
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed asymmetric hydrophosphorylation of alkynes: facile access to P-stereogenic phosphinates†‡
Zhiping
Yang
ab,
Xiaodong
Gu
b,
Li-Biao
Han
 c and
Jun (Joelle)
Wang
*b
c and
Jun (Joelle)
Wang
*b
aHarbin Institute of Technology, Harbin 150080, China
bShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: wang.j@sustech.edu.cn
cNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki 305-8565, Japan
First published on 29th June 2020
Abstract
Despite the importance of P-chiral organophosphorus compounds in asymmetric catalysis, transition metal-catalyzed methods for accessing P-chiral phosphine derivatives are still limited. Herein, a catalytic enantioselective method for the synthesis of P-stereogenic alkenylphosphinates is developed through asymmetric hydrophosphorylation of alkynes. This process is demonstrated for a wide range of racemic phosphinates and leads to diverse P-stereogenic alkenylphosphinates directly.
Introduction
P-Chiral organophosphorus compounds are broadly utilized as synthetic building blocks of bioactive molecules1 and have served as an important class of chiral ligands that have significantly contributed to metal-catalyzed2 and organocatalytic3 transformations. P-stereogenic phosphinates are important molecules in medicinal and synthetic chemistry. For example, arylphosphinosugars have received continuous attention and demonstrated powerful activities on human cancer cell line panels.4 However, P-chiral organophosphorus compounds are less studied due to their synthetic challenges, compared with chiral phosphine ligands where planar or point chirality is presented in the carbon framework.Despite the importance of P-stereogenic phosphinates, general and efficient methods for their preparation are rather rare. Traditionally, enantioenriched P-chiral phosphorus compounds are achieved through the use of chiral reagents or auxiliary-assisted transformations, using menthol or chiral amino alcohol, for example.5 Recently, a variety of examples involving metal-catalyzed asymmetric processes through desymmetrization of prochiral phosphorus compounds have emerged.6–11 Dialkynylphosphine oxides are the typical examples for constructing P-stereogenic phosphine oxides,6 and the first desymmetrization of dialkynylphosphine oxides was reported by using Rh(I)-catalyzed cycloaddition.6a Desymmetrization of divinylphosphine oxides7 and phospholene oxides8 was also well-developed to construct P-stereogenic centers. Several elegant examples of inter- or intramolecular Pd-catalyzed enantioselective C–H arylation of phosphinamides, phosphonates and phosphine oxides were disclosed independently by Duan, Tang, Ma, Xu and Han.9 Soon after, Cramer reported Rh-catalyzed desymmetric alkynylation of phosphinamides with alkynes10a,b and Ir-catalyzed arylation and amination of phosphine oxides.10c,d Very recently, Zhang presented an asymmetric P–C cross-coupling for the efficient synthesis of P-stereogenic phosphine oxides catalyzed by Pd and their Xiao-phos.11 Nevertheless, there have been only two desymmetrization examples reported for the enantioselective synthesis of P-stereogenic phosphinates. In 2009, Hoveyda and Gouverneur reported a molybdenum-catalyzed asymmetric ring-closing metathesis to obtain P-stereogenic phosphinates (Scheme 1, eqn (2a)).12 In 2019, Trost showed the desymmetrization of phosphinic acids by stereoselectively alkylating one of the enantiotopic oxygens through Pd-catalyzed asymmetric allylic alkylation to give P-stereogenic phosphinates with diversified substituents (Scheme 1, eqn (2b)).13 Therefore, it is desirable to develop other new methods for the synthesis of multifunctional P-stereogenic phosphinates.
