
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ Raman study of the photoinduced behavior of dye molecules on TiO2(hkl) single crystal surfaces†
Sheng-Pei
Zhang
a,
Jia-Sheng
Lin
a,
Rong-Kun
Lin
a,
Petar M.
Radjenovic
a,
Wei-Min
Yang
a,
Juan
Xu
b,
Jin-Chao
Dong
*a,
Zhi-Lin
Yang
 a,
Wei
Hang
*a,
Zhong-Qun
Tian
*a and
Jian-Feng
Li
*a
a,
Wei
Hang
*a,
Zhong-Qun
Tian
*a and
Jian-Feng
Li
*a
aDepartment of Physics, State Key Laboratory of Physical Chemistry of Solid Surfaces, iChEM, College of Chemistry and Chemical Engineering, College of Energy, College of Materials, Xiamen University, Xiamen 361005, China. E-mail: Li@xmu.edu.cn; jinchaodong1209@126.com; weihang@xmu.edu.cn; zqtian@xmu.edu.cn
bFujian Province Key Laboratory of Modern Analytical Science and Separation Technology, College of Chemistry, Chemical Engineering and Environment, Minnan Normal University, Zhangzhou 363000, China
First published on 17th April 2020
Abstract
In dye-sensitized solar cells (DSSCs), the TiO2/dye interface significantly affects photovoltaic performance. However, the adsorption and photoinduced behavior of dye molecules on the TiO2 substrate remains unclear. Herein, shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) was used to study the adsorption and photoinduced behavior of dye (N719) molecules on different TiO2(hkl) surfaces. On TiO2(001) and TiO2(110) surfaces, the in situ SHINERS and mass spectrometry results indicate S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage in the anchoring groups of adsorbed N719, whereas negligible bond cleavage occurs on the TiO2(111) surface. Furthermore, DFT calculations show the stability of the SC anchoring group on three TiO2(hkl) surfaces in the order TiO2(001) < TiO2(110) < TiO2(111), which correlated well with the observed photocatalytic activities. This work reveals the photoactivity of different TiO2(hkl) surface structures and can help with the rational design of DSSCs. Thus, this strategy can be applied to real-time probing of photoinduced processes on semiconductor single crystal surfaces.
C bond cleavage in the anchoring groups of adsorbed N719, whereas negligible bond cleavage occurs on the TiO2(111) surface. Furthermore, DFT calculations show the stability of the SC anchoring group on three TiO2(hkl) surfaces in the order TiO2(001) < TiO2(110) < TiO2(111), which correlated well with the observed photocatalytic activities. This work reveals the photoactivity of different TiO2(hkl) surface structures and can help with the rational design of DSSCs. Thus, this strategy can be applied to real-time probing of photoinduced processes on semiconductor single crystal surfaces.
Dye-sensitized solar cells (DSSCs) have attracted much interest due to their versatility, low cost, and ease of fabrication into flexible devices.1 However, to date, the highest certified power conversion efficiency (PCE) recorded for a DSSC under simulated sunlight illumination is only 11.9%,2 while the theoretical maximum PCE is over 30%.3 Additionally, the long-term stability of DSSCs is very important for practical applications. DSSCs function via a charge injection mechanism between closely interacting dye/semiconductor molecules.4 Studying the adsorption and photoinduced behavior of N719 molecules on TiO2 surfaces can reveal the fundamental reaction processes and help improve the conversion efficiency and lifetime of DSSCs.5
Previously, there have been some research studies on the adsorption configuration and electronic properties of N719 dye molecules at TiO2 surfaces. Grätzel et al. concluded, based on Fourier transform infrared spectroscopy (FTIR), that N719 was anchored on the TiO2 surface in a bridged configuration via two carboxyl groups.4a They further showed with density functional theory (DFT) calculations that deprotonation of the terminal carboxylic groups cause the excitation energy to increase.6 In addition, Tian et al. investigated the Ag@TiO2/N719 interface under a series of potential controls using in situ Raman spectroscopy and found that the SCN ligand participates in N719 adsorption on the surface of aggregated TiO2 nanoparticles,7 while Lund et al. employed high-performance liquid chromatography (HPLC) and ultraviolet-visible spectroscopy (UV/Vis) to determine the photoinduced degradation and regeneration rates for N719 at the TiO2 surface. Depending on the rates achieved by the DSSC, they calculated operating times as high as 20 years or as low as 0.5 years.5 However, most of these studies only focused on polycrystalline TiO2 substrate surfaces where it is difficult to study the structure–activity relationship of the substrate. For a better understanding of the reaction processes, investigating atomically flat single crystals with well-defined surface structures as well as electronic and optical properties can act as a bridge between experimental studies and theoretical simulations.
