
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteric and stereoscopic disulfide construction for cross-linkage via N-dithiophthalimides†‡
Wen-Chao
Gao
 ab,
Jun
Tian
b,
Yu-Zhu
Shang
b and
Xuefeng
Jiang
*ac
ab,
Jun
Tian
b,
Yu-Zhu
Shang
b and
Xuefeng
Jiang
*ac
aShanghai Key Laboratory of Green Chemistry and Chemical Process, Department of Chemistry, East China Normal University, Shanghai, 200062, P. R. China. E-mail: xfjiang@chem.ecnu.edu.cn
bCollege of Biomedical Engineering, Taiyuan University of Technology, Taiyuan, 030024, P. R. China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai, 200032, P. R. China
First published on 20th March 2020
Abstract
Disulfide bonds are a significant motif in life and drug-delivery systems. In particular, steric hindrance and stereoscopic disulfide linkers are closely associated with the stability of antibody–drug conjugates, which affects the potency, selectivity, and pharmacokinetics of drugs. However, limited availability and diversity of tertiary thiols impede the construction of steric and stereoscopic disulfides for cross-linkage in biochemistry and pharmaceuticals. Through modulating the mask effect of disulfurating reagents, we develop a facile and robust strategy for construction of diverse steric and stereoscopic disulfides via N-dithiophthalimides. The practical cross-linkage of biomolecules including amino acids, saccharides, and nucleosides with different drugs and fluorescent molecules is successfully established through hindered disulfide linkers.
Introduction
Development of targeted delivery conjugates, such as antibody–drug conjugates (ADCs)1,2 and small molecule–drug conjugates (SMDCs),3 has become a promising direction in modern drug development. The nature of linkers between targeting moieties and drugs plays the key role in the therapeutic efficacy of drug conjugates.4 As a commonly used linker, disulfides can be cleaved via an intracellular thiol-redox process for procedural drug release.5 Thus, surroundings of disulfide linkers significantly affect the potency, selectivity, and pharmacokinetics of conjugates.6,7 Due to the embedded cavity and chiral environment in cellular redox systems, the stability of disulfide linkers mainly relies on the adjacent steric and stereoscopic effects (Scheme 1a).8 Clinically approved antibody–drug conjugates such as Mylotarg and Besponsa demonstrated that