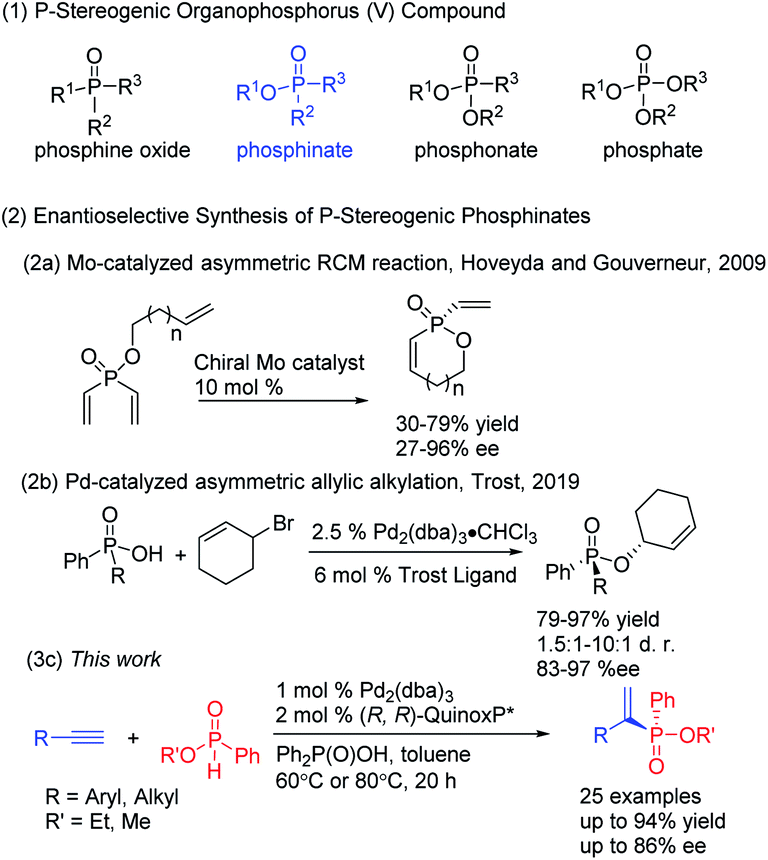 | ||
| Scheme 1 Enantioselective synthesis of P-stereogenic phosphinates. | ||
Pd-catalyzed addition of an H–P(O)R1R2 to alkynes is one of the most straightforward and atom-efficient approaches for the construction of a C–P bond.14 The first Pd-catalyzed addition of (RO)2P(O)H to alkynes was reported by Tanaka and Han to give the corresponding alkenylphosphonates.14a Later, they developed a similar oxidative addition using (RP)-menthyl-phenylphosphinate to give enantiomerically pure P-chiral alkenylphosphinates with retention of configuration at phosphorus.14b Though Han and co-workers reported a comprehensive study on the generality, scope, limitations, and mechanism of the palladium-catalyzed hydrophosphorylation of alkynes recently,14c the catalytic enantioselective hydrophosphorylation of alkynes with phosphinates is still not reported, given more than 22 years have passed since the first Pd-catalyzed hydrophosphorylation was reported. In 2006, Gaumont reported the Pd-catalyzed asymmetric hydrophosphination of alkynes with phosphine–boranes, only 70% conversion and 42% enantiomeric excess were obtained.15 In 2018, Dong reported the hydrophosphinylation of 1,3-dienes to afford chiral allylic phosphine oxides with high enantio- and regiocontrol.16 Herein, we disclose the first catalytic enantioselective hydrophosphorylation reaction of alkynes with phosphinates, which provides a highly efficient approach to prepare chiral alkenylphosphinates with P-chirality.
Results and discussion
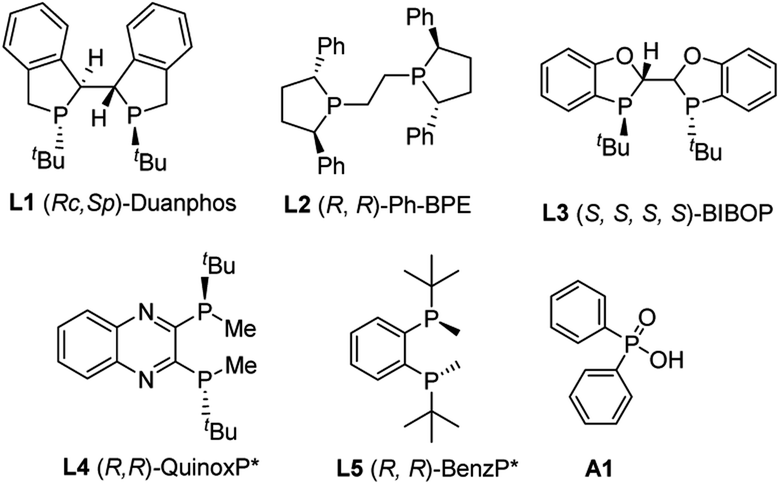
To begin the investigation, phenylacetylene 1a and ethyl phenylphosphinate 2a were chosen as the model substrates. Various types of ligands were initially evaluated, and most bidentate bisphosphine ligands with P chirality worked well in this transformation. When Duanphos L1 was used as the ligand, the reaction proceeded smoothly to afford alkenylphosphinate 3aa in 70% yield with 70% ee (Table 1, entry 1). (R,R)-Ph-BPE exhibited poor reactivity and enantioselectivity (Table 1, entry 2). (S,S,S,S)-BIBOP L3 showed good reactivity, yet no product enantioselectivity was observed (Table 1, entry 3). To our delight, (R,R)-QuinoxP* L4 afforded 3aa in 70% yield with 83% ee (Table 1, entry 4). A similar ligand (R,R)-benzP* L5 gave 86% ee but with poor yield (Table 1, entry 5). A higher reaction temperature was required to allow the reaction to reach completion when Pd(dba)2 was used (Table 1, entry 6). A brief survey of solvents revealed that THF, 1,4-dioxane and DCE resulted in inferior yields and enanatioselectivities (Table 1, entries 7–9). Unlike phosphinic acid and secondary phosphine oxide,17 phosphinate 2a was not able to be easily racemized by base or transition metals. Thus, it is difficult for phosphinate to realize the dynamic kinetic resolution. Then, a kinetic resolution process was desired (for details, see the ESI‡). However, when 1 equiv. of phosphinate was used, the product 3aa was obtained in 50% yield with 55% ee and the (R)-2a was recovered in 40% yield with 61% ee at 60 °C (the S factor is only 6) (Table 1, entry 10). Optimization of the ratio of 1a/2a was performed to enhance enantioselectivity of 3aa. When 4 equiv. 