Surface-enhanced Raman spectroscopy (SERS) is a non-destructive and ultrasensitive technique that has become a powerful surface analysis tool.8 Unfortunately, conventional SERS techniques cannot be applied to single crystal surfaces because they lack sufficient surface plasmon resonance (SPR).9 In 2010, our group invented the shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS)10 technique for obtaining enhanced surface Raman signals, irrespective of the surface and with single crystal surface compatibility. Shell-isolated nanoparticles (SHINs) act as surface Raman signal enhancers without detrimentally affecting the external environment or reaction processes due to their inert silica shell coating.11 Since then, important catalytic reactions, such as the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) with their intermediate species, as well as the structure of interfacial water at single-crystal electrode surfaces have been observed using in situ SHINERS.12
Herein, the adsorption and photoinduced behavior of N719 molecules on three TiO2(hkl) surfaces (rutile TiO2(001), TiO2(110), and TiO2(111)) under 405 nm laser irradiation were investigated with in situ SHINERS. Combined with DFT simulations, the TiO2(hkl) facet effect and the intrinsic photocatalytic mechanism were elucidated and provide a promising method for studying photoinduced surface processes in real-time.
In this system, N719 dye molecules were assembled on rutile TiO2(hkl) surfaces and SHINs were deposited on top and then transferred to a Raman cell filled with acetonitrile (without N719 in solution). During in situ Raman measurements, the cell was illuminated with two different wavelength lasers, 638 nm and 405 nm (6600 mW cm−2), for detecting Raman signals and exciting the photoinduced reactions, respectively (Fig. 1a). Besides, the power density of the 405 nm laser is 66 times higher than one sun condition (100 mW cm−2), so the photoinduced reaction speed of N719 on the TiO2 surface will be much higher due to more photo-generated carriers being produced under 405 nm laser illumination. Therefore, it is more likely for us to obtain the Raman signal in the current research. Fig. 1b shows the TEM image of two adjacent SHINs. The enhancement factor of SHINs on the TiO2 surfaces was assessed using three-dimensional finite-difference time-domain (3D-FDTD) simulations (Fig. 1c). Plasmonic coupling dictates the Raman enhancement factor and is shown to be stronger between adjacent SHINs (55 nm dia. Au core @ 2 nm thick SiO2 shell) than between SHINs and the rutile TiO2(hkl) substrate; however N719 molecules were only adsorbed on the TiO2 single crystal surface. Therefore, all N719 Raman signals measured herein must originate from the electromagnetic field enhancement between the SHINs and the substrate which had a calculated enhancement factor of about 103 times (Fig. 1c). Importantly, compared to a strategy with no SHINs (Fig. S1†), this SHINERS strategy provides enough enhancement for detection of surface N719.
