. [Be4(NH2)6(NH3)4](CN)2·4NH3 (1d) crystallizes isopointal to the chloride and bromide compound, under the assumption that the center of gravity of the cyanide anions occupy the Wyckoff positions of the halide anions. [Be4(NH2)6(NH3)4](SCN)2·3NH3 (1e) crystallizes in the monoclinic space group P21/c (14) and [Be4(NH2)6(NH3)4](N3)2·6NH3 (1f) crystallizes in the monoclinic space group C2/c (15) quite similar to 1c. Projections of the unit cells of 1a–f are shown in Fig. S1–S7 of the ESI.†
After the extraction of several single crystals from the reaction tubes of 1a–f at −78 °C under argon atmosphere, the tubes were sealed again and thawed to ambient temperature. The remaining ammonia was released at ambient temperature, whereby the crystals turned opaque due to the loss of their crystal ammonia. The remains of the single crystals where used for IR and Raman spectroscopy as well as X-ray powder diffraction. It turned out that they were all amorphous. Their IR and Raman spectra are similar to each other and are depicted in ESI Fig. S14 and S17–S21.† They show ammine and amide bands as well as the characteristic bands of CN−, SCN− and N3−, respectively.
We did not observe further aggregation to larger clusters in the course of these reactions, even when a significantly larger excess of beryllium metal was used or the reaction times were prolonged. Therefore, cation 1 is the final product of the oxidation of beryllium in ammonia in presence of ammono acids like ammonium salts NH4X (X = Cl, Br, I, CN, SCN, N3) or beryllium halides. When [Be4(NH2)6(NH3)4]X2 (X = Cl, Br, I) and M(NH2) (M = Na, K) were reacted in liquid ammonia at ambient temperature, we were not able to isolate a distinct compound other than the starting materials. Thus, a substitution of the ammine through amide ligands to synthesize Be(NH2)2 or a [Be4(NH2)10]2− anion, analogous to [Be4(OH)10]2−, seems not feasible under the applied conditions.25
2.1 Speciation in solution
To gain more insight into the solution speciation and to find out if the [Be4(NH2)6(NH3)4]2+ cation is also stable in solution or if it is formed exclusively in solid state, the remains of the single crystals of 1a–f were flame sealed together with approx. 0.2 mL of liquid ammonia in thick-walled NMR tubes. Their NMR spectra were recorded at ambient temperature. Thereby, we only observed signals for the iodide compounds, as the others remained in solid state with no observable solubility in NH3. 1c and 1c′ exhibit a 9Be NMR signal at 4.5 ppm (ω1/2 = 4.4 Hz). In their 1H NMR spectrum a singlet at −0.49 ppm (ω1/2 = 10.9 Hz) corresponds to the amido ligands while no signal for the corresponding ammine ligands is observed. As evident from the single crystal data, the Be–NH3 bond is much weaker than the Be–[NH2]− bond. Therefore, we assume that there is a vivid exchange between ammine ligands and ammonia that is fast on the NMR timescale and the averaged signal is almost equal to that of ammonia due to the huge excess of free NH3.Based on the obtained spectra of 1c additional NMR experiments were performed to find out whether intermediates between known [Be(NH3)4]2+ and 1 exist. Accordingly 0.25–4 eq. of Be were reacted with 1 eq. of BeI2 in liquid ammonia (see also ESI Table S1†). For this, the flame sealed NMR tubes with the corresponding Be/BeI2 mixtures in liquid NH3 were stored for several days in a sealed container to ensure that the reactions were complete and the generated beryllium species were in equilibrium with each other. This also ensured that the NMR tubes could withstand the pressure of the formed dihydrogen, which could otherwise have led to the explosion of the NMR tubes inside the spectrometer. All recorded 1H and 9Be NMR spectra are shown in Fig. S10.† We observed a singlet at 4.3 ppm (ω1/2 = 60.3 Hz) in the 1H NMR spectrum and a singlet at 3.3 ppm (ω1/2 = 2.3 Hz) in the 9Be NMR spectrum when BeI2 was dissolved in liquid NH3, which is assigned to [Be(NH3)4]2+ in accordance to the literature.28 Exchange spectroscopy (EXSY) revealed interchange between coordinated ammine ligands and the solvent (ESI Fig. S12†). Since the Be–NH3 bond in 1 is significantly weaker than in [Be(NH3)4]2+, as evident from the Be–N distances in the solid state, ammine ligand dissociation in 1 should be easier. This explains why no signal corresponding to Be-bound ammine was observed in the 1H NMR spectrum of 1.
If beryllium metal was added step by step, three additional signals emerged in the 9Be NMR spectra at 3.7, 4.1 and 4.5 ppm, two of which (3.7 and 4.1 ppm) plus the signal of [Be(NH3)4]2+ disappeared when 3 eq. of Be were added. The only remaining signal corresponded to cation 1. Addition of more Be did not alter the spectra and unreacted metal remained in the NMR tubes. This is in accordance with our observations in the previous section. The two signals appearing in the interim at 3.7 (ω1/2 = 4.3 Hz) and 4.1 ppm (ω1/2 = 5.7 Hz), which are clearly noticeable in the 9Be NMR spectra of 0.25 eq. and 0.5 eq. Be with 1 eq. BeI2 in NH3 (Fig. 3) correlate with two intermediates on the way to cation 1. The corresponding 1H NMR spectra showed signals of one beryllium coordinated ammine at 3.69 ppm and one beryllium coordinated amide at 0.28 ppm in solution, which belong to the two intermediates respectively. However based on these spectra we could not identify the nature of these species.