gem-dimethyl-hindered disulfide linkers enhanced the stability of conjugates in internal circulation and the selectivity of payload delivery in cytosol.2 Furthermore, these effects were found in anti-cancer natural products9 and peptides10 for the expression of cytoxicity and receptor-selectivity as well.11,12 Therefore, efficient construction of steric and stereoscopic disulfides for molecule cross-linkage is urgently demanded. Although substitution of tertiary thiols with preactivated S-partners is conventionally employed for the synthesis of steric disulfides (Scheme 1b),13 adjacent variation of hindrance, stereocenter construction and molecule-cross-linkage remain underdeveloped.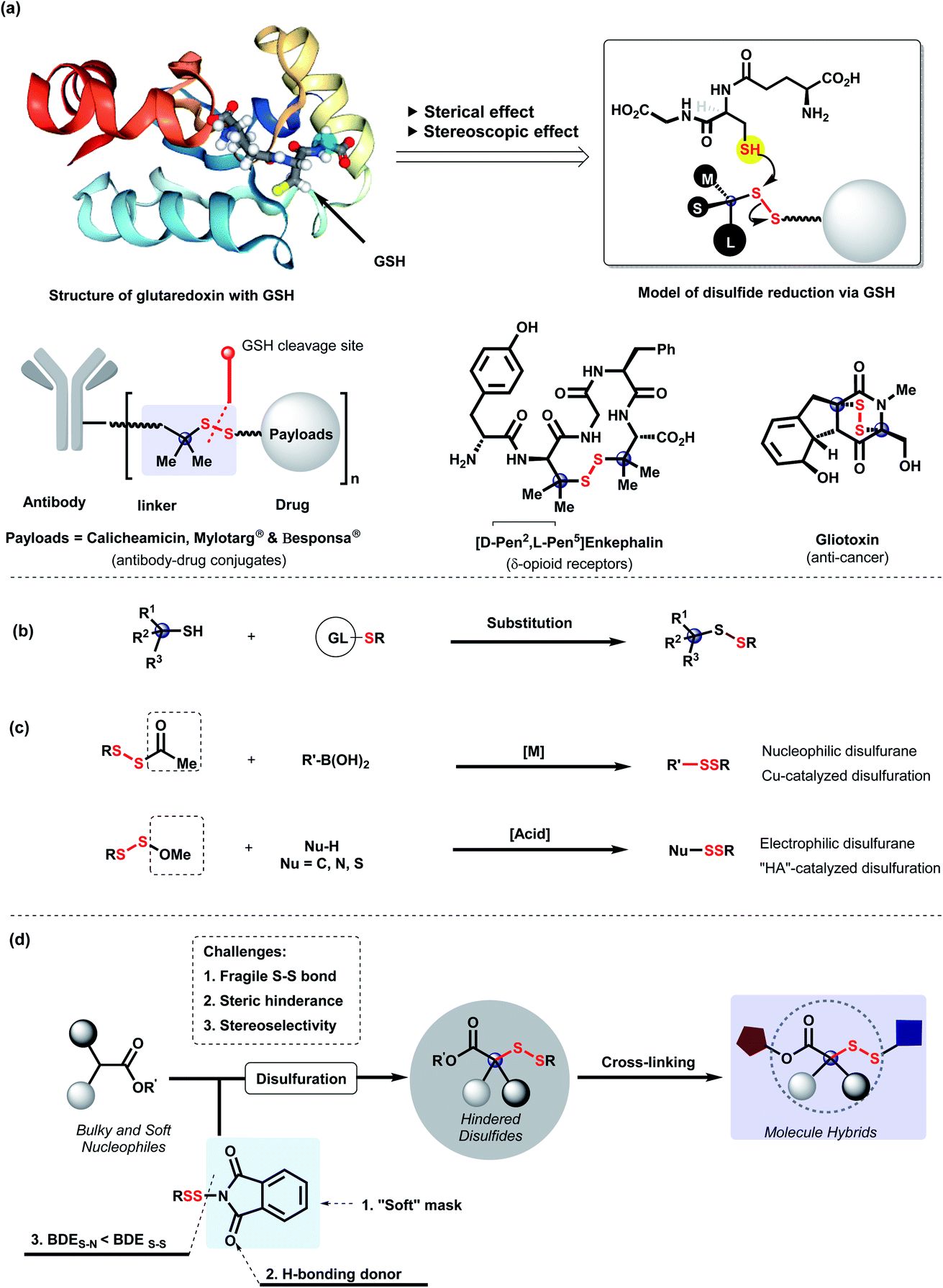 | ||
| Scheme 1 (a) Significance of steric and stereoscopic disulfdies. (b) Substitution of tertiary thiol. (c) Disulfuration via mask strategy. (d) Disulfuration for steric and stereoscopic disulfide construction. | ||
Direct disulfuration via designed disulfurating reagents has become a better choice due to their straightforward facility for unsymmetrical disulfide and late-stage modification. To meet diverse requirements, masked disulfurating reagents (RSSAc and RSSOMe) were developed by us to incorporate RSS moieties into multifarious systems (Scheme 1c).14,15 Considering the great importance of steric and stereoscopic disulfides, we are intrigued by disulfuration of α-branched esters to access diverse hindered disulfides. Nevertheless, steric hindrance of α-branched esters is the first barrier for disulfuration. Furthermore, due to the flexibility and volatility of disulfuryl groups16 and the relatively longer distance between substituents on disulfides and the adjacent carbon center, three-dimensional induction of disulfides is confronted with great challenge. We envisage that disulfurating reagents, N-dithiophthalimides with “soft” mask phthalimide,17–19 enable the facilitation of the disulfuration of branched soft nucleophiles based on the hard and soft acid and base (HSAB) model.20 Moreover, the carbonyl groups from imide are capable of serving as H-bonding donors to assist the chiral induction.21 Herein, we disclose a straightforward disulfurating strategy for construction of steric and stereoscopic disulfides via N-dithiophthalimides, which is a splendid cross-linkage of various functional molecules for molecule hybrids (Scheme 1d).
