
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical α-addition involved electrooxidative [3 + 2] annulation of phenols and electron-deficient alkenes†
Qiang
Zhao
,
Ji-Kang
Jin
,
Jie
Wang
,
Feng-Lian
Zhang
* and
Yi-Feng
Wang
 *
*
Hefei National Laboratory for Physical Sciences at the Microscale, Center for Excellence in Molecular Synthesis of CAS, Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China. E-mail: zfl9@ustc.edu.cn; yfwangzj@ustc.edu.cn
First published on 23rd March 2020
Abstract
An electrooxidative [3 + 2] annulation of phenols and electron-deficient alkenes for the synthesis of C3-functionalized 2-aryl-2,3-dihydrobenzofuran derivatives was achieved. The ring construction starts by a unique α-addition of carbon radicals derived from anodic oxidation of phenols to electron-deficient alkenes. The subsequent anodic oxidation of the resulting alkyl radical intermediates followed by trapping with the phenolic hydroxy group assembles the 2,3-dihydrobenzofuran core. Such a pathway enables the installation of various electrophilic functionalities including alkoxycarbonyl, alkylaminocarbonyl, trifluoromethyl, and cyano groups at the C-3 of the 2,3-dihydrobenzofuran framework, which is unattainable by other intermolecular reactions. The application of this method for a rapid synthesis of a bioactive natural product is demonstrated.
Introduction
2,3-Dihydrobenzofurans constitute the core skeletons of a number of natural products and bioactive molecules.1 In particular, 2-aryl-2,3-dihydrobenzofuran-3-carboxylic acid derivatives have shown important biological and medicinal applications (Fig. 1a).2 Therefore, the development of methods for practical and environmentally friendly synthesis is of great significance. So far, methods to assemble such a 2-aryl-3-carboxyl-2,3-dihydrobenzofuran framework have been mainly limited to intramolecular annulation reactions (Fig. 1b),3 including carbene C–H insertion reactions of alkylated phenols,4 radical Witkop photocyclization/elimination/addition cascades,5 alkylation via C–H bond activation,6 oxidative [3 + 2] cyclization,7 and hydrogenation of benzofurans.8 Notably, many approaches suffered from multiple-step synthesis of pre-functionalized starting materials, limited substrate scope, and the use of precious transition metal catalysts or stoichiometric amounts of strong oxidants. To achieve green and more step- and atom-economical synthesis, the exploration of new protocols based on intermolecular reactions of readily accessible starting materials is highly desirable.9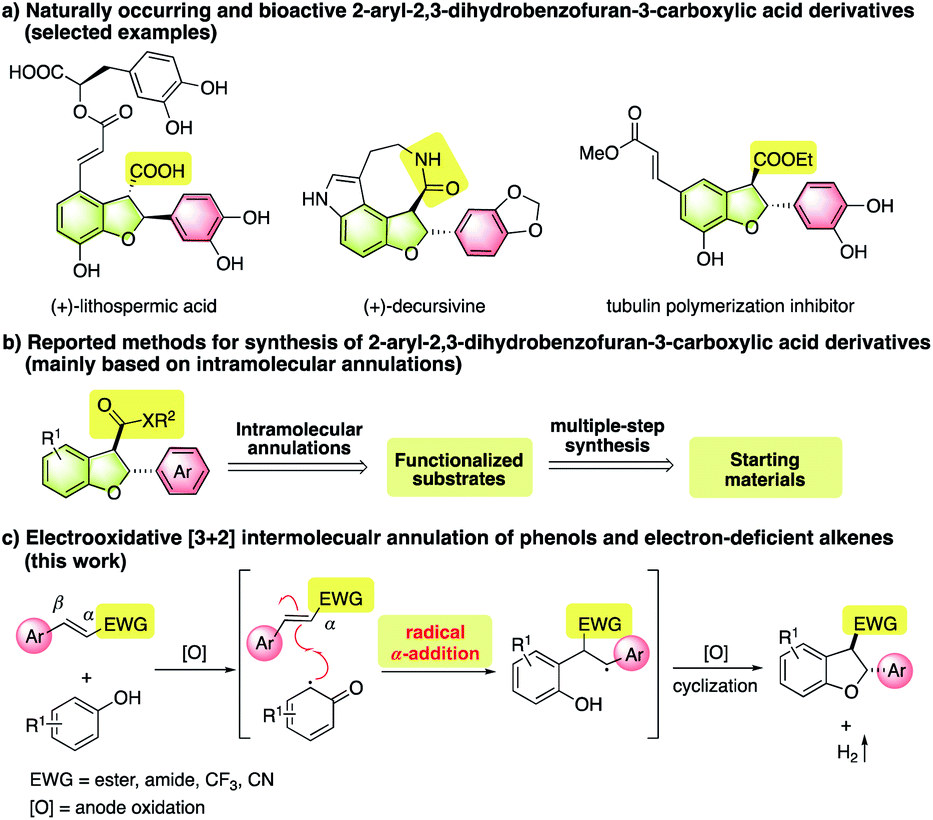 | ||
| Fig. 1 Synthesis of 2-aryl-2,3-dihydrobenzofuran-3-carboxylic acid derivatives and analogous. | ||
Oxidative [3 + 2] annulation of easily available phenols and alkenes presents an appealing strategy to assemble the 2,3-dihydrobenzofuran framework.10 For example, the pioneering work by Swenton11 and Chiba12 demonstrated successful anodic cycloaddition reactions. However, most reported reactions rely on the use of electron-rich alkenes as electron donor components that can either trap electron-demanding intermediates10a–d,11,12 or serve as reductive partners to promote oxidative transformations.10e–g In contrast, alkenes bearing electron-withdrawing groups have rarely been used as viable substrates most likely due to the unmatched electronic nature. Considering that a wide variety of electron-poor alkenes are readily accessible, the success of reliable annulation methods would enable economical and