
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical synthesis and immunological evaluation of new generation multivalent anticancer vaccines based on a Tn antigen analogue†
Carlo
Pifferi
 ab,
Ane
Ruiz-de-Angulo
b,
David
Goyard
a,
Claire
Tiertant
a,
Nagore
Sacristán
b,
Diego
Barriales
c,
Nathalie
Berthet
a,
Juan
Anguita
*cd,
Olivier
Renaudet
*a and
Alberto
Fernández-Tejada
*bd
ab,
Ane
Ruiz-de-Angulo
b,
David
Goyard
a,
Claire
Tiertant
a,
Nagore
Sacristán
b,
Diego
Barriales
c,
Nathalie
Berthet
a,
Juan
Anguita
*cd,
Olivier
Renaudet
*a and
Alberto
Fernández-Tejada
*bd
aDépartement de Chimie Moléculaire, Université Grenoble Alpes, UMR 5250, CNRS, 38000 Grenoble, France. E-mail: olivier.renaudet@ujf-grenoble.fr
bChemical Immunology Lab, CIC bioGUNE, Biscay Science and Technology Park, Building 801A, 48160 Derio, Spain. E-mail: afernandeztejada@cicbiogune.es
cInflammation and Macrophage Plasticity Lab, CIC bioGUNE, Biscay Science and Technology Park, Building 801A, 48160 Derio, Spain. E-mail: janguita@cicbiogune.es
dIkerbasque, Basque Foundation for Science, Maria Diaz de Haro 13, 48009 Bilbao, Spain
First published on 8th April 2020
Abstract
Tumor associated carbohydrate antigens (TACAs), such as the Tn antigen, have emerged as key targets for the development of synthetic anticancer vaccines. However, the induction of potent and functional immune responses has been challenging and, in most cases, unsuccessful. Herein, we report the design, synthesis and immunological evaluation in mice of Tn-based vaccine candidates with multivalent presentation of the Tn antigen (up to 16 copies), both in its native serine-linked display (Tn-Ser) and as an oxime-linked Tn analogue (Tn-oxime). The high valent vaccine prototypes were synthesized through a late-stage convergent assembly (Tn-Ser construct) and a versatile divergent strategy (Tn-oxime analogue), using chemoselective click-type chemistry. The hexadecavalent Tn-oxime construct induced robust, Tn-specific humoral and CD4+/CD8+ cellular responses, with antibodies able to bind the Tn antigen on the MCF7 cancer cell surface. The superior synthetic accessibility and immunological properties of this fully-synthetic vaccine prototype makes it a compelling candidate for further advancement towards safe and effective synthetic anticancer vaccines.
Introduction
Cancer immunotherapy approaches based on synthetic vaccines containing tumor-associated carbohydrate antigens (TACAs) represent a topic of high interest.1–3 Anticancer vaccines have been conceived as innovative therapeutic agents for inclusion in multi-therapy settings in order to elicit anti-tumor immunity in patients with prior surgical resection of a primary tumor. This approach has the potential of preventing, or at least prolonging the time to recurrence at the metastatic sites, while exhibiting reduced toxicity compared to cytotoxic treatments such as radio- and chemotherapy.4 Active immunotherapy educates the immune system of a patient to recognize tumor-associated antigens displayed on the vaccine construct and to trigger an immune response selectively directed towards malignant cells.5,6 Extraordinary advances in Immunology and Synthetic Chemistry have led to the development of subunit vaccines based on tumor-associated glycans for the generation of more effective carbohydrate-directed immune responses.7–10 The administration of carbohydrate antigens alone in a vaccine formulation can only weakly activate B-cells, resulting in production of low-affinity IgM antibodies and short-living plasma cells. This is because, apart from a few exceptions,11 carbohydrates are T-independent antigens that are not able to associate with MHC molecules and activate helper T-cells (TH) to provide the required signals for B-cell stimulation. This, in turn, results in impaired antibody isotype-switching (to high-affinity IgG antibodies) and deficient production of both long-living plasma cells and memory B-cells.12,13 As a consequence, conjugate-vaccine design relies on the hapten-carrier effect, whereby helper T-cell epitopes are included in the form of a protein carrier (semi-synthetic vaccines)14–16 or as discrete TH-epitopes (fully-synthetic vaccines), either directly connected to B-cell epitopes through linker moieties or grafted onto non-immunogenic scaffolds or nanoparticles.17–19Aberrantly glycosylated versions of the protein mucin-1 (MUC1) are overexpressed in most human epithelial cancers,20,21 making this glycoprotein a target of high interest for both diagnostic and immunotherapeutic applications.22 Cancer-related MUC1 tandem repeats display truncated carbohydrate moieties at O-glycosylation sites, exposing glycan epitopes such as the Tn antigen that are normally hidden in mucins expressed in non-transformed cells. The Tn antigen consists of an N-acetyl-D-galactosamine (GalNAc) unit α-O-linked to the serine (Ser) or threonine (Thr) of a peptide backbone,23,24 and is overexpressed in approximately 90% of breast carcinomas,25 and in 70–90% of colon, bladder, cervix, ovary, stomach and prostate cancers.26–28 As such, it represents an excellent target for vaccine development, and we and other research groups have focused efforts on the design of anticancer vaccine candidates based on the Tn antigen.29–38 The inherent low immunogenicity of carbohydrate antigens is even more critical in TACAs, as they are self-derived antigens and therefore not prone to being recognized as foreign molecules by the immune system.39,40 Thus, a fundamental challenge for TACA-based vaccines involves the ability to produce functional, isotype-switched, IgG antibodies that are able to recognize native antigens expressed on cancer cells and selectively promote their clearance. Advances in the chemical-immunology field have provided evidence that through an accurate structural design, it is possible to develop effective TACA-based anticancer vaccines with promising preclinical outcomes.10,41–48 To overcome important limitations associated with protein–hapten conjugates,49–51 fully synthetic vaccine approaches are being developed that enable the assembly of structurally defined and easily characterizable constructs via modular chemical strategies.15,52–55 In this context, we report herein the design, synthesis and immunological evaluation of unprecedented fully synthetic vaccines with high Tn-antigen valency that are able to elicit robust and functional immune responses in mice.
Results and discussion
Design and synthesis
Since 2005, some of us have pioneered the use of cyclic Regioselectively Addressable Functionalized Templates (RAFTs) as designed scaffolds for the presentation of B- and T-cell epitopes in synthetic vaccine prototypes.18,56,57 The RAFT core consists of a cyclic decapeptide featuring two Pro–Gly residues as β-turn inducers, which stabilize its conformation in solution and provide a relatively rigid structure where lysines' side chains point towards two spatially opposite directions, thus defining an upper and a lower domain.58 In these early prototypes, four units of the Tn antigen moiety were grafted onto these scaffolds by using oxime ligation, a versatile chemoselective reaction that enabled the straightforward preparation of tetravalent structures by coupling deprotected aminooxy-glycan moieties without the need for activating agents.59,60 While tetravalent vaccine constructs were immunogenic,56,59,61–63 we reasoned that higher carbohydrate valency would enhance recognition properties due to the multivalent effect, leading to increased B-cell receptor (BCR) clustering,64–66 which represents an early and key step that impacts downstream immune signaling, including B–T cell communication.67,68 Therefore, to go one step further and improve the immunological properties and potency of early prototypes, we developed a new set of fully synthetic Tn-based vaccine candidates displaying a 4-fold increase in B-cell epitope ratio as well as additional T cell epitopes for more efficient immune responses (Fig. 1a).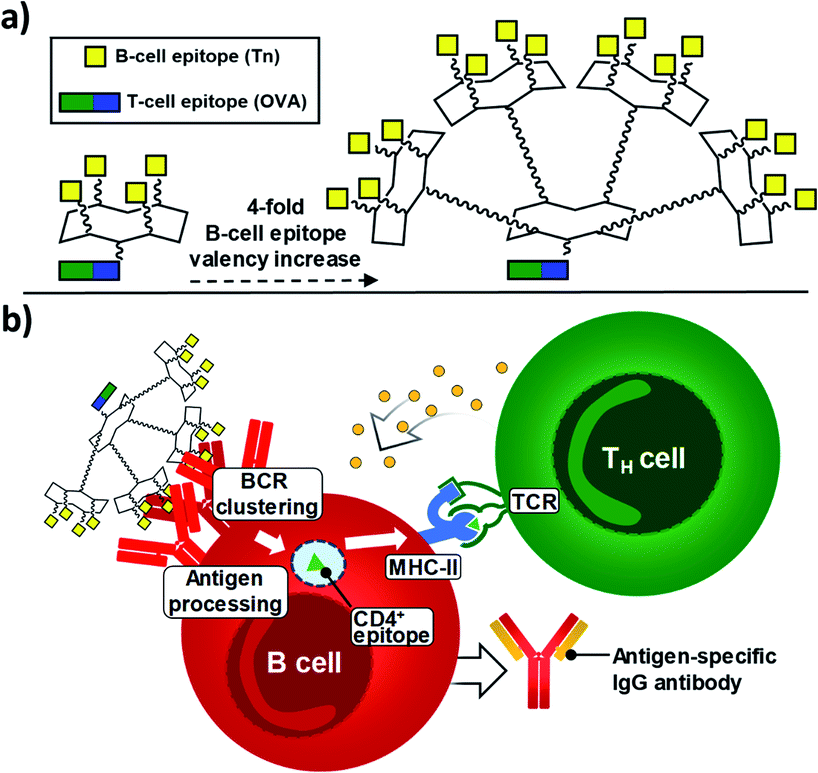 | ||
| Fig. 1 (a) New generation of fully-synthetic vaccines embedded with a 4-fold B-cell epitope ratio over the entire construct. (b) Early, critical events for the efficient production of antigen-specific, isotype-switched IgG involve an efficient clustering of B-cell receptors (BCRs). | ||
Thus, multivalency-driven recognition of carbohydrate antigens by BCRs followed by receptor-mediated internalization of the vaccine construct incorporating a CD4+ epitope would allow antigen-presenting B cells to load the TH epitope onto MHC-II molecules on the cell surface. Direct B-T cell contact via T-cell receptor (TCR) on CD4+ T lymphocytes would provide bidirectional signaling, ultimately leading to B-cell proliferation and differentiation (Fig. 1b).69–71
Initially, we generated a small series of multivalent glycodendrimers as B-cell epitope carriers (Scheme 1, compounds 2, 5–8) to investigate whether a higher order multivalency could be beneficial to antibody binding in an anti-Tn mAb interaction assay (Fig. 2).74 We first synthesized Tn-based glycodendrimers 2 and 5, in which GalNAc units are α-O-linked to the Ser hydroxyl groups (referred to as “Tn-Ser” along the text), as in the native Tn antigen. Starting from intermediate 1 (see the ESI, Scheme S1†), bearing four protected Tn-Ser residues, global O-acetyl and Fmoc removal gave tetravalent Tn-Ser glycodendrimer 2 (Scheme 1a). Meanwhile, functionalization of the free amino group of the lower lysine side chain in 1 with Boc-aminooxyacetic acid N-hydroxysuccinimide ester (Boc-Aoa-NHS),75 followed by base-mediated deprotection of the four GalNAc-Ser residues afforded intermediate 3.