Results and discussion
Collective N-phthalimide-based sulfurating reagent library
Although successful protocols for N-dithiophthalimide (PhthSSR) synthesis have been reported,18,19 moisture-sensitive PhthSCl makes scalable preparation and application burdensome. A revised protocol affords a solid scale-up procedure: pretreatment of thiols with N,N′-thiobisphthalimide (1.5 equiv.) in 1,2-dichloroethane at 80 °C afforded PhthSSR (1) in moderate to excellent yields on a 2–3 mmol scale (Table 1). The structure of phthalimide-based disulfurating reagents was further confirmed by the X-ray analysis of 1a. A variety of aromatic substituents, including electron-donating and -withdrawing groups on the phenyl ring (1a and 1b), naphthyl (1c and 1d), and thienyl (1e) rings, were well tolerated in this reaction. Besides, diverse aliphatic disulfurating reagents including substituted benzyl (1f and 1g), tert-butyl (1h), cyclohexyl (1i), alkenyl (1j) and alkynyl (1k) groups could be smoothly constructed. Furthermore, phthalimide-based disulfurating reagents containing cross-linking groups (carboxyl, hydroxyl, borono and hydroxysuccinimide) were successfully prepared in high yields under the standard conditions (1l–q). The compatibility of various linkage-containing groups provides an excellent basis for molecule conjugation and hybridization.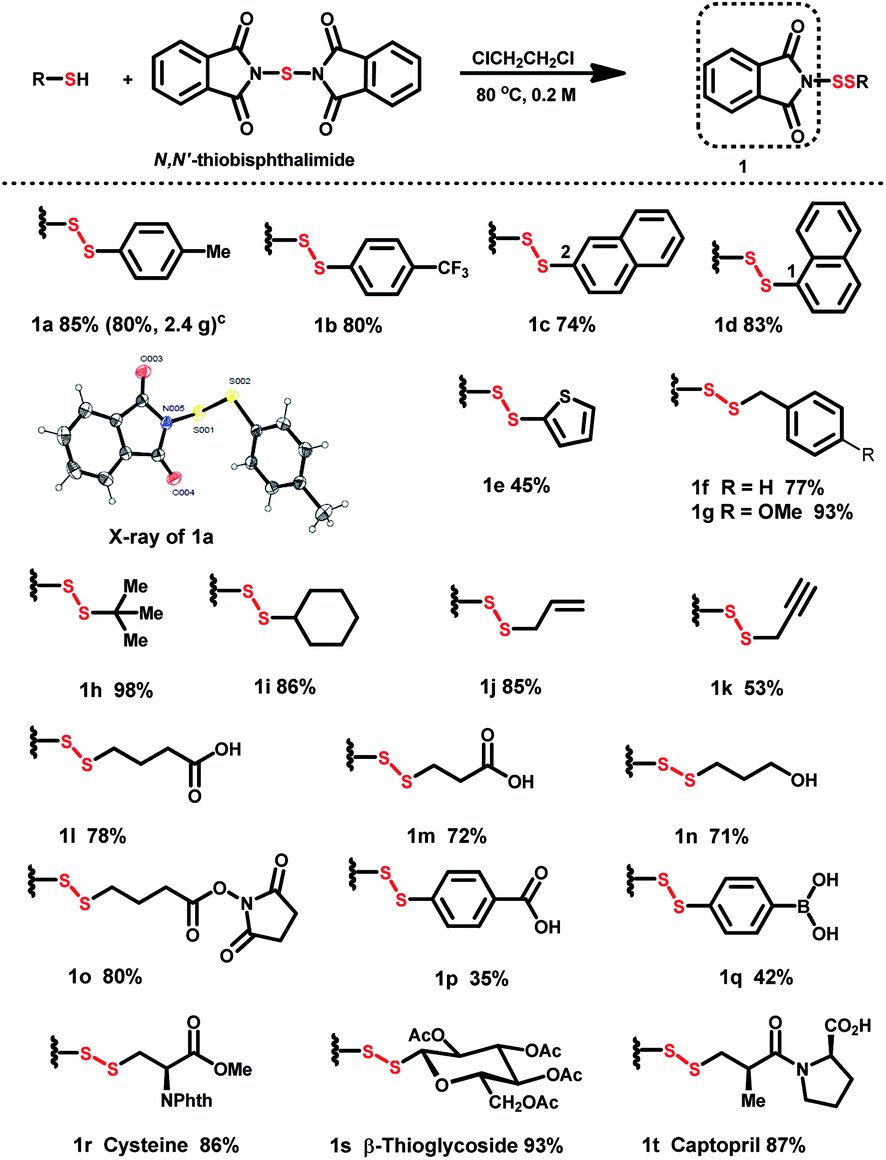
Thiol-containing biomolecules such as cysteine, β-thioglycoside, and anti-hypertension drug captopril were converted to the desired disulfurating reagents in high yields (1r–t). Notably, all these N-dithiolphathalimides can be stored at room temperature (25 °C) for 4–6 months without any detectable deterioration.