diverse synthesis of structurally elaborate products. More importantly, many electrophilic motifs, such as carbonyl groups, could be easily converted to various functionalities, which would enrich 2,3-dihydrobenzofuran libraries for further biological and medicinal applications. To this end, developing conceptually distinct pathways is of great significance. Herein, we report an unprecedented electrooxidative [3 + 2] annulation between phenols and electron-deficient alkenes towards the synthesis of structurally novel C3-functionalized 2-aryl-2,3-dihydrobenzofuran derivatives (Fig. 1c). In this process, the key step is a radical α-addition of carbon radicals derived from single-electron oxidation of phenols to electron-poor alkenes.
In radical addition reactions, a radical species usually prefers to undergo β-addition to electron-deficient alkenes owing to the strong inductive effect of the electron-withdrawing group or the external Lewis acid activation.13 For substrates bearing a β-radical stabilizing group, such as an aryl functionality, α-addition is possible to be the main pathway,14 in particular for the case where the ensuing step is driven by an energetically highly favorable process.15 Very recently, our group disclosed an interesting radical α-addition of NHC–boryl radicals to electron-poor alkenes.16 Mechanistic studies revealed that such α-addition was actually kinetically and thermodynamically feasible, especially for β-aryl substituted ones. Stimulated by this finding, we posited a radical α-addition involved oxidative annulation of phenols and electron-deficient alkenes. Meanwhile, as a green and oxidant-free synthetic tool,17 an electrochemical reaction was considered as the preferred choice to promote this assumed process. As depicted in Fig. 1c, single-electron oxidation of phenols on the anode could easily take place,18 and the resulting carbon radicals were expected to undergo α-addition to electron-deficient alkenes. The subsequent single-electron oxidation of the resulting radical intermediates followed by cyclization would provide a 2,3-dihydrobenzofuran framework. At the same time, hydrogen gas would be released as the sole byproduct by the reduction of protons at the cathode.
Results and discussion
We commenced our study by investigating the reaction of 4-methoxyphenol (1a) and cinnamate 2a in an undivided cell equipped with two electrodes. To our delight, the proposed annulation product 3aa was isolated in 82% yield using n-Bu4NBF4 as the electrolyte and 1,1,1,3,3,3-hexafluoroisopropyl alcohol (HFIP)/CH2Cl2 as co-solvents at 10 mA constant current for 2 h (Table 1, entry 1). The solvent effect was examined. It was found that a comparable yield was obtained when HFIP was used as the sole solvent (entry 2), while the employment of CF3CH2OH/CH2Cl2 (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) led to a decreased yield (entry 3). Moreover, replacing HFIP with (CH3)3CHOH resulted in a dramatic decrease in the reaction efficiency (entry 4). Using n-Bu4NPF4 as the electrolyte instead of n-Bu4NBF4 led to a lower yield (entry 5). As for the electrode material used, replacing the graphite rod cathode with a platinum plate one could also give high product yield (entry 6). Importantly, the reaction could be carried out under atmospheric conditions while maintaining a good efficiency (entry 7). The control experiment showed that no reaction occurred without electric current (entry 8), verifying the electron transfer mechanism of this process.