2a was used, the best yield and ee were obtained (Table 1, entry 4 vs. entries 10–13). When the amount of ethyl phenylphosphinate 2a was increased to 6 equiv., the yield was reduced which might due to the coordinative saturation of the palladium center by the excess amount of 2a, and hence resulted in catalyst deactivation (Table 1, entries 12 and 13). Omitting Ph2P(O)OH resulted in a reduced yield, but a little enhanced product enanatioselectivity (Table 1, entry 14 vs. 4). Thus, the optimal reaction conditions were toluene at 60 °C with 1 mol% Pd2(dba)3, 2 mol% (R,R)-QuinoxP*, and 4 mol% phosphinic acid.|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | Ligand | Solvent | Temp (°C) | Yield (%) | ee (%) |
| a Reaction conditions: 1 mol% Pd2(dba)3, 2 mol% ligand, and 4 mol% Ph2P(O)OH in 1 mL toluene were stirred for 10 min in an argon atmosphere. 0.25 mmol alkynes and 1.0 mmol ethyl phenylphosphinate were added, and the mixture was stirred at the indicated temperature. Isolated yields. Determined by HPLC analysis. b 2 mol% Pd(dba)2 was used instead of 1 mol% Pd2(dba)3. c 1 equiv. ethyl phenylphosphinate was used. d 3 equiv. ethyl phenylphosphinate was used. e 6 equiv. ethyl phenylphosphinate was used. f 2 mL toluene was used. g Without Ph2P(O)OH. | |||||
| 1 | L1 | Toluene | 60 | 70 | 70 |
| 2 | L2 | Toluene | 60 | Trace | — |
| 3 | L3 | Toluene | 60 | 85 | 7 |
| 4 | L4 | Toluene | 60 | 70 | 83 |
| 5 | L5 | Toluene | 60 | 11 | 86 |
| 6b | L4 | Toluene | 80 | 85 | 76 |
| 7 | L4 | THF | 60 | 68 | 75 |
| 8 | L4 | Dioxane | 60 | 54 | 81 |
| 9 | L4 | DCE | 60 | Trace | — |
| 10c | L4 | Toluene | 60 | 50 | 55 |
| 11d | L4 | Toluene | 60 | 59 | 78 |
| 12e | L4 | Toluene | 60 | 34 | 85 |
| 13e,f | L4 | Toluene | 60 | 44 | 86 |
| 14g | L4 | Toluene | 60 | 60 | 84 |

With these optimized conditions in hand, the reaction scope was next examined (Table 2). It was found that a large range of alkynes was applicable in this reaction system. The aryl alkynes substituted with electron-donating groups (MeO, Me, Et, n-pent, and t-Bu) (3ba–3ga, 3la, and 3ma) or electron-withdrawing groups (Cl, F, Br, and CF3) (3ha–3ka, and 3na) at the para- or meta-positions were all well-tolerated and gave satisfactory results. However, substrates with sterically demanding alkynes gave diminished enantioselectivity (3da, 91% yield, 30% ee). The arene rings having the –CN or –NO2 groups did not give the desired products. Substrates with thiophene or pyridine moieties worked well under these reaction conditions, providing the desired products 3oa and 3pa with moderate yield and good ee. Internal alkynes, diphenylacetylene, also gave moderate yield and ee (3qa, 69% yield, 61% ee). It was also demonstrated that aliphatic alkynes were functional and gave slightly decreased yields and good ee (3ra–3ta). Phosphinate reacted with a terminal alkyne prior to a disubstituted alkyne to give the enyne 3ta. As shown in Table 2, it was found that this transformation tolerated a variety of alkynes, including heteroaromatic alkynes, aliphatic alkynes and internal alkynes.
