
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron porphyrin catalysed light driven C–H bond amination and alkene aziridination with organic azides†
Yi-Dan
Du
 ac,
Cong-Ying
Zhou‡
a,
Wai-Pong
To
a,
Hai-Xu
Wang
a and
Chi-Ming
Che
*abc
ac,
Cong-Ying
Zhou‡
a,
Wai-Pong
To
a,
Hai-Xu
Wang
a and
Chi-Ming
Che
*abc
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, China. E-mail: cmche@hku.hk
bHKU Shenzhen Institute of Research & Innovation, Shenzhen, China
cShanghai-Hong Kong Joint Laboratory in Chemical Synthesis, Shanghai Institute of Organic Chemistry, 354 Feng Lin Road, Shanghai, China
First published on 17th April 2020
Abstract
Visible light driven nitrene transfer and insertion reactions of organic azides are an attractive strategy for the design of C–N bond formation reactions under mild reaction conditions, the challenge being lack of selectivity as a free nitrene reactive intermediate is usually involved. Herein is described an iron(III) porphyrin catalysed sp3 C–H amination and alkene aziridination with selectivity by using organic azides as the nitrogen source under blue LED light (469 nm) irradiation. The photochemical reactions display chemo- and regio-selectivity and are effective for the late-stage functionalization of natural and bioactive compounds with complexity. Mechanistic studies revealed that iron porphyrin plays a dual role as a photosensitizer and as a catalyst giving rise to a reactive iron–nitrene intermediate for subsequent C–N bond formation.
Introduction
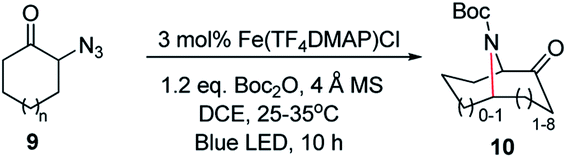
The development of efficient methods for selective C–N bond formation under mild reaction conditions is important in organic synthesis owing to the ubiquity of amino groups in bioactive natural products and pharmaceuticals. Nitrene C–H insertion and alkene aziridination are powerful methods for installing amino groups into organic molecules.1 In this regard, there is burgeoning interest in using organic azides as the nitrene source as only nitrogen gas is produced as a by-product and an external oxidant is not required. However, nitrene transfer reactions of organic azides, especially for intermolecular reactions, often require elevated temperature, which may limit the substrate scope and product selectivity, as well as increase the chance for competitive side reactions. Photochemical activation of organic azides offers a potential solution to this issue. As an example, Yoon and co-workers found that irradiation of azidoformates and alkenes with visible light in the presence of [Ir(ppy)2(dttbpy)]PF6 led to aziridination products at room temperature.2a König and co-workers employed [Ru(bpy)3](Cl)2 as a photosensitizer to achieve sp2 C–H amidation of electron-rich heteroaromatics with benzoyl azides under visible light irradiation.2c These reactions proceeded via a free nitrene intermediate generated through energy transfer from triplet excited states of transition metal sensitizers to organic azides. While free nitrenes react efficiently with organics, their high reactivity and short lifetime present significant hurdles for the reactions with relatively unreactive sp3 C–H bonds or multi C–H bonds in a selective manner. As metal–nitrenes are envisioned to display higher selectivity than free nitrenes, photochemical generation of reactive metal–nitrene species from organic azides would be appealing. We are interested in the studies of Newcomb and co-workers on the generation of reactive iron–oxo species for C–H hydroxylation through light irradiation of iron porphyrins in the presence of ClO4−.3 A collaborative study by Martin-Diaconescu, Neese, Roithova, Bill, Lloret-Fillol, Costas and their co-workers reported the generation of iron–nitrido species by photolysis of an iron–azide complex bearing a pentadentate aminopyridine ligand.4 Abu-Omar and co-workers revealed the first examples of mononuclear imido complexes of manganese(V) and chromium(V) corroles under photolytic or thermal conditions.5 We conceive that metalloporphyrins with labile axial coordination sites are potential appealing catalysts for visible-light driven nitrene C–H insertion and aziridination with organic azides,6 in which metalloporphyrin acts as the photosensitizer and at the same time captures free nitrene to give a reactive metal–nitrene/imido intermediate. Herein is described a light induced intermolecular sp3 C–H bond amination and alkene aziridination with organic azides and intramolecular sp3 C–H bond amination of alkyl azides catalysed by iron porphyrin (Scheme 1). Robert and co-workers reported an iron(III) porphyrin that catalyses light-induced CO2 reduction to CO.7a Draksharapu, Gruden, Browne and co-workers reported a non-heme iron photocatalyst for light driven aerobic oxidation of methanol.7b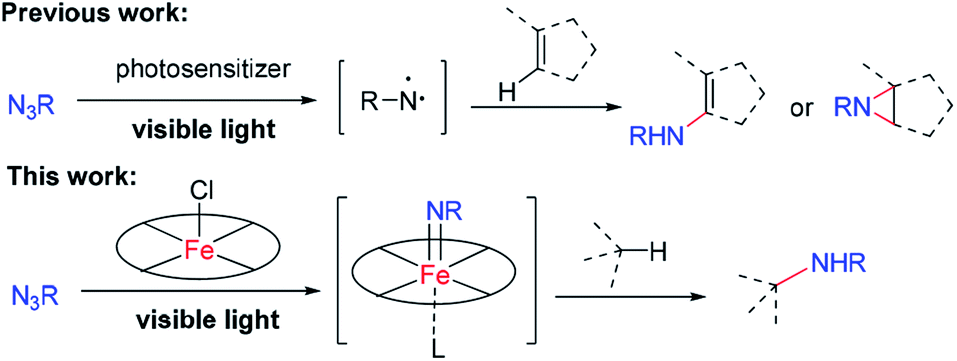 | ||
| Scheme 1 C–H amination with organic azides by iron porphyrin photocatalysis. | ||
Results and discussion
Reaction conditions and mechanism of iron-porphyrin photocatalysis
