
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient alkene hydrosilation with bis(8-quinolyl)phosphine (NPN) nickel catalysts. The dominant role of silyl-over hydrido-nickel catalytic intermediates†
Jian
Yang‡
a,
Verònica
Postils‡
 bcd,
Michael I.
Lipschutz
a,
Meg
Fasulo
a,
Christophe
Raynaud
b,
Eric
Clot
b,
Odile
Eisenstein
*be and
T. Don
Tilley
*a
bcd,
Michael I.
Lipschutz
a,
Meg
Fasulo
a,
Christophe
Raynaud
b,
Eric
Clot
b,
Odile
Eisenstein
*be and
T. Don
Tilley
*a
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: tdtilley@berkeley.edu
bICGM, Université de Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: odile.eisenstein@umontpellier.fr
cInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus Montilivi, Girona, 17071, Spain
dKimika Fakultatea, Euskal Herriko Unibertsitatea, PK 1072, Donostia, 20080, Spain
eHylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, Oslo N-0315, Norway
First published on 24th April 2020
Abstract
A cationic nickel complex of the bis(8-quinolyl)(3,5-di-tert-butylphenoxy)phosphine (NPN) ligand, [(NPN)NiCl]+, is a precursor to efficient catalysts for the hydrosilation of alkenes with a variety of hydrosilanes under mild conditions and low catalyst loadings. DFT studies reveal the presence of two coupled catalytic cycles based on [(NPN)NiH]+ and [(NPN)NiSiR3]+ active species, with the latter being more efficient for producing the product. The preferred silyl-based catalysis is not due to a more facile insertion of alkene into the Ni–Si (vs. Ni–H) bond, but by consistent and efficient conversions of the hydride to the silyl complex.
Introduction
The hydrosilation of alkenes is a widely practiced catalytic transformation, and is of great importance for the production of organosilanes and materials based on poly(siloxane)s.1–6 For industrial applications, the most common and active hydrosilation catalysts are based on platinum, and to a lesser degree other precious metals. Due to the high cost associated with such metals, there is increasing interest in utilization of less expensive first-row transition metal catalysts for this transformation.7,8 There has been notable progress toward this goal,9–12 including the pioneering work of Brookhart and coworkers on a cationic Cp*Co system for the catalytic hydrosilation of 1-hexene with Et3SiH.9 More recently, Weix, Holland and coworkers have described an effective cobalt catalyst system for anti-Markovnikov hydrosilation of alkenes.10 Also, Chirik et al. have described neutral bis(imino)pyridine iron bis(dinitrogen) complexes as catalyst precursors for the hydrosilation of a variety of alkenes with PhSiH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and some tertiary silanes.12
11 and some tertiary silanes.12
Catalytic systems based on nickel would seem promising given the congeneric relationship of this metal to platinum.7,8,13,14 For example, nickel(II) and nickel(0) complexes such as Ni(PPh3)2Cl2 and [{Ni(η-CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSiMe2)2O}2{μ-(η-CH2CHSiMe2)2O}] exhibit activity toward hydrosilation, but are associated with harsh reaction conditions or limited substrate scope, and usually involve extensive dehydrosilation and/or silane redistribution as processes that compete with hydrosilation.13–15 Indenyl nickel complexes have been reported by Zargarian and coworkers, for the regioselective hydrosilation of styrene with PhSiH3.16,17 In 2012, this laboratory reported the rapid and selective (>95%) anti-Markovnikov hydrosilation of 1-octene with Ph2SiH2, with the two-coordinate Ni(II) amido Ni[N(SiMe3)DIPP]2 as catalyst.18 In recent years, a number of new nickel catalyst precursors have been reported, including examples featuring chelating, pincer, or redox-active ligands as well as a Ni-MOF species.19–29 The catalytic systems reported by Chirik24 and Hu28 demonstrate the use of commercially relevant alkoxysilanes in hydrosilations.
CHSiMe2)2O}2{μ-(η-CH2CHSiMe2)2O}] exhibit activity toward hydrosilation, but are associated with harsh reaction conditions or limited substrate scope, and usually involve extensive dehydrosilation and/or silane redistribution as processes that compete with hydrosilation.13–15 Indenyl nickel complexes have been reported by Zargarian and coworkers, for the regioselective hydrosilation of styrene with PhSiH3.16,17 In 2012, this laboratory reported the rapid and selective (>95%) anti-Markovnikov hydrosilation of 1-octene with Ph2SiH2, with the two-coordinate Ni(II) amido Ni[N(SiMe3)DIPP]2 as catalyst.18 In recent years, a number of new nickel catalyst precursors have been reported, including examples featuring chelating, pincer, or redox-active ligands as well as a Ni-MOF species.19–29 The catalytic systems reported by Chirik24 and Hu28 demonstrate the use of commercially relevant alkoxysilanes in hydrosilations.
In general, the development of generally effective and useful alkene hydrosilations using first-row metals such as manganese, iron, cobalt and nickel is still at an early stage of development. Notably, a challenging impediment concerns the lack of mechanistic information for hydrosilations with such metals, which thwarts attempts to rationally design and discover more efficient catalysts by optimizing electronic and structural properties that promote preferred mechanistic pathways. A prominent mechanistic issue relates to the tendency of first-row metals to undergo one-electron processes, unlike more effective heavier metal catalysts that promote bond-making and -breaking via two-electron oxidative addition and reductive elimination as in the well-known Chalk–Harrod mechanism.1–6 Thus, some attention has focused on attempts to develop new mechanisms for hydrosilation that may not require two-electron processes, such as those based on silylene complex intermediates,30 redox active ligands,24,29 and the electrophilic activation of silanes.31
In an approach to discovery of new first-row metal catalysts for alkene hydrosilation, this laboratory is investigating tridentate ligands containing a central, strong donor atom. Such ligands are expected to promote stability for the catalytic centre, and high reactivity toward substrate activations.32–35 Previous studies centered on bis(8-quinolyl)silyl (NSiN) complexes of platinum, which were found to be unusually active toward Si–H bond oxidative addition.36,37 In the work described here, the central anionic silyl group of the NSiN ligand is replaced by a neutral phosphorous donor. The resulting NPN ligand combines a soft, strong donor and two harder, labile quinolyl groups in a chelating scaffold expected to promote catalytic bond activations.38–45 Cationic nickel complexes of the NPN ligand bis(8-quinolyl)(3,5-di-tert-butylphenoxy)phosphine (1) are shown to be highly effective, general catalysts for hydrosilations of alkenes. Importantly, these catalysts appear to operate by an unusual mechanism that has important implications for the use of first-row metals in hydrosilation and related catalytic reactions. In particular, a DFT investigation shows that the key mechanistic steps in this catalysis involve non-oxidative, concerted cycloadditions of Si–H with Ni–C bonds that proceed through four-centered transition states. A notable aspect to this mechanism is that the preferred hydrosilation pathway is associated with alkene insertion into a Ni–Si (rather than a Ni–H) bond, even though this step is inherently less favorable. It is the subsequent, rate-determining regioselective addition of the Si–H to the Ni–C bond that controls this preference.