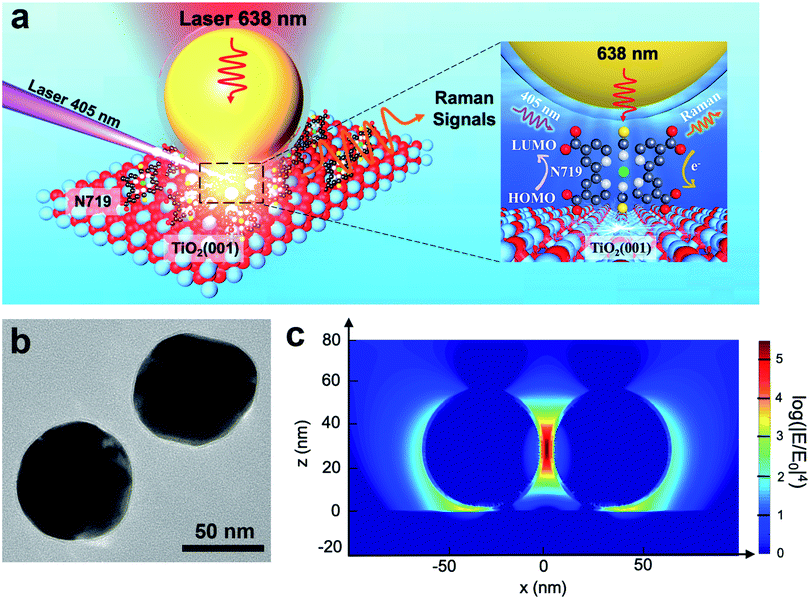 | ||
| Fig. 1 (a) Schematic diagram of using a SHINERS nanoantenna to study the photoinduced reaction of N719 illuminated with two lasers on TiO2. The inset shows electron transfer from the 405 nm laser excited N719 molecules to the TiO2 single crystal surface and the 638 nm laser used to detect the changes in excited the molecules. (b) TEM image of 55 nm Au@2 nm SiO2 SHINs. (c) 3D-FDTD simulation of the enhancement hotspot of a SHIN dimer with a nanogap of 2 nm on rutile TiO2. | ||
In Fig. 2a, three Raman bands are visible on all pristine rutile TiO2(hkl) surfaces at around 238 cm−1, 448 cm−1 and 610 cm−1 assigned to the multi-phonon, Eg, and A1g modes of the Ti–O–Ti bond vibrations, respectively,13 and their relative peak intensities (I238 cm−1, I448 cm−1 and I610 cm−1) are clearly surface facet dependent. For the N719 molecule (see Fig. S2† for the chemical structure), there are two SCN binding groups attached to the Ru atom, both of which share the highest occupied molecular orbital (HOMO) and the 2,2-bipyridyl-4,4-dicarboxylic ligand contains the lowest unoccupied molecular orbital (LUMO) of the molecule. In the UV-vis absorption spectrum of N719 dissolved in ethanol (Fig. S3†), N719 has two clear adsorption peaks. The first peak, located at around 389 nm, is associated with a mixture of ligand-to-ligand charge transfer (LLCT) and metal-to-ligand charge transfer (MLCT) processes, while the second peak at 532 nm is related to low energy MLCT processes within N719 molecules.14 The I610 cm−1/I448 cm−1 is the same after adding SHINs on the TiO2(001) surface (Fig. S4†), but interestingly, after N719 is adsorbed at the surface, the I610 cm−1/I448 cm−1 on all three rutile TiO2(hkl) surfaces changed. In Fig. 2b, I610 cm−1/I448 cm−1 intensities decreased on TiO2(111) and TiO2(001) surfaces and increased on the TiO2(110) surface. For N719 molecules chemisorbed on rutile TiO2(hkl) surfaces, the different N719 anchoring modes result in different interactions of Ti–O–Ti (Fig. 2b),15 so the ratios of I610 cm−1/I448 cm−1 differ between the three TiO2(hkl) surfaces after N719 adsorption, manifesting as a change in Raman peak intensities.11a
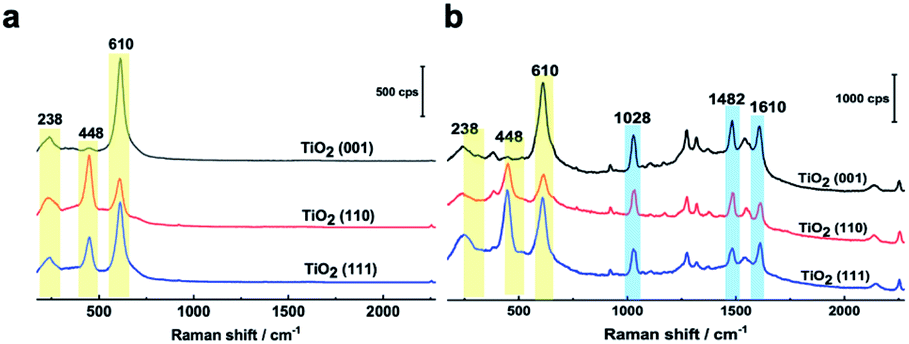 | ||
| Fig. 2 (a) SHINER spectra of rutile TiO2(001), TiO2(110), and TiO2(111) surfaces in acetonitrile without and (b) with N719 molecules adsorbed. | ||