At the outset, we examined a panel of iron porphyrins for light driven C–H amination of indane 1a with 3,5-bis(trifluoromethyl)phenyl azide 2a (Scheme 2). The reaction was conducted at 25–35 °C with blue LED (469 nm) irradiation. Fe(TTP)Cl, Fe(p-Cl-TPP)Cl, Fe(TMP)Cl, Fe(p-F-TPP)Cl, Fe(TDCPP)Cl, Fe(TDCDMAP)Cl, Fe(F20TPP)Cl and Fe(TF4DMAP)Cl gave C–H amination product 3a in 12–99% yields with Fe(TF4DMAP)Cl8 being the most effective one (99% yield in 24 h; 93% yield in 15 h; Table S1 in the ESI†). No reaction was observed in the absence of the iron porphyrin catalyst or blue LED irradiation revealing that both the iron porphyrin catalyst and light are indispensable in the photochemical C–H amination reaction. The photo-stability of Fe(TF4DMAP)Cl and Fe(F20TPP)Cl in deaerated DCE was examined. As portrayed in Fig. S1 and S2,† respectively, the UV-visible absorption spectra of these two complexes remained unchanged upon blue LED (469 nm) irradiation for at least 18 hours. An iron(II) porphyrin mono(dialkylcarbene) complex, Fe(F20TPP)(Ad) (Ad = 2-adamantylidene),9 also catalysed the reaction to give 3a in 98% yield. [Fe(F20TPP)]2(μ-O), which was slowly converted to Fe(F20TPP)Cl upon light irradiation (Fig. S3†), is another effective catalyst affording 3a in 96% yield. Other Ru, Mn, Co and Ir porphyrins were less effective, giving 3a in 15% to 82% yields along with by-product 3,5-bis(trifluoromethyl)aniline 4a in 8–50% yields. Other iron complexes supported by non-porphyrin N4/N5 ligands failed to give the desired C–H amination product under the same reaction conditions. In the absence of molecular sieves, the product yield dropped to 30%. The use of free porphyrin H2TF4DMAP or [Ru(bpy)3](Cl)2·6H2O as the photosensitizer furnished 3a in 10% and 15% yields, respectively. When Rh2(OAc)4 was used as the catalyst, no reaction was observed under the same conditions. The photochemical reaction could also take place to give 3a in 93% yield under thermal conditions with Fe(TF4DMAP)Cl as the catalyst but at a reaction temperature of 120 °C. When the reaction was performed under irradiation of a green LED (530 nm) for 36 h, 3a was obtained in 25% yield.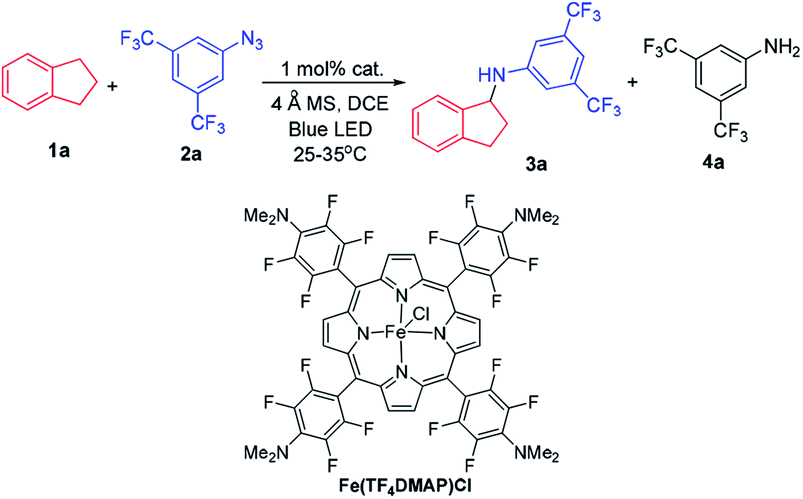 | ||
| Scheme 2 C–H amination of indane 1a with azide 2a and the structure of Fe(TF4DMAP)Cl. | ||
To investigate whether the metal–nitrene intermediate was involved in this light driven C–H amination, several experiments were conducted. [1] The kinetic isotope effect (KIE) was examined by the reaction of ethylbenzene, d10-ethylbenzene and arylazide 2a under the photocatalytic conditions (Scheme 3A). A KIE value of 6.5 was obtained. This KIE value in the range of 5–11 observed in transition metal porphyrin catalysed sp3 C–H amination is supportive of a metal–nitrene intermediate that reacts with the C–H bond via a H-atom abstraction mechanism.10 Bettinger and co-workers found that a free nitrene (R2BN) generated from the photolysis of azidoboranes can undergo intermolecular insertion into a C–H bond of cyclohexane. The KIE value was measured to be 1.35.10d [2] To examine if the reaction involves a radical chain mechanism, the quantum yields (QY) of these photo-chemical reactions have been estimated by using potassium ferrioxalate as a chemical actinometer. It was found that the QY for the reaction of styrene and 2a was 12% while that of indane and 2a was 5.5%. A lower QY of 3.5% was found with Fe(3,5-DitBu-Chenphyrin)Cl11 instead of Fe(TF4DMAP)Cl as the photocatalyst. Since the QY values of these reactions were less than 100%, the photochemical reaction is less likely to involve a radical chain mechanism.12 [3] The cross-over reaction of indane with 2a and 2,3,4,5,6-pentafluoroaniline gave C–H amination products 3a and 3c in 70% and 15% yield, respectively, suggesting a stepwise mechanism (Scheme 3B). [4] When the photochemical C–H amination of indane with arylazide 2a was performed with the chiral Fe(D4-por*)Cl (H2(D4-por*) = meso-tetrakis-{(1R,4S,5S,8R)-1,2,3,4,5,6,7,8-octahydro-1,4:5,8-dimethano-anthracen-9-yl}-porphyrin) catalyst, the C–H aminated product 3a was obtained in 82% yield and with 20% ee (Scheme 4). With Fe(D4-por*)Cl as the catalyst, the reaction of styrene with TsN3 led to aziridine 11d in 49% yield and with 34% ee. This result is comparable to that in our previous work in which the stoichiometric reaction of styrene with Ru(D4-por*)(NTs)2 gave 11d with 27% ee.13 [5] MALDI-MS analysis of the stoichiometric reaction of Fe(TF4DMAP)Cl and azide 2a showed a m/z signal at 1355.1475, corresponding to the iron–nitrene/imido formulation (Fig. S4†). [6] The aziridination of alkenes and 2a under photocatalytic conditions displayed good stereospecificity, giving cis-aziridines from cis-alkenes and trans-aziridines from trans-alkenes in up to 90% yield (Scheme 5), which favours the involvement of a metal–nitrene intermediate rather than a free triplet nitrene. [7] We attempted to detect if any short-lived species would be generated from the iron(III) porphyrin chloride complexes (Fe(TF4DMAP)Cl and Fe(F20TPP)Cl) upon photo-excitation in the presence of azide 2a by nanosecond time-resolved absorption spectroscopy. A transient signal of spectral change at 350–420 nm was observed 1 μs after laser flash and decayed back to the initial base line after 25 μs; such a signal was not observed for photoexcitation of the iron porphyrin complex in the absence of azide 2a under the same conditions. But as the signal was close to the laser excitation wavelength of 355 nm and relatively weak, it would be difficult to make any conclusive statement. These results altogether suggest that in the iron porphyrin catalysed light driven C–H amination, a reactive metal–nitrene/imido species was generated in the course of photolysis.