Results and discussion
Catalyst synthesis
The new ligand 1 was obtained via bis(8-quinolyl)(dimethylamino)phosphine in moderate yield by lithiation of 8-bromoquinoline followed by addition of 0.5 equiv. of (Me2N)PCl2. Treatment of this synthetic intermediate with 3,5-di-tert-butylphenol in refluxing toluene provided ArtBuO-NPN (1) in 77% yield. Reaction of 1 with Ni(DME)Cl2 (DME = dimethoxyethane) in dichloromethane afforded an insoluble product characterized as the dichloride (ArtBuO-NPN)NiCl2 (2, Scheme 1). Abstraction of chloride from 2 with 1.1 equiv. of Li(OEt2)3B(C6F5)4 in bromobenzene-d5 generated the corresponding, presumably solvated cationic nickel monochloride complex in situ. In a preliminary screen, addition of tert-butylethylene (90 equiv.) and Ph2SiH2 (114 equiv.) to this solution cleanly yielded the hydrosilation product Ph2SiHCH2CH2tBu within 1 h at 23 °C. | ||
| Scheme 1 Synthesis of NPN ligand and Ni complexes. | ||
This finding motivated a more detailed study of (ArtBuO-NPN)Ni cationic complexes as hydrosilation catalysts, and attempts to isolate the cationic nickel complexes (Scheme 1, X = B(C6F5)4, 3a; X = SbF6, 3b) via treatment of 2 with a slight excess of Li(OEt2)3B(C6F5)4 or AgSbF6.§ While multiple attempts to obtain X-ray quality crystals of 3a inevitably produced oily material, the crystal structure of 3b revealed a dimeric structure in the solid state (Fig. 1). A rather unusual structural feature of 3b is coordination of the “weakly coordinating” SbF6 anion, characterized by a short Ni–F distance of 2.262(4) Å. The Sb–F distance associated with this interaction is 1.904(4) Å, about 0.03 Å longer than the average terminal Sb–F bond.46,47
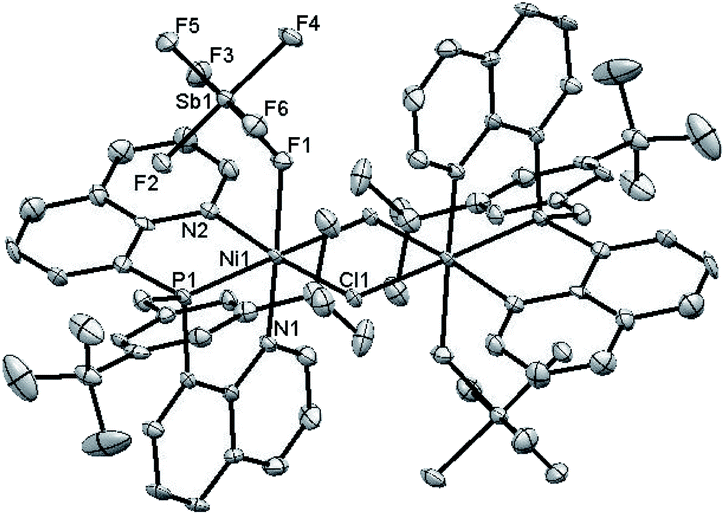 | ||
| Fig. 1 ORTEP drawing of the X-ray crystal structure of 3b. Hydrogen atoms and C6H5F are omitted for clarity. Key bond distances (Å): Ni(1)–N(1) = 2.092(6), Ni(1)–N(2) = 2.070(7), Ni(1)–P(1) = 2.347(2), Ni(1)–F(1) = 2.262(4), Sb(1)–F(1) = 1.904(4), Sb(1)–F(2) = 1.873(5), Sb(1)–F(3) = 1.877(5), Sb(1)–F(4) = 1.874(5), Sb(1)–F(5) = 1.867(5), Sb(1)–F(6) = 1.869(5). | ||
Catalytic hydrosilation
Complexes 3a,b were investigated as precatalysts for the hydrosilation of alkenes. While 3b exhibits modest activities for hydrosilations of cyclopentene with Ph2SiH2 (ca. 26% conversion after 18 h at 23 °C with 0.56 mol% Ni loading), its [B(C6F5)4] anion analogue 3a is much more effective and results in complete conversion within 1 h (Table 1, entries 1 and 2). The lower activity of the SbF6− complex is presumably due to the stronger association of this anion with the Ni center, making a coordination site for substrates less available. Secondary hydrosilanes are the most efficient substrates for hydrosilations catalyzed by 3a. Reactions were generally carried out in bromobenzene-d5, but fluorobenzene, chlorobenzene and 1,2-dichlorobenzene are also suitable solvents. Results of typical catalytic runs are listed in Table 1. Yields were determined by 1H-NMR spectroscopy via integration against an added internal standard and the identities of the products were further confirmed by GC-MS analysis. With Ph2SiH2 as the substrate, complete hydrosilation of tert-butylethylene, cyclopentene, 4-methyl-1-pentene, and 1-hexene proceeded to completion at 23 °C within 1 h with 0.56 mol% Ni loading (entries 2–5). The hydrosilation product distribution appears to depend on the steric properties of the alkene substrates. In general, the mono-hydrosilation product is dominant but especially with less hindered alkenes, small quantities of the double-hydrosilation product are produced (entries 2, 4 and 5). Thus, the relative reactivities of alkene substrates for double-hydrosilation follow the trend: 1-hexene > 4-methyl-1-pentene > cyclopentene > tert-butylethylene. Note that with the sterically encumbered alkene tert-butylethylene, the mono-hydrosilation product Ph2SiHCH2CH2tBu is exclusively formed (entry 3).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ni (mol%) | Olefin | Silane | t (h) | Yieldc, % |
|
a Hydrosilation catalysis was carried out using 3a, except entry 1 which involves 3b.
b General conditions: entries 1–6 and 9, silanes (1.27 equiv.), 23 °C, C6D5Br; entries 8 and 10–16, silanes (1.27 equiv.), 23 °C, o-C6H4Cl2.
c Yields were determined by NMR spectroscopy by integrating the product signals against an internal standard, and the identities of the products were confirmed by GC-MS. Where more than one hydrosilation product is generated, yields represent the combination of both products.
d In addition to Ph2SiH(cyclopentyl), Ph2Si(cyclopentyl)2 (<5%) was also formed.
e Hydrosilation product distribution: Ph2SiH(CH2)3CH(CH3)2 and Ph2Si[(CH2)3CH(CH3)2]2 (5.3:1 ratio).
f Hydrosilation product distribution: Ph2SiH(CH2)5CH3 and Ph2Si[(CH2)5CH3]2 (3:1 ratio).
g The reaction was carried out without solvent; hydrosilation product distribution: Ph2SiH(CH2)5CH3 and Ph2Si[(CH2)5CH3]2 (7.3:1 ratio).
h Small amounts (ca. 8%) of unidentified products in addition to the hydrosilation product were observed.
i In addition to the expected hydrosilation product, small amounts of cyclopentane and other unidentified silicon-containing products were observed.