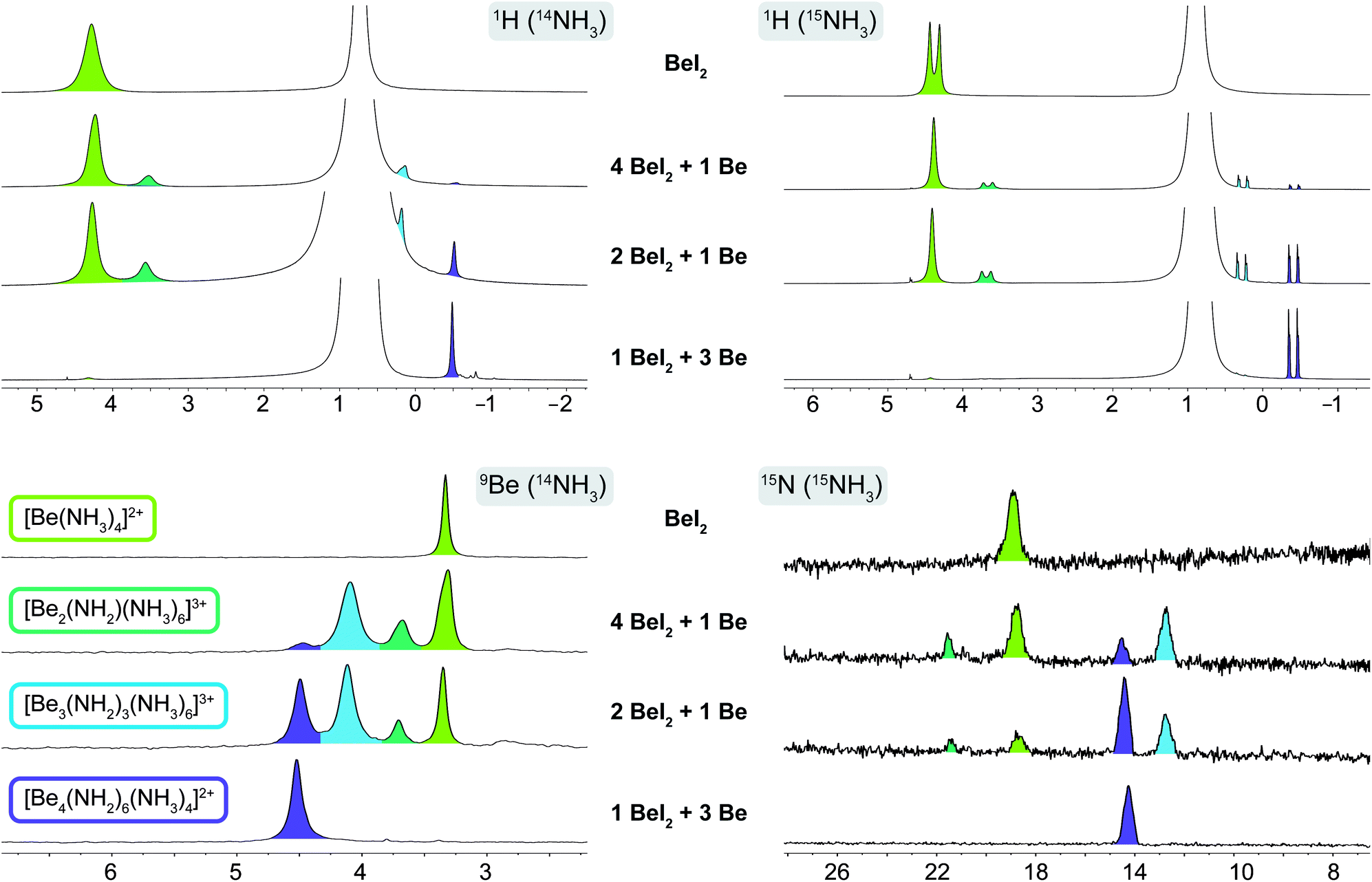 | ||
| Fig. 3 1H, 9Be & 15N NMR spectra of four Be/BeI2 mixtures in liquid 14NH3 and 15NH3 respectively. Species are assigned via color code. Chemical shift δ is given in ppm. | ||
To obtain more information on these intermediates, four of the NMR experiments were repeated in 15N enriched ammonia (99.9% 15N). The recorded 1H and 15N NMR spectra in 15NH3 are shown in Fig. 3 (for 9Be NMR spectra in 15NH3 see ESI Fig. S9†). In the 9Be spectrum of [Be(15NH3)4]2+ we do not observe 1JBeN coupling, which is presumably due to exchange between the ammine ligands and 15NH3. This exchange is also observed in the 9Be NMR spectrum of 1. Therefore, we do not observe a 1JBeN coupling towards the ammine ligands. However, the observation of a quartet at 4.6 ppm (q, 1JBeN = 5.3 Hz) in the 9Be spectrum (ESI Fig. S9†) clearly shows that three amido ligands are bound to each beryllium nucleus. This proves the existence of cation [Be4(15NH2)6(15NH3)4]2+ in solution. However, this coupling is not observed in the proton decoupled 15N NMR spectrum, since the resolution is to low to resolve the expected septet caused by coupling to two beryllium nuclei. Only the signal for the [NH2]− ligands of the [Be4(15NH2)6(15NH3)4]2+ cation is observed at 14.3 ppm (ω1/2 = 19.7 Hz), while the ammine ligands exhibit fast exchange with free ammonia. Therefore, the chemical shift of the averaged signal is again almost equal to that of ammonia. The 15N NMR signal of the [Be(15NH3)4]2+ cation emerges at 18.9 ppm (ω1/2 = 27.8 Hz). For the intermediates only two signals at 12.8 ppm (ω1/2 = 22.0 Hz) and 21.6 ppm (ω1/2 = 9.6 Hz) are present. The 1H NMR spectra in 15NH3 show doublets at −0.40 ppm (d, 1JNH = 57.8 Hz) for [Be4(15NH2)6(15NH3)4]2+ and at 4.38 ppm (d, 1JNH = 67.40 Hz) for [Be(15NH3)4]2+. However, the 1JNH coupling of the latter collapses as soon as beryllium metal is introduced. Surprisingly, the 1JNH coupling of the intermediate signals at 0.28 ppm (d, 1JNH = 56.4 Hz) and 3.69 ppm (d, 1JNH = 59.46 Hz) persists. The reason for these observations might be caused by proton exchange between [NH2]− and NH3. Therefore, the 1JNH coupling of the weaker bound NH3 collapses as soon as [NH2]− ions are present in solution.