|
|
|---|
| a Conditions: 1 mol% Pd2(dba)3, 2 mol% ligand L4, and 4 mol% Ph2P(O)OH in 1 mL toluene were stirred for 10 min in an argon atmosphere. 0.25 mmol alkynes 1b–1t and 1.0 mmol 2a were added, and the mixture was stirred at 60 °C for 20 h. Isolated yields. Determined by HPLC analysis. b 80 °C was used instead. |
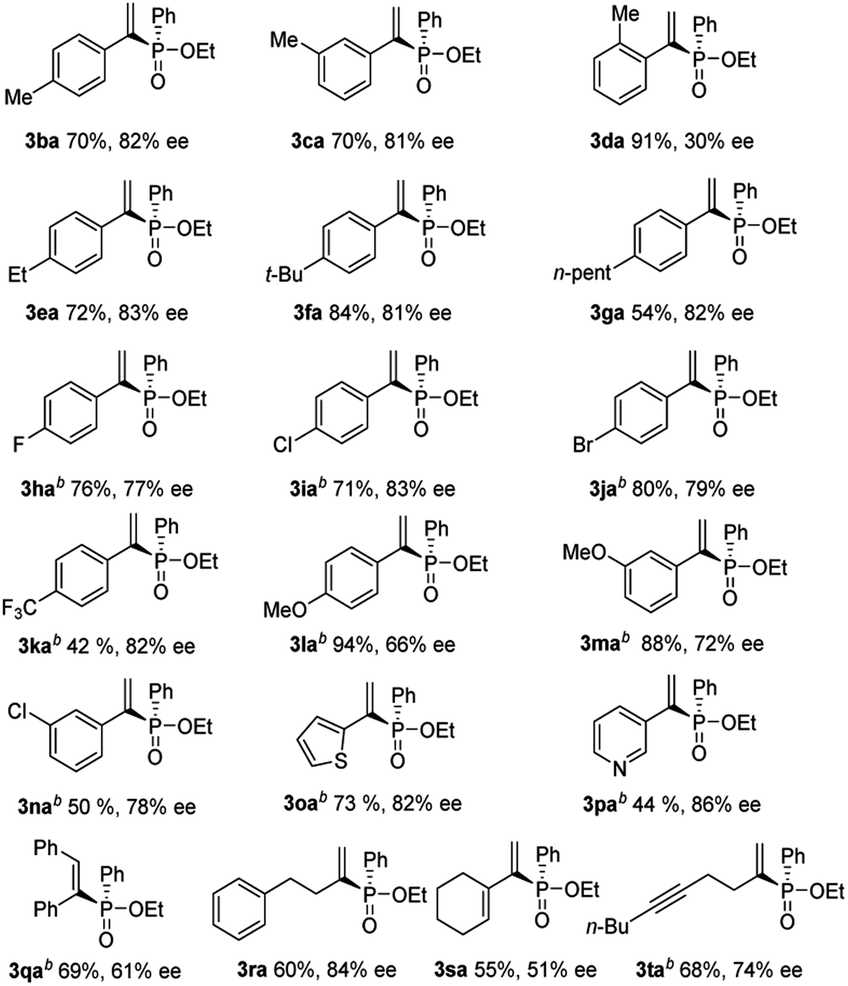
|

Encouraged by the results obtained from alkynes, we further attempted to expand this catalytic system with various H-phosphinates to obtain alkenylphosphonates (Table 3). Substrates with methyl ester or propyl ester were also subjected to hydrophosphorylation and the corresponding alkenylphosphonates (3ab and 3ac) were formed in moderate yields and enantioselectivities. A substrate with isopropyl ester gave decreased yield (3ad), only 32% yield, and slightly decreased enantioselectivity, 73% ee. The phenylphosphinate with Me on the arene ring only gave the product 3ae in 47% yield and 37% ee at a higher temperature. Compound 3fb with the t-Bu group was obtained in decreased yield compared to 3fa. When secondary phosphine oxides were tested under similar reaction conditions, the hydrophosphorylation product 3af was formed with 28% yield in 54% ee.