|
|||||
| 1 | 0.56 | Cyclopentene | Ph2SiH2 | 18 | 26 |
| 2d | 0.56 | Cyclopentene | Ph2SiH2 | 1 | 98 |
| 3 | 0.56 |
t
BuCHCH2 |
Ph2SiH2 | 1 | 97 |
| 4e | 0.56 | 4-methyl-1-pentene | Ph2SiH2 | 1 | 96 |
| 5f | 0.56 | 1-hexene | Ph2SiH2 | 1 | 98 |
| 6f | 0.10 | 1-hexene | Ph2SiH2 | 17 | 97 |
| 7g | 0.23 | 1-hexene | Ph2SiH2 | 3 | 98 |
| 8 | 0.56 | Cyclopentene | PhMeSiH2 | 0.5 | 92 |
| 28 | 98 | ||||
| 9 | 0.56 |
t
BuCHCH2 |
Et2SiH2 | 26 | 99 |
| 10h | 0.56 | 4-methyl-1-pentene | Ph2MeSiH | 1.5 | 98 |
| 11 | 1.0 | Cyclopentene | Ph2MeSiH | 2 | 98 |
| 12 | 0.56 |
t
BuCHCH2 |
Ph2MeSiH | 2 | 98 |
| 13 | 0.56 | Cyclopentene | R2MeSiH (R = Me3SiO) | 1.5 | 98 |
| 14 | 3.0 | Cyclopentene | Ph2ClSiH | 42 | 90 |
| 65 | 95 | ||||
| 15i | 0.56 | Cyclopentene | PhSiH3 | 20 | 22 |
| 41 | 30 | ||||
| 16i | 0.56 | Cyclopentene | MesSiH3 | 1.1 | 75 |
Further reduction of the catalyst loading proved possible, as demonstrated by use of 0.1 mol% Ni loading to achieve the hydrosilation of 1-hexene in 17 h at 23 °C (ca. 970 turnovers; entry 6). Hydrosilation reactions may also be carried out in neat 1-hexene and complete reaction occurred within 3 h at 23 °C at a catalyst loading of 0.23 mol% (entry 7). Other secondary hydrosilanes, such as PhMeSiH2 and Et2SiH2, were also effective for this reaction (entries 8 and 9).
Attempts to hydrosilylate cyclopentene with tertiary silanes in bromobenzene, with 3a as catalyst, often led to decomposition of the catalyst and low conversions, presumably due to reaction of active nickel species with aryl bromide. More effective catalysis with tertiary silanes is observed with less reactive solvents, such as 1,2-dichlorobenzene or chlorobenzene (entries 10–14). In the presence of 0.56 mol% catalyst, the hydrosilation of 4-methyl-1-pentene with Ph2MeSiH proceeds to completion in less than 1.5 h (entry 10). Cyclopentene and tert-butylethylene were readily hydrosilylated with Ph2MeSiH (entries 11 and 12). At a Ni loading of 0.56 mol%, the complete hydrosilation of cyclopentene with the commercially relevant silane (Me3SiO)2MeSiH occurred within 1.5 h at 23 °C (entry 13). Hydrosilation of cyclopentene with Ph2SiClH is relatively slow, requiring 65 h for approximately 95% conversion at 3.0 mol% of Ni loading (entry 14).
The catalytic activity of 3a was compared to that of some simple Ni(II) complexes and their [B(C6F5)4]− salts to evaluate the enhancement to hydrosilation afforded by the ArtBuO-NPN ligand. The hydrosilation of 4-methyl-1-pentene with Ph2MeSiH (conditions identical to Table 1, entry 10) was attempted using Ni(DME)Cl2, (Ph3P)2NiCl2, and their respective [B(C6F5)4]− salts (generated in situ) as precatalysts. Only the (Ph3P)2NiCl2 + Li[B(C6F5)4] catalyst precursor resulted in any observable hydrosilation product, and only in 4% yield. These results, along with comparisons to those cited above,13–20,22–29 indicate that the ArtBuO-NPN complex 3a is exceptionally active as a nickel-based hydrosilation catalyst.
Hydrosilations catalyzed by 3a are generally less efficient for primary (vs. secondary) silanes. Entry 15 shows that hydrosilation of cyclopentene with PhSiH3 occurred at a 0.56 mol% Ni loading but with only 30% conversion after 41 h at 23 °C. Interestingly, under identical reaction conditions hydrosilation with the bulkier silane MesSiH3 (Mes = 2,4,6-C6H2Me3) is much more rapid (entry 16). The origin of this unusual selectivity is still unclear but may relate to the crowded nature of the transition states for silane activation (vide infra).
The mechanism of hydrosilation catalyzed by paramagnetic 3a has been challenging to probe experimentally, given the (presumably) multi-step transformation of [(NPN)NiCl]+ to the active catalyst (vide infra), and a lack of success in isolating complexes from catalytic or stoichiometric reactions. Also, it has not been possible to identify metal-containing species from the complex NMR spectra of reaction mixtures; however, reaction of 3a with Ph2SiH2 results in rapid formation of Ph2SiHCl, and sharp NMR resonances suggest that several diamagnetic nickel species are present. Also, monitoring catalytic reactions by 1H-NMR spectroscopy revealed the formation of Ph2SiHCl during catalysis. These results suggest that a nickel hydride is formed as an intermediate; however, such a species could not be identified by NMR spectroscopy. Also, a catalytic reaction with diphenylsilane-d2 and tert-butylethylene with a Ni loading of 1.0 mol% yielded an approximately 1:1.6 ratio of Ph2SiDCHDCH2tBu and Ph2SiDCH2CHDtBu (Scheme 2). This implies a rapid and reversible insertion/deinsertion step involving a nickel hydride, such as [(NPN)NiH]+, in an “off-cycle” process or prior to elimination of the hydrosilation product. Future mechanistic studies will involve exploratory synthetic efforts to obtain isolable, single-site catalytic species, primarily hydride and silyl derivatives of the [(NPN)Ni]+ fragment. To gain more mechanistic information, a DFT study was carried out.
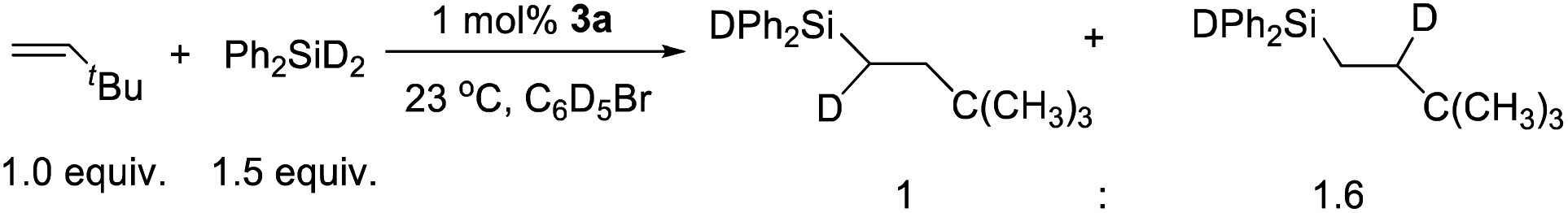 | ||
| Scheme 2 tert-Butylethylene hydrosilation with diphenylsilane-d2 catalyzed by 3a. | ||
Computational studies
To gain a better understanding of the mechanism of this catalytic hydrosilation, DFT studies were carried out with a dispersion corrected hybrid functional and an implicit model for the solvent (1,2-dichlorobenzene). Full computational details are given in the ESI.† The Ni complexes with the pincer ligand, labelled as (NPN), and the substrates (cyclopentene and PhSiH3) were represented in full. These substrates were selected because they are associated with smaller conformational diversity, though they are not the most reactive, and therefore allow manageable computation times. Although PhSiH3 is not one of the better substrates in this catalysis, we presume that this results from side reactions of the smaller silane, leading to condensed and inactive nickel–silicon species formed via multiple Si–H activations in the silane. The latter trend is well known for reactions of silanes with nickel complexes.48–52 Exploration of potential energy surfaces revealed that numerous, slightly different, pathways give the same products. For these pathways, the intermediates and transition states differ by subtle changes in the structures of the NPN ligand, which can be folded or flat and which can be κ3- or κ2-coordinated. Only the preferred pathway is presented and brief mention of other pathways is made when appropriate.Catalyst generation likely occurs by reaction of the cationic catalyst precursor, {[(NPN)NiCl]2}2+, with the silane substrate. This is indicated by experimental observations that implicate reaction of this paramagnetic dinuclear nickel chloride with a hydrosilane to produce a putative diamagnetic nickel complex (vide supra). The nickel product would appear to be the hydride [(NPN)NiH]+, and though nickel hydrides are now well established,53,54 cationic nickel hydrides of this type are more unusual but have precedence.55–59 In evaluating the energetics of this reaction, it was assumed that the starting chloride complex dissociates to a monomer that possesses a more accessible and thus more reactive Ni–Cl bond. Exchange of the silane Si–H and catalyst precursor Ni–Cl bonds gives the observed chlorosilane PhSiH2Cl and presumably the hydride [(NPN)NiH]+. This exchange reaction is accompanied by a spin crossover, since the precatalyst is paramagnetic while the active catalyst species appears to be diamagnetic. In addition, the computational studies are based on the reasonable assumption that the catalytic cycle occurs on a singlet electronic surface, especially since interaction between the nickel and the substrates should stabilize closed-shell electronic states. Validation of a functional via comparison with experimental data is currently not possible given the lack of key crystallographic data. Test calculations with several functionals indicated that the singlet electronic state is relatively more preferred for [(NPN)NiH]+ in comparison to [(NPN)NiCl]+, as expected from the stronger ligand field in the former. In view of these considerations, the hybrid functional PBE0, considered as reliable for determination of reaction pathways, was selected.60,61
The Cl/H exchange reaction involving monomeric [(NPN)NiCl]+ and PhSiH3 is similar to the related generation of an (indenyl)nickel hydride, by reaction of the corresponding chloride with PhSiH3.62 On the singlet electronic surface, the reaction is endoergic (ΔG = 11 kcal mol−1) and requires a Gibbs activation energy of 19.8 kcal mol−1 relative to the separated reagents (ESI Fig. S4†). These energetics do not account for any stabilization of the hydride (e.g., by dimerization or facile reactions with species present in solution, such as the silane or olefin substrates present under catalytic conditions, as developed below). The nickel hydride was therefore considered to be the initial reactive species and the origin of energies was therefore [(NPN)NiH]+, corrected by inclusion of the appropriate substrates for mass balance.