Sterically bulky disulfides
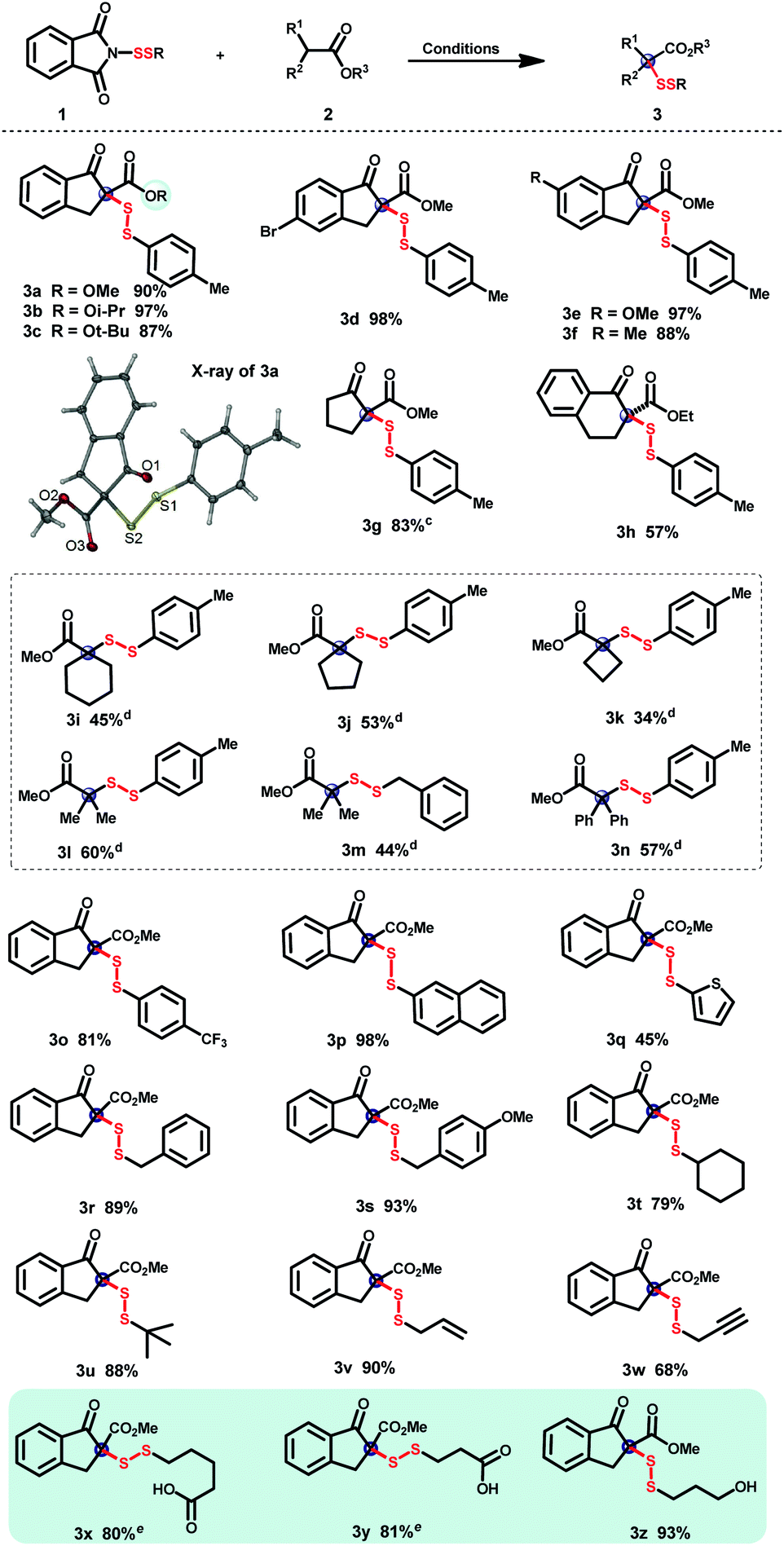
With the large-scale N-dithiophthalimide library in hand, the construction of quaternary α-disulfuryl β-keto esters was consequently explored. As shown in Table 2, disulfuration of various β-keto esters was first examined using DMAP as a catalyst. Diverse scales of ester groups were tolerated in the current disulfuration reaction (3a–c), whose structure was further confirmed by the X-ray analysis of 3a. Five-membered ring β-keto esters with electron-donating and -withdrawing groups on the phenyl ring were well compatible in this transformation affording the corresponding products in excellent yields (3d–f). Aliphatic cyclopentanone-derived β-keto ester was also successfully transformed into the desired product 3g in good yield. As for the β-keto ester bearing a six-membered-ring, the product 3h was obtained in 57% yield. Unfortunately, acyclic 1,3-dicarbonyl substrates such as 2-methyl-ethylbenzoylacetate, 3-methylpentane-2,4-dione, or 2-methyl-diethylmalonate failed to afford the desired steric disulfides. Notably, various gem-substituent-hindered disulfides could be readily afforded with silyl ketene acetals as nucleophiles.22 Incorporation of disulfuryls into silyl ketene acetals with spiro-rings such as spirocyclohexyl (3i), spirocyclopentyl (3j), and spirocyclobutyl (3k) is delightly compatible, providing a rapid protocol for spiro-hindered disulfide synthesis. Moreover, not only gem-dimethyl-hindered disulfides (3l and 3m), but also gem-diphenyl-hindered one (3n) could be well constructed through this disulfuration reaction in moderate yields. A broad range of N-dithiophthalimides, regardless of electron-donating or -withdrawing groups on the phenyl ring, efficiently transformed the disulfur motifs into the desired products (3o–s). Aliphatic disulfurating reagents, bearing adjacent tertiary and quaternary carbon centers (3t and 3u), could be transformed into the desired disulfide products with bilateral hindrance efficiently. The successful incorporation of the allylic disulfuryl motif demonstrated the good compatibility of the C–C double bond (3v). Disulfides with the terminal C–C triple bond (3w) could be also tolerated in this transformation, enabling the further linkage via the click reaction.23 Notably, free carboxyl (3x and 3y) and hydroxyl (3z) were well compatible, which provided more chances for further molecule cross-linkage.| a Reaction conditions: β-keto ester (0.1 mmol), N-dithiophthalimide (0.12 mmol) and DMAP (0.02 mmol) in CH2Cl2 (1 mL) were stirred at room temperature for 12–24 h. b Isolated yields. c TMS enol ethers were used instead of the corresponding β-keto esters. d Reaction conditions: TMS ketene acetal (0.2 mmol), N-dithiophthalimide (0.4 mmol), and DMAP (0.04 mmol) in THF (2 mL) were stirred at room temperature for 8–12 h. e 0.1 mmol of DMAP were used. |
|---|
|
Sterically bulky disulfide cross-linkage
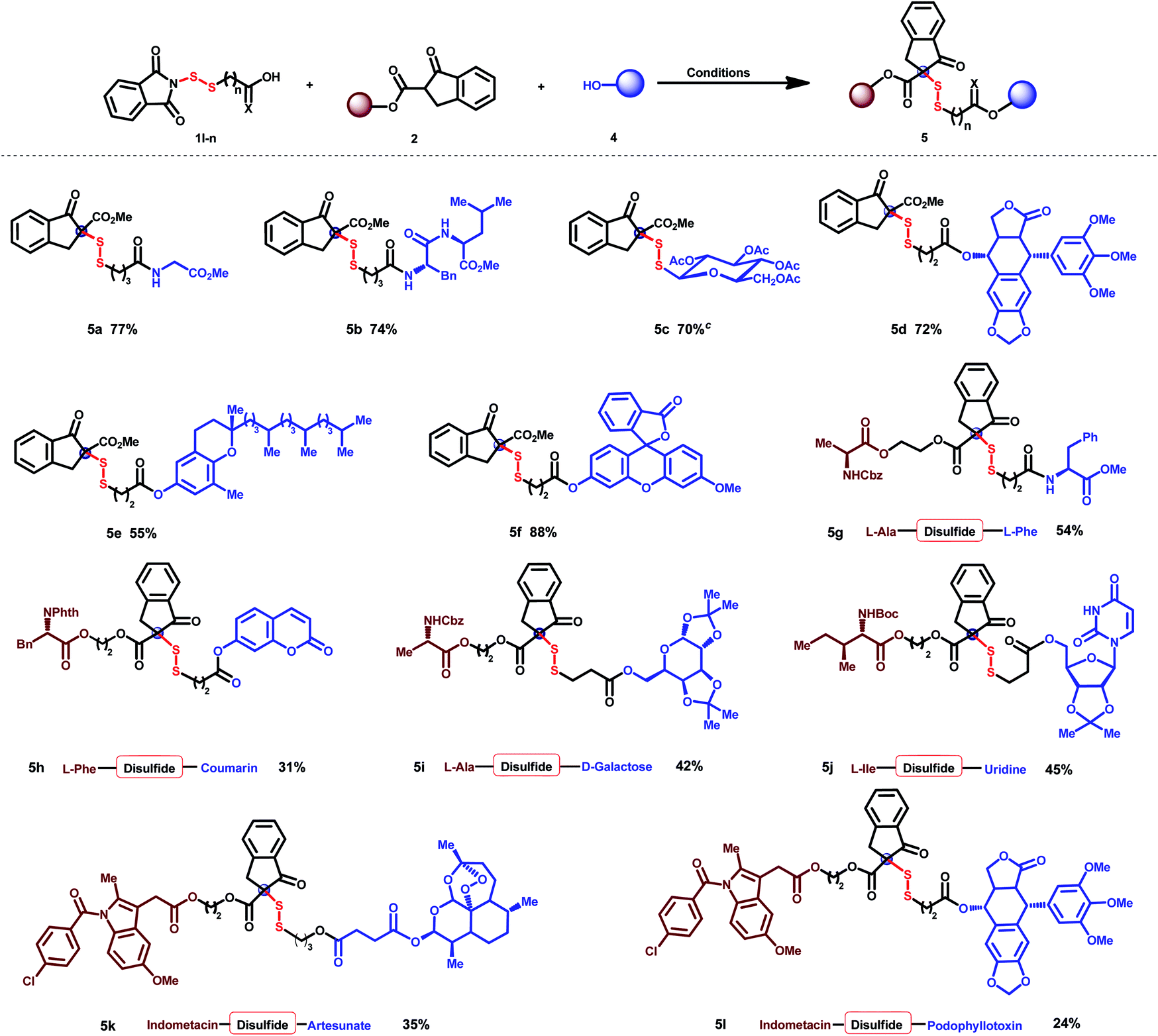
To further demonstrate the practicality of the present method, steric disulfides were comprehensively applied for molecule hybridization (Table 3).24 Biomolecules such as amino acids (5a), dipeptides (5b), and β-thiosaccharides (5c) can be efficiently linked with β-keto esters via this steric disulfide. The anti-cancer drug podophyllotoxin (5d), antioxidation natural product δ-tocopherol (5e), and molecule probe fluorescein (5f) were efficiently linked with β-keto esters through one-pot disulfuration and esterification. Hybridization of diverse amino acids (5g), amino acid with fluorescent molecules (5h), amino acid with saccharide (5i), and amino acid