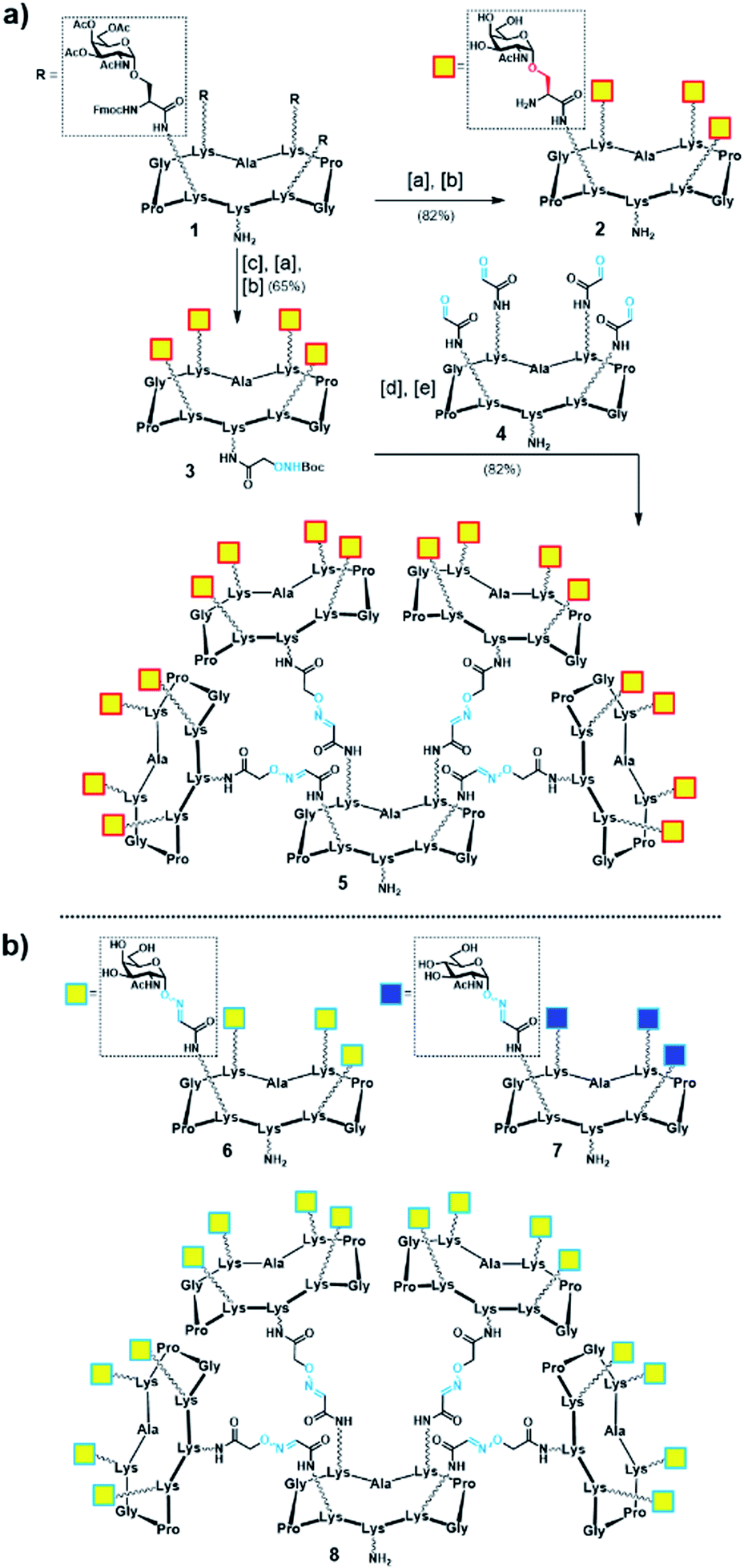 | ||
Scheme 1 (a) Synthesis of tetravalent and hexadecavalent Tn-Ser glycodendrimers 2 and 5. Reagents and conditions: [a] 5% piperidine in CH3CN, r.t., 15 min; [b] NaOMe/MeOH (pH ≈ 10), r.t., 20 min; [c] Boc-Aoa-NHS (1.2 eq.), DIPEA (2.0 eq.), CH3CN/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 20 min; [d] TFA/CH2Cl2 (1:1), r.t., 30 min; [e] 4 (ref. 72) (0.17 eq.), 0.1% TFA in H2O, 37 °C, 45 min. (b) Tn-oxime glycodendrimers 6 (ref. 61) and 8 (ref. 73), and tetravalent GlcNAc-oxime control 7 (ref. 73). :1), 20 min; [d] TFA/CH2Cl2 (1:1), r.t., 30 min; [e] 4 (ref. 72) (0.17 eq.), 0.1% TFA in H2O, 37 °C, 45 min. (b) Tn-oxime glycodendrimers 6 (ref. 61) and 8 (ref. 73), and tetravalent GlcNAc-oxime control 7 (ref. 73). | ||
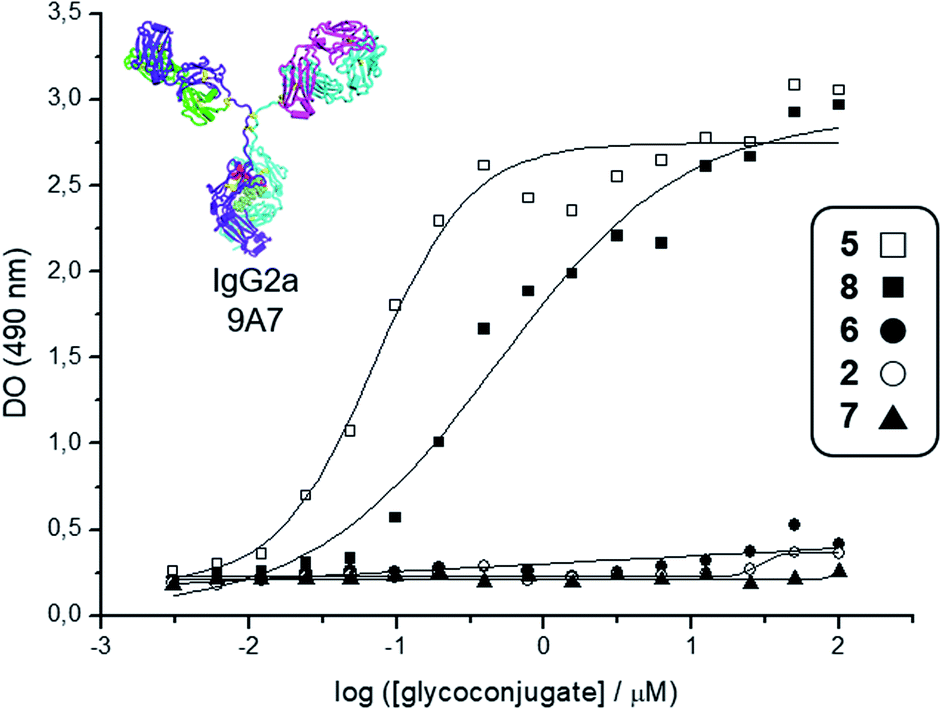 | ||
| Fig. 2 Interaction curves of the anti-Tn mAb 9A7 (ref. 74) with glycodendrimers 5 (□), 8 (■), 6 (●), 2 (○), and 7 (▲). ELISA plates were coated with decreasing concentrations of glycodendrimers, starting from 100 μM. Results expressed as in absorbance values at 490 nm. | ||
Deprotection of the Boc-aminooxy group on 3 and subsequent oxime ligation with core scaffold 4,72 which displays four α-oxo-aldehyde groups,76 provided hexadecavalent Tn-Ser glycodendrimer 5 in four steps from intermediate 1 following a convergent strategy involving unprotected moieties (see the ESI, Scheme S4†). Conversely, the Tn-analogue glycodendrimer 8, in which the GalNAc units are attached via oxime linkages (referred to as “Tn-oxime” along the text), was synthesized in a divergent fashion by grafting the aminooxy-GalNAc moiety onto a hexadecavalent, α-oxo-aldehyde functionalized scaffold as the last step in the route.73 With our set of compounds in hand featuring hexadecavalent Tn-Ser/oxime structures (Scheme 1, compounds 5 and 8, respectively) and their tetravalent analogues (Scheme 1, compounds 2 and 6 (ref. 61), respectively), we then performed direct interaction assays to evaluate their ability to be recognized by anti-Tn antibodies (anti-Tn mAb clone 9A7).74 Tetravalent construct bearing GlcNAc-oxime residues (Scheme 1, compound 7 (ref. 73)) was used as a negative control (Fig. 2).