|
|
|---|
| a Conditions: 1 mol% Pd2(dba)3, 2 mol% ligand L4, and 4 mol% Ph2P(O)OH in 1 mL toluene were stirred for 10 min in an argon atmosphere. 0.25 mmol alkynes 1a and 1f and 1.0 mmol phenylphosphinate 2a–2d or phenyphosphine oxide 2e were added, and the mixture was stirred at 60 °C for 20 h. Isolated yields. Determined by HPLC analysis. b 80 °C was used instead. |
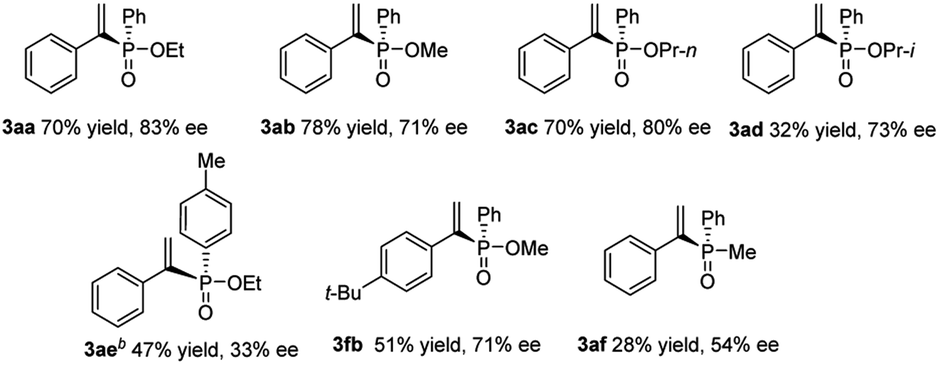
|

To evaluate the synthetic potential of the current catalytic system, a gram–scale reaction between phenylacetylene and ethyl phenylphosphinate was performed, and the product 3aa was furnished in 68% yield and 82% ee (Scheme 2a). Further synthetic transformations of the hydrophosphorylation products were also illustrated. Compounds 4 and 5 were prepared through a Suzuki–Miyaura coupling without loss of enantiopurity (Scheme 2b). The absolute configuration of the phosphinate product 3ja was confirmed as R-configuration by X-ray crystallography of its derivative 5.18 The Heck-type reaction of aryl diazonium salts with alkenylphosphinate 3aa led to cis-stilbenes 3qa with excellent stereoselectivity without loss of chirality (Scheme 2c, 99% yield and 82% ee) which could make up for the moderate yield and ee of 3qa obtained by direct hydrophosphorylation of diphenylacetylene (Table 2). To construct 1,2-biphosphine derivative, the addition of HPPh2 to product 3aa was achieved by copper(I)-catalyzed conjugate hydrophosphination to give biphosphine derivative 6 in 92% yield and 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr without loss of enantiopurity (Scheme 2d, 79% ee for both diastereomers).19
:1 dr without loss of enantiopurity (Scheme 2d, 79% ee for both diastereomers).19
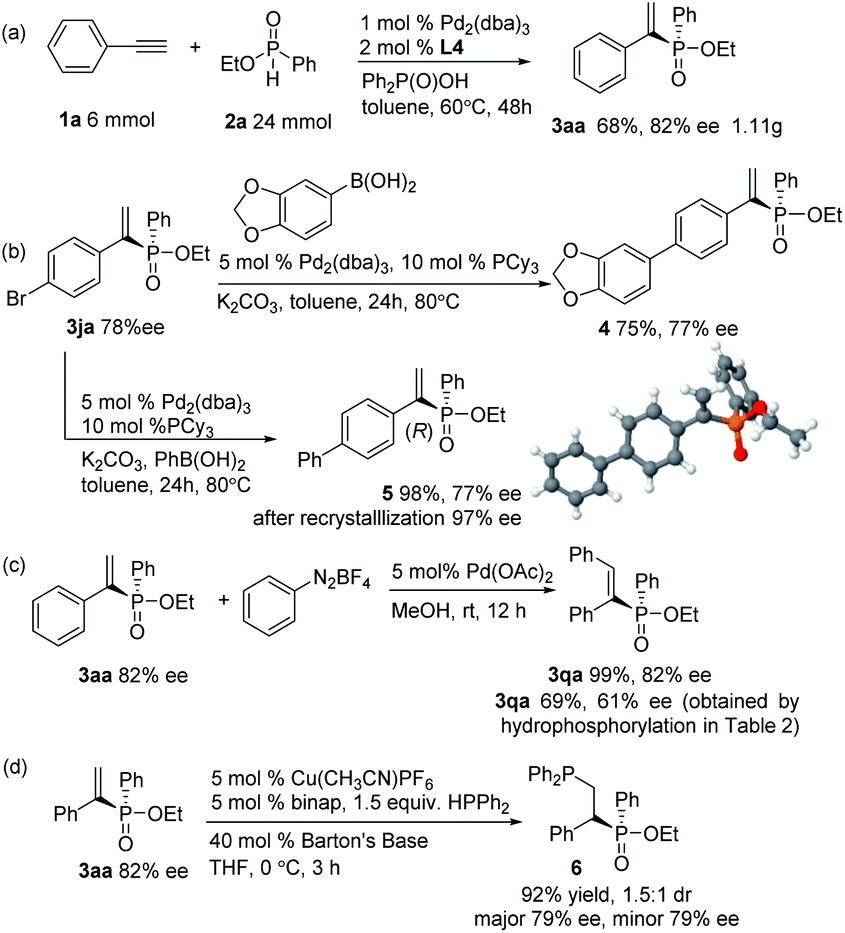 | ||
| Scheme 2 Gram-scale version of the reaction and synthetic transformation of the hydrophosphorylation products. | ||
A deuterium-labelling experiment has been conducted by using D-P(O)(OMe)Ph as the starting material,20 giving the product 3ab (42%), D1-3ab (50%) and a small amount of D2-3ab (6%) in which two deuteriums were incorporated at the terminal alkene carbon atoms (Scheme 3). This result suggested that the oxidative addition, hydropalladation, and ligand exchange are reversible. On the basis of the deuterium labeling experiment and previous report,14 