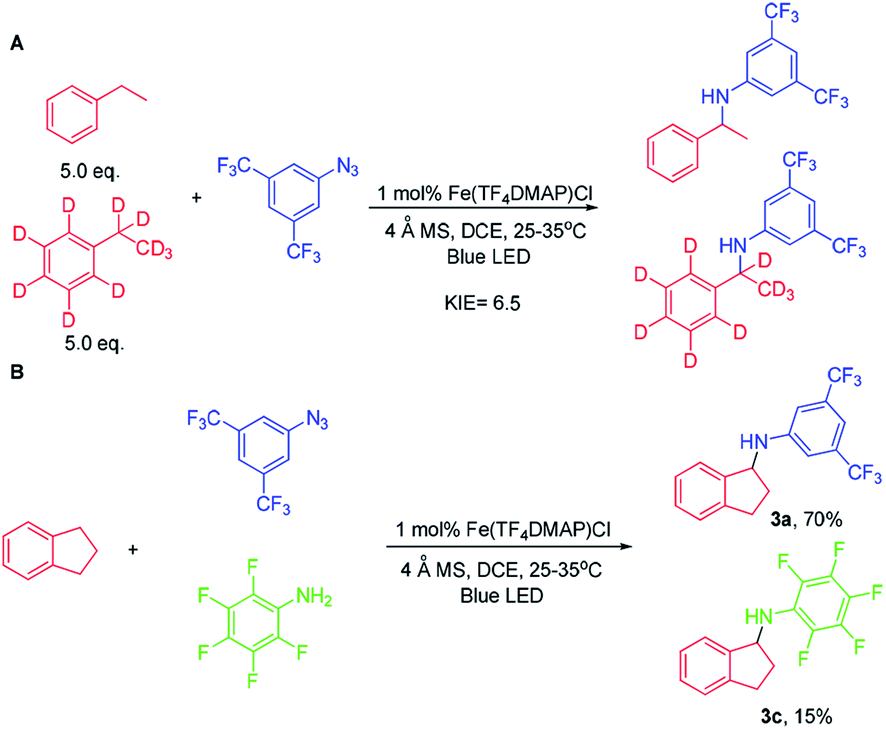 | ||
| Scheme 3 Mechanistic study. | ||
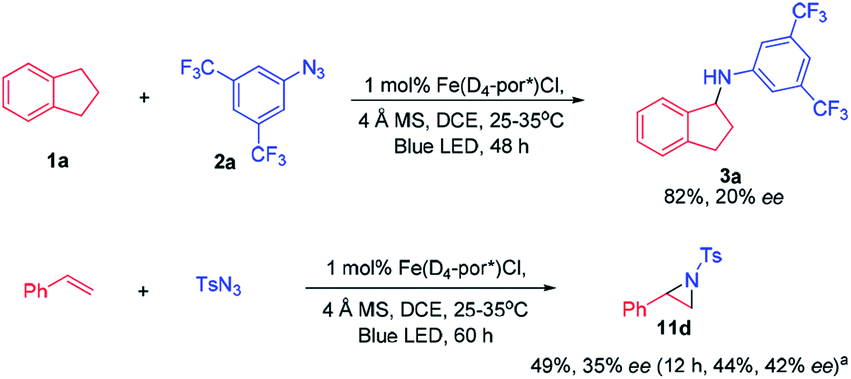 | ||
| Scheme 4 Asymmetric C–H amination and aziridination catalysed by chiral iron porphyrin. a 5 mol% catalyst was used. | ||
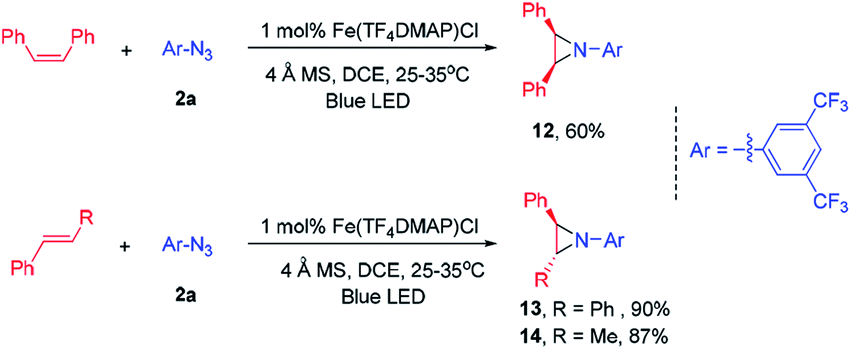 | ||
| Scheme 5 Stereospecific aziridination of alkenes and arylazide 2a catalysed by Fe(TF4DMAP)Cl. | ||
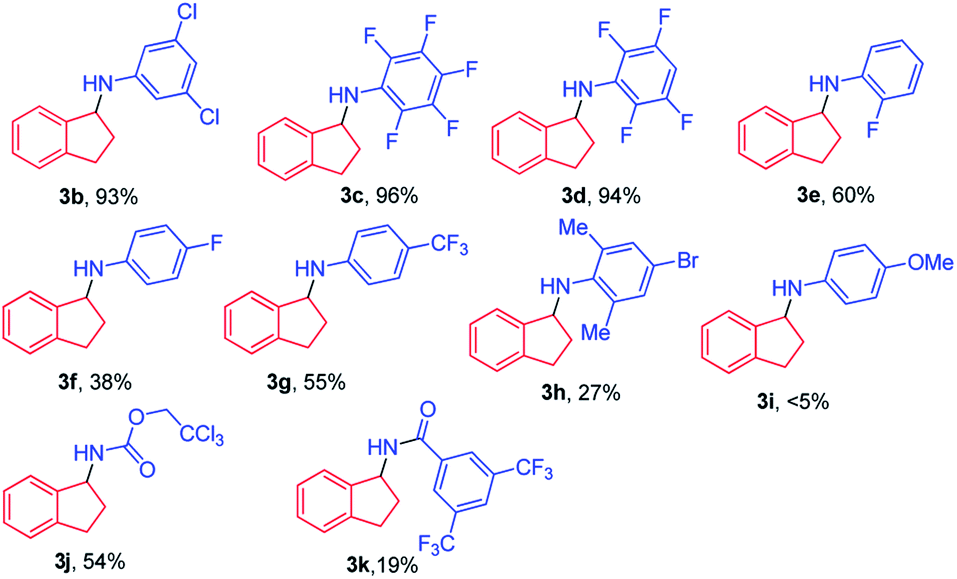
With the optimal conditions, the scope of organic azides for the iron porphyrin-catalysed light driven sp3 C–H amination was examined by using indane as the substrate. As shown in Table 1, electron-withdrawing substituents (CF3, F and Cl) of aryl azides facilitated the intermolecular C–H amination leading to a high yield of products (93–96%, 3b–3d). For aryl azides having only one electron-withdrawing substituent, C–H amination products were obtained in moderate yields (27–60%, 3e–3h). The electron-donating substituent OMe was found to disfavour the reaction resulting in poor product yield (<5%, 3i). It is worth noting that azidoformate Troc-N3 was also reactive in the photocatalytic C–H amination giving 3j in 54% yield. The use of 3,5-bis(trifluoromethyl)benzoyl azide under photocatalytic conditions led to poor yield of the C–H amination product 3k. No reaction was observed when tosyl azide, diphenyl phosphorylazide or benzylazide was used as the nitrene source.
|
|
|---|