:4) led to a decreased yield (entry 3). Moreover, replacing HFIP with (CH3)3CHOH resulted in a dramatic decrease in the reaction efficiency (entry 4). Using n-Bu4NPF4 as the electrolyte instead of n-Bu4NBF4 led to a lower yield (entry 5). As for the electrode material used, replacing the graphite rod cathode with a platinum plate one could also give high product yield (entry 6). Importantly, the reaction could be carried out under atmospheric conditions while maintaining a good efficiency (entry 7). The control experiment showed that no reaction occurred without electric current (entry 8), verifying the electron transfer mechanism of this process.
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | 3aa yieldb |
| a Reaction conditions: graphite rod anode, graphite rod cathode, constant current = 10 mA. 2a (0.3–0.4 mmol), 1a (1.5 equiv.), n-Bu4NBF4 (1.0 equiv.), solvent (10 mL), rt, 2 h, under N2. b NMR yields using 1,1,2,2-tetrachloroethane as the internal standard. c Isolated yield. | ||
| 1 | None | 83% (82%)c |
| 2 | HFIP instead of HFIP/CH2Cl2 (6:4) |
76% |
| 3 | CF3CH2OH instead of HFIP | 65% |
| 4 | (CH3)2CHOH instead of HFIP | 9% |
| 5 | n-Bu4NPF6 instead of n-Bu4NBF4 | 58% |
| 6 | Platinum plate cathode instead of graphite rod cathode | 88%c |
| 7 | Under air | 80%c |
| 8 | No electric current | NR |
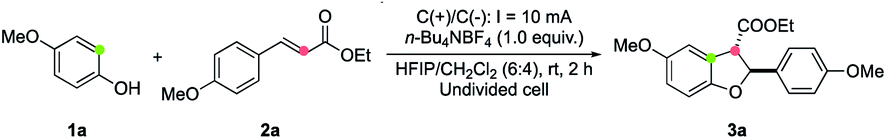
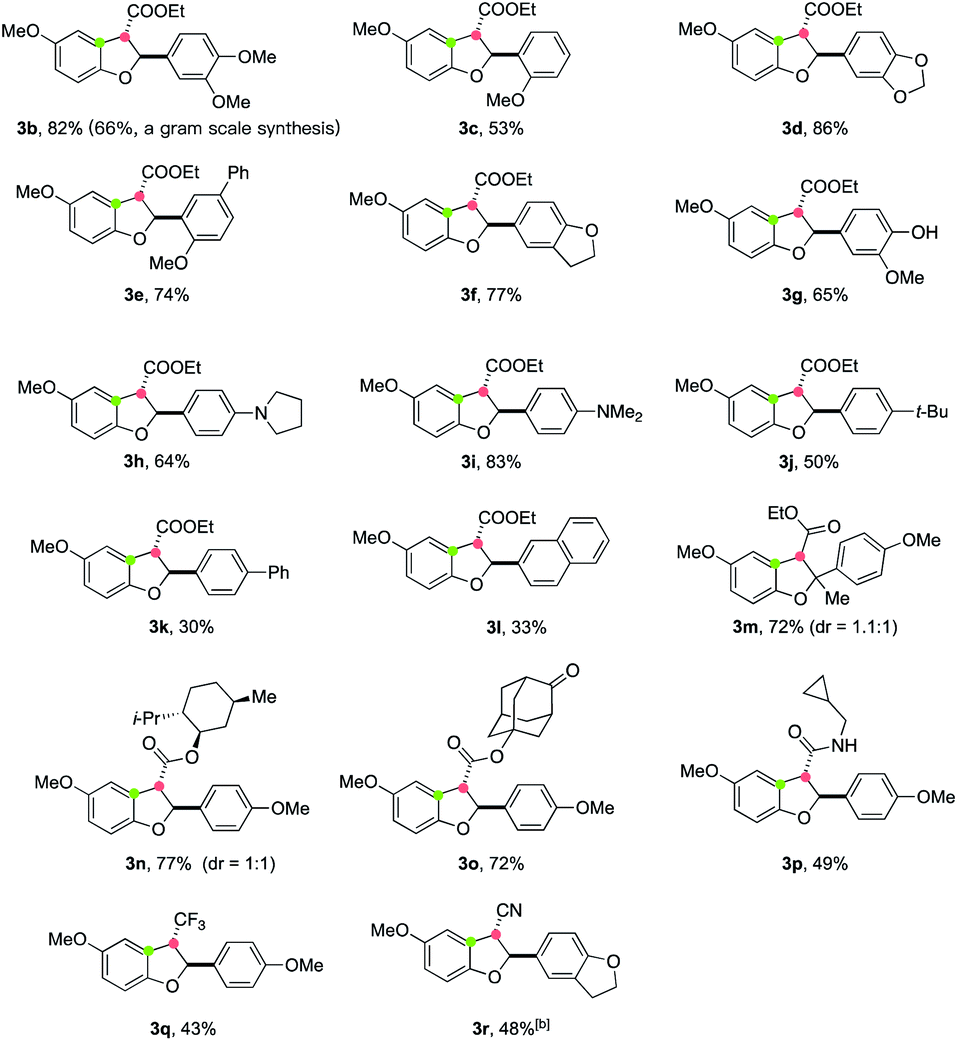
Using the optimized reaction conditions, the scope of electron-deficient alkenes in this electrooxidative [3 + 2] annulation was first evaluated (Table 2). A broad range of β-aryl-α,β-unsaturated esters could be converted to cyclized products in good yields (3b–3f). A gram scale synthesis of 3b was achieved in 66% yield, demonstrating the practicability of this electrochemical protocol. Notably, a free-phenol containing substrate 2g was also a viable substrate, furnishing the desired annulation product 3g in 65% yield without the detection of any dimerization product of 2g.19 Moreover, no further oxidation of this phenol product to engage in a second cyclization was observed. This protocol was also applicable for the construction of alkylamino-substituted products (3h and 3i). However, the reaction of 2j–2l, where the aryl ring bears less