The reaction of [(NPN)NiH]+ with the silane PhSiH3 is calculated as an essentially thermoneutral reaction (ΔG = −0.6 kcal mol−1) that yields [(NPN)NiSiH2Ph]+ and H2 (central reaction equilibrium of Scheme 3) via a transition state with ΔG‡ of 6.9 kcal mol−1 relative the energy reference. However, this thermoneutrality corresponds to a situation where H2 remains in solution. This reaction should be driven more strongly toward formation of the silyl complex by the poor solubility of H2.63
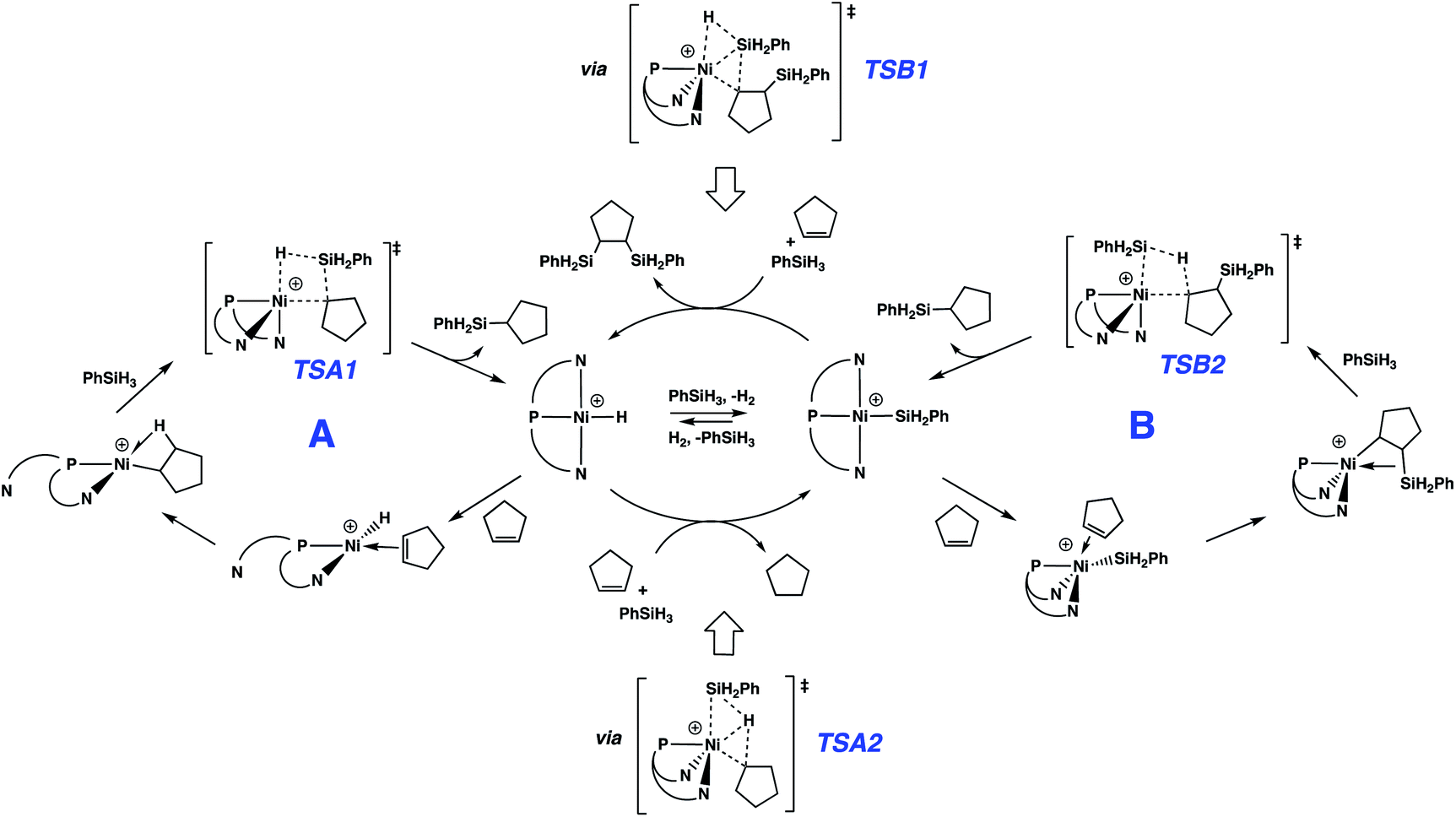 | ||
| Scheme 3 Proposed mechanisms for nickel-catalyzed hydrosilation of alkenes. | ||
Reactions of the cationic nickel hydride initially with cyclopentene and then with PhSiH3 provide a viable catalytic cycle for hydrosilation (Fig. 2 and cycle A of Scheme 3). As shown in Fig. 2, this reaction sequence begins with coordination of cyclopentene to the nickel centre, assisted by decoordination of a quinolyl group to give a square planar alkene hydride complex with a dangling group (κ2-NPN) and the alkene double bond perpendicular to the Ni coordination plane. Pathways of higher energy for alkene binding were obtained for the fully coordinated κ3-NPN ligand (see ESI, Page S28†). Insertion of the cyclopentene π-bond into the Ni–H bond is achieved with a low Gibbs energy barrier (ΔG‡ = 4.6 kcal mol−1, relative to the π-complex, i.e. −2.3 kcal mol−1 relative to the energy reference) and yields a cyclopentyl complex stabilized by a β-agostic C–H bond trans to a quinolyl. During this step the NPN ligand remains κ2-coordinated. The formation of this cyclopentyl intermediate is favoured by thermodynamics (ΔG = −13.9 kcal mol−1). Coordination of the silane to the cyclopentyl intermediate (for which the loss of entropy dominates the enthalpy of coordination) can occur with two orientations of the Si–H bond relative to the Ni–cyclopentyl bond. If the silyl group is closer to the cyclopentyl ligand, a silyl group migrates and a silylcyclopentane is obtained along with the nickel hydride. If the hydrogen atom of the Si–H bond is closer to the cyclopentyl ligand, a hydrogen atom migrates and a cyclopentane and a nickel silyl complex are formed. These two reactions are concerted and the Gibbs energy profiles of Fig. 2 show that the migration of H is preferred over that of the silyl group (TSA2 is lower than TSA1 by 2.4 kcal mol−1) and the corresponding products (cyclopentane and nickel silyl) are energetically preferred by ΔG = 8.7 kcal mol−1 over the silylcyclopentane and nickel hydride. These kinetic and thermodynamic factors have significant consequences for the reaction mechanism for hydrosilation. Importantly, the hydride should rapidly exit catalytic cycle A by way of alkane formation and production of the corresponding silyl complex. Indeed, small amounts of alkane, consistent with this reactivity, are observed in catalytic reaction mixtures (vide supra). Note that, as mentioned above, the hydride also has another pathway to the silyl complex by direct reaction with the silane and elimination of H2 (Scheme 3). As well, the silyl complex has a viable pathway back to the hydride, via insertion of alkene to give a β-silyl alkyl complex that will react with silane to generate a 1,2-bis(silyl)alkane in addition to the nickel-hydride (vide infra).