with nucleoside (5j) was all successfully established, providing possibilities to modify protein structures, improve pharmacological efficacy and overcome drug resistance.25,26 Non-steroidal anti-inflammatory drug indomethacin could be successfully cross-linked with antimalarial drug artesunate (5k) and anti-cancer drug podophyllotoxin (5l), which may serve as a dual-mode for the treatment of diseases.| a Conditions: 1 (0.1 mmol), 2a (0.1 mmol), and DMAP (1.1 equiv.) in CH2Cl2 (2 mL) were stirred at room temperature for 12 h; then 4 (1 equiv.), HOBt (1.5 equiv.), EDCI (1.5 equiv.) and DIPEA (2.5 equiv.) were added, and the mixture was stirred at room temperature overnight. b Isolated yields. c Conditions: 1s (0.12 mmol), 2a (0.1 mmol) and DMAP (0.02 mmol) in CH2Cl2 (1 mL) were stirred at room temperature for 12 h. |
|---|
|
Stereoscopic disulfide construction
To create the three-dimensional environment for discrimination and recognition of biomolecules, construction of stereoscopic disulfides was subsequently explored. The strong coordinating properties of unstable disulfuryl groups obstruct the chiral induction with metal catalysts.27 Considering the existence of H-bonding acceptors in both β-keto esters and N-phthalimides,28 chiral organic molecules based on cinchona alkaloids were tested as a catalyst for the desired enantioselective induction.29,30 The best result was obtained when the disulfuration was performed in dimethoxyethane (DME) with (DHQD)2PHAL as a catalyst (Table 4).31 Both aromatic and aliphatic disulfur motifs could be transformed into the desired disulfides in high yields with good to excellent enantioselectivities (6a–f). Notably, the enantiomeric excess of 6c could be improved to 98% after recrystallization once. The absolute configuration R of the stereogenic carbon center in disulfuryl products was determined by X-ray crystal structure analysis of the optically active product 6d.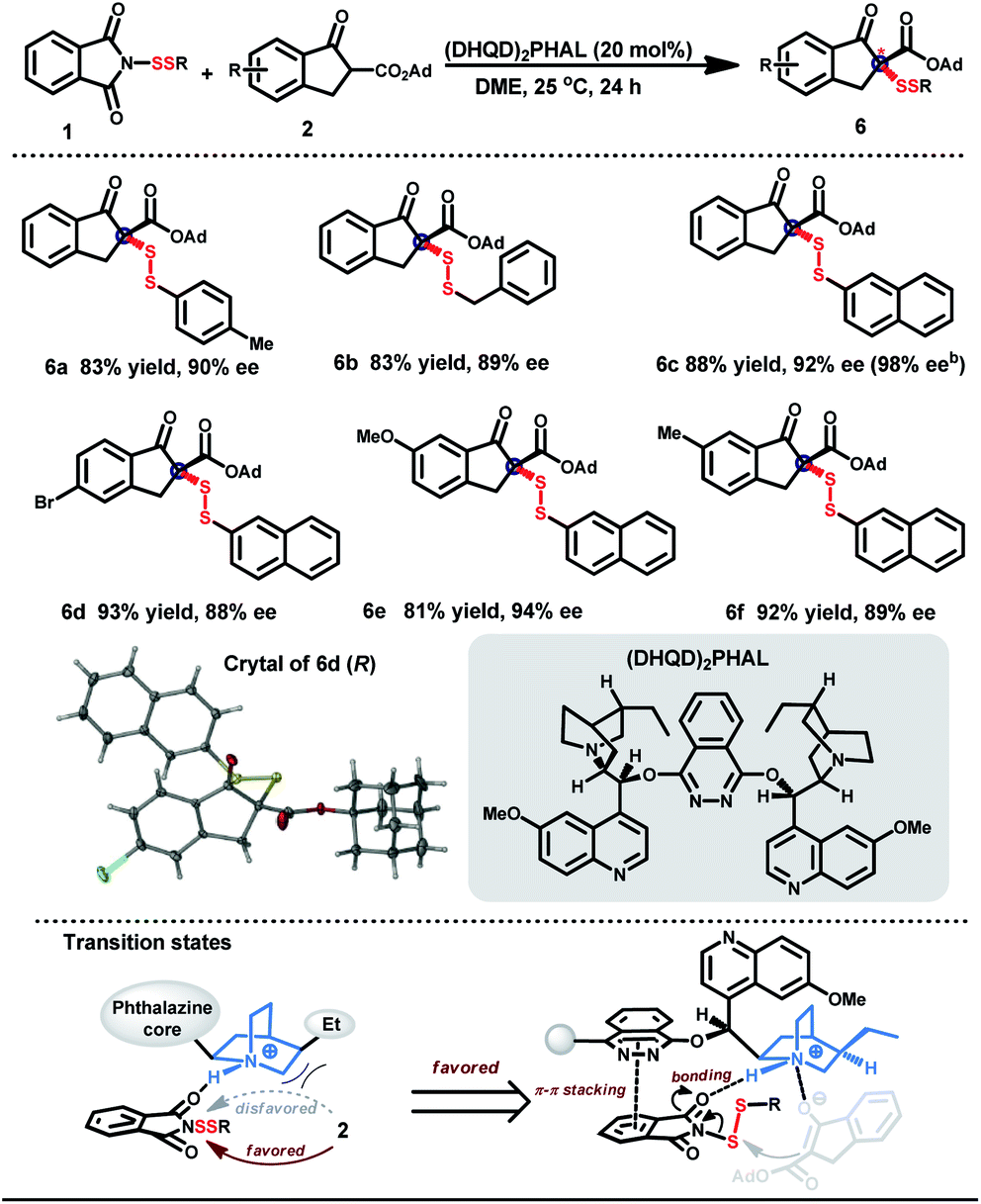