electron-donating groups, led to decreased yields (for 3j–3l) most likely due to the inferior efficiency for the second single-electron oxidation to generate carbon cation intermediates. Remarkably, the presence of an additional methyl group at the β-carbon of 2 (R3 = Me) did not retard the annulation, forming product 3m containing a tetrasubstituted carbon center20 in 72% yield, albeit with low diastereoselectivity. This annulation method also allowed for the incorporation of alcohol-containing functional molecules, such as menthol (3n) and 5-hydroxy-2-adamantanone (3o), onto the 2,3-dihydrobenzofuran framework. Importantly, other electron-withdrawing groups, such as alkylaminocarbonyl (3p), trifluoromethyl (3q), and cyano (3r) moieties, could also be installed at the C-3 position from annulation reactions of the corresponding electron-deficient alkenes.
|
|
|---|
|
a Standard reaction conditions: graphite rod anode, graphite rod cathode, constant current = 10 mA. 2 (0.3–0.4 mmol), 1a (1.5 equiv.), n-Bu4NBF4 (1.0 equiv.), HFIP/CH2Cl2 (6:4, 10 mL), rt, 2 h, under N2.
b The reaction was conducted with a constant cell potential of +2.5 V in HFIP/CH2Cl2 (7:3, 10 mL) for 10 h at rt.
|
|
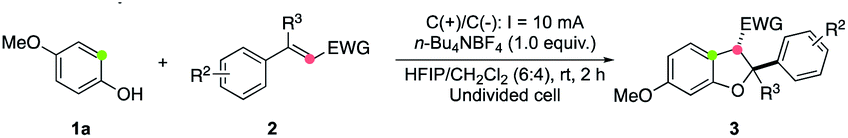
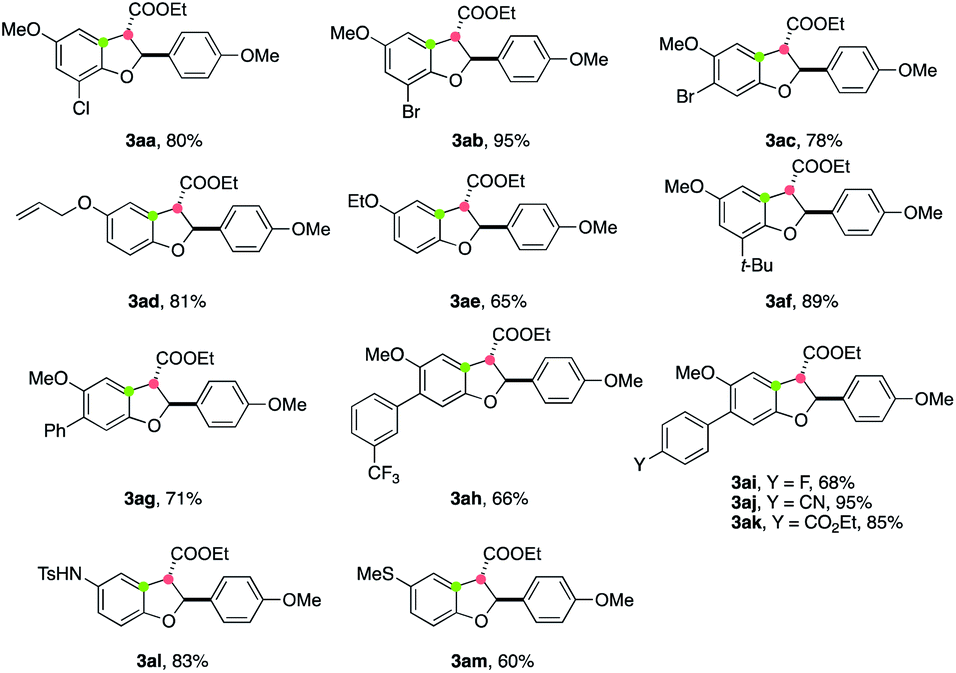
Next, we turned our attention to study the applicability of phenol components in this annulation reaction. The reactions between a variety of substituted phenols 1 and cinnamate 2a worked well, delivering the corresponding annulated products 3 in moderate to good yields (Table 3). Various functional groups, including chloride (3aa), bromide (3ab, 3ac), simple alkene (3ad), alkoxyl (3ae), and alkyl (3af) could be tolerated. The 2,3-trans stereochemistry of 3ac was confirmed by X-ray crystallographic analysis.21 A range of 6-aryl-substituted products bearing CF3, F, CN, and CO2Et motifs (3ag–3ak) were accessed in good yields. Furthermore, TsNH- and MeS-substituted phenols were capable of participating in this electrooxidative annulation, providing cyclized products 3al and 3am in synthetically useful yields.
|
|
|---|
|
a Standard reaction conditions: graphite rod anode, graphite rod cathode, constant current = 10 mA. 2a (0.3–0.4 mmol), 1 (1.5 equiv.), n-Bu4NBF4 (1.0 equiv.), HFIP/CH2Cl2 (6:4, 10 mL), rt, 2 h, under N2.