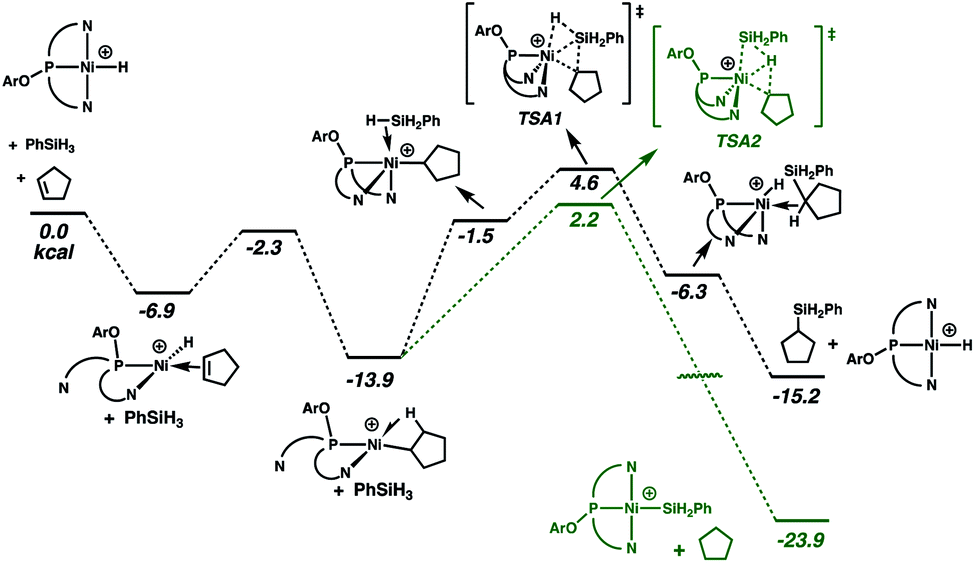 | ||
| Fig. 2 Gibbs energy profile (kcal mol−1) for cycle A of the hydrosilation catalyzed by [(NPN)NiH]+. | ||
Especially given the accumulation of the nickel silyl complex [(NPN)NiSiH2Ph]+ in the reaction sequences described above, its role as a potential hydrosilation catalyst was investigated. In fact, catalysis with this species (Fig. 3 and cycle B of Scheme 3) was found to involve a pathway that is lower in energy than that of cycle A (see Fig. S5 and S6 in ESI† for detailed numerical values). This is due to the fact that the catalytic cycle initiated by the silyl complex starts at −23.9 kcal mol−1 relative to the energy reference, due to the exoergicity of the hydrogenation of cyclopentene as described in Fig. 2. Qualitatively, the elementary steps of cycle B are similar to those of cycle A. Coordination of cyclopentene to the nickel silyl is marginally exoergic and occurs without decoordination of a quinolyl group. The transition state for insertion of the alkene into the Ni–Si bond is only 6.2 kcal mol−1 above that of the alkene complex. This insertion step yields an alkyl ligand with the silyl group in the β position, and results in a relatively short Ni⋯Si contact (2.66 Å) indicative of a β-agostic C(β)–Si(γ) bond, as has been observed for numerous alkyl complexes.64–69 This silyl-substituted alkyl intermediate reacts with PhSiH3, to yield either the silyl or the hydride complex. As expected, the energetically more favourable transition state corresponds to a transfer of H (TSB2) and leads directly to the hydrosilation product Ph(C5H11)SiH2 with regeneration of the silyl catalyst (cycle B of Scheme 3). The transition state (TSB1) for the silyl transfer is 2 kcal mol−1 higher in energy. Thus, the release of the 1,2-bis(silyl)alkane and the hydride [(NPN)NiH]+ is both kinetically and thermodynamically (11.9 kcal mol−1) disfavoured. Consistent with earlier results, the net effect of the competing reactions is that the silyl species should accumulate in favour of the hydride. In addition, the major organic product (determined by cycle B) is the hydrosilation product, in good agreement with the experimental evidence.