While hexadecavalent Tn-Ser compound 5 exhibited the strongest binding, the interaction curve of the Tn-oxime glycodendrimer 8 indicated that this multivalent system can serve as an effective analogue of the native Tn antigen. In contrast, tetravalent Tn-containing compounds 2 and 6 showed absorbance values close to the baseline, comparable to those of negative control 7. These initial results prompted us to pursuit the synthesis of the complete vaccine structures based on selected glycodendrimers 5 and 8 for immunogenicity studies in mice. In addition to the hexadecavalent vaccine prototypes, the corresponding tetravalent vaccine constructs derived from 2 and 6 were also synthesized to evaluate the impact of antigen valency in the elicited immune response.
To complete the design of our synthetic vaccine prototypes, we incorporated T helper CD4+ and CD8+ epitopes from ovalbumin77–80 (OVA) into the previous glycodendrimers to generate potent immune responses with strong and long-lasting production of IgG antibodies against the T-cell independent Tn carbohydrate antigen. OVA323–339 CD4+ T helper and OVA257–264 CD8+ T cell epitopes were synthesized “in-line” incorporating a cysteine residue at the C-terminus for further chemoselective conjugation to the core cyclopeptide scaffold (Scheme 2). The synthesis started with protected peptide sequence 9, which was obtained using standard Fmoc-based automated solid phase peptide synthesis (SPPS) on a Rink amide resin. Treatment with a TFA/TIS/H2O (96:2:2) cocktail resulted in the removal of all acid-labile side-chain protecting groups, with concomitant cleavage from the resin, affording peptide 10 in a 41% overall yield.
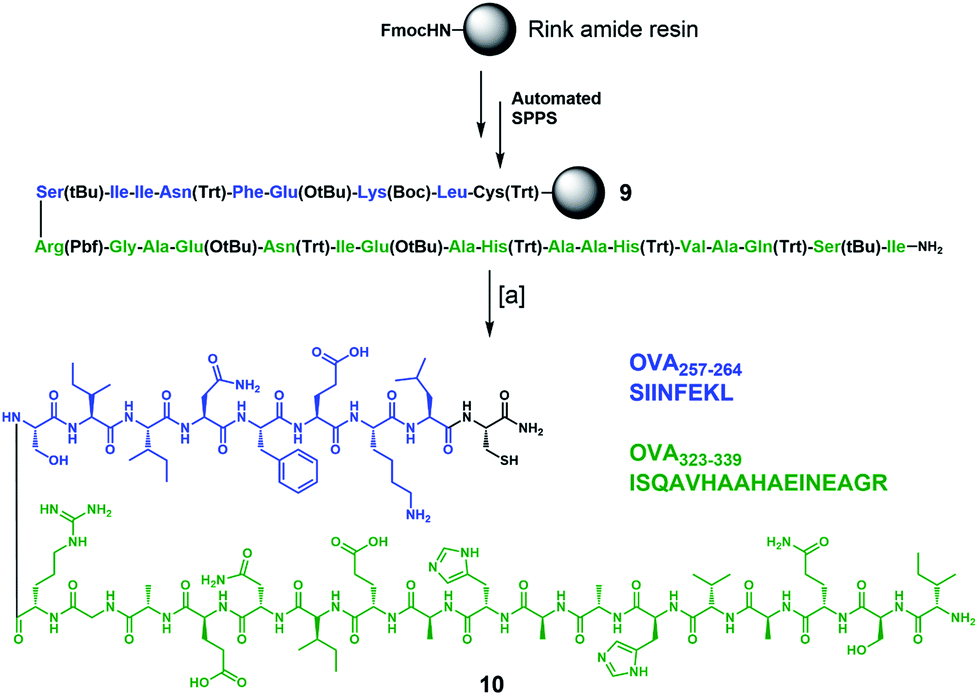 | ||
| Scheme 2 Synthesis of immunostimulant peptide 10 containing in-line CD4+ and CD8+ epitopes from OVA. Reagents and conditions: [a] TFA/TIS/H2O (96:2:2), r.t., 3 h (×3 cycles), 41% overall yield. | ||
We first focused our efforts on the synthesis of the “native-like” Tn-Ser vaccine prototypes based on glycodendrimers 2 and 5. Starting from building block 1 (see Scheme 1), which displays protected Tn-Ser peripheral residues and a free amino group of the lysine of the scaffold, coupling with the NHS ester of Boc-[S-(3-nitro-2-pyridinesulfenyl)]-cysteine (Boc-Cys(NPys)-NHS) in DMF provided intermediate 11 (Scheme 3a).81,82 Taking advantage of the thiol activating nature of the NPys group, Boc removal (1:1 TFA/CH2Cl2) from the cysteine residue, followed by disulfide bridge formation with cysteine-containing peptide 10 gave OVA-functionalized intermediate 12. Finally, global deprotection under Zemplén conditions (NaOMe/MeOH, pH ≈ 10) with concomitant removal of the O-acetyl and Fmoc protecting groups provided the tetravalent vaccine candidate 13 (Scheme 3a). The same modular strategy was then applied to the synthesis of the hexadecavalent Tn-Ser vaccine construct based on glycodendrimer 5 (Scheme 1). Hexadecavalent intermediate 14, obtained by convergent attachment of four copies of protected Tn-Ser peripheral glycodendrimer S5 to the α-oxo-aldehyde-bearing central scaffold 4 through oxime linkages (see the ESI, Scheme S6†), was functionalized at its lower domain with OVA peptide 10via disulfide bridge formation by using the three-step procedure described above (Scheme 3b). The resulting Tn-Ser hexadecavalent construct 15, however, was found to be unstable to the various deprotection conditions used to remove the O-acetyl and Fmoc moieties (see the ESI, Scheme S8†), which presumably affected the internal oxime linkages, failing to afford the fully-deprotected vaccine candidate 16.