we proposed a mechanistic pathway for this catalysis. Chiral palladium complex A is formed from Pd2(dba)3 and (R,R)-QuinoxP*. It is proposed that oxidative addition of the O–H bond of Ph2P(O)OH to palladium triggers the reaction to produce the internal palladium intermediate B. The hydropalladation of alkynes takes place first to give an internal alkenylpalladium C by Markovnikov addition. Subsequent ligand exchange of this complex C with phosphinate 2a gives the internal phosphorylpalladium intermediate D. A reduced yield was observed in the absence of Ph2P(O)OH. Thus, an alternative pathway is also possible in which the intermediate E is generated directly by the oxidative addition of the P–H bond of 2a to palladium. Then hydropalladation of alkynes takes place to give the same intermediate D. Finally, reductive elimination gives the desired alkenylphosphinate product 3aa and regenerates the active chiral palladium complex A.
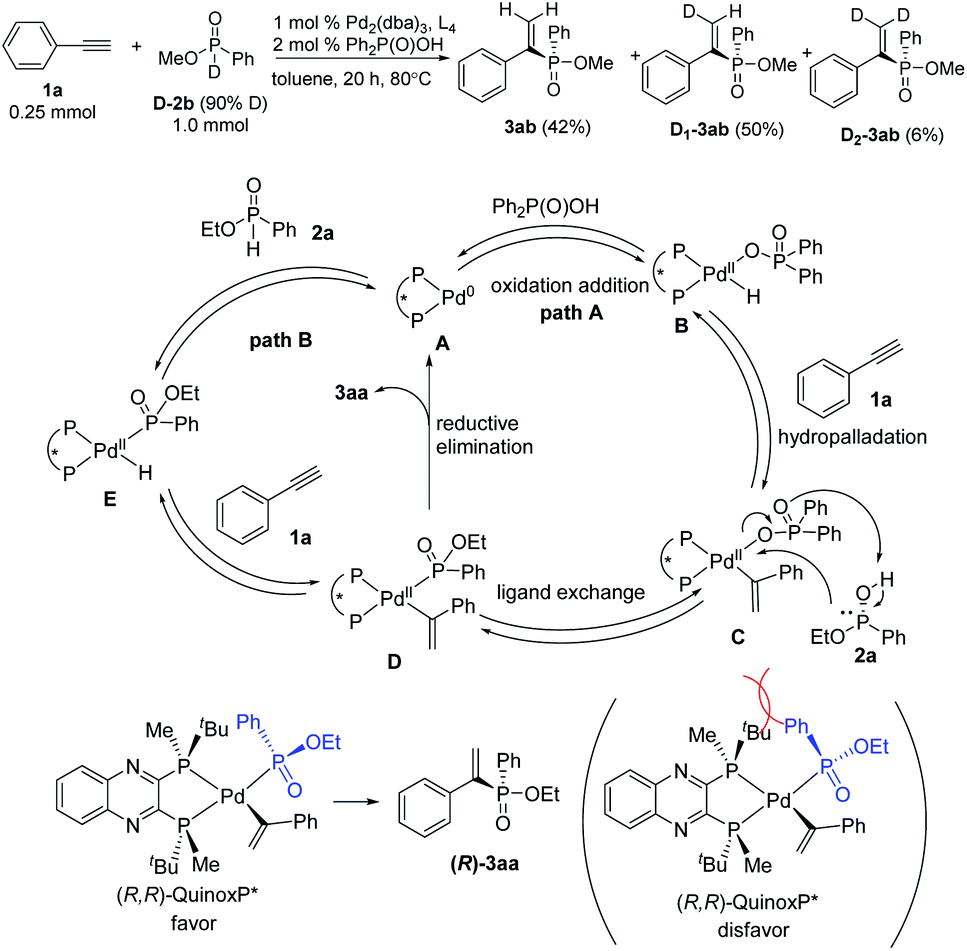 | ||
| Scheme 3 Deuterium labeling experiment, the proposed mechanism and the stereochemical pathway. | ||
Conclusions
In summary, we have developed an efficient method to synthesize alkenylphosphinates with P-chirality through the first Pd-catalyzed enantioselective hydrophosphorylation of alkynes, showing that this hydrophosphorylation reaction is a powerful and practical approach for the preparation of these valuable P-stereogenic organophosphorus compounds. Studies on further application of these chiral organophosphorus compounds are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the National Natural Science Foundation of China (NSFC 21971102) and Guangdong Innovative Program (2019BT02Y335) for financial support. Z. Yang acknowledges a short-term student training partially supported by AIST.Notes and references
- (a) G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704 CrossRef CAS PubMed; (b) H. K. Zbigniew, H. K. Marcin, D. Jozef and V. S. Chris, Curr. Org. Chem., 2011, 15, 2015 CrossRef; (c) A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955 CrossRef CAS PubMed.
- (a) M. Benaglia and S. Rossi, Org. Biomol. Chem., 2010, 8, 3824 RSC; (b) S. J. Connon, Angew. Chem., Int. Ed., 2006, 45, 3909 CrossRef CAS PubMed; (c) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (d) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (e) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344 CrossRef CAS PubMed; (f) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed.