For N719 adsorbed at different rutile TiO2(hkl) surfaces, both 1482 cm−1 and 1610 cm−1 peaks, belonging to the CN and CC stretching vibrations of the bipyridyl ring, respectively,16 are surface facet dependent (Fig. 2b). For TiO2(110), the 1482 cm−1 peak intensity (I1482 cm−1) is obviously stronger than the 1610 cm−1 peak intensity (I1610 cm−1), whereas on TiO2(111), the I1482 cm−1 is weaker than I1610 cm−1, and both peak intensities are similar on TiO2(001). This can be attributed to the different rutile single crystal surfaces having different N719 adsorption configurations, giving rise to different bond enhancements.11a,17
SHINERS was then employed to study, in situ, the adsorption and photoinduced behavior of N719 molecules on the TiO2(001) surface under 405 nm laser illumination. Fig. S5† shows Raman spectra of TiO2(111) with and without 405 nm laser illumination and shows no observable interference peaks. This lack of Raman shifts under 405 nm illumination can be further verified using the formula in Fig. S6.† Furthermore, in Fig. S7,† the Raman signals for rutile TiO2(110) without N719 adsorbed in acetonitrile were observed under extended illumination times (50 min). And peaks associated with the rutile TiO2(110) substrate are not changed, indicating that 405 nm laser illumination does not change the TiO2(hkl) substrate spectral peaks. As shown in Fig. 3a and b, as the illumination time increases, the Raman peaks of the adsorbed N719 molecules gradually weaken, while the rutile TiO2 peaks increase significantly. Considering these spectral trends over time (Fig. 3b), we tentatively ascribe this trend to desorption of N719 molecules from the TiO2(001) surface as opposed to conformational changes in the N719 adsorption angle. Additionally, the Raman peak of the SCN group at around 2136 cm−1 blue shifts to 2146 cm−1 with increasing illumination time, indicating that as the N719 surface coverage decreases the N719 molecules adsorb on the TiO2(001) surface via the S-terminal of the NCS group, corresponding with previous studies.7,18.
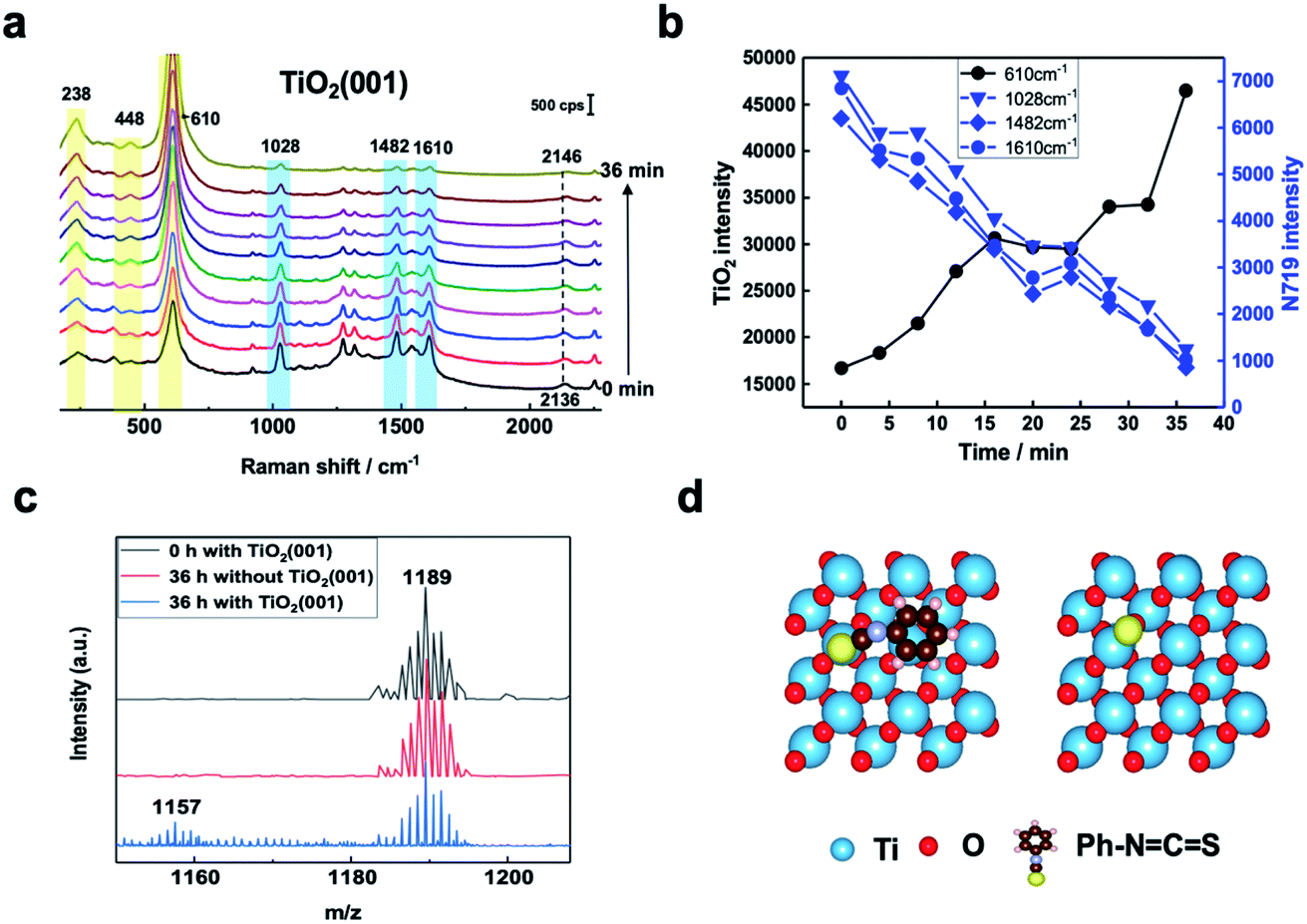 | ||
| Fig. 3 (a) SHINER Spectra of N719 adsorbed at TiO2(001) in acetonitrile under 405 nm laser illumination, spectra collected every 4 minutes. (b) Plot of changes in significant N719 (blue lines and points) and TiO2 (black line and points) spectral peak intensities with time. (c) Mass spectra of a 5 × 10−4 M N719 ethanol solution containing TiO2(001) under 405 nm laser illumination after 0 h and 36 h, and without TiO2(001) after 36 hours. (d) Schematic of the optimized structures of the Ph–NCS group and S atom adsorbed on rutile TiO2(001). | ||