| a Reactions were run under argon with 0.5 mmol of the azide, 5.0 mmol of indane, 5 × 10−3 mmol of the catalyst (1 mol% with respect to the azide) and 120 mg 4 Å MS in 2.0 mL of anhydrous DCE in a 10.0 mL sealed tube. The tube was irradiated with a blue LED at 25–35 °C. |
|
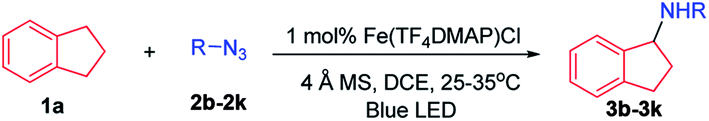
With azide 2a as the nitrene source, the scope of hydrocarbons was examined. As shown in Table 2, azide 2a underwent nitrene insertion into the benzylic C–H bonds of a range of substrates including ethylbenzene, tetralin, diphenylmethane, fluorene, xanthene, and isochroman to give the amination products in good to high yields (5a–5l). For substituted ethylbenzenes, both electron-donating and -withdrawing groups (4-methoxy, 4-methyl, and 4-chloride) led to high product yields (5g–5j). When isopentylbenzene which bears both benzylic and tertiary C–H bonds was used as the substrate, the reaction took place regio-selectively at the benzylic C–H bond over the tertiary C–H bond (5k).
|
|
|---|
| a Reactions were run under argon with 0.5 mmol of the azide, 5.0 mmol of the substrates, 5 × 10−3 mmol of the catalyst (1 mol% with respect to the azide) and 120 mg 4 Å MS in 2.0 mL of anhydrous DCE in a 10.0 mL sealed tube. The tube was irradiated with a blue LED at 25–35 °C. b 5 mol% catalyst was used. |
|
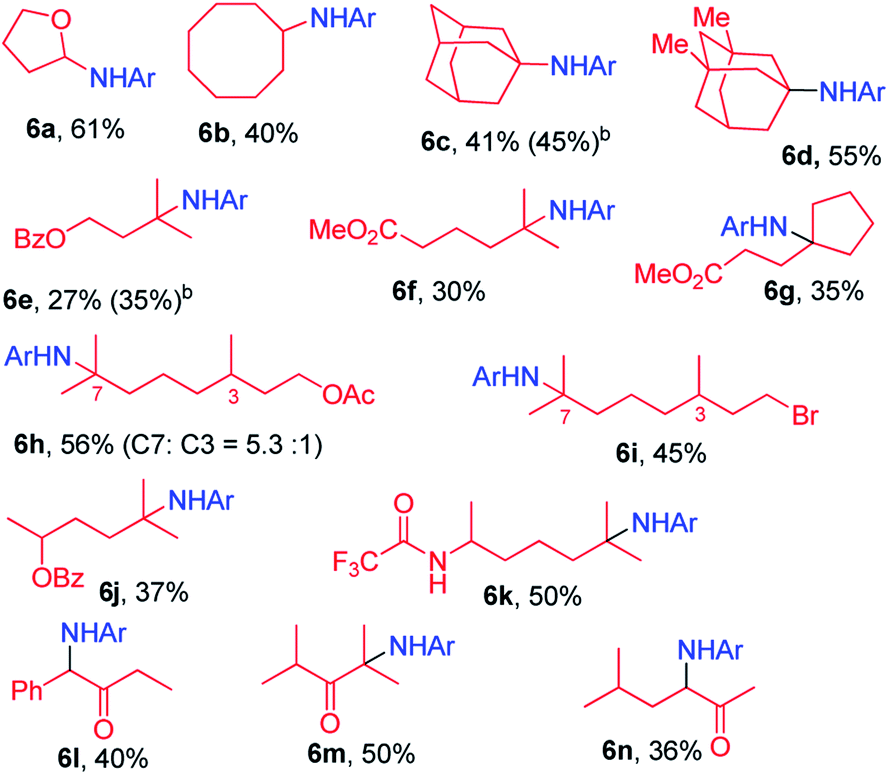
|
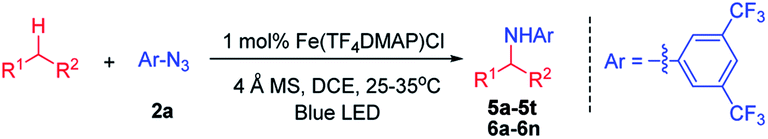
In the case of methyl (S)-2-(4-isobutylphenyl)propanoate which has two electronically different benzylic C–H bonds, the C–H amination took place at the more electron-rich C–H bond (5l).