To evaluate, whether anionic beryllium amide species were present in solution, we dissolved K[Be(NH2)3]24,29 in 15NH3 and examined it NMR spectroscopically. This compound shows one singlet at 12.2 ppm (ω1/2 = 12.0 Hz) in the 9Be NMR spectrum. This huge down-field shift compared to the species described above, shows that the trigonal planar coordination around the Be nucleus is retained in solution. In the 1H and 15N NMR spectra no signal other than that of ammonia emerges (Fig. S11†). This suggests that, in contrast to the species observed above, the amido ligands are bound weakly. Dissociated amido ligands then easily deprotonate ammonia, which leads to an averaged signal for all amido ligands and ammonia. This low Be–NH2 bond strength is presumably caused by the negative charge of the complex ion. This should also be the reason why [Be(NH2)3]− remains three-coordinated in the presence of a huge excess of NH3. Thus, we can exclude the presence of anionic beryllium amide species during the formation of complex cation 1.
Based on these observations we propose a plausible reaction pathway for the formation of 1 from beryllium metal and [Be(NH3)4]2+ (Scheme 2). At first ammono acidic [Be(NH3)4]2+ reacts with one eq. Be under H2 formation. This generates one free amido ligand and μ2-[NH2]− bridged dinuclear [Be2(NH2)(NH3)6]3+ (2). 2 would still have strongly bound ammine ligands, which should not exchange with free NH3. Therefore, the respective signals at 3.69 and 21.6 ppm in the 1H and 15N NMR spectra are assigned to the ammine ligands of 2. This is supported by the large 1JNH coupling constant, which is close to the one observed for [Be(NH3)4]2+ and is indicative for a stronger N–H bond than in [NH2]−. Accordingly the 9Be NMR signal at 3.7 ppm is also ascribed to 2. We assume that the 1H NMR signal of the μ2-[NH2]− group is covered by the solvent signal, while the respective 15N NMR signal is too weak and broad for observation. This is also supported by quantum chemistry, which gives Be–NH2 distances of 1.76 Å for 2, which are longer than in 1.
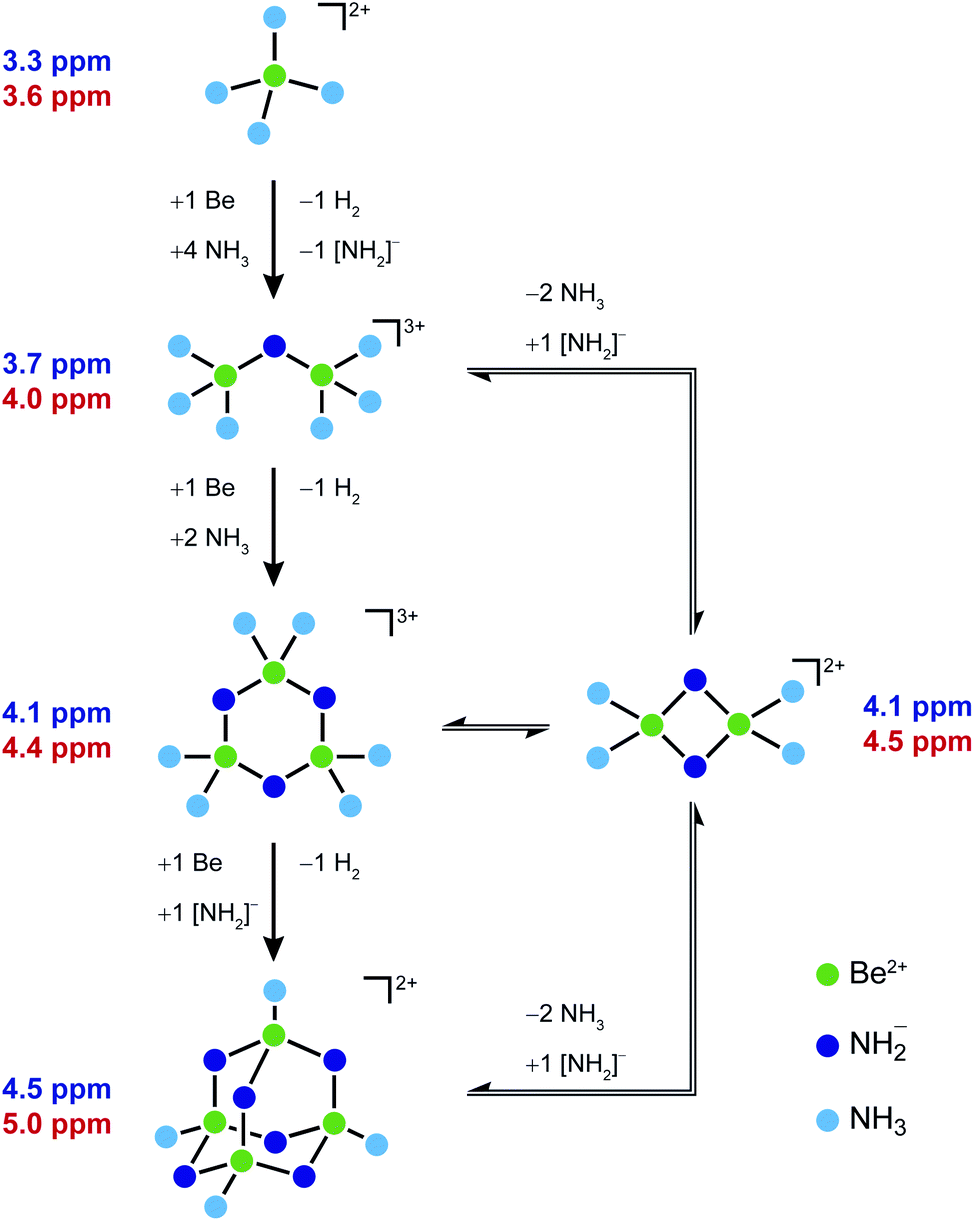 | ||
| Scheme 2 Reaction pathway from [Be(NH3)4]2+ to 1via three intermediates observed in the NMR spectra (Fig. 3). The assignment is based on their experimental (purple) and calculated (red) 9Be NMR chemical shifts. | ||