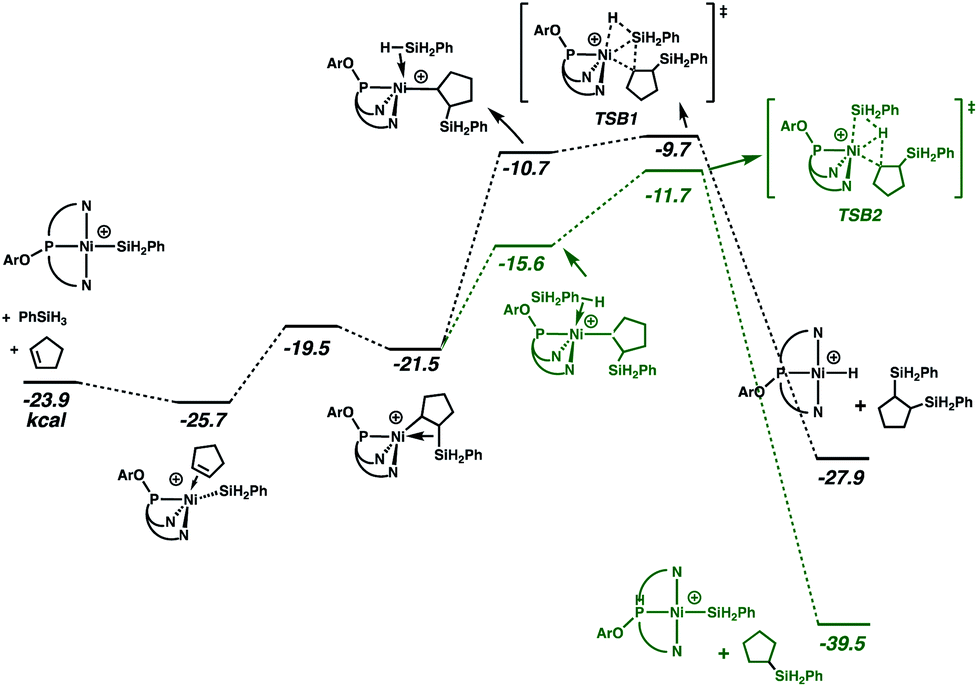 | ||
| Fig. 3 Gibbs energy profile (kcal mol−1) for cycle B of the hydrosilation catalyzed by [(NPN)NiSiH2Ph]+. | ||
Discussion of the mechanism
Globally, all reactions have rather low energies of activation suggesting that in this catalytic system, both the hydride and silyl complexes of Ni are very reactive. One reason for the high reactivity could relate to the substantial flexibility and hemilability of the NPN ligand. For instance, the solid state structure of the dinuclear precatalyst shows a strongly folded NPN ligand with an angle between the two N–Ni–P planes of around 90°, and similar features are obtained for computed structures of κ3-NPN pincer complexes. However, the NPN pincer can be significantly flattened as it is in (NPN)NiH+ and (NPN)NiSiH2Ph+, which have fold angles of 162° and 170°, respectively. Additional proof of the importance of the flexibility of the NPN ligand in facilitating the reaction is evidenced by the fact that various isomeric forms, relatively close in energy, were found for several minima and transition states leading to the same product. Thus, in the case of the hydride, the lowest transition state for cyclopentene coordination is accompanied by a κ3- to κ2-change in coordination mode; however, transition states with fully coordinated κ3-NPN were located at slightly higher energies. In contrast, the preferred transition state for cyclopentene coordination to the silyl has a κ3-NPN ligand and higher-energy transition states have the NPN ligand with a coordination intermediate between κ2 and κ3.The 1,2 addition of the silane to the Ni-cyclopentyl cation, to form cyclopentane and the Ni–silyl complex or the silyl-cyclopentane and hydride complex (Fig. 2), occurs in a concerted manner without formation of any intermediate. The calculations indicate that the transition state for hydrogen transfer is systematically preferred over silyl transfer although the difference in energy between the two transition states is only 2 kcal mol−1. Similar features have been obtained for the hydrosilation of olefins by lanthanide complexes.70–73 However, the 4-centre transition state for a metathesis reaction with a d0 metal is calculated to be planar, whereas TSA2 is butterfly-shaped with the transferring H atom closest to Ni (1.460 Å) and about midway between Si (1.700 Å) and the cyclopentyl carbon (1.673 Å). Similar features are observed for TSB2 (Ni⋯H: 1.455 Å, Si⋯H: 1.769 Å, C⋯H: 1.645 Å). To understand these unusual features, an NBO analysis is presented for TSB2 (Scheme 3 and Fig. 4), which is associated with formation of the major product. The second-order perturbation and NLMO analyses reveal a 3-centre 2-electron interaction involving Ni (9%), Si (37%) and H (54%), which describes a donation of the Si–H bond into a 4s Ni orbital. In addition, this Ni–H–Si orbital (mostly located on H–Si) acts as a strong donor to the Ni–C(cyclopentyl) σ* orbital. This electron delocalization induces a concerted cleavage of the Si–H and Ni–C bonds, and formation of the Ni–Si and C–H bonds. The NBO analysis does not attribute a bond to the Ni⋯H interaction, but indicates a very weak donation from a Ni lone pair to H. Consequently, there is no oxidative addition of the Si–H bond in this step. This is also supported by the NBO charges (+0.85e for Si, 0.00e for H, 0.49e for Ni and −0.46e for C), which suggest a rather high density on Ni that is not compatible with oxidation.
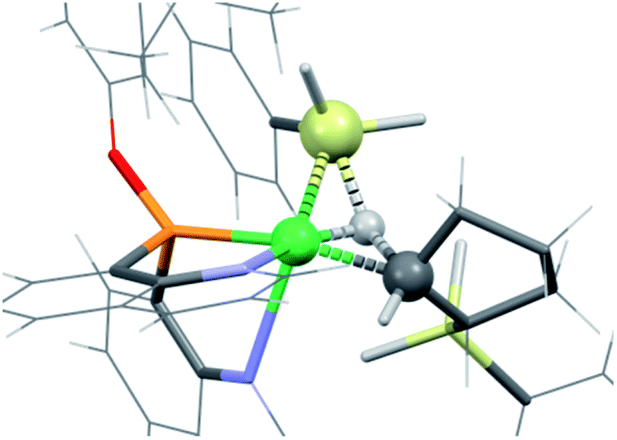 | ||
| Fig. 4 Transition state TSB2 for reaction of PhSiH3 with [(NPN)Ni(C5H9)SiH2Ph]+. The atoms involved in the reaction (Si, H, Ni and C) are represented by spheres (green for Ni, yellow for Si, white for H and black for C). Other atoms are represented by wireframe. | ||
Using a Bader atoms-in-molecule analysis, Vastine and Hall proposed that a general bonding continuum exists among the variously named “oxidative addition”, “σ-bond metathesis” and “metal-assisted metathesis” reactions.74,75 Ess, Goddard and Periana used an energy decomposition analysis (ALMO-EDA) to classify such C–H activation reactions as electrophilic, ambiphilic or nucleophilic on the basis of the net direction of charge transfer energy stabilization.76 The NBO method used in this work is related to the ALMO-EDA because it identifies the donor and acceptor and ranks the associated stabilization energy. Within this scheme, it is very clear that the Si–H bond acts as a strong donor and that the stabilization energy associated with donation of the Ni–C bond is an order of magnitude smaller. There is thus significant electronic reorganization within the Ni orbitals, which is necessary for cleaving the Ni–C bond and forming the Ni–Si bonds. We suggest that the reaction should be viewed as electrophilic σ-bond metathesis within the classification of Goddard et al.76 This is also compatible with the large exoergicity of the transformation, which is in part driven by transformation of a weak Si–H into a strong C–H bond (an energy gain of ∼20 kcal mol−1). Donation of the Si–H bond to nickel is actually already present in the σ(Si–H) complexes of PhSiH3 with [(NPN)Ni(C5H9)SiH2Ph]+, which was located as an energetically high-lying minimum before the transition state TSB2 (Fig. 3 and Fig. S6 in ESI†). This highlights the role of σ-complexes in 1,2-addition to metal–carbon σ-bonds.77,78 Additionally, it is interesting to note that the non-planarity of this TSB2 transition state suggests that a variety of 4-centre transition state structures with similar electronic characteristics are possible. Finally, the lack of an oxidizing Ni⋯H interaction at the transition state is not solely due to the expected difficulty in oxidizing a Ni(II) cationic centre, as a similar absence of redox character has been suggested for σ-bond exchanges involving Ni(0) centres,79,80 but this does not extend to heavier metals of Group 10.81
The two catalytic cycles of Scheme 3 are quite similar and are based on reactions of analogous hydride (cycle A) or silyl (cycle B) complexes. In A, the silyl group migrates to an alkyl ligand, while in B, hydride migrates to a silyl-substituted alkyl ligand. Thus, both cycles lead to the hydrosilation product. Each of these cycles is associated with a transition state variant differing only by the way the Si–H bond adds across the Ni–C bond, with A proceeding viaTSA2 and B utilizing TSB1. This is consistent with the well-known trend for hydrogen migration being energetically preferred over silyl migration. Also, for the hydride intermediate, it is more efficient to undergo silylation viaTSA2, which converts the hydride to the silyl complex, further favouring cycle B. A microkinetic study using the calculated activation energies confirms the strongly dominant formation of the alkylsilane over that of the alkane or the bis(silyl)alkane (see full description of the microkinetic model in the ESI†).82
Interestingly, the unusual preference for hydrosilation catalysis promoted by a silyl complex, in the presence of the corresponding hydride, does not arise from preferential insertion of the olefin into the Ni–Si bond. Alkene insertion into a M–Si bond has been experimentally observed for various metals, but on the basis of experiments83 and calculations84 (notably for platinum), it is thought that this step generally requires a higher energy relative to analogous insertions into M–H bond. This difference has been used to conclude that the Chalk–Harrod mechanism for hydrosilation with platinum occurs via alkene insertion into Pt–H.1–6 In fact, for the system described here, the calculations are consistent with the known preference for insertion into the Ni–H bond. The preference for the catalytic cycle based on the nickel silyl catalyst arises from the fact that the hydride complex quickly exits its cycle due to the kinetic and thermodynamic preference for formation of the silyl complex, by way of alkane elimination (Fig. 2 and Scheme 3).