|
|
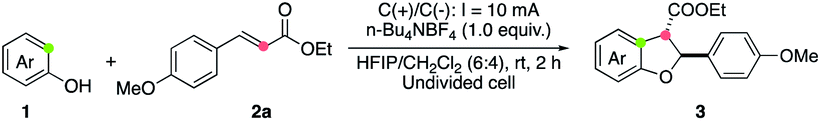
The synthetic utility of this electrooxidative [3 + 2] annulation protocol was demonstrated by a rapid synthesis of 3′,4-di-O-methylcedrusin (4), which is a natural product22 with a broad spectrum of bioactivity.2a,23 As depicted in Fig. 2, the annulation of 1n and 2b took place smoothly under the standard reaction conditions, affording 3bn in 42% yield. The following reduction of two ester groups with LiAlH4 provided product 4 directly in 76% yield. It should be noted that a commonly used method to assemble 4 relies on a four-step synthesis,24 including an oxidative annulation using stoichiometric amounts of oxidants to construct the 2,3-dihydrobenzofuran skeleton. The present two-step procedure offers a more straightforward and oxidant-free route, thereby enjoying more advantages in economical and green synthesis.
 | ||
| Fig. 2 Synthesis of 3′,4-di-O-methylcedrusin. | ||
To test the reaction mechanism of this oxidative annulation, several mechanistic studies have been performed. As illustrated in Fig. 3a, when 1o was subjected to the standard annulation reaction condition, its dimerization product 5 was isolated in 87% yield. This suggests that radical I should be generated by anodic oxidation, while its addition to electron rich 1o would proceed much faster than its addition to 2a,25 thereby affording dimer 5 without the detection of the cyclization product. DFT calculations were then carried out to gain deeper insights into the specific α-regioselectivity of the radical addition step (Fig. 3b).26 The addition of radical II to the α-position of 2a requires a much lower activation free energy than addition to the β-position (TS-a-I +24.0 kcal mol−1versusTS-b-I +30.4 kcal mol−1). Besides, the resulting Int-a-II (−0.6 kcal mol−1) is more stable than Int-b-II (+3.8). Furthermore, the subsequent single-electron oxidation of Int-a-II to a carbon cation should be an energetically favorable process, owing to the strong stabilization from the adjacent para-methoxyphenyl group. As a result, the α-addition pathway is thermodynamically and kinetically more favored than the β-addition pathway, thereby ensuring exclusive radical α-regioselectivity.