2 reacts with a further eq. of Be under H2 evolution to trinuclear [Be3(NH2)3(NH3)6]3+ (3a). This complex constitution is supported by numerous examples of six-membered beryllium heterocycles.28,30–333a should already have relatively weak Be–NH3 bonds, which leads to exchange between the coordinated ammine and free NH3 in solution. Therefore, no ammine signal is expected in the NMR spectra. On the other hand the μ2-[NH2]− groups should be strongly bound, like in 1. This is also supported by the computational Be–NH2 distance of 1.73 Å which is almost identical to the corresponding distances calculated for 1. The [NH2]− moieties of 3a account for the signals at 0.28 and 12.8 ppm in the 1H and 15N NMR spectra, respectively. Accordingly the 9Be NMR signal at 4.1 ppm belongs to 3a. We can not exclude that 3a is in equilibrium with the dinuclear cation [Be2(NH2)2(NH3)4]2+ (3b), since this would exhibit the same NMR signals and there are also some examples of four-membered beryllium heterocycles.18,34–36 However, we regard 3a the more probable intermediate since reaction with one eq. Be and the free amido ligand, which was generated in the first step, results in the formation of 1 without further byproducts. Scheme 2 shows all plausible species, which are assigned to the signals in the NMR spectra in Fig. 3.
We also investigated the reaction pathway and NMR chemical shifts presented in Scheme 2 using quantum chemical methods (computational details for the DFT-PBE0/def2-TZVP calculations are available in the ESI†). The optimized geometry of cation 1 shows excellent agreement with the available experimental structural data. In the case of cation 1 of 1a, the predicted Be–NH2 distance is 1.72 Å which compares very well with the experimental range of 1.703(16)–1.73(1) Å. For the longer Be–NH3 distances, the predicted value 1.82 Å is also in line with the experimental range of 1.78(2)–1.807(2) Å. The predicted 9Be NMR shifts shown in Scheme 2 strongly support the experimental assignment of the NMR signals. The predicted NMR shifts are slightly overestimated compared to the experimental values, but the overestimation is very systematic and the relative trends of the experimental and computational NMR shifts are in good agreement.
2.2 Reactivity
Since the ammine ligands of 1 are only weakly bound to the beryllium cations, we were curious if they could by substituted by other N-donor solvents and whether this would break the adamantyl-shaped cation and give access to more beryllium amido compounds. So, [Be4(NH2)6(NH3)4]I2, which showed a reasonable solubility in ammonia, was treated with various solvents.The suspension of 1c′ in pyridine resulted in a vivid evolution of ammonia and a mucilaginous precipitate. The suspension was heated to 80 °C for several days. Thereby, column shaped crystals grew in the upper part of the reaction tube, while considerable amounts of precipitate remained unchanged and no signals were observed in the 9Be NMR spectra of that solution. The single crystals were isolated for X-ray diffraction and IR spectroscopy and consist of [OBe8(NH2)12(py)4]I2·5py (4), whose cation is depicted in Fig. 4.