Conclusions
This study identifies a readily accessible and potentially tunable nickel-based catalytic system for the hydrosilation of alkenes. The catalysis is exceptionally efficient, exhibits high activity towards a broad spectrum of hydrosilanes, and involves an abundant transition metal. These results therefore raise interesting questions regarding the influence of structural modifications of the catalyst on hydrosilation activities and selectivities.A DFT computational study indicates that catalysis with the precatalyst [(ArOtBu–NPN)NiCl]22+ (3) involves two possible catalytic cycles, using Ni-hydride or Ni–silyl catalysts, but the preferred pathway involves an alkene insertion into a Ni–Si bond and low energy barriers for the fundamental steps. The low-energy profiles are likely associated with the high flexibility of the NPN ligand, manifested in its ability to readily cycle between κ3- and κ2-coordination, and “fold” to vary bond angles. In the two possible catalytic cycles, the rate-determining step is the 1,2-addition of silane to the Ni–alkyl bond and not the insertion of the olefin into the Ni–H or Ni–Si bonds. This product-forming step involves a favoured transition state with H adding to the alkyl ligand and formation of a Ni–Si bond (H in the β position of the 4-membered transition state). Other bond activation processes are possible, including those of a catalytic cycle for hydrosilation by the nickel hydride, but the consistent positioning of hydrogen β to the metal centre in concerted 4-membered transition states results in rapid exit of the hydride from its catalytic cycle, with formation of the silyl complex. Thus, the preferred catalysis based on the silyl complex is not due to a more facile insertion of alkene into the Ni–Si (vs. Ni–H) bond, but by persistent, efficient conversions of the hydride to the silyl complex. Also, analysis of the electronic structure of the 1,2-addition of the Si–H bond to the Ni–alkyl complex shows that no redox process has occurred at the metal and that the electronic density of the Si–H bond serves to cleave the Ni–C sigma bond. Thus, the reaction is best seen as an electrophilic σ-bond metathesis following the classification proposed by Goddard et al.76 The key transition state has a folded, “butterfly” structure, and as such is very different from the planar structures associated with σ-bond metathesis reactions,85 demonstrating that planarity of the 4-centered transition state is not a requirement for this type of reaction.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Dow Corning is gratefully acknowledged for financial support of the experimental aspects of this research. The authors further wish to acknowledge the National Science Foundation for partial support, under grants CHE-1566538 and CHE-1954808. O. E. was partially supported by the Research Council of Norway through its Centres of Excellence Scheme Project 262695. V. P. R. thanks MINECO for the Grant BES-2012-052801 and the fellowship EEBB-I-14-07908, which paid for her stay at the University of Montpellier. V. P. R also wishes to acknowledge the financial support from the University of the Basque Country (UPV/EHU) under PIC216-18 and ESPDOC18/85. Richard Taylor, Binh Nguyen and Misha Tzou (Dow Corning) are thanked for valuable discussions. O. E. is grateful to the Miller foundation which has fostered this international collaboration.Notes and references
- B. Marciniec, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer Netherlands, Dordrecht, First edn, 2009, vol. 1 Search PubMed.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef.
- B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS.
- I. Ojima, Z. Li and J. Zhu, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Ltd, Chichester, UK, First edn, 198AD, pp. 1687–1792 Search PubMed.
- L. N. Lewis, J. Stein, Y. Gao, R. E. Colborn and G. Hutchins, Platinum Met. Rev., 1997, 41, 66–79 CAS.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS.
- J. V Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef PubMed.
- M. Brookhart and B. E. Grant, J. Am. Chem. Soc., 1993, 115, 2151–2156 CrossRef CAS.
- C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244–13247 CrossRef CAS PubMed.
- S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed.
- A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567–570 CrossRef CAS PubMed.
- Y. Kiso, M. Kumada, K. Tamao and M. Umeno, J. Organomet. Chem., 1973, 50, 297–310 CrossRef CAS.
- Y. Kiso, M. Kumada, K. Maeda, K. Sumitani and K. Tamao, J. Organomet. Chem., 1973, 50, 311–318 CrossRef CAS.
- H. Maciejewski, B. Marciniec and I. Kownacki, J. Organomet. Chem., 2000, 597, 175–181 CrossRef CAS.
- F.-G. Fontaine, R.-V. Nguyen and D. Zargarian, Can. J. Chem., 2003, 81, 1299–1306 CrossRef CAS.
- Y. Chen and D. Zargarian, Can. J. Chem., 2009, 87, 280–287 CrossRef CAS.
- M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC.
- L. Benítez Junquera, M. C. Puerta and P. Valerga, Organometallics, 2012, 31, 2175–2183 CrossRef.
- A. Kuznetsov and V. Gevorgyan, Org. Lett., 2012, 14, 914–917 CrossRef CAS PubMed.
- Z. Zhang, L. Bai and X. Hu, Chem. Sci., 2019, 10, 3791–3795 RSC.
- V. Srinivas, Y. Nakajima, W. Ando, K. Sato and S. Shimada, Catal. Sci. Technol., 2015, 5, 2081–2084 RSC.
- J. Mathew, Y. Nakajima, Y.-K. Choe, Y. Urabe, W. Ando, K. Sato and S. Shimada, Chem. Commun., 2016, 52, 6723–6726 RSC.
- I. Pappas, S. Treacy and P. J. Chirik, ACS Catal., 2016, 6, 4105–4109 CrossRef CAS.
- T. J. Steiman and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 6104–6110 CrossRef CAS PubMed.
- I. Buslov, J. Becouse, S. Mazza, M. Montandon-Clerc and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 14523–14526 CrossRef CAS PubMed.
- I. Buslov, S. C. Keller and X. Hu, Org. Lett., 2016, 18, 1928–1931 CrossRef CAS PubMed.
- I. Buslov, F. Song and X. Hu, Angew. Chem., Int. Ed., 2016, 55, 12295–12299 CrossRef CAS PubMed.
- C. L. Rock and R. J. Trovitch, Dalton Trans., 2019, 48, 461–467 RSC.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- M. C. Lipke, A. L. Liberman-Martin and T. D. Tilley, Angew. Chem., Int. Ed., 2017, 56, 2260–2294 CrossRef CAS PubMed.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS PubMed.
- D. Morales-Morales and C. M. Jensen, The chemistry of pincer compounds, Elsevier, Amsterdam, The Netherlands, First edn, 2007 Search PubMed.
- M. D. Fryzuk, Can. J. Chem., 1992, 70, 2839–2845 CrossRef CAS.