This photochemical C–H amination is also applicable to allylic C–H bonds. The reaction of cyclohexa-1,4-diene with 2a gave the allylic C–H amination product 5m in 70% yield. Allylbenzene gave a mixture of C–H aminated isomeric products (5na and 5nb). The reaction of 1-(allyloxy)-4-(prop-1-en-2-yl)benzene proceeded with high chemo- and regio-selectivity to give 5o in 35% yield. When 2,3-dimethylbuta-1,3-diene was subjected to photocatalysis, a 7-membered heterocyclic product 5p was isolated in 78% yield. The reaction also took place at the allylic C–H bond of α,β-unsaturated aldehydes and α,β-unsaturated ketones in good product yields (5q–5s). When cyclic 2-norbornene was used, bridged secondary C–H amination product 5t was obtained in 52% yield with an endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo ratio of 3:1 along with the aziridination product in 12% yield. No allylic C–H amination product was observed.
:exo ratio of 3:1 along with the aziridination product in 12% yield. No allylic C–H amination product was observed.
We next examined the photocatalytic C–H amination of tetrahydrofuran and unactivated C–H bonds, the functionalization of which is a challenge. Tetrahydrofuran was reactive under photocatalytic conditions giving the amination product 6a in 61% yield (Table 2). The reaction of cyclooctane with 2a proceeded smoothly to give 6b in 40% yield. When the substrate contains tertiary, secondary and primary C–H bonds, the photocatalytic amination preferentially occurred at the tertiary C–H bond over the secondary and primary C–H bonds (6c–6g). We also investigated the regioselectivity of the photochemical C–H amination of substrates bearing multiple tertiary C–H centers. When dihydrocitronellyl acetate or dihydrocitronellyl bromide was subjected to the photochemical C–H amination, the reaction occurred preferentially at the tertiary C–H bond remote from the electron-withdrawing group (acetate or bromo, 6h or 6i). This regioselectivity could be attributed to the deactivation of the C(3)–H bond by the electron-withdrawing group.
To our surprise, the α-C–H bond of ketone, which is generally thought to be deactivated by a carbonyl moiety, could also be aminated. Treatment of 1-phenylbutan-2-one with 2a and a catalytic amount of iron porphyrin at room temperature under blue LED irradiation afforded α-C–H amination product 6l in 40% yield. Likewise, 2,4-dimethylpentan-3-one and 5-methylhexan-2-one gave α-C–H aminated products 6m and 6n in 50% and 36% yields, respectively. No reaction was found when methyl 2-phenylacetate was used. These results suggest that 2a might react with the enol form of ketone to afford an aziridine which underwent ring-opening to give the final α-C–H amination product.14
Intramolecular C–H amination of alkyl azides
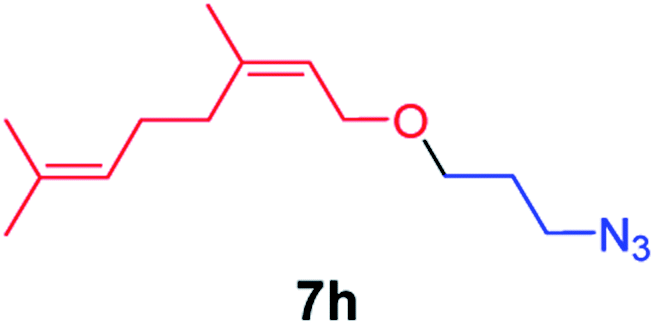
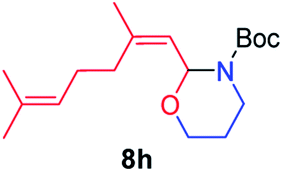
We next used photocatalysis of intramolecular C–H amination of alkyl azides15,16 to form imidazolidines,17 a versatile class of intermediates in organic synthesis. As shown in Table 3, a variety of N-containing alkyl azides underwent C–H amination to give the corresponding imidazolidines in 45–98% yield. It is noteworthy that when 1-(2-azidoethyl)indoline was subjected to photolysis, an indole product was obtained in 95% yield presumably derived from ring-opening of the imida-zolidine product (entry 7). For the reaction of O-containing alkyl azide 7h, a 1,3-oxazinane derivative 8h was obtained in 55% yield (entry 8).|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%) |
| a Reactions were run under argon with 0.3 mmol of the azide, Boc2O (0.36 mmol), 3 × 10−3 mmol of the catalyst (1 mol% with respect to the azide) and 120 mg 4 Å MS in 2.0 mL of anhydrous DCE in a 10.0 mL sealed tube. The tube was irradiated with a blue LED at 25–35 °C. Isolated yields are shown. b [Fe(F20TPP)Cl] was used as the catalyst. Yields were determined by 1H NMR spectroscopy. | |||
| 1 |
|
|
75 |