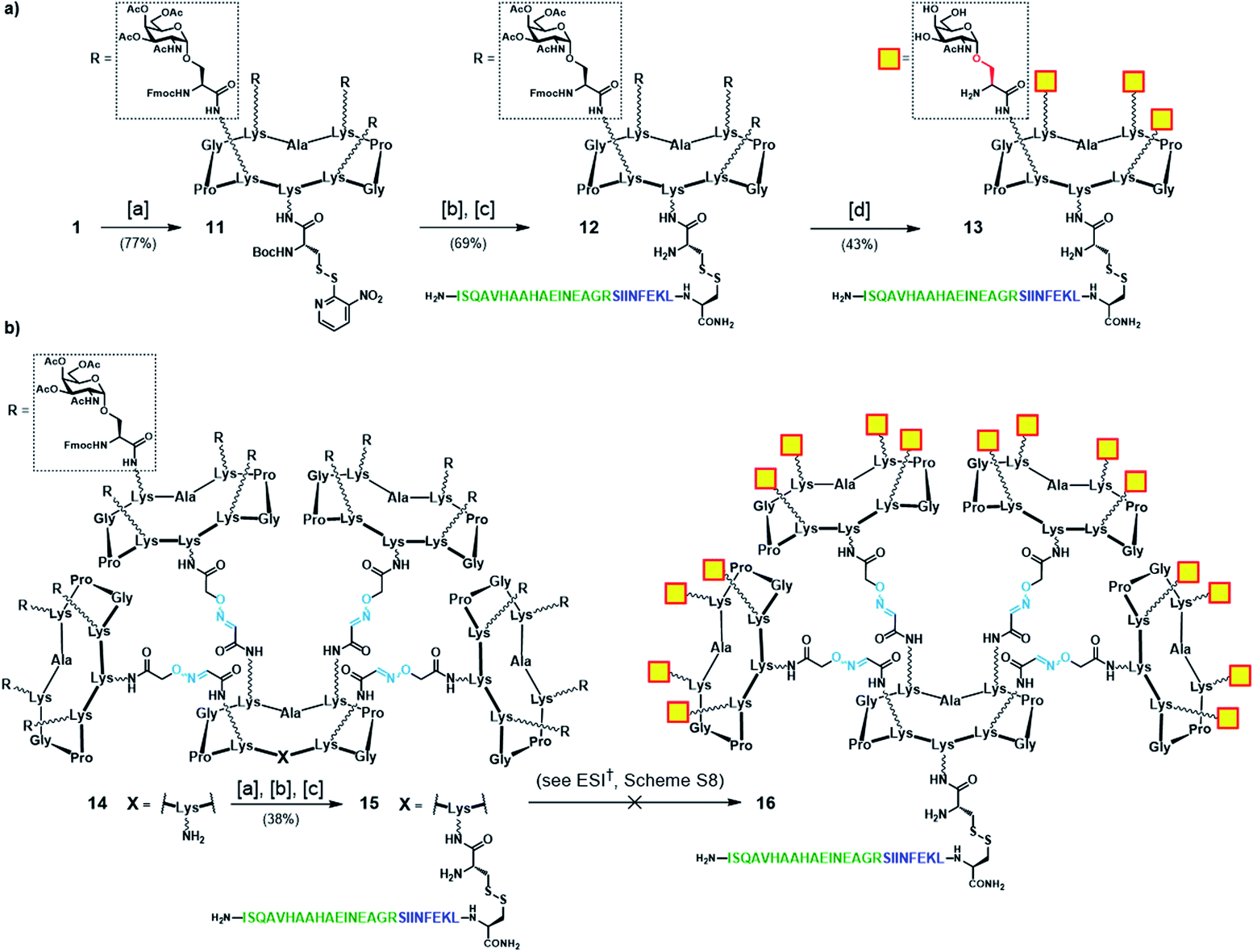 | ||
| Scheme 3 (a) Synthetic strategy for tetravalent Tn-Ser vaccine candidate 13. (b) Synthetic strategy towards hexadecavalent Tn-Ser vaccine candidate 16 (see the ESI, Scheme S8†). Reagents and conditions: [a] Boc-Cys(NPys)NHS (1.5–1.6 eq.), DIPEA (1.5 eq.), DMF, r.t., 30 min; [b] TFA/CH2Cl2 (1:1), r.t., 30 min; [c] 10 (1.1–1.2 eq.), DMF/NaOAc buffer (2:1, pH 4.5, 40 mM), r.t., 30 min. | ||
To address this problem, we designed an alternative strategy towards a modified vaccine construct (19) in which the internal oxime linkages were replaced with 1,4-triazoles (Scheme 4). Since this modification involves the “internal” part of the final glycosylated structure and not the external B-cell epitope display, the binding ability of the construct should not be affected. In contrast to the synthetic route towards 16, the new strategy allows for a more convergent assembly via late-stage CuAAC and also enables O-acetyl and Fmoc removal from the Tn-Ser moiety at an earlier stage in the synthesis. In the event, CuAAC reaction between a slight excess of fully-deprotected peripheral glycodendrimer 17 (see the ESI, Scheme S9†) equipped with an alkyne handle on the lower domain and OVA-functionalized, azide-bearing core scaffold 18 (see the ESI, Scheme S10†) was carried out in a degassed DMF/PBS mixture using our previously reported protocol in the presence of copper(II) sulfate, THPTA and sodium ascorbate.83 Hexadecavalent Tn-Ser vaccine construct 19 was thus obtained in 75% yield after RP-HPLC purification.
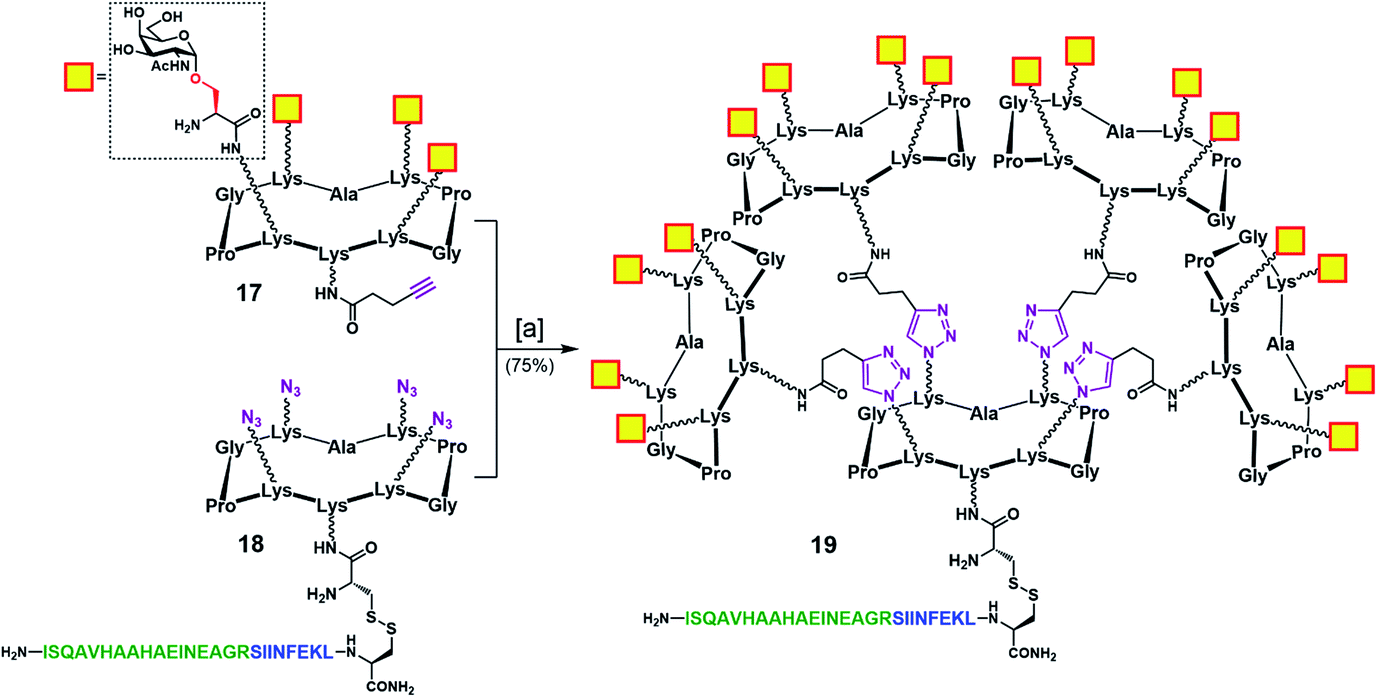 | ||
| Scheme 4 Convergent assembly of hexadecavalent Tn-Ser vaccine candidate 19 with internal triazole linkers. Reagents and conditions: [a] 18, 17 (5.0 eq.), THPTA (10 eq.), CuSO4 (cat.), Na ascorbate (30 eq.), DMF, PBS (pH 7.4, 10 mM), r.t., 2 h. | ||
With Tn-Ser vaccines 13 and 19 in hand, we next directed our efforts towards the synthesis of the Tn-oxime vaccine candidates. Unlike for the Tn-Ser constructs, the presence of a single free amino acid (Lys on the lower domain of the scaffold) in glycodendrimers 6 and 8 enabled the use of a versatile divergent strategy involving late-stage chemoselective functionalization at this position followed by installation of the T cell OVA epitopes in the last step of the route. Thus, starting from glycodendrimer 6 (see Scheme 1b), vaccine construct 20 was synthesized in 53% yield over three steps via Boc-Cys(Npys) installation, and disulfide bridge formation with OVA peptide 10 (Scheme 5). Following this three-step sequence, vaccine candidate 21 was obtained analogously from hexadecavalent glycodendrimer 8, although its poor solubility in DMF required the use of a DMF/PBS mixture (1:1, pH 7.4) to achieve the coupling of the Boc-Cys(NPys) residue with a satisfying 41% yield after RP-HPLC purification (Scheme 5).