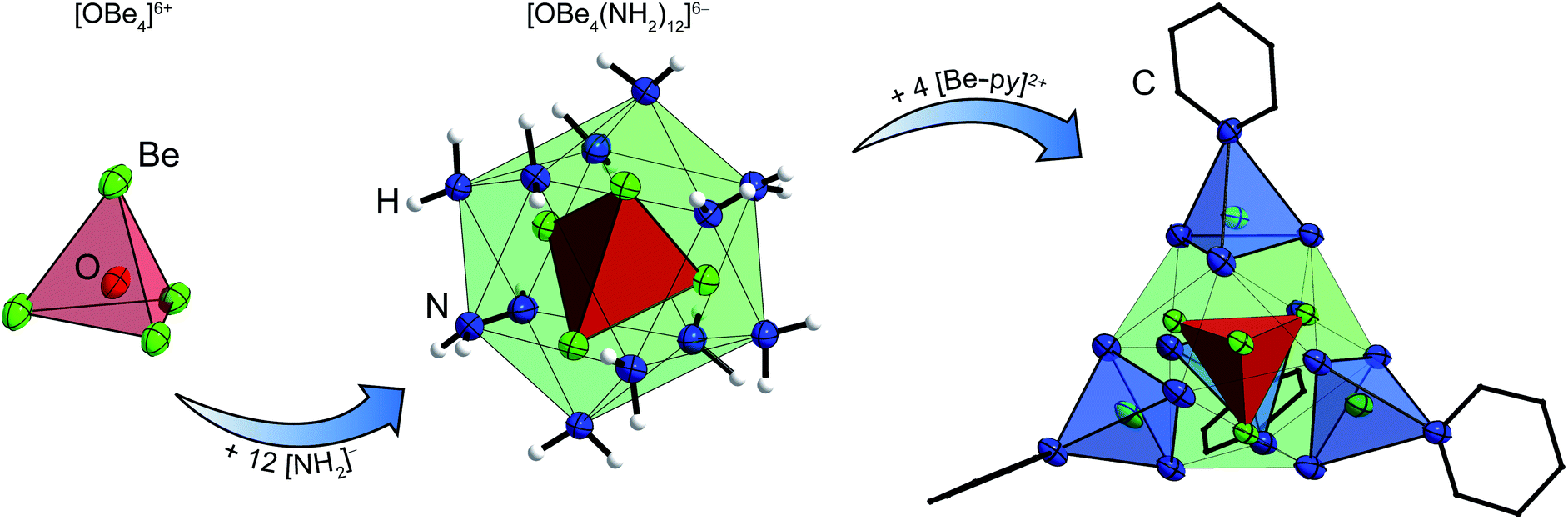 | ||
| Fig. 4 Build-up of the molecular structure of the [Be8O(NH2)12(py)4]2+ cation of 4. Ellipsoids are depicted at 70% probability at 100 K. Hydrogen atoms are depicted with arbitrary radii. In the structure on the right carbon atoms are shown as wire frame and hydrogen atoms are omitted for clarity. | ||
This beryllium-oxy-amide cluster contains a central [OBe4]6+ unit with Be–O atomic distances of 1.671(4) & 1.678(4) Å, equal to those in beryllium-oxy-carboxylates.37,38 Besides the beryllium-oxy-carboxylates there is also an oxy-nitrate39 and an oxy-carbonate known.40 Cation 4 is the first oxy-amide of beryllium that has the [OBe4]6+ core and only the second described beryllium-oxy-amide. The other is a linear beryllium-oxy-amide anion ([OBe2(NH2)4]2−), in which the Be–O atomic distance of 1.502(2) Å is significantly shorter than in 4.24 The [OBe4]6+ core in 4 is surrounded by twelve amido ligands which form a distorted icosahedral anionic fragment: [OBe4(NH2)12]6−. The Be–N atomic distances towards the central beryllium atoms, that are coordinated by the μ4-oxido ligand and three amido ligands each, range from 1.733(4) to 1.770(4) Å which tends to be longer than the corresponding distances in 1. Cation 4 is completed by four [Be–py]2+ fragments (py = pyridine), that are arranged tetrahedrally around the icosahedral [OBe4(NH2)12]6− fragment. The outer four beryllium atoms are coordinated by three μ2-amido ligands and one pyridine molecule each. For those beryllium atoms the Be–N atomic distances towards the amido ligands are 1.690(4)–1.702(4) Å, which is significantly shorter than the corresponding distances towards the inner beryllium atoms and also shorter than the Be–N atomic distances in 1 by tendency. The Be–N atomic distances towards the pyridine ligands are 1.828(4) & 1.836(4) Å, therefore much longer than those reported for Be–py bonds in BeCl2(py)2 (1.741(5) & 1.758(5) Å)17 and [Be(py)4]2+ (1.723(8)–1.738(8) Å).41 The complex cation of 4 is most comparable to compounds in which a tetrahedral [OZn4]6+ or [OCo4]6+ unit is coordinated by twelve μ2-2-aminoethanethiolate ligands, which in turn coordinate to four additional metal cations in total.42–44 However, to the best of our knowledge there is no directly comparable compound known. Additionally this cluster proves that a [Be4O]6+ core can be stabilized without bidentate O-donor ligands. The μ4-O2− anion in 4 most likely originated from water that was present in insufficiently dried pyridine according to Scheme 3. Apart from the obtained single crystals a considerable amount of amorphous precipitate was left in the reaction tube from which we were not able to determine the composition.
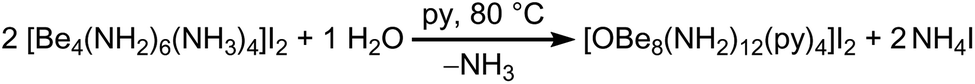 | ||
| Scheme 3 Reaction of 1c′ in pyridine (py) with traces of H2O at 80 °C. | ||
In the case of the treatment of 1c′ with NEt3 no reaction was observed and 1c′ remained unchanged even after the mixture was heated to 100 °C for several days. For ethylenediamine we observed a vivid evolution of ammonia as soon as it came into contact with 1c′ and an amorphous, voluminous precipitate was formed. Attempts to recrystallize the precipitate failed and no signals were observable via9Be NMR spectroscopy. When 1c′ was treated with aniline, the applied powder increased in volume without gas evolution. When this suspension was subsequently heated to 100 °C for several weeks single crystals of [Be(NH3)4]I2 grew on the glass wall while the suspension turned light brown. This means that cation 1 is decomposed by aniline under the formation of [Be(NH3)4]I2. However, we could not determine the composition of the remaining brown precipitate. Like for ethylenediamine no 9Be NMR signal was observed.