| 2 |
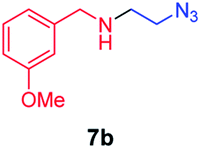
|
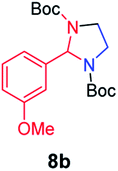
|
70 |
| 3 |
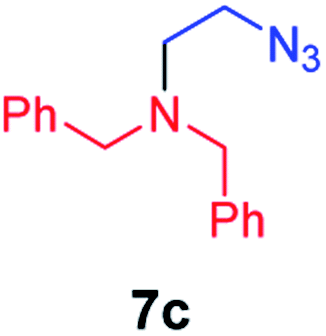
|
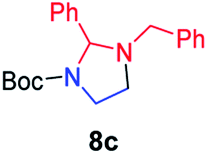
|
78 |
| 4 |
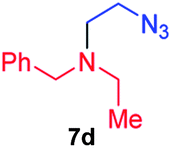
|
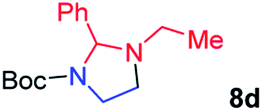
|
61; 30b |
| 5 |
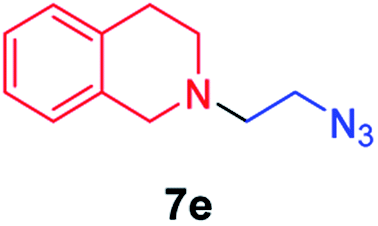
|
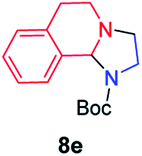
|
45 |
| 6 |
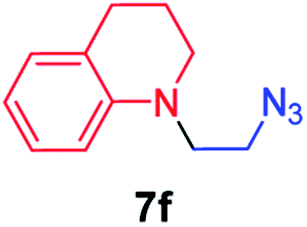
|
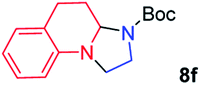
|
98 |
| 7 |
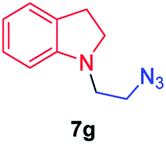
|
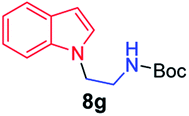
|
95 |
| 8 |
|
|
55 |
Intramolecular C–H amination of α-azidoketones
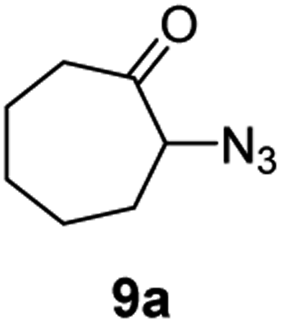
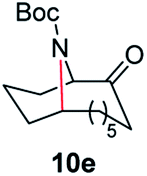
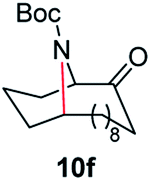
A nitrogen bridged bicyclic moiety is prevalent in naturally occurring alkaloids.18 This valuable scaffold can be constructed by our iron-catalysed light driven C–H amination of α-azidoketones. As shown in Table 4, various medium-sized cyclic α-azidoketones underwent intramolecular C–H amination to give the corresponding nitrogen bridged bicyclic compounds in 42–85% yields (entry 1–6). It is noteworthy that 2-azidocyclohexan-1-one failed to give the intramolecular C–H amination product, suggesting that the ring strain affected the reactivity.|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv. (%) | Yield (%) |
| a Reactions were run under argon with 0.3 mmol of the azide, Boc2O (0.36 mmol), 9 × 10−3 mmol of the catalyst (3 mol% with respect to the azide) and 120 mg 4 Å MS in 2.0 mL of anhydrous DCE in a 10.0 mL sealed tube. The tube was irradiated with a blue LED at 25–35 °C. Isolated yields are shown in the brackets. | ||||
| 1 |
|
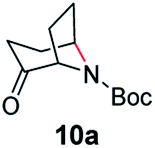
|
70 | 46 (42) |
| 2 |
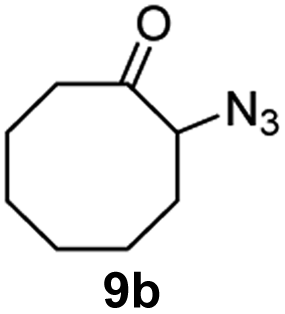
|
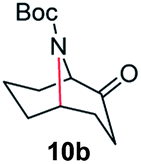
|
100 | 90 (80) |
| 3 |
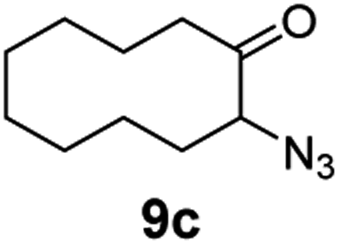
|
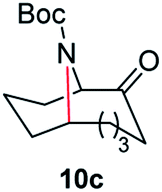
|
100 | 92 (85) |
| 4 |
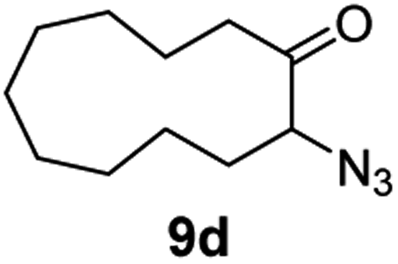
|
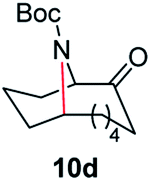
|
100 | 82 (77) |
| 5 |
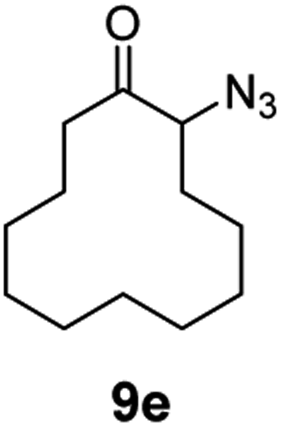
|
|
100 | 84 (77) |
| 6 |
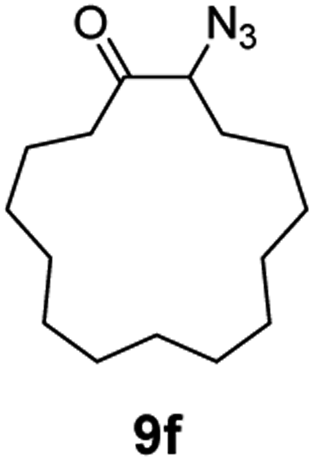
|
|
100 | 90 (82) |
Derivatization of natural products
As photocatalysis can be performed under mild conditions and with high regio- and chemo-selectivity, we conceive that this methodology has the potential to selectively aminate the C–H bond of other complex natural products which possess various C–H bonds and functional groups. We first examined the C–H amination of Ac-cholesterol that contains a total of 48 C–H bonds, 22 of which are secondary, 7 tertiary and 4 allylic (Scheme 6A). Under the photochemical conditions, Ac-Cholesterol underwent amination exclusively at the allylic C–H bond remote from the electron-withdrawing group -OAc in 77% isolated yield with a dr ratio of 4.5:1. Fe(F20TPP)Cl also catalysed the light-driven amination of Ac-cholesterol with 52% product yield. Similar to cholesterol, the reaction of Bz-diosgenin gave allylic C–H bond aminated product 16 in 60% isolated yield with a dr ratio of 3:1 when 2b was used as the nitrene source (Scheme 6B). Amination of the C–H bond adjacent to the oxygen atom was not observed. When 2a was used as the nitrene source, the corresponding amination product was obtained in 78% yield with a dr ratio of 7:1. Treatment of an estrone derivative (Scheme 6C) with 2a and Fe(TF4DMAP)Cl under photocatalytic conditions gave an allylic amination product 18 in 79% yield. Likewise, a double allylic amination of a progesterone derivative (Scheme 6D) was observed with 28% yield. When (−)-ambroxide was subjected to photolysis, the C–H amination occurred at the C–H bond adjacent to the O atom giving imine product 20 in 23% yield with a recovery of 45% starting material (Scheme 6E). Treatment of (+)-nootkatone with 2a under photochemical conditions gave an aziridination product in 30% yield.