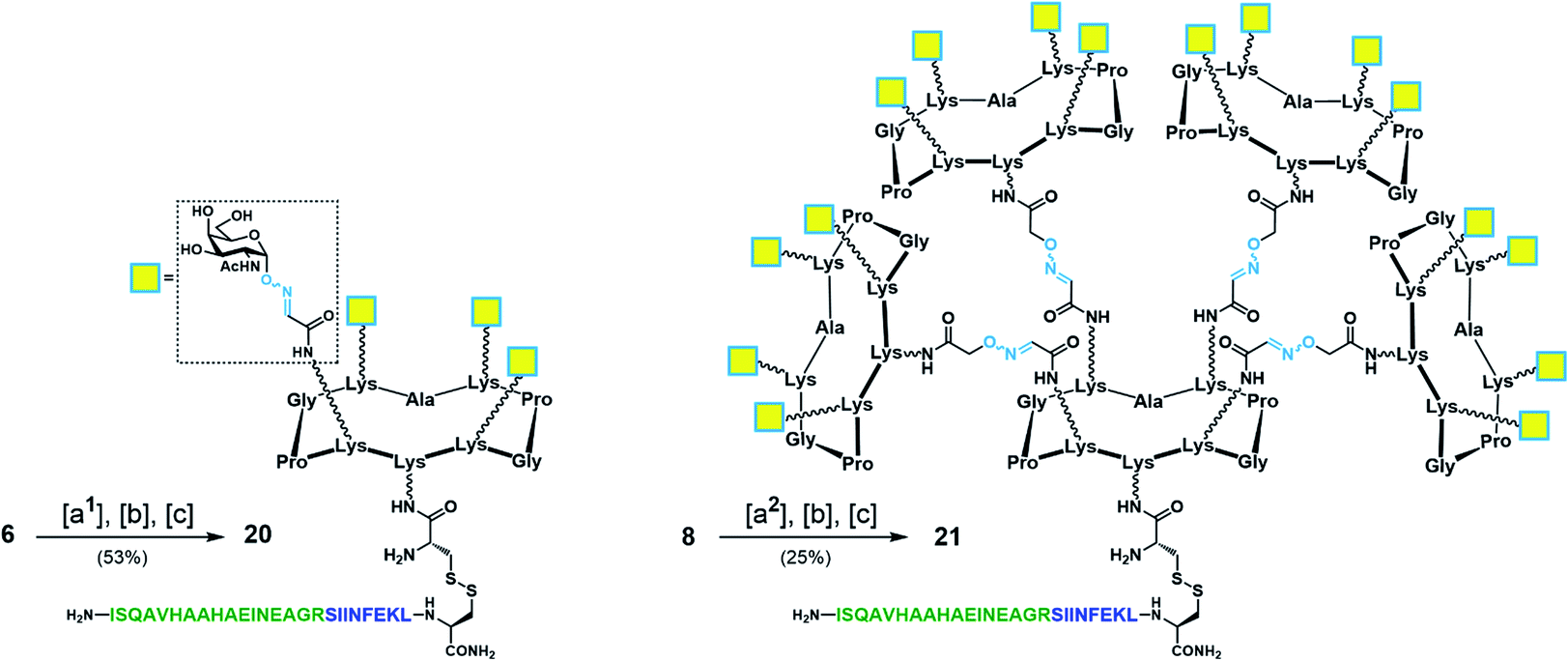 | ||
| Scheme 5 Synthesis of tetravalent and hexadecavalent Tn-oxime vaccine candidates 20 and 21. Reagents and conditions: [a1] Boc-Cys(NPys)NHS (1.5–1.6 eq.), DIPEA (1.5 eq.), DMF, r.t., 30 min, 74% yield; [a2] Boc-Cys(NPys)NHS (1.5 eq.), DMF/PBS (pH 7.4, 10 mM) (1:1), 60 min, 41% yield; [b] TFA/CH2Cl2 (1:1), r.t., 30 min; [c] 10 (1.1–1.2 eq.), DMF/NaOAc buffer (pH 4.5, 40 mM) (1:1), r.t., 20 min. | ||
Immunological evaluation
With the synthetic vaccine candidates in hand, we next evaluated their ability to elicit immune responses. Groups of five C57BL/6 mice were immunized subcutaneously three times every two weeks with each vaccine construct (50 μg dose) (Group A: hexadecavalent Tn-oxime 21; Group B: hexadecavalent Tn-Ser 19; Group C: tetravalent Tn-oxime 20; Group D: tetravalent Tn-Ser 13) in combination with the saponin QS-21 as an adjuvant84 (20 μg). In addition, a group of control mice (Group E) were immunized with compound 21 without adjuvant (Fig. 3).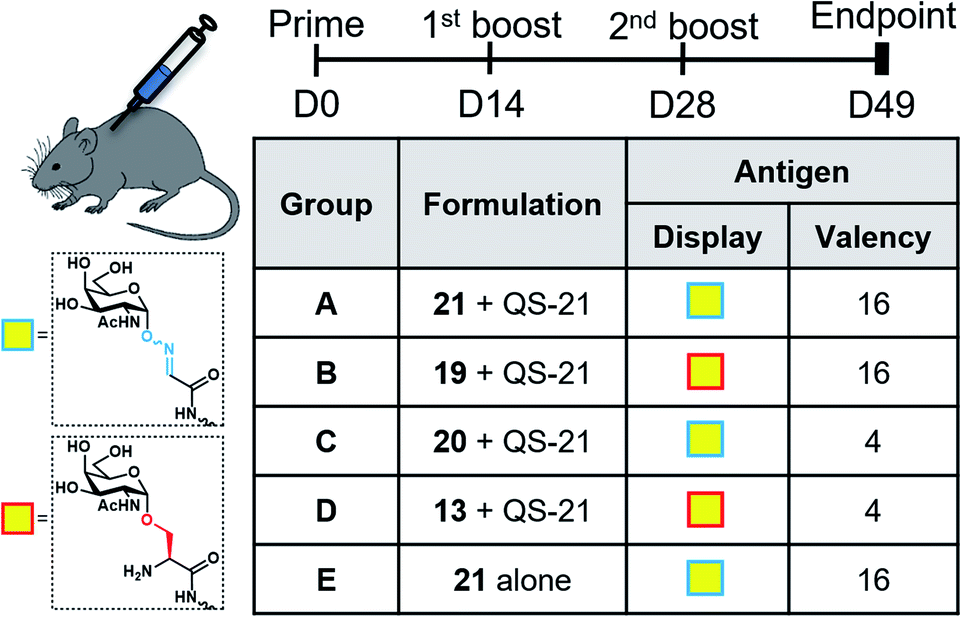 | ||
| Fig. 3 Immunization schedule and vaccination groups. | ||
Three weeks after the last immunization, blood was collected for serological analysis and the mice were sacrificed to assess cellular immunity. Notably, no toxic side effects (e.g. local inflammation, systemic reactions, mouse weight loss or death) were observed over the course of the immunizations (data not shown), indicating the non-toxicity of the synthetic vaccine constructs. First, we evaluated the ability of the constructs to generate antibody responses and of the antisera to bind the Tn-antigen in different presentation modes [native Tn-Ser residues (5, 2) and unnatural Tn-oxime analogues (8, 6), as well as in higher (5, 8) and lower (2, 6) valency, see Scheme 1]. Microtiter plates were coated with the corresponding Tn glycodendrimers lacking the OVA peptide and the total anti-Tn IgG levels in blood sera were detected by ELISA. Group A mice, immunized with hexadecavalent oxime-linked Tn construct 21 in combination with QS-21, exhibited the highest IgG levels against its glycodendrimer counterpart 8 (Fig. 4a). Moreover, this group was the only one in which all five mice were able to generate humoral responses. In contrast, Group B (hexadecavalent Tn-Ser 19 + QS-21) showed variable but lower IgG levels, while Groups C (20 + QS-21) and D (13 + QS-21) in which mice were immunized with the corresponding tetravalent constructs, exhibited IgG levels similar to the no adjuvant control (Group E). Interestingly, IgG antibodies produced by Group A mice (hexadecavalent Tn-oxime 21 + QS-21) were also able to recognize hexadecavalent Tn-Ser glycodendrimer 5 (Fig. 4b), showing therefore no clear preference for the native or unnatural Tn presentation on the scaffold. However, antisera from these mice (Group A) were less efficient in binding the Tn antigen presented through low-valency glycodendrimers Tn-oxime 6 and Tn-Ser 2 (Fig. 4c and d). Therefore, increased Tn antigen valency was found to be crucial for high-affinity binding of the IgG antibodies to the Tn antigen construct. On the other hand, while Group B mice showed antisera (IgG antibodies) able to bind both high-valency glycodendrimers 8 and 5 (Fig. 4a and b), their levels were considerably lower than those exhibited by Group A, and were not able to bind tetravalent glycodendrimers 6 and 2 (Fig. 4c and d). In these assays, tetravalent compounds 20 and 13 (Groups C and D, respectively) elicited IgG antibody levels similar to the no adjuvant Group E, highlighting the importance of the increased multivalency of vaccine candidates 21 and 19 to generate potent humoral responses. In addition to the presence of the OVA CD4+ T helper epitope, co-administration of QS-21 as an adjuvant was found to be essential for antibody class-switching and elicitation of IgG antibodies, with IgM antibody levels being negligible at the last time point (see the ESI, Fig. S45†).85 Antibody titration using oxime-linked glycodendrimer 8 for coating was carried out for the two groups showing the highest OD values, i.e. Group A (mice immunized with hexadecavalent Tn-oxime 21 + QS-21) and B (mice vaccinated with hexadecavalent Tn-Ser 19 + QS-21) (Fig. 5).