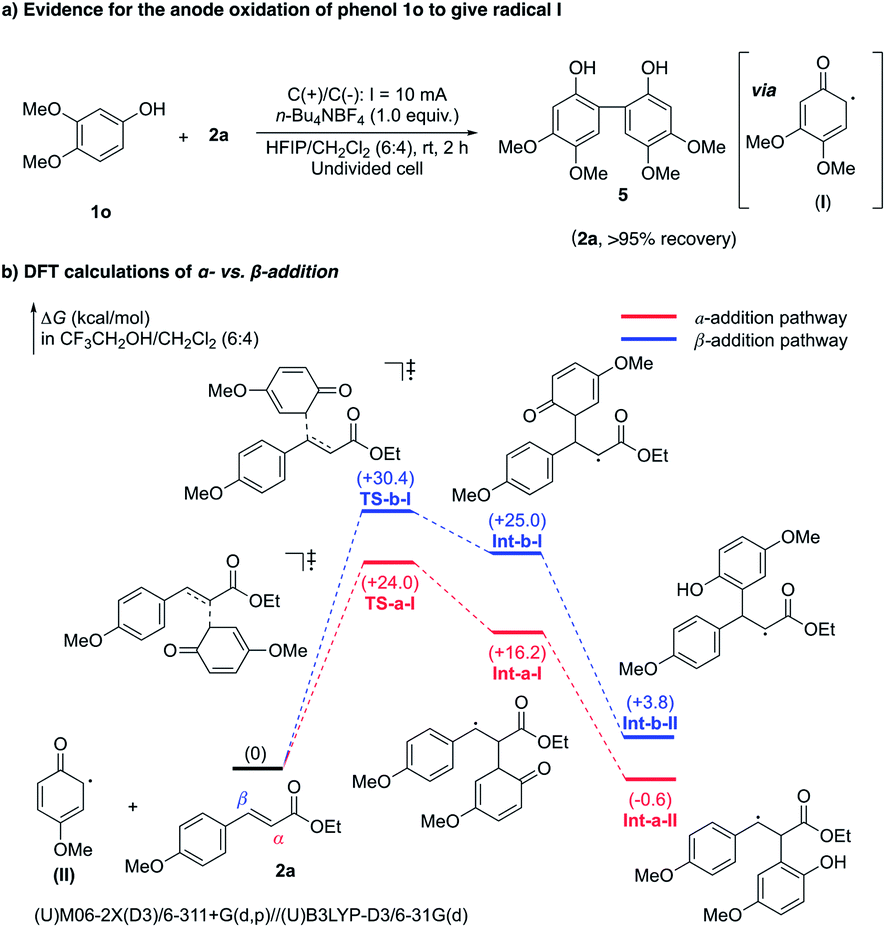 | ||
| Fig. 3 Mechanistic studies. | ||
Conclusions
In summary, we have developed a practical and green electrooxidative [3 + 2] annulation of phenols and electron-deficient alkenes. This protocol enables the construction of C3-functionalized 2,3-dihydrobenzofuran derivatives from simple starting materials. These products are difficult to access using the reported oxidative annulation methods. The key to success lies in a kinetically and thermodynamically favorable regioselective α-addition of carbon radicals to electron-deficient alkenes. Such specific α-regioselectivity as well as the electrooxidative annulation method may inspire the design of new radical approaches for further synthetic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21672195, 21702201, and 21971226), the Fundamental Research Funds for the Central Universities (WK2060190082), and the University of Science and Technology of China. The numerical calculations in this study have been done on the supercomputing system in the Supercomputing Center of the University of Science and Technology of China.Notes and references
- (a) S. Apers, A. Vlietinck and L. Pieters, Phytochem. Rev., 2003, 2, 201–217 CrossRef; (b) G. Q. Shi, J. F. Dropinski, Y. Zhang, C. Santini, S. P. Sahoo, J. P. Berger, K. L. MacNaul, G. Zhou, A. Agrawal, R. Alvaro, T.-Q. Cai, M. Hernandez, S. D. Wright, D. E. Moller, J. V. Heck and P. T. Meinke, J. Med. Chem., 2005, 48, 5589–5599 CrossRef PubMed; (c) E. D. Coy, L. E. Cuca and M. Sefkow, Bioorg. Med. Chem. Lett., 2009, 19, 6922–6925 CrossRef PubMed.