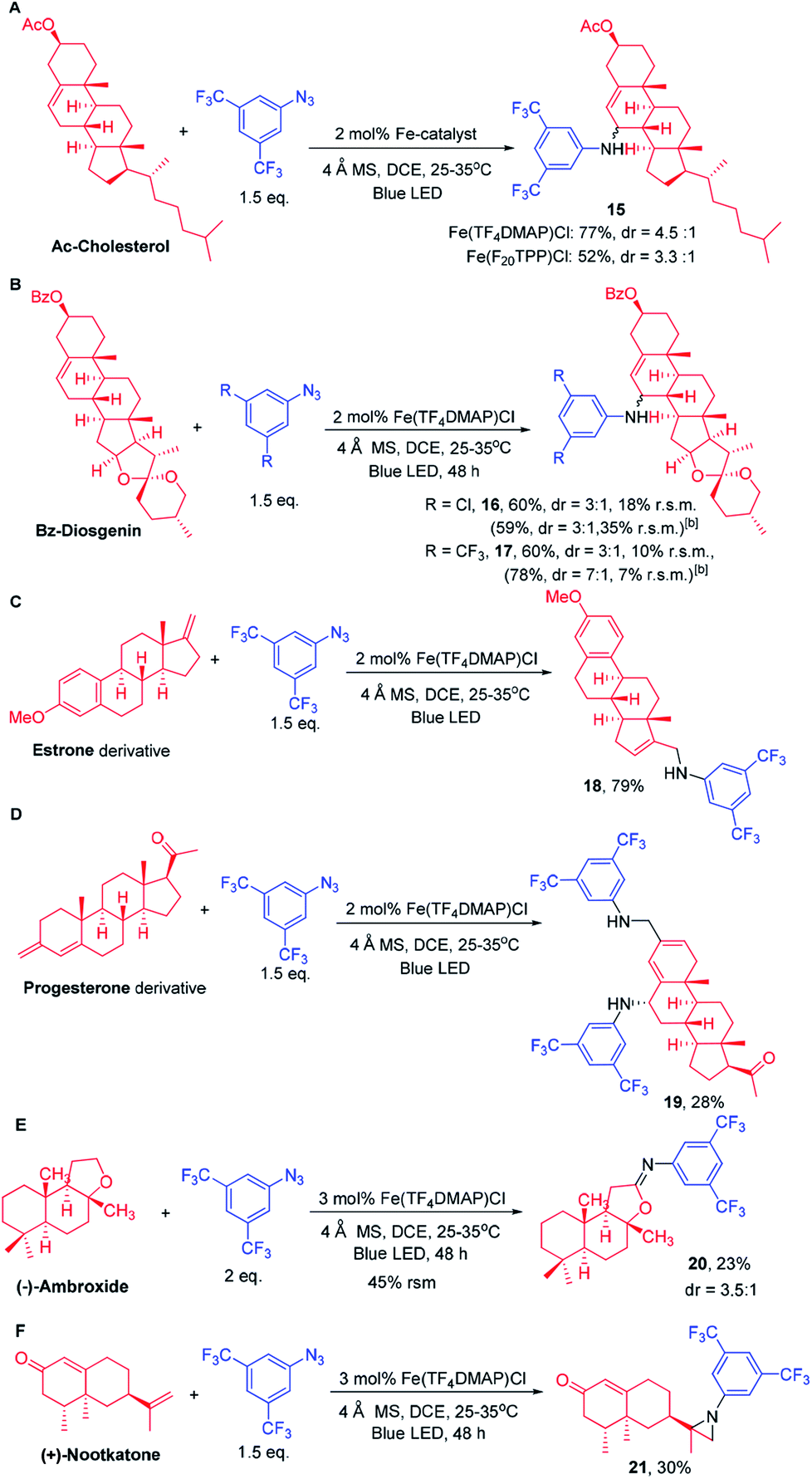 | ||
| Scheme 6 Late-stage diversification of complex molecules via [Fe(TF4DMAP)Cl]-catalysed C–H amination. [a] The reaction was conducted with 2 mol% catalyst and irradiated for 48 h. [b] The reaction was conducted with 5 mol% catalyst and irradiated for 24 h. r.s.m. = recovery of starting material. | ||
Conclusions
In summary, we have demonstrated that the iron porphyrin Fe(TF4DMAP)Cl is an efficient catalyst for selective intermolecular C–H amination and alkene aziridination of organic azides and intramolecular sp3 C–H bond amination of alkyl azides under blue LED light irradiation. Mechanistic study revealed that Fe(TF4DMAP)Cl played a dual role in the reactions acting as a photosensitizer and as a catalyst to form a metal–nitrene intermediate for subsequent C–N bond formation. A variety of sp3 C–H bonds can be aminated in good to high yields and with excellent regioselectivity. The reactivity trend of C–H bonds for the photocatalytic amination was found to be benzylic ≈ allylic > tertiary > secondary > primary C–H bond. The method is also effective for selective late stage C–H amination of complex natural products. The photochemical reaction herein developed has several eco-friendly advantages including: (1) using organic azides as aminating agents, (2) using catalysts of biocompatible and earth abundant iron, and (3) using blue LED light as the energy source. Nonetheless the use of DCE as the solvent is not a desirable feature and studies on using an environmentally benign solvent system to replace DCE are needed to further improve this iron porphyrin catalysed photochemical amination reaction. The chiral Fe(D4-por*)Cl can catalyse the light driven C–H amination and alkene aziridination with moderate enantioselectivity. To our knowledge, this work represents the first examples of visible light driven sp3 C–H amination and alkene aziridination with organic azides by iron porphyrin photocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC 91856203 and 21472159), Basic Research Program-Shenzhen Fund (JCYJ20170412140251576 and JCYJ20180508162429786), Hong Kong Research Grants Council General Research Fund (17301817 and 17303815) and “Laboratory for Synthetic Chemistry and Chemical Biology” funded by the Health@InnoHK of Innovation and Technology Commission, Hong Kong Special Administrative Region.Notes and references
- Selected recent reviews: (a) B. Darses, R. Rodrigues, L. Neuville, M. Mazurais and P. Dauban, Chem. Commun., 2017, 53, 493 RSC; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed; (c) T. Uchida and T. Katsuki, Chem. Rec., 2014, 14, 117 CrossRef CAS PubMed; (d) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed; (e) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 RSC; (f) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926 RSC; (g) J. W. W. Chang, T. M. U. Ton and P. W. H. Chan, Chem. Rec., 2011, 11, 331 CrossRef CAS PubMed; (h) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC.