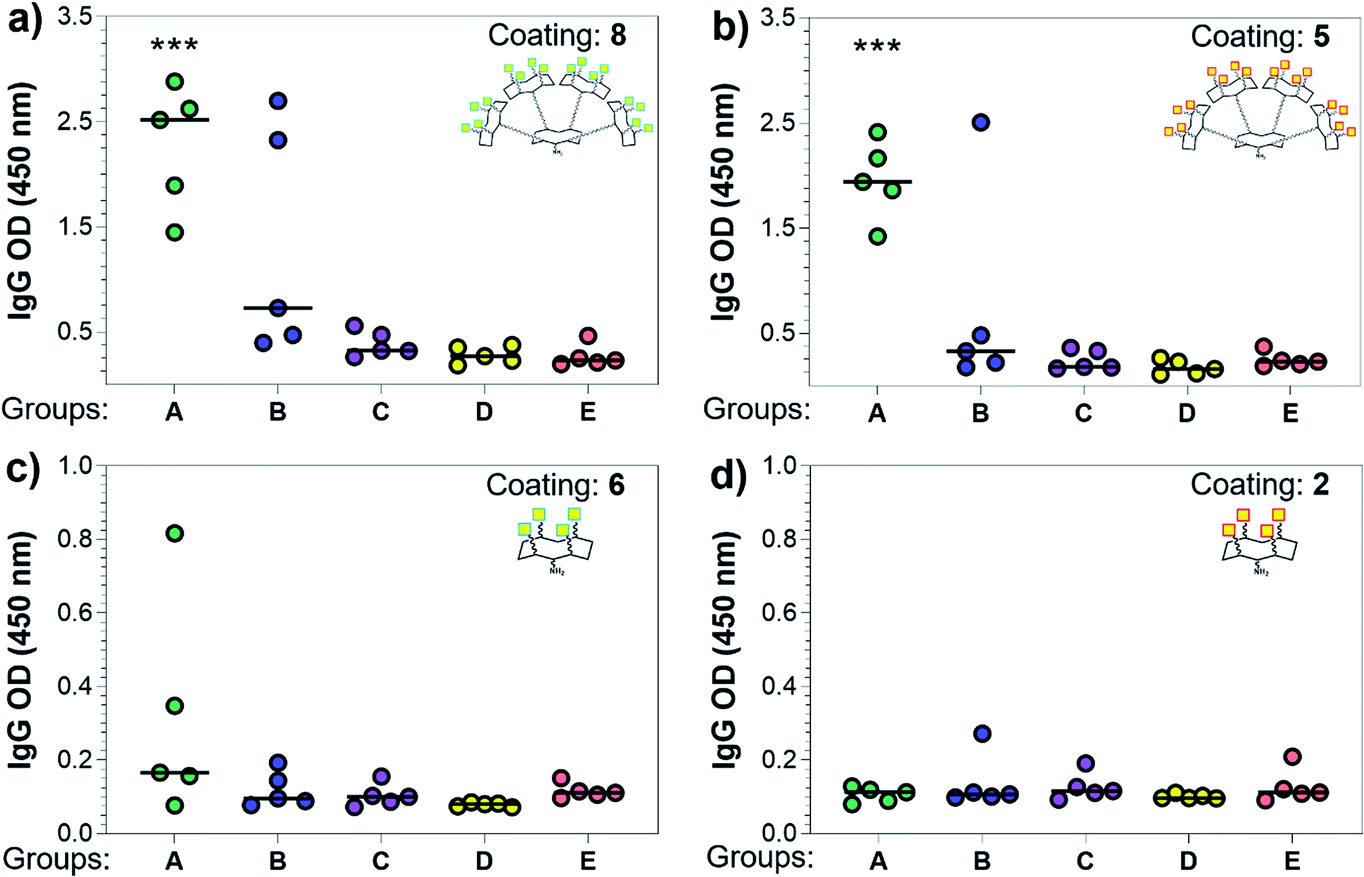 | ||
| Fig. 4 Analysis of total IgG antibodies elicited against the synthetic glycodendrimers. Microtiter plates were coated with the four different glycosylated scaffolds lacking the OVA epitopes: (a) 8, (b) 5, (c) 6, and (d) 2 (see Scheme 1); and the antisera from mice A to E were probed. Statistical significance compared to no-adjuvant control group (Group E) was assessed using two-tailed unpaired Student's t-test: ***p < 0.001. | ||
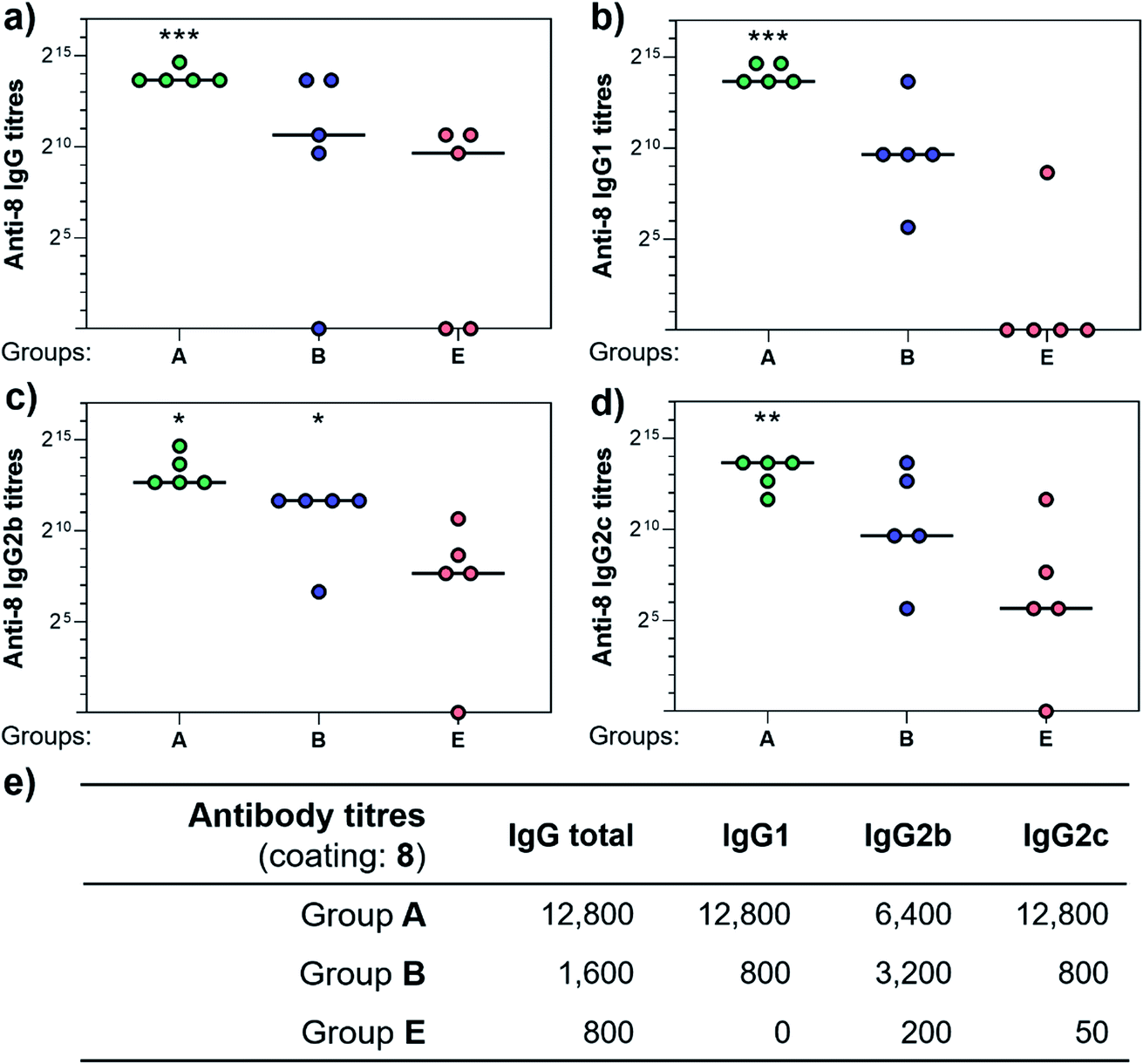 | ||
| Fig. 5 Comparison of antibody titers against hexadecavalent Tn-oxime glycodendrimer 8. Microtiter plates were all coated with 8 and antisera from group A, B, and E were analyzed for (a) total IgG, (b) IgG1, (c) IgG2b, and (d) IgG2c levels. (e) Table showing antibody titers median values for groups of five mice. Statistical significance compared to no-adjuvant control (Group E) by two-tailed unpaired Student's t-test: *P < 0.05, **P < 0.01, ***P < 0.001. | ||
Antibody subtyping of the anti-8 IgG isotypes revealed that Group A mice showed not only high total IgG titers but also elevated levels of the IgG1, IgG2b and IgG2c antibody subtypes. Interestingly, the IgG2c antibody titers in this group were as high as those observed for IgG1 antibodies, suggesting that 21 in combination with QS-21 elicits a balanced Th1/Th2 immune response (Fig. 5e). The IgG2c subtype is of particular interest since it is associated with potent antitumor effect such as complement- and antibody-dependent cell toxicity in mice.86 In contrast, IgG antibody titers from Group B mice were not significantly higher than those of the no adjuvant control Group E, and subtyping of these antibodies revealed a bias towards the IgG2b subclass. These results indicate that hexadecavalent vaccine construct 21 bearing oxime-linked Tn antigen analogue is more efficient than its native Tn-Ser counterpart 19 in generating potent humoral responses, and was therefore selected for further immunological studies.