Mass spectrometry was used to examine photoinduced reaction products. In Fig. 3c, the produced ion of m/z 1157 appears after 36 hours under 405 nm laser illumination which corresponds to the loss of an S atom from N719. This confirms bond cleavage of the SC bond in the SCN group adsorbed on the TiO2(001) surface under 405 nm laser illumination producing a [RuII(H2dcbpy)2(NCS)(CN)] species.19 For comparison, the adsorption behavior of the NCS functional group of a Ph–NCS species on different rutile TiO2(hkl) surfaces was simulated using DFT. This also allowed for the reaction energy change due to the SC double bond cleavage to be directly calculated. The calculated structures of the Ph–NCS group and S atom adsorbed on rutile TiO2(001) are shown in Fig. 3d. The SC dissociation energy on TiO2(001) was found to be lower than that on TiO2(111) and TiO2(110) surfaces (Fig. S8†). Thus, the TiO2(001) peak intensities (238 cm−1, 448 cm−1, and 610 cm−1) increase as the illumination time increases due to increasing SC bond cleavage resulting in N719 desorption.20
To further determine the facet effect, the adsorption and photoinduced behavior of N719 was also systemically studied on TiO2(111) and TiO2(110) with in situ SHINERS. In Fig. 4a, the time-dependent Raman spectra of N719 at the TiO2(110) surface are similar to those at the TiO2(001) surface (Fig. 3a), with N719 Raman intensities decreasing with increasing illumination times; however the trend is less pronounced. Meanwhile, the Raman peak shift of the SCN group around 2143 cm−1 is also small (∼6 cm−1). In contrast, in Fig. 4b, the N719 Raman peak intensities gradually increase with time; meanwhile the rutile TiO2(111) peaks around 236 cm−1, 446 cm−1 and 610 cm−1 significantly weaken. The resulting decrease in the HOMO–LUMO gap in the adsorption complex produces a species that is more strongly Raman resonant with the 638 nm laser, resulting in the observed higher peak intensities.21 In addition, The SCN peak is more stable on the TiO2(111) surface during the photoinduced reaction processes and does not shift with time. Furthermore, comparing the mass spectra between surfaces, less of the [RuII(H2dcbpy)2(NCS)(CN)] photoinduced degradation product is produced on TiO2(111) than on the TiO2(001) and TiO2(110) surfaces (Fig. S9†), indicating that N719 adsorption is more stable on TiO2(111). Interestingly, the SCN complex has different Raman peak shifts in the order TiO2(111) (0 cm−1) < TiO2(110) (6 cm−1) < TiO2(001) (10 cm−1), which correlates well with the photoinduced stability of the SCN group on three rutile TiO2(hkl) surfaces, and the DFT simulated thermodynamic stability of the SC functional group. Thus, the important effect of TiO2 structural on N719 molecules adsorbed on three different rutile TiO2(hkl) surfaces was elucidated.