Conclusions
In summary, an efficiently and generally applicable protocol for the construction of steric and stereoscopic disulfides has been developed via disulfuration of α-branched esters with N-dithiophthalimides as disulfurating reagents. Steric disulfides with subtle variation of adjacent hindered groups are comprehensively accessed with high functional-group tolerance, facilitating the structural regulation of disulfuryl linkage. Enantioselective construction of stereoscopic disulfides is also realized, which can provide chiral disulfide candidates for biological recognition. With design and synthesis of steric disulfide linkers, hybridization of various biomolecules, drugs and fluorescent tags is well linked by reversibly fractured disulfides, which provides synthetic basis for mimic of life processes and pharmacological therapy. Efforts toward the drug discovery of steric and stereoscopic disulfides are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support provided by the National Key Research and Development Program of China (2017YFD0200500), NSFC (21971065, 21722202, 21672069, and 21901179 for W.-C. Gao), S&TCSM of Shanghai (Grant 18JC1415600), Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, and the National Program for Support of Top-notch Young Professionals, China Postdoctoral Science Foundation (No. 2019M651424) and the Key R.& D. Program of Shanxi Province (No. 201803D421093).Notes and references
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef PubMed.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef PubMed.
- N. Krall, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2013, 52, 1384–1402 CrossRef CAS PubMed.