- (a) S. O. Scholz, E. P. Farney, S. Kim, D. M. Bates and T. P. Yoon, Angew. Chem., Int. Ed., 2016, 55, 2239 CrossRef CAS PubMed; (b) E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793 CrossRef CAS PubMed; (c) E. Brachet, T. Ghosh, I. Ghosh and B. König, Chem. Sci., 2015, 6, 987 RSC; (d) P. Bellotti, J. Brocus, F. E. Orf, M. Selkti, B. König, P. Belmont and E. Brachet, J. Org. Chem., 2019, 84, 6278 CrossRef CAS PubMed; (e) Y. Zhang, X. Dong, Y. Wu, G. Li and H. Lu, Org. Lett., 2018, 20, 4838 CrossRef CAS PubMed; (f) S. Zhu, A. Pathigoola, G. Lowe, D. A. Walsh, M. Cooper, W. Lewis and H. W. Lam, Chem. - Eur. J., 2017, 23, 17598 CrossRef CAS PubMed.
- Z. Pan, Q. Wang, X. Sheng, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 2009, 131, 2621 CrossRef CAS PubMed.
- G. Sabenya, L. Lázaro, I. Gamba, V. Martin-Diaconescu, E. Andris, T. Weyhermüller, F. Neese, J. Roithova, E. Bill, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2017, 139, 9168 CrossRef CAS PubMed.
- N. Y. Edwards, R. A. Eikey, M. I. Loring and M. M. Abu-Omar, Inorg. Chem., 2005, 44, 3700 CrossRef CAS PubMed.
- (a) S. Fantauzzi, A. Caselli and E. Gallo, Dalton Trans., 2009, 5434 RSC; (b) B. J. Anding and L. K. Woo, Handbook of porphyrin science, World Scientific, Singapore, 2012, vol. 21, ch. 100 (An overview of Metalloporphyrin-Catalyzed Carbon and Nitrogen Group Transfer Reactions) Search PubMed.
- (a) H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74 CrossRef CAS PubMed; (b) J. Chen, S. Stepanovic, A. Draksharapu, M. Gruden and W. R. Browne, Angew. Chem., Int. Ed., 2018, 57, 3207 CrossRef CAS PubMed.
- Y.-D. Du, Z.-J. Xu, C.-Y. Zhou and C.-M. Che, Org. Lett., 2019, 21, 895 CrossRef CAS PubMed.
- H.-X. Wang, Q. Wan, K.-H. Low, C.-Y. Zhou, J.-S. Huang, J.-L. Zhang and C.-M. Che, Chem. Sci., 2020, 11, 2243 RSC.
- (a) S.-M. Au, J.-S. Huang, W.-Y. Yu, W.-H. Fung and C.-M. Che, J. Am. Chem. Soc., 1999, 121, 9120 CrossRef CAS; (b) S. K.-Y. Leung, W.-M. Tsui, J.-S. Huang, C.-M. Che, J.-L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629 CrossRef CAS PubMed; (c) P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2019, 23, 7945 CrossRef PubMed; (d) M. Filthaus, L. Schwertmann, P. Neuhaus, R. W. Seidel, I. M. Oppel and H. F. Bettinger, Organometallics, 2012, 31, 3894 CrossRef CAS.
- S. Zhu, J. A. Perman and X. P. Zhang, Angew. Chem., Int. Ed., 2008, 47, 8460 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- J.-L. Liang, J.-S. Huang, X.-Q. Yu, N. Zhu and C.-M. Che, Chem.–Eur. J., 2002, 8, 1563 CrossRef CAS.
- (a) T. M. U. Ton, C. Tejo, D. L. Y. Tiong and P. W. H. Chan, J. Am. Chem. Soc., 2012, 134, 7344 CrossRef CAS PubMed; (b) K. Tokumasu, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2016, 138, 2664 CrossRef CAS PubMed.
- (a) E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed; (b) D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 1983 CrossRef CAS PubMed; (c) D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599 CrossRef CAS PubMed.
- K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947 CrossRef CAS PubMed.
- G. S. de Carvalho, R. M. Dias, F. R. Pavan, C. Q. Leite, V. L. Silva, C. G. Diniz, D. T. de Paula, E. S. Coimbra, P. Retailleau and A. D. da Silva, Med. Chem., 2013, 9, 351 CrossRef CAS PubMed.
- (a) G. A. Cordell, M. L. Quinn-Beattie and N. R. Farnsworth, Phytother. Res., 2001, 15, 183 CrossRef CAS PubMed; (b) S. Wonnacott and T. Gallagher, Mar. Drugs, 2006, 4, 228 CrossRef CAS; (c) C. R. Edwankar, R. V. Edwankar, J. R. Deschamps and J. M. Cook, Angew. Chem., Int. Ed., 2012, 51, 11762 CrossRef CAS PubMed; (d) Z.-J. Zhang, J. Yang, J. He, X.-D. Wu, L.-D. Shao, Y. Li, S.-X. Huang, R.-T. Li and Q.-S. Zhao, Tetrahedron Lett., 2014, 55, 6490 CrossRef CAS; (e) J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of products. See DOI: 10.1039/d0sc00784f |
| ‡ Present address: College of Chemistry and Materials Science, Jinan University, Guangzhou, China |
| This journal is © The Royal Society of Chemistry 2020 |