- (a) L. Pieters, S. Van Dyck, M. Gao, R. Bai, E. Hamel, A. Vlietinck and G. Lemière, J. Med. Chem., 1999, 42, 5475–5481 CrossRef PubMed; (b) R.-W. Jiang, K.-M. Lau, P.-M. Hon, T. C. W. Mak, K.-S. Woo and K.-P. Fung, Curr. Med. Chem., 2005, 12, 237–246 CrossRef; (c) S. Tewtrakul, H. Miyashiro, N. Nakamura, M. Hattori, T. Kawahata, T. Otake, T. Yoshinaga, T. Fujiwara, T. Supavita, S. Yuenyongsawad, P. Rattanasuwon and S. Dej-Adisai, Phytother Res., 2003, 17, 232–239 CrossRef PubMed; (d) H. Zhang, S. Qiu, P. Tamez, G. T. Tan, Z. Aydogmus, N. V. Hung, N. M. Cuong, C. Angerhofer, D. Doel Soejarto, J. M. Pezzuto and H. H. S. Fong, Pharm. Biol., 2002, 40, 221–224 CrossRef.
- (a) F. Bertolini and M. Pineschi, Org. Prep. Proced. Int., 2009, 41, 385–418 CrossRef; (b) Z. Chen, M. Pitchakuntla and Y. Jia, Nat. Prod. Rep., 2019, 36, 666–690 RSC.
- (a) W. Kurosawa, T. Kan and T. Fukuyama, Synlett, 2003, 1028–1030 Search PubMed; (b) W. Kurosawa, T. Kan and T. Fukuyama, J. Am. Chem. Soc., 2003, 125, 8112–8113 CrossRef; (c) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef; (d) Y. Koizumi, H. Kobayashi, T. Wakimoto, T. Furuta, T. Fukuyama and T. Kan, J. Am. Chem. Soc., 2008, 130, 16854–16855 CrossRef; (e) H. Wang, G. Li, K. M. Engle, J.-Q. Yu and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6774–6777 CrossRef.
- (a) M. Mascal, K. V. Modes and A. Durmus, Angew. Chem., Int. Ed., 2011, 50, 4445–4446 CrossRef; (b) H. Qin, Z. Xu, Y. Cui and Y. Jia, Angew. Chem., Int. Ed., 2011, 50, 4447–4449 CrossRef; (c) W. Hu, H. Qin, Y. Cui and Y. Jia, Chem.–Eur. J., 2013, 19, 3139–3147 CrossRef.
- S. J. O'Malley, K. L. Tan, A. Watzke, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 13496–13497 CrossRef.
- (a) D. Sun, Q. Zhao and C. Li, Org. Lett., 2011, 13, 5302–5305 CrossRef; (b) K. Liang, J. Yang, X. Tong, W. Shang, Z. Pan and C. Xia, Org. Lett., 2016, 18, 1474–1477 CrossRef PubMed.
- (a) J. Fischer, G. P. Savage and M. J. Coster, Org. Lett., 2011, 13, 3376–3379 CrossRef; (b) L. Qin, D.-D. Vo, A. Nakhai, C. D. Andersson and M. Elofsson, ACS Comb. Sci., 2017, 19, 370–376 CrossRef.
- R. P. Pandit, S. T. Kim and D. H. Ryu, Angew. Chem., Int. Ed., 2019, 58, 13427–13432 CrossRef PubMed.
- For selected recent examples, see: (a) T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 11056–11059 CrossRef PubMed; (b) T. Tomakinian, R. Guillot, C. Kouklovsky and G. Vincent, Angew. Chem., Int. Ed., 2014, 53, 11881–11885 CrossRef; (c) K. C. Nicolaou, S. M. Dalby, S. Li, T. Suzuki and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2009, 48, 7616–7620 CrossRef CAS PubMed; (d) C. Chan, C. Li, F. Zhang and S. J. Danishefsky, Tetrahedron Lett., 2006, 47, 4839–4841 CrossRef CAS; (e) K. Liu, S. Tang, P. Huang and A. Lei, Nat. Commun., 2017, 8, 775 CrossRef; (f) N. Denizot, A. Pouilhès, M. Cucca, R. Beaud, R. Guillot, C. Kouklovsky and G. Vincent, Org. Lett., 2014, 16, 5752–5755 CrossRef CAS; (g) R. Beaud, R. Guillot, C. Kouklovsky and G. Vincent, Angew. Chem., Int. Ed., 2012, 51, 12546–12550 CrossRef CAS.