 | ||
| Fig. 4 (a) SHINER Spectra of N719 adsorbed on a rutile TiO2(110) surface with 405 nm laser illumination. The right shows the DFT simulated optimized structures of the Ph–NCS group (top) and S atom (below) adsorbed on rutile TiO2(110). (b) SHINER Spectra of N719 molecules adsorbed on a rutile TiO2(111) surface with 405 nm laser illumination. Spectra were collected every 4 minutes. The right shows the DFT simulated optimized structures of the Ph–NCS group (top) and S atom (below) on rutile TiO2(111). | ||
In summary, using in situ SHINERS and DFT simulations, SCN was found to be the main adsorption group of the N719 dye molecule at different rutile TiO2(hkl) surfaces and to play a determining role in its stability under 405 nm laser illumination. Importantly, different rutile TiO2(hkl) surfaces were also shown to have obvious facet effects on N719 molecular adsorption and photoinduced behavior. Under illumination, the photoinduced process caused the N719 molecules to desorb from TiO2(110) and TiO2(001) surfaces. Desorption was shown to occur via bond cleavage of the SC bond using mass spectrometry. In addition, DFT simulations showed the TiO2(001) surface to have the highest reaction activity for catalyzing SC double bond cleavage, while the TiO2(111) surface had the lowest reaction activity and was the most stable single crystal surface for adsorbed N719 molecules. These results indicate that N719 molecules will have long-term stability adsorbed on TiO2(111), providing important guidance for the design of practical DSSCs. Our research has elucidated the mechanism of DSSC interfacial reaction and demonstrates the universality of this approach to study the photoinduced reaction on semiconductor single crystal surfaces.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21902137, 21775127, 21703180, 21802111, and 21427813), the Fundamental Research Funds for the Central Universities (20720190044), the China Postdoctoral Science Foundation (2019M652250), and the China Postdoctoral Innovation Talent Support Program (BX20190184).Notes and references
- (a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef; (b) M. L. Parisi, S. Maranghi and R. Basosi, Renewable Sustainable Energy Rev., 2014, 39, 124 CrossRef CAS; (c) A. Fakharuddin, R. Jose, T. M. Brown, F. Fabregat-Santiago and J. Bisquert, Energy Environ. Sci., 2014, 7, 3952 RSC; (d) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- M. A. Green, Y. Hishikawa, E. D. Dunlop, D. H. Levi, J. Hohl-Ebinger and A. W. Y. Ho-Baillie, Prog. Photovoltaics Res. Appl., 2018, 26, 659 CrossRef.
- M. Shakeel Ahmad, A. K. Pandey and N. Abd Rahim, Renewable Sustainable Energy Rev., 2017, 77, 89 CrossRef CAS.
- (a) M. K. Nazeeruddin, R. Humphry-Baker, P. Liska and M. Grätzel, J. Phys. Chem. B, 2003, 107, 8981 CrossRef CAS; (b) N. Takeda and B. A. Parkinson, J. Am. Chem. Soc., 2003, 125, 5559 CrossRef CAS PubMed; (c) Q. Q. Zuo, Y. L. Feng, S. Chen, Z. Qiu, L. Q. Xie, Z. Y. Xiao, Z. L. Yang, B. W. Mao and Z. Q. Tian, J. Phys. Chem. C, 2015, 119, 18396 CrossRef CAS; (d) L. Q. Xie, D. Ding, M. Zhang, S. Chen, Z. Qiu, J. W. Yan, Z. L. Yang, M. S. Chen, B. W. Mao and Z. Q. Tian, J. Phys. Chem. C, 2016, 120, 22500 CrossRef CAS.
- F. Nour-Mohhamadi, S. D. Nguyen, G. Boschloo, A. Hagfeldt and T. Lund, J. Phys. Chem. B, 2005, 109, 22413 CrossRef CAS PubMed.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Gratzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- Z. Qiu, M. Zhang, D. Y. Wu, S. Y. Ding, Q. Q. Zuo, Y. F. Huang, W. Shen, X. D. Lin, Z. Q. Tian and B. W. Mao, Chemphyschem, 2014, 14, 2217 CrossRef PubMed.