- K. Tsuchikama and Z. An, Protein Cell, 2018, 9, 33–46 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- R. Y. Zhao, S. D. Wilhelm, C. Audette, G. Jones, B. A. Leece, A. C. Lazar, V. S. Goldmacher, R. Singh, Y. Kovtun, W. C. Widdison, J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2011, 54, 3606–3623 CrossRef CAS.
- G. Saito, J. A. Swansonb and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS.
- J. D. Sadowsky, T. H. Pillow, J. Chen, F. Fan, C. He, Y. Wang, G. Yan, H. Yao, Z. Xu, S. Martin, D. Zhang, P. Chu, J. Cruz-Chuh, A. O'Donohue, G. Li, G. D. Rosario, J. He, L. Liu, C. Ng, D. Su, G. D. L. Phillips, K. R. Kozak, S.-F. Yu, K. Xu, D. Leipold and J. Wai, Bioconjugate Chem., 2017, 28, 2086–2098 CrossRef CAS.
- A. Marion, M. Groll, D. H. Scharf, K. Scherlach, M. Glaser, H. Sievers, M. Schuster, C. Hertweck, A. A. Brakhage, I. Antes and E. M. Huber, ACS Chem. Biol., 2017, 12, 1874–1882 CrossRef CAS.
- H. I. Mosberg, R. Hurst, V. J. Hruby, K. Gee, H. I. Yamamura, J. J. Galligan and T. F. Burks, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 5871–5874 CrossRef CAS.
- N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC.
- J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079–1087 CrossRef CAS.
- Z. Pei, C. Chen, J. Chen, J. Cruz-Chuh, R. Delarosa, Y. Deng, A. Fourie-O'Donohue, I. Figueroa, J. Guo, W. Jin, S. C. Khojasteh, K. R. Kozak, B. Latifi, J. Lee, G. Li, E. Lin, L. Liu, J. Lu, S. Martin, C. Ng, T. Nguyen, R. Ohri, G. L. Phillips, T. H. Pillow, R. K. Rowntree, N. J. Stagg, D. Stokoe, S. Ulufatu, V. A. Verma, J. Wai, J. Wang, K. Xu, Z. Xu, H. Yao, S.-F. Yu, D. Zhang and P. S. Dragovich, Mol. Pharmaceutics, 2018, 15, 3979–3996 CrossRef CAS.
- X. Xiao, M. Feng and X. Jiang, Angew. Chem., Int. Ed., 2016, 55, 14121–14125 CrossRef CAS.
- X. Xiao, J. Xue and X. Jiang, Nat. Commun., 2018, 9, 2191 CrossRef.
- M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef.
- D. N. Harpp, K. Steliou and T. H. Chan, J. Am. Chem. Soc., 1978, 100, 1222–1228 CrossRef.
- K. C. Nicolaou, R. Li, Z. Lu, E. N. Pitsinos, L. B. Alemany, M. Aujay, C. Lee, J. Sandoval and J. Gavrilyuk, J. Am. Chem. Soc., 2018, 140, 12120–12136 CrossRef.
- J. Zou, J. Chen, T. Shi, Y. Hou, F. Cao, Y. Wang, X. Wang, Z. Jia, Q. Zhao and Z. Wang, ACS Catal., 2019, 9, 11426–11430 CrossRef.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef.
- P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef.
- A. A. Kislukhin, V. P. Hong, K. E. Breitenkamp and M. G. Finn, Bioconjugate Chem., 2013, 24, 684–689 CrossRef.
- M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203–219 CrossRef.
- A. Çapcı, M. M. Lorion, H. Wang, N. Simon, M. Leidenberger, M. C. B. Silva, D. R. M. Moreira, Y. Zhu, Y. Meng, J. Y. Chen, Y. M. Lee, O. Friedrich, B. Kappes, J. Wang, L. Ackermann and S. B. Tsogoeva, Angew. Chem., Int. Ed., 2019, 58, 13066–13079 CrossRef.
- L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996–4028 CrossRef.
- Z. Qiao, J. Wei and X. Jiang, Org. Lett., 2014, 16, 1212–1215 CrossRef.
- T. Govender, P. I. Arvidsson, G. E. M. Maguire, H. G. Kruger and T. Naicker, Chem. Rev., 2016, 116, 9375–9437 CrossRef.
- X. Wang, T. Yang, X. Cheng and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 12860–12864 CrossRef.
- T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856–12859 CrossRef.
- For details of condition optimization, see the ESI‡.
Footnotes |
| † Dedicated to Prof. Eric Block for his 78th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedure, crystal data, and copies of NMR and HPLC spectra. CCDC 1975995–1975997. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01060j |
| This journal is © The Royal Society of Chemistry 2020 |