- (a) G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101 CrossRef CAS PubMed; (b) P. W. N. M. v. Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077 CrossRef PubMed; (c) X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272 CrossRef CAS PubMed; (d) P. J. Guiry and C. P. Saunders, Adv. Synth. Catal., 2004, 346, 497 CrossRef CAS; (e) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (f) Y.-M. He, Y. Feng and Q.-H. Fan, Acc. Chem. Res., 2014, 47, 2894 CrossRef CAS PubMed; (g) M. Dutartre, J. Bayardon and S. Juge, Chem. Soc. Rev., 2016, 45, 5771 RSC.
- (a) J. Grembecka, A. Mucha, T. Cierpicki and P. Kafarski, J. Med. Chem., 2003, 46, 2641 CrossRef CAS PubMed; (b) H.-J. Cristau, J. Monbrun, J. Schleiss, D. Virieux and J.-L. Pirat, Tetrahedron Lett., 2005, 46, 3741 CrossRef CAS.
- (a) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906 CrossRef CAS PubMed; (b) O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS PubMed; (c) E. Bergin, C. T. O'Connor, S. B. Robinson, E. M. McGarrigle, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS PubMed; (d) B. D. Vineyard, W. S. Knowles, M. J. Sabacky, G. L. Bachman and D. J. Weinkauff, J. Am. Chem. Soc., 1977, 99, 5946 CrossRef CAS; (e) O. Korpiun, R. A. Lewis, J. Chickos and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4842 CrossRef CAS; (f) Z. S. Han, H. Wu, Y. Xu, Y. Zhang, B. Qu, Z. Li, D. R. Caldwell, K. R. Fandrick, L. Zhang, F. Roschangar, J. J. Song and C. H. Senanayake, Org. Lett., 2017, 19, 1796 CrossRef CAS PubMed; (g) Z. S. Han, L. Zhang, Y. Xu, J. D. Sieber, M. A. Marsini, Z. Li, J. T. Reeves, K. R. Fandrick, N. D. Patel, J.-N. Desrosiers, B. Qu, A. Chen, D. M. Rudzinski, L. P. Samankumara, S. Ma, N. Grinberg, F. Roschangar, N. K. Yee, G. Wang, J. J. Song and C. H. Senanayake, Angew. Chem., Int. Ed., 2015, 54, 5474 CrossRef CAS PubMed; (h) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS PubMed; (i) T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740 CrossRef PubMed; (j) E. J. Corey, Z. Chen and G. J. Tanoury, J. Am. Chem. Soc., 1993, 115, 11000 CrossRef CAS; (k) S. Juge and J. P. Genet, Tetrahedron Lett., 1989, 30, 2783 CrossRef CAS.