- S. M. Nie and S. R. Emery, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- (a) J. F. Li, Y. J. Zhang, A. V. Rudnev, J. R. Anema, S. B. Li, W. J. Hong, P. Rajapandiyan, J. Lipkowski, T. Wandlowski and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 2400 CrossRef CAS PubMed; (b) J. F. Li, Y. J. Zhang, S. Y. Ding, R. Panneerselvam and Z. Q. Tian, Chem. Rev., 2017, 117, 5002 CrossRef CAS PubMed; (c) Y. H. Wang, M. M. Liang, Y. J. Zhang, S. Chen, P. Radjenovic, H. Zhang, Z. L. Yang, X. S. Zhou, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2018, 57, 11257 CrossRef CAS PubMed.
- (a) J. C. Dong, X. G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z. L. Yang, D. Y. Wu, J. M. Feliu, C. T. Williams, Z. Q. Tian and J. F. Li, Nat. Energy, 2019, 4, 60 CrossRef CAS; (b) J. C. Dong, M. Su, V. Briega-Martos, L. Li, J. B. Le, P. Radjenovic, X. S. Zhou, J. M. Feliu, Z. Q. Tian and J. F. Li, J. Am. Chem. Soc., 2020, 142, 715 CrossRef CAS PubMed; (c) N. Bodappa, M. Su, Y. Zhao, J. B. Le, W. M. Yang, P. Radjenovic, J. C. Dong, J. Cheng, Z. Q. Tian and J. F. Li, J. Am. Chem. Soc., 2019, 141, 12192 CrossRef CAS PubMed; (d) C. Y. Li, J. B. Le, Y. H. Wang, S. Chen, Z. L. Yang, J. F. Li, J. Cheng and Z. Q. Tian, Nat. Mater., 2019, 18, 697 CrossRef CAS PubMed.
- S. K. Parayil, R. J. Psota and R. T. Koodali, Int. J. Hydrogen Energy, 2013, 38, 10215 CrossRef CAS.
- (a) K. E. Lee, M. A. Gomez, T. Regier, Y. Hu and G. P. Demopoulos, J. Phys. Chem. C, 2011, 115, 5692 CrossRef CAS; (b) H. Rensmo, S. Lunell and H. Siegbahn, J. Photochem. Photobiol., A, 1998, 114, 117 CrossRef CAS; (c) A. El-Shafei, M. Hussain, A. Atiq, A. Islam and L. Han, J. Mater. Chem., 2012, 22, 24048 RSC.
- J. G. Ma, C. R. Zhang, J. J. Gong, B. Yang, H. M. Zhang, W. Wang, Y. Z. Wu, Y. H. Chen and H. S. Chen, J. Chem. Phys., 2014, 141, 234705 CrossRef PubMed.
- K. E. Lee, M. A. Gomez, S. Elouatik and G. P. Demopoulos, Langmuir, 2010, 26, 9575 CrossRef CAS PubMed.
- (a) A. G. Thomas and K. L. Syres, Chem. Soc. Rev., 2012, 41, 4207 RSC; (b) R. B. Jaculbia, H. Imada, K. Miwa, T. Iwasa, M. Takenaka, B. Yang, E. Kazuma, N. Hayazawa, T. Taketsugu and Y. Kim, Nat. Nanotechnol., 2020, 15, 105 CrossRef CAS PubMed.
- J. Singh, A. Gusain, V. Saxena, A. K. Chauhan, P. Veerender, S. P. Koiry, P. Jha, A. Jain, D. K. Aswal and S. K. Gupta, J. Phys. Chem. C, 2013, 117, 21096 CrossRef CAS.
- P. T. Nguyen, P. E. Hansen and T. Lund, Sol. Energy, 2013, 88, 23 CrossRef CAS.
- C. Pérez León, L. Kador, B. Peng and M. Thelakkat, J. Phys. Chem. B, 2006, 110, 8723 CrossRef PubMed.
- S. Ben-Jaber, W. J. Peveler, R. Quesada-Cabrera, E. Cortes, C. Sotelo-Vazquez, N. Abdul-Karim, S. A. Maier and I. P. Parkin, Nat. Commun., 2016, 7, 12189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00588f |
| This journal is © The Royal Society of Chemistry 2020 |