In case of MeCN, 1c′ seemed also insoluble. But when the suspension was heated to 90 °C for several days, the solution turned yellow and the starting material was completely recrystallized into column shaped colorless single crystals of bis(N-acetimidoylacetamidinato-N,N′)-beryllium(II) (Be(C4H8N3)2) (5) in the tetragonal space group P42212 (94). Its molecular structure is depicted in Fig. 5 in which the beryllium cation is tetrahedrally coordinated by four nitrogen atoms of two N-acetimidoylacetamidinate anions. Here, the Be–N atomic distance of 1.6897(17) Å is shorter than that in BeBr2(MeCN)2 (1.713(4) Å)45 but in the range of those in Be(NMe2)2 (1.57–1.75 Å)46 and a beryllium 2-(2-pyridyl)indole compound with Be–N atomic distances of 1.665(5)–1.753(6) Å.47 The isoelectronic Be(acac)2 complex shows the same coordination around the Be2+ cation but exhibits much shorter Be–O atomic distances of 1.607(3)–1.626(3) Å.48
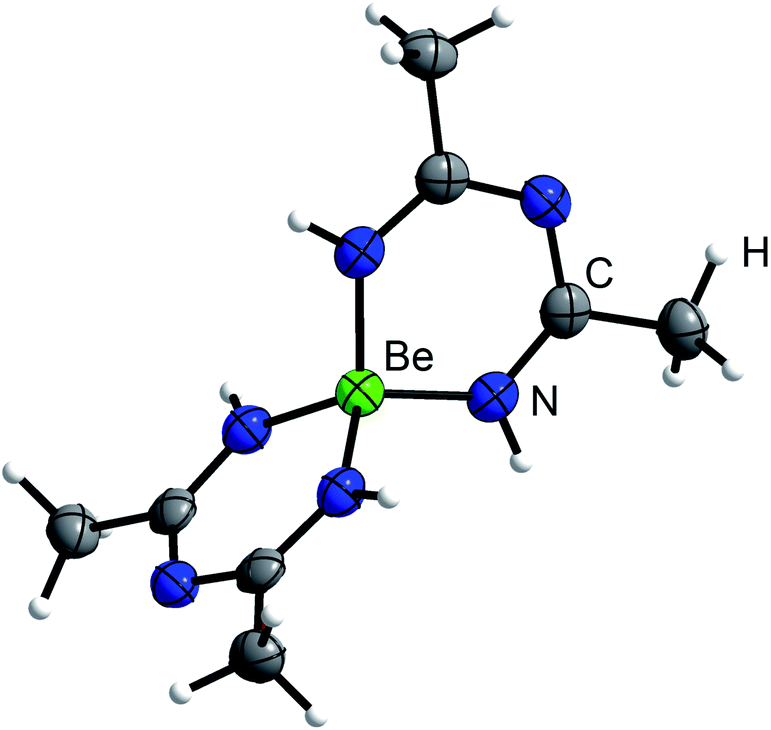 | ||
| Fig. 5 Molecular structure of 5. Ellipsoids are depicted at 70% probability at 100 K. Hydrogen atoms are depicted with arbitrary radii. | ||
The formation of the N-acetimidoylacetamidinate anion through the condensation of nitriles in alkaline solutions is well known, especially in the presence of Ni2+ cations that form similar complexes, but with a square planar coordination around the Ni atoms.49–51 In analogy to this hydroxy-induced mechanism,49,51 we assume that in the case of 5 an amide anion starts the coupling according to Scheme 4. From this follows the over all reaction equation depicted in Scheme 5.
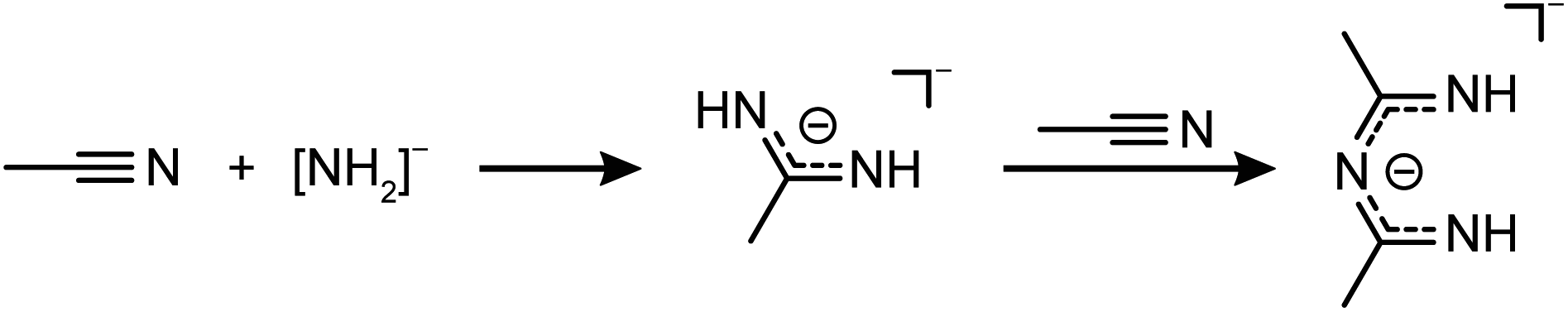 | ||
| Scheme 4 Amide induced coupling of acetonitrile into N-acetimidoylacetamidinate. | ||
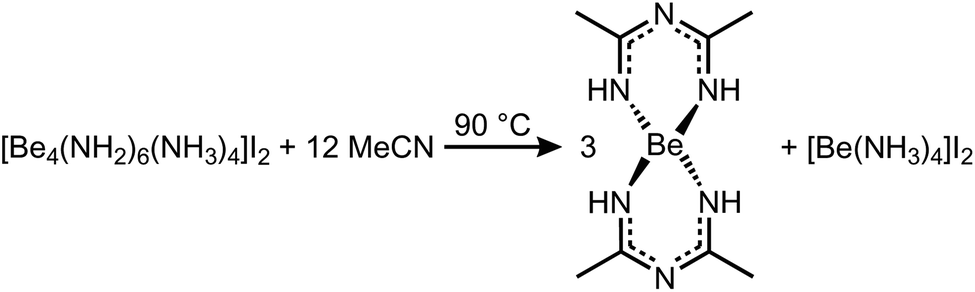 | ||
| Scheme 5 Reaction of 1c′ in acetonitrile at 90 °C. | ||
Single crystals of 5 were dissolved in CDCl3. The recorded NMR spectra showed compound 5 exclusively (Fig. S13†) with a singlet at 2.5 (ω1/2 = 5.7 Hz) in the 9Be NMR spectrum. However, the N–H protons were not observed in the 1H NMR spectrum. We assume that due to the coordination to the beryllium cation, their signals are extremely broadened. This broadening is also noticeable for the C–H protons. Additionally the N–H protons should be relatively acidic. Therefore, H–D-exchange with CDCl3 might also be the cause for the absence of the signals in the 1H NMR spectrum. Especially, since the N–H bands are clearly visible in the IR spectrum of 5 (Fig. S15†).