- (a) B. D. Gates, P. Dalidowicz, A. Tebben, S. Wang and J. S. Swenton, J. Org. Chem., 1992, 57, 2135–2143 CrossRef CAS; (b) M. L. Kerns, S. M. Conroy and J. S. Swenton, Tetrahedron Lett., 1994, 35, 7529–7532 CrossRef.
- K. Chiba, M. Fukuda, S. Kim, Y. Kitano and M. Tada, J. Org. Chem., 1999, 64, 7654–7656 CrossRef.
- (a) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377–10441 CrossRef; (b) P. Renaud and M. Gerster, Angew. Chem., Int. Ed., 1998, 37, 2562–2579 CrossRef.
- (a) F. Gu, W. Huang, X. Liu, W. Chen and X. Cheng, Adv. Synth. Catal., 2018, 360, 925–931 CrossRef; (b) S. Martinet, A. Méou and P. Brun, Eur. J. Org. Chem., 2009, 2306–2311 CrossRef CAS.
- (a) G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491–3495 CrossRef CAS; (b) H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881–1884 CrossRef CAS; (c) S. Martinet, A. Méou and P. Brun, Eur. J. Org. Chem., 2009, 2306–2311 CrossRef CAS; (d) Z. Li, Z. Cui and Z.-Q. Liu, Org. Lett., 2013, 15, 406–409 CrossRef CAS; (e) P. Xu, A. Abdukader, K. Hu, Y. Cheng and C. Zhu, Chem. Commun., 2014, 50, 2308–2310 RSC.
- (a) S.-C. Ren, F.-L. Zhang, A.-Q. Xu, Y. Yang, M. Zheng, X. Zhou, Y. Fu and Y.-F. Wang, Nat. Commun., 2019, 10, 1934 CrossRef; (b) Y.-S. Huang, J. Wang, W.-X. Zheng, F.-L. Zhang, Y.-J. Yu, M. Zheng, X. Zhou and Y.-F. Wang, Chem. Commun., 2019, 55, 11904–11907 RSC.
- (a) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef PubMed; (b) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef; (c) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef PubMed; (d) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef; (e) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292–301 CAS; (f) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (g) Z.-W. Hou, Z.-Y. Mao and H.-C. Xu, Synlett, 2017, 28, 1867–1872 CrossRef CAS.
- (a) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (b) P. Feng, G. Ma, X. Chen, X. Wu, L. Lin, P. Liu and T. Chen, Angew. Chem., Int. Ed., 2019, 58, 8400–8404 CrossRef CAS; (c) S. Tang, S. Wang, Y. Liu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737–4741 CrossRef CAS; (d) R. Mei, J. Koeller and L. Ackermann, Chem. Commun., 2018, 54, 12879–12882 RSC; (e) A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef CAS; (f) A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef CAS; (g) J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef PubMed; (h) Z. Shi, Y. Wang, B. Tian and M. Ding, Chem.–Eur. J., DOI:10.1002/chem.201904750.
- M.-A. Constantin, J. Conrad and U. Beifuss, Green Chem., 2012, 14, 2375–2379 RSC.
- X. S. Liang, R. D. Li and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58, 13885–13889 CrossRef CAS.
- CCDC 1968793 contains the supplementary crystallographic data for this paper.
- L. Pieters, T. De Bruyne, M. Claeys, A. Vlietinck, M. Calomme and D. vanden Berghe, J. Nat. Prod., 1993, 56, 899–906 CrossRef CAS PubMed.
- (a) S. Apers, D. Paper, J. Bürgermeister, S. Baronikova, S. Van Dyck, G. Lemière, A. Vlietinck and L. Pieters, J. Nat. Prod., 2002, 65, 718–720 CrossRef CAS PubMed; (b) S. V. Miert, S. V. Dyck, T. J. Schmidt, R. Brun, A. Vlietinck, G. Lemière and L. Pieters, Bioorg. Med. Chem., 2005, 13, 661–669 CrossRef PubMed.
- G. Lemière, M. Gao, A. De Groot, R. Dommisse, J. Lepoivre, L. Pieters and V. Buss, J. Chem. Soc., Perkin Trans. 1, 1995, 1775–1779 RSC.
- A. Kirste, G. Schnakenburg and S. R. Waldvogel, Org. Lett., 2011, 13, 3126–3129 CrossRef CAS PubMed.
- For caculation details and references, see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1968793. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01078b |
| This journal is © The Royal Society of Chemistry 2020 |