- (a) G. Nishida, K. Noguchi, M. Hirano and K. Tanaka, Angew. Chem., Int. Ed., 2008, 47, 3410 CrossRef CAS PubMed; (b) Y. Zheng, L. Guo and W. Zi, Org. Lett., 2018, 20, 7039 CrossRef CAS PubMed; (c) Y. Zhang, F. Zhang, L. Chen, J. Xu, X. Liu and X. Feng, ACS Catal., 2019, 9, 4834 CrossRef CAS.
- Z. Wang and T. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 1702 CrossRef CAS PubMed.
- (a) K. M.-H. Lim and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 8122 CrossRef CAS PubMed; (b) Z. Pakulski and K. M. Pietrusiewicz, Tetrahedron: Asymmetry, 2004, 15, 41 CrossRef CAS; (c) F. de Azambuja, R. C. Carmona, T. H. D. Chorro, G. Heerdt and C. R. D. Correia, Chem. - Eur. J., 2016, 22, 11205 CrossRef CAS PubMed.
- (a) Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS PubMed; (b) L. Liu, A.-A. Zhang, Y. Wang, F. Zhang, Z. Zuo, W.-X. Zhao, C.-L. Feng and W. Ma, Org. Lett., 2015, 17, 2046 CrossRef CAS PubMed; (c) G. Xu, M. Li, S. Wang and W. Tang, Org. Chem. Front., 2015, 2, 1342 RSC; (d) Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS PubMed; (e) Y.-H. Chen, X.-L. Qin, J. Guan, Z.-J. Du and F.-S. Han, Tetrahedron: Asymmetry, 2017, 28, 522 CrossRef CAS; (f) Y. Lin, W.-Y. Ma, Q.-Y. Sun, Y.-M. Cui and L.-W. Xu, Synlett, 2017, 28, 1432 CrossRef CAS; (g) Z. Li, Z.-Q. Lin, C.-G. Yan and W.-L. Duan, Organometallics, 2019, 38, 3916 CrossRef CAS.
- (a) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS PubMed; (b) Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981 RSC; (c) Y.-S. Jang, L. Wozniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS PubMed; (d) Y.-S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088 CrossRef CAS PubMed.
- Q. Dai, W. Li, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 20556 CrossRef CAS PubMed.
- J. S. Harvey, S. J. Malcolmson, K. S. Dunne, S. J. Meek, A. L. Thompson, R. R. Schrock, A. H. Hoveyda and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 762 CrossRef CAS PubMed.
- B. M. Trost, S. M. Spohr, A. B. Rolka and C. A. Kalnmals, J. Am. Chem. Soc., 2019, 141, 14098 CrossRef CAS PubMed.
- (a) L.-B. Han and M. Tanaka, J. Am. Chem. Soc., 1996, 118, 1571 CrossRef CAS; (b) L. B. Han, C. Q. Zhao, S. Y. Onozawa, M. Goto and M. Tanaka, J. Am. Chem. Soc., 2002, 124, 3842 CrossRef CAS PubMed; (c) T. Chen, C.-Q. Zhao and L.-B. Han, J. Am. Chem. Soc., 2018, 140, 3139 CrossRef CAS.
- B. Join, D. Mimeau, O. Delacroix and A.-C. Gaumont, Chem. Commun., 2006, 3249 RSC.
- S.-Z. Nie, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 16450 CrossRef CAS PubMed.
- (a) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS PubMed; (b) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, J. Am. Chem. Soc., 2019, 141, 16584 CrossRef CAS PubMed.
- CCDC 1977604 [for 5] contain the supplementary crystallographic data for this paper.
- W.-J. Yue, J.-Z. Xiao, S. Zhang and L. Yin, Angew. Chem., Int. Ed., 2020, 59, 2 CrossRef.
- C. Li, Q. Wang, J.-Q. Zhang, J. Ye, J. Xie, Q. Xu and L.-B. Han, Green Chem., 2019, 21, 2916 RSC.
Footnotes |
| † Dedicated to Professor Albert S. C. Chan on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1977604. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01049a |
| This journal is © The Royal Society of Chemistry 2020 |