3 Conclusion
The multinuclear complex cation [Be4(NH2)6(NH3)4]2+ (1) is readily obtained when an excess of beryllium metal is reacted with NH4X (X = Cl, Br, I, CN, SCN, N3) or beryllium halides in liquid ammonia. Under evolution of dihydrogen, compounds 1a–f are formed as cuboctahedral crystals at ambient temperature. Except for the iodide compounds they show poor solubility in NH3. Therefore, only 1c were examined NMR spectroscopically in liquid 14NH3 and 15NH3. 1 is generated from [Be(NH3)4]2+via two intermediates and retains its adamantyl shaped structure in solution. The NMR signals of the intermediates were assigned to the most likely beryllium species using computational methods. The successive build-up of the tetranuclear cation 1 from [Be(NH3)4]2+via a dimer [Be2(NH2)(NH3)6]3+ and a ring-shaped trimer [Be3(NH2)3(NH3)6]3+ emerged as the most plausible reaction path.To determine whether the ammine ligands can be substituted or higher aggregated beryllium amido clusters can be obtained, compounds 1a–c were reacted with alkali metal amides in liquid ammonia at ambient temperature. From these reactions no distinct compounds could be isolated other than the starting materials. Treatment of 1c′ with various N-donor solvents in order to substitute the ammine ligands through other N-donor ligands resulted in the formation of Be(C4H8N3)2 (5) and octa-nuclear oxy-amide 4. 5 was formed by the amide promoted coupling of acetonitrile into N-acetimidoylacetamidinate that subsequently coordinated to Be2+ and is similar to the isoelectronic Be(acac)2. 4 was obtained when 1c′ was treated with pyridine. Single crystal diffraction revealed the formation of the complex cation [Be8O(NH2)12(py)4]2+, which is the first example of a cationic beryllium-oxy-amide. While its [OBe4]6+ core unit is well known in the chemistry of beryllium, an icosahedral coordination around this core by twelve anionic ligands or the expansion into an octa-nuclear beryllium cluster is unprecedented and is a completely new structural motif in coordination chemistry.
Our results show that multi-nuclear beryllium complex cations can easily be generated in liquid ammonia. Therefore, NH3 seems to be indeed the most promising solvent to obtain beryllium complexes with bio-relevant molecules. Investigations into this direction are currently ongoing and will be presented in due course. Furthermore, there are – to the best of our knowledge – no known NH2-bridged multinuclear compounds of the other alkaline earth metals, zinc or aluminium.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. R. B. thanks Prof. F. Kraus for moral and financial support as well as the provision of laboratory space. The DFG is gratefully acknowledged for financial support (BU2725/5-1 & BU2725/8-1). A. J. K thanks CSC – the Finnish IT Center for Science for computational resources.Notes and references
- O. Kumberger and H. Schmidbaur, Chem. Unserer Zeit, 1993, 27, 310–316 CrossRef CAS.
- L. C. Perera, O. Raymond, W. Henderson, P. J. Brothers and P. G. Plieger, Coord. Chem. Rev., 2017, 352, 264–290 CrossRef CAS.
- T. M. McCleskey, V. Buchner, R. W. Field and B. L. Scott, Rev. Environ. Health, 2009, 24, 75–115 CAS.
- A. P. Fontenot, Ann. Am. Thorac. Soc., 2018, 15, S81–S85 CrossRef PubMed.
- H. Schmidbaur, Coord. Chem. Rev., 2001, 215, 223–242 CrossRef CAS.
- G. Jander, Die Chemie in wasserähnlichen Lösungsmitteln, Springer-Verlag, Berlin, Göttingen, Heidelberg, 1949 Search PubMed.
- M. Müller and M. R. Buchner, Chem. Commun., 2019, 55, 13649–13652 RSC.
- T. S. Keizer, N. N. Sauer and T. M. McCleskey, J. Inorg. Biochem., 2005, 99, 1174–1181 CrossRef CAS PubMed.
- M. Müller, F. Pielnhofer and M. R. Buchner, Dalton Trans., 2018, 47, 12506–12510 RSC.
- B. L. Scott, Z. Wang, B. L. Marrone and N. N. Sauer, J. Inorg. Biochem., 2003, 94, 5–13 CrossRef CAS PubMed.
- B. L. Scott, T. M. McCleskey, A. Chaudhary, E. Hong-Geller and S. Gnanakaran, Chem. Commun., 2008, 2837–2847 RSC.
- S. De, G. Sabu and M. Zacharias, Phys. Chem. Chem. Phys., 2020, 22, 799–810 RSC.
- R. C. Haddon, S. V. Chichester and J. H. Marshall, Tetrahedron, 1986, 42, 6293–6300 CrossRef CAS.
- M. Müller and M. R. Buchner, Angew. Chem., Int. Ed., 2018, 57, 9180–9184 CrossRef PubMed.
- M. Schmidt and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 1998, 53, 1294–1300 CAS.
- F. Kraus, M. B. Fichtl and S. A. Baer, Z. Naturforsch., B: J. Chem. Sci., 2008, 64, 257–262 Search PubMed.
- M. P. Dressel, S. Nogai, R. J. F. Berger and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 173–182 CAS.
- M. Müller and M. R. Buchner, Inorg. Chem., 2019, 58, 13276–13284 CrossRef PubMed.
- H. Jacobs and B. Nöcker, Z. Anorg. Allg. Chem., 1993, 619, 73–76 CrossRef CAS.
- H. Jacobs and K. Jänichen, J. Less-Common Met., 1990, 159, 315–325 CrossRef CAS.
- T. M. M. Richter, S. LeTonquesse, N. S. A. Alt, E. Schlücker and R. Niewa, Inorg. Chem., 2016, 55, 2488–2498 CrossRef CAS PubMed.
- B. Fröhling and H. Jacobs, Z. Anorg. Allg. Chem., 1997, 623, 1103–1107 CrossRef.
- H. Jacobs, J. Birkenbeul and D. Schmitz, J. Less-Common Met., 1982, 85, 79–86 CrossRef CAS.
- M. Müller and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 483–489 Search PubMed.
- H. Schmidbaur, M. Schmidt, A. Schier, J. Riede, T. Tamm and P. Pyykkö, J. Am. Chem. Soc., 1998, 120, 2967–2968 CrossRef CAS.
- H. Jacobs, Z. Anorg. Allg. Chem., 1976, 427, 1–7 CrossRef CAS.
- E. Spundflasche, H. Fink and H. Seifert, Z. Anorg. Allg. Chem., 1995, 621, 1723–1726 CrossRef CAS.
- F. Kraus, S. A. Baer, M. R. Buchner and A. J. Karttunen, Chem.–Eur. J., 2012, 18, 2131–2142 CrossRef CAS PubMed.
- M. G. B. Drew, J. E. Goulter, L. Guémas-Brisseau, P. Palvadeau, J. Rouxel and P. Herpin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2579–2582 CrossRef.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2010, 636, 1516–1521 CrossRef.
- D. Naglav, B. Tobey, C. Wölper, D. Bläser, G. Jansen and S. Schulz, Eur. J. Inorg. Chem., 2016, 2424–2431 CrossRef CAS.
- R. Puchta, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2009, 635, 1196–1199 CrossRef CAS.
- R. Puchta, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2011, 637, 67–74 CrossRef CAS.
- M. Bayram, D. Naglav, C. Wölper and S. Schulz, Organometallics, 2017, 36, 467–473 CrossRef CAS.
- A. Paparo and C. Jones, Chem.–Asian J., 2019, 14, 486–490 CrossRef CAS PubMed.
- M. Müller and M. R. Buchner, Chem.–Eur. J., 2019, 25, 11147–11156 CrossRef PubMed.
- A. Tulinsky, C. R. Worthington and F. Pignataro, Acta Crystallogr., 1959, 12, 623–626 CrossRef CAS.
- R. J. F. Berger, M. A. Schmidt, J. Juselius, D. Sundholm, P. Sirsch and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 979–989 CAS.
- M. J. Haley, S. C. Wallwork, B. Duffin, N. Logan and C. C. Addison, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 829–830 CrossRef.
- M. Dahm and A. Adam, Z. Anorg. Allg. Chem., 2000, 626, 494–501 CrossRef CAS.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2010, 636, 515–517 CrossRef.
- K.-I. Okamoto, T. Konno and J. Hidaka, J. Chem. Soc., Dalton Trans., 1994, 533–537 RSC.
- K.-I. Okamoto, M. Matsumoto, Y. Miyashita, N. Sakagami, J. Hidaka and T. Konno, Inorg. Chim. Acta, 1997, 260, 17–26 CrossRef CAS.
- T. Konno, K.-I. Okamoto and J. Hidaka, Inorg. Chem., 1994, 33, 538–544 CrossRef CAS.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2010, 636, 1438–1440 CrossRef.
- J. L. Atwood and G. D. Stucky, J. Am. Chem. Soc., 1969, 91, 4426–4430 CrossRef CAS.
- S.-F. Liu, Q. Wu, H. L. Schmider, H. Aziz, N.-X. Hu, Z. Popović and S. Wang, J. Am. Chem. Soc., 2000, 122, 3671–3678 CrossRef CAS.
- S. Onuma and S. Shibata, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1181–1183 CrossRef.
- S. V. Kryatov, A. Y. Nazarenko, M. B. Smith and E. V. Rybak-Akimova, Chem. Commun., 2001, 1174–1175 RSC.
- J. P. Wikstrom, A. S. Filatov and E. V. Rybak-Akimova, Chem. Commun., 2010, 46, 424–426 RSC.
- G. Thiele, B. Wagner and S. Dehnen, Eur. J. Inorg. Chem., 2015, 5329–5334 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, characterization data, extended crystallographic data, selected NMR, IR and Raman spectra. CCDC 1982295–1982303. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01112f |
| This journal is © The Royal Society of Chemistry 2020 |