- M. Stradiotto, K. L. Fujdala and T. D. Tilley, Chem. Commun., 2001, 1200–1201 RSC.
- P. Sangtrirutnugul and T. D. Tilley, Organometallics, 2007, 26, 5557–5568 CrossRef CAS.
- P. A. Aguirre, C. A. Lagos, S. A. Moya, C. Zúñiga, C. Vera-Oyarce, E. Sola, G. Peris and J. C. Bayón, Dalton Trans., 2007, 5419–5426 RSC.
- W.-H. Sun, Z. Li, H. Hu, B. Wu, H. Yang, N. Zhu, X. Leng and H. Wang, New J. Chem., 2002, 26, 1474–1478 RSC.
- T. Suzuki, Bull. Chem. Soc. Jpn., 2004, 77, 1869–1876 CrossRef CAS.
- P. Wehman, H. M. A. van Donge, A. Hagos, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Organomet. Chem., 1997, 535, 183–193 CrossRef CAS.
- T. Suzuki, K. Kashiwabara and J. Fujita, Bull. Chem. Soc. Jpn., 1995, 68, 1619–1626 CrossRef CAS.
- H. A. Hudali, J. V. Kingston and H. A. Tayim, Inorg. Chem., 1979, 18, 1391–1394 CrossRef CAS.
- J. Flapper, H. Kooijman, M. Lutz, A. L. Spek, P. W. N. M. van Leeuwen, C. J. Elsevier and P. C. J. Kamer, Organometallics, 2009, 28, 1180–1192 CrossRef CAS.
- A. Kermagoret, F. Tomicki and P. Braunstein, Dalton Trans., 2008, 2945–2955 RSC.
- W. H. Hersh, J. Am. Chem. Soc., 1985, 107, 4599–4601 CrossRef CAS.
- R. Bröchler, I. H. T. Sham, M. Bodenbinder, V. Schmitz, S. J. Rettig, J. Trotter, H. Willner and F. Aubke, Inorg. Chem., 2000, 39, 2172–2177 CrossRef PubMed.
- R. Beck and S. A. Johnson, Organometallics, 2012, 31, 3599–3609 CrossRef CAS.
- R. Beck and S. A. Johnson, Organometallics, 2013, 32, 2944–2951 CrossRef CAS.
- S. Shimada, M. L. N. Rao, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2001, 40, 213–216 CrossRef CAS PubMed.
- D. Schmidt, T. Zell, T. Schaub and U. Radius, Dalton Trans., 2014, 43, 10816 RSC.
- R. C. Handford, P. W. Smith and T. D. Tilley, Organometallics, 2018, 37, 4077–4085 CrossRef CAS.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373–8426 CrossRef CAS PubMed.
- J. Wenz, A. Kochan, H. Wadepohl and L. H. Gade, Inorg. Chem., 2017, 56, 3631–3643 CrossRef CAS PubMed.
- L. Sacconi, A. Orlandini and S. Midollini, Inorg. Chem., 1974, 13, 2850–2859 CrossRef CAS.
- M. J. Sgro and D. W. Stephan, Organometallics, 2012, 31, 1584–1587 CrossRef CAS.
- R. Cariou, T. W. Graham and D. W. Stephan, Dalton Trans., 2013, 42, 4237–4239 RSC.
- T. Steinke, B. K. Shaw, H. Jong, B. O. Patrick and M. D. Fryzuk, Organometallics, 2009, 28, 2830–2836 CrossRef CAS.
- I. Morales-Becerril, M. Flores-Álamo, A. Tlahuext-Aca, A. Arévalo and J. J. García, Organometallics, 2014, 33, 6796–6802 CrossRef CAS.
- A. Mahler, B. G. Janesko, S. Moncho and E. N. Brothers, J. Chem. Phys., 2018, 148, 244106 CrossRef PubMed.
- S. Dohm, A. Hansen, M. Steinmetz, S. Grimme and M. P. Checinski, J. Chem. Theory Comput., 2018, 14, 2596–2608 CrossRef CAS PubMed.
- Y. Chen, C. Sui-Seng, S. Boucher and D. Zargarian, Organometallics, 2005, 24, 149–155 CrossRef CAS.
- E. Brunner, J. Chem. Eng. Data, 1985, 30, 269–273 CrossRef CAS.
- L. Perrin, L. Maron, O. Eisenstein and M. F. Lappert, New J. Chem., 2003, 27, 121–127 RSC.
- M. P. Conley, G. Lapadula, K. Sanders, D. Gajan, A. Lesage, I. del Rosal, L. Maron, W. W. Lukens, C. Copéret and R. A. Andersen, J. Am. Chem. Soc., 2016, 138, 3831–3843 CrossRef CAS PubMed.
- P. D. Bolton, E. Clot, N. Adams, S. R. Dubberley, A. R. Cowley and P. Mountford, Organometallics, 2006, 25, 2806–2825 CrossRef CAS.
- H. Fan, B. C. Fullmer, M. Pink and K. G. Caulton, Angew. Chem., Int. Ed., 2008, 47, 9112–9114 CrossRef CAS PubMed.
- E. J. Stoebenau and R. F. Jordan, J. Organomet. Chem., 2006, 691, 4956–4962 CrossRef CAS.
- J. Arnold, T. D. Tilley, A. L. Rheingold and S. J. Geib, Organometallics, 1987, 6, 473–479 CrossRef CAS.
- L. Perrin, L. Maron, O. Eisenstein and T. D. Tilley, Organometallics, 2009, 28, 3767–3775 CrossRef CAS.
- N. Barros, O. Eisenstein and L. Maron, Dalton Trans., 2010, 39, 10749–10756 RSC.
- N. Barros, O. Eisenstein and L. Maron, Dalton Trans., 2010, 39, 10757–10767 RSC.
- J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed.
- B. A. Vastine and M. B. Hall, J. Am. Chem. Soc., 2007, 129, 12068–12069 CrossRef CAS PubMed.
- B. A. Vastine and M. B. Hall, Coord. Chem. Rev., 2009, 253, 1202–1218 CrossRef CAS.
- D. H. Ess, W. A. Goddard and R. A. Periana, Organometallics, 2010, 29, 6459–6472 CrossRef CAS.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed.
- J. Guihaumé, S. Halbert, O. Eisenstein and R. N. Perutz, Organometallics, 2012, 31, 1300–1314 CrossRef.
- J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098–13101 CrossRef CAS PubMed.
- S. Tang, O. Eisenstein, Y. Nakao and S. Sakaki, Organometallics, 2017, 36, 2761–2771 CrossRef CAS.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- A. K. Roy and R. B. Taylor, J. Am. Chem. Soc., 2002, 124, 9510–9524 CrossRef CAS PubMed.
- S. Sakaki, N. Mizoe, M. Sugimoto and Y. Musashi, Coord. Chem. Rev., 1999, 190–192, 933–960 CrossRef CAS.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: It contains the experimental details (synthesis and characterization), the computational details, the microkinetic study and the calculated energies of all species discussed, the Gibbs energy profiles for the Ni–H and Ni–silyl catalytic cycles (including competing pathways) and additional chemical reactions. The coordinates of all calculated species are provided as an. xyz file directly readable by Mercury. The crystallographic data of 3b is registered as CCDC 1981795. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00997k |
| ‡ These authors contributed equally. |
| § 3a can also be generated in situ via treatment of 3b with 2.1 equiv. of Li(OEt2)3B(C6F5)4. See ESI† for details. |
| This journal is © The Royal Society of Chemistry 2020 |