We then evaluated the ability of the antisera to recognize the Tn antigen in a native context and analyzed the binding of the vaccine-induced serum antibodies to a human cancer cell line (MCF7) expressing the natural Tn antigen by using fluorescence microscopy. Notably, IgG antibodies elicited by immunization with compound 21 plus QS-21 (Group A mice) were able to specifically bind Tn-expressing MCF7 cells. Moreover, indirect immunofluorescence images showed high signal coming from sera from Group A mice, whereas antisera from mice immunized with 21 without QS-21 (Group E) showed no Tn-specific IgG binding (Fig. 6a). Antibody binding of sera from mice Group A showed broad membrane surface localization (Fig. 6b). In contrast, although compound 21 alone (Group E) was able to elicit IgG antibodies to a small extent (Fig. 5e), these antibodies were not able to specifically recognize natural Tn antigen expressed in this tumor cell. These results highlight that antibodies elicited by the Tn-oxime analogue vaccine construct 21 coadministered with QS-21 are functional and able to recognize the native Tn antigen expressed on the cancer cell surface, confirming the tumor specificity of the generated antibody responses.
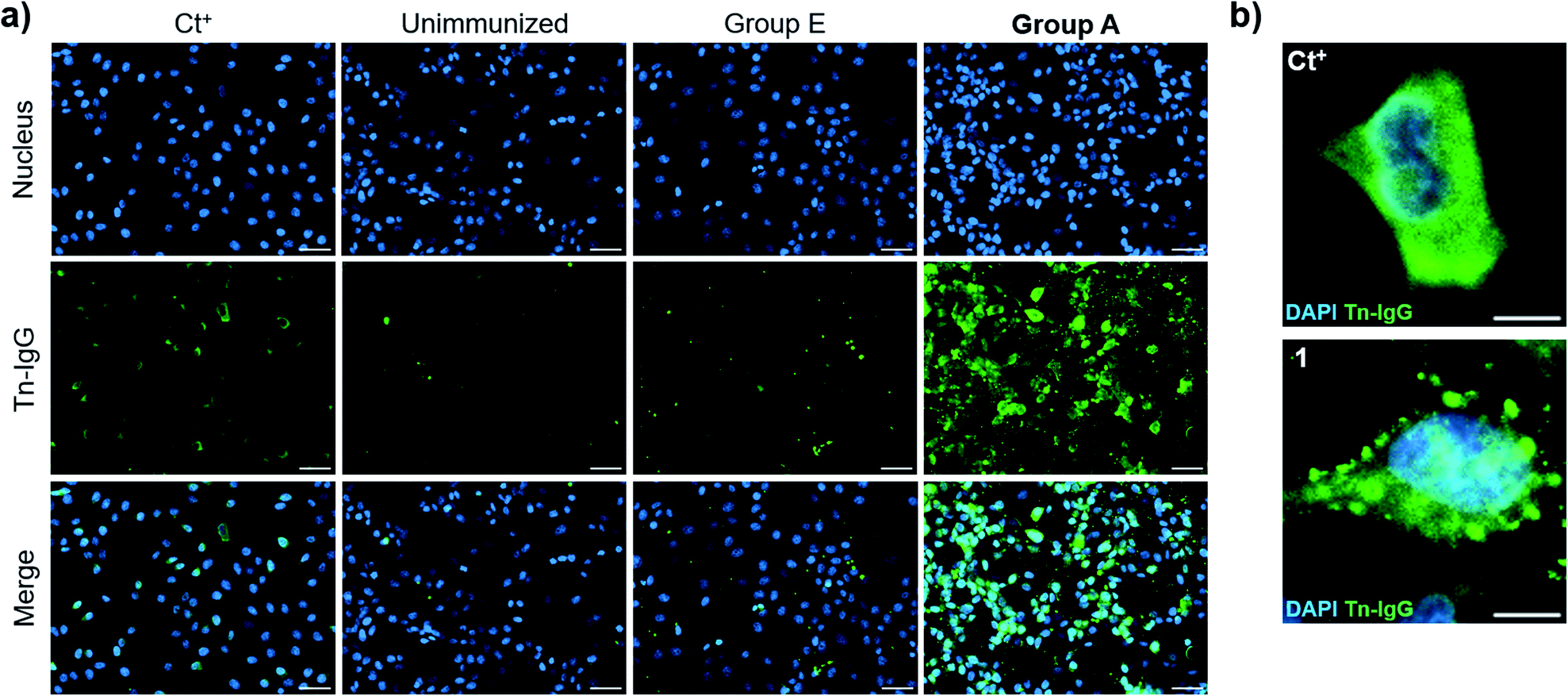 | ||
| Fig. 6 Cell surface reactivity of mouse antisera against native Tn antigen-expressed on the surface of human breast cancer cell line, MCF7. MCF7 cells were incubated with antisera from mice of Group A and IgG antibody binding was analyzed by indirect immunofluorescence. (a) Fluorescence images of positive control (Ct+), sera from non-immunized mice (unimmunized), sera from no-adjuvant control Group E and sera from Group A mice, respectively; scale bar = 50 × 10−6 m. (b) Detailed region of interest (ROI) of positive control and Group A sera; scale bar = 10 × 10−6 m. | ||
Next, we also investigated the cellular immune responses elicited by Tn-oxime construct 21 by testing its ability to specifically activate T cells. At the time of sacrifice (three weeks after the last immunization, day 49), whole splenocytes were harvested and assayed for T cell restimulation in the presence of full-length OVA protein. After 48 h stimulation, splenocytes were analyzed for activation markers using flow cytometry by assessing the percentage of CD4+CD44high (ref. 87 and 88) and CD8+CD107a+ (ref. 89) in the restimulated splenocyte pools. Notably, vaccine construct 21 in combination with QS-21 activated CD4+ as well as CD8+ T cells after stimulation with the specific antigen, confirming the immunogenicity of compound 21 for both B- and T-cells (Fig. 7). Therefore, construct 21 stands out as a potent and safe synthetic Tn analogue vaccine candidate that, coadministered with QS-21, is able to induce robust humoral and cellular immune responses without toxic side effects.
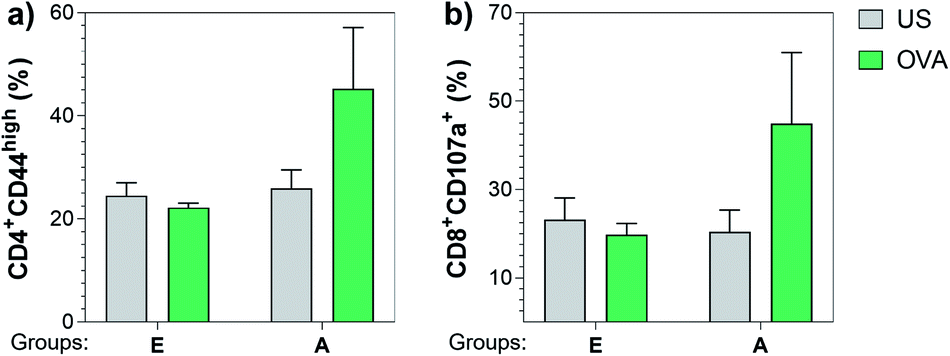 | ||
| Fig. 7 Construct 21 co-administered with QS-21 (Group A) was able to specifically activate CD4+ and CD8+ T cells upon re-stimulation with full length OVA protein. Three weeks after the last immunization, splenocytes were harvested and stimulated with 10 μg mL−1 of OVA. After incubating for 48 h, cells were analyzed for (a) CD4+CD44high and (b) CD8+CD107a+ T cells by flow cytometry. Data are presented as a mean ± SEM of five mice per group. US = unstimulated splenocytes (negative control). | ||
Conclusions
In summary, we report the design, synthesis and immunological evaluation of fully synthetic hexadecavalent Tn-based vaccine candidates with a 4-fold increased carbohydrate epitope ratio compared to earlier generation vaccines. These Tn-based constructs were designed as higher-valency B cell-targeting modules to address the need for an effective BCR clustering to boost B cell activation and antibody production. The synthesis of the hexadecavalent Tn-Ser construct resulted especially challenging due to late-stage global deprotection issues that resulted in cleavage of the internal oxime linkages and product decomposition. Instead, synthetic access to an alternative Tn-Ser construct (19, Scheme 4) was achieved by replacing the four internal oxime linkers with triazole groups, enabling a convergent assembly of the modules using CuAAC click chemistry. To circumvent the challenges associated to the native Tn-Ser vaccine candidates, as a more practical chemical solution we generated more synthetically accessible vaccine constructs based on Tn analogues displaying the Tn antigen via oxime linkages. Thus, we developed a divergent and streamlined strategy whereby convenient functionalization of the hexadecavalent Tn-oxime glycodendrimer 8 was carried out in a three-step late-stage sequence to give the final vaccine structure 21 (Scheme 5). In mouse immunization studies with QS-21 as an adjuvant, hexadecavalent Tn-oxime and Tn-Ser constructs 21 and 19 elicited higher IgG antibody responses than their tetravalent analogues 20 and 13, confirming the high-valency benefit of our new-generation vaccines. Between the hexadecavalent constructs, Tn-oxime vaccine candidate 21 elicited the highest IgG antibody titers, consistently higher than those generated by Tn-Ser construct 19, and were also the most efficient in binding the Tn antigen in both presentation modes, i.e. oxime-linked Tn (8) and Tn-Ser (5). Moreover, IgG subtyping of the vaccine-induced antibodies showed different patterns for Group A and Group B sera. Immunization with 21 (plus QS-21, Group A) elicited high IgG1 and IgG2c isotype titers, whereas vaccination with 19 (plus QS-21, Group B) revealed a bias towards the IgG2b subtype (Fig. 5). Notably, antibodies generated by Group A mice were also able to bind Tn-expressing MCF7 cancer cells as assessed by fluorescence microscopy (Fig. 6), confirming the tumoral specificity of the produced antibodies. Furthermore, vaccine construct 21 was also able to activate CD4+ and CD8+ cellular responses, as assessed by flow cytometry analysis of OVA-restimulated splenocytes (Fig. 7). In conclusion, we have developed a synthetically accessible new-generation vaccine candidate (21) based on a multivalent oxime-linked Tn-antigen analogue that showed no toxicity and was able to generate potent and functional humoral and cellular immune responses. The superiority of this fully synthetic Tn-based vaccine construct 21, in terms of both its synthetic accessibility and immunological properties, makes it a promising candidate for further development towards cancer immunotherapy applications.Ethical statement
Animals were cared for and handled in compliance with the Guidelines for Accommodation and Care of Animals (European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes) and internal guidelines. Mice were housed in standard cages and fed on a standard diet ad libitum. All the experimental procedures were approved by the appropriate local authorities. The CIC bioGUNE animal facility is fully accredited by AAALAC International.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Research Council (ERC-2016-STG-716878 “ADJUV-ANT VACCINES” to A. F.-T.; ERC-2014-COG-647938 “LEGO” to O.R.), Spanish Ministry of Science, Innovation and Universities (CTQ2017-87530-R, RYC-2015-17888 to A. F.-T.; RTI2018-096494-B-100, SAF2015-65327-R to J. A.; Severo Ochoa Accreditation SEV-2016-0644 to CIC bioGUNE) is gratefully acknowledged. O. R. thanks support from the CNRS, the Université Grenoble Alpes, the ICMG FR 2607, the French ANR project Glyco@Alps (ANR-15-IDEX-02), the Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003). A. F.-T. thanks Raquel Fernández for inspiration.Notes and references
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed
.
- I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS PubMed
- D. Feng, A. S. Shaikh and F. Wang, ACS Chem. Biol., 2016, 11, 850–863 CrossRef CAS PubMed
- C. Guo, M. H. Manjili, J. R. Subjeck, D. Sarkar, P. B. Fisher and X. Y. Wang, Adv. Canc. Res., 2013, 119, 421–475 CAS
- O. J. Finn, Nat. Rev. Immunol., 2003, 3, 630–641 CrossRef CAS PubMed
- I. Melero, G. Gaudernack, W. Gerritsen, C. Huber, G. Parmiani, S. Scholl, N. Thatcher, J. Wagstaff, C. Zielinski, I. Faulkner and H. Mellstedt, Nat. Rev. Clin. Oncol., 2014, 11, 509–524 CrossRef CAS PubMed
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS PubMed
- L. Cipolla, F. Peri and C. Airoldi, Adv. Anticancer Agents Med. Chem., 2008, 8, 92–121 CrossRef CAS PubMed
- T. Buskas, P. Thompson and G. J. Boons, Chem. Commun., 2009, 5335–5349 RSC
- R. M. Wilson and S. J. Danishefsky, J. Am. Chem. Soc., 2013, 135, 14462–14472 CrossRef CAS
- B. A. Cobb and D. L. Kasper, Cell. Microbiol., 2005, 7, 1398–1403 CrossRef CAS PubMed
- B. Treanor, Immunology, 2012, 136, 21–27 CrossRef CAS PubMed
- T. Kurosaki, K. Kometani and W. Ise, Nat. Rev. Immunol., 2015, 15, 149–159 CrossRef CAS PubMed
- P. Costantino, R. Rappuoli and F. Berti, Expet Opin. Drug Discov., 2011, 6, 1045–1066 CrossRef CAS PubMed
- F. Peri, Chem. Soc. Rev., 2013, 42, 4543–4556 RSC
- F. Micoli, R. Adamo and P. Costantino, Molecules, 2018, 23(6), 1451 CrossRef PubMed
- J. M. Silva, M. Videira, R. Gaspar, V. Préat and H. F. Florindo, J. Controlled Release, 2013, 168, 179–199 CrossRef CAS PubMed
- C. Pifferi, N. Berthet and O. Renaudet, Biomater. Sci., 2017, 5, 953–965 RSC
- C. Colombo, O. Pitirollo and L. Lay, Molecules, 2018, 23, 1712 CrossRef PubMed
- S. Nath and P. Mukherjee, Trends Mol. Med., 2017, 20, 332–342 CrossRef PubMed
- S. K. Lau, L. M. Weiss and P. G. Chu, Am. J. Clin. Pathol., 2004, 122, 61–69 CrossRef
- K. Imai, T. Hayashi, T. Suwa, Y. Makiguchi, F. Itoh, Y. Hinoda and T. Takahashi, Int. Congr. Ser., 2001, 1223, 125–133 CrossRef CAS
- O. Prokop and G. Uhlenbruck, Med. Welt, 1969, 46, 2515–2519 CAS
- Y. Cao, A. Merling, U. Karsten, S. Goletz, M. Punzel, R. Kraft, G. Butschak and R. Schwartz-Albiez, Int. J. Cancer, 2008, 123, 89–99 CrossRef CAS PubMed
- G. F. Springer, P. R. Desai and I. Banatwala, Naturwissenschaften, 1974, 61, 457–458 CrossRef CAS PubMed
- G. F. Springer, J. Mol. Med., 1997, 75, 594–602 CrossRef CAS PubMed
- G. F. Springer, Science, 1984, 224, 1198–1206 CrossRef CAS PubMed
- P. R. Desai, Transfus. Med. Rev., 2000, 14, 312–325 CrossRef CAS PubMed
- T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem. Int. Ed., 2011, 50, 1770–1791 CrossRef CAS PubMed
- R. Lo-Man, S. Vichier-Guerre, R. Perraut, E. Dériaud, V. Huteau, L. BenMohamed, O. M. Diop, P. O. Livingston, S. Bay and C. Leclerc, Cancer Res., 2004, 64, 4987–4994 CrossRef CAS PubMed
- R. A. De Silva, Q. Wang, T. Chidley, D. K. Appulage and P. R. Andreana, J. Am. Chem. Soc., 2009, 131, 9622–9623 CrossRef CAS PubMed
- R. A. De Silva, D. K. Appulage, H. Pietraszkiewicz, K. R. Bobbitt, J. Media, J. J. Shaw, F. A. Valeriote and P. R. Andreana, Cancer Immunol. Immunother., 2012, 61, 581–585 CrossRef CAS PubMed
- S. Götze, D. C. K. Rathwell, P. H. Seeberger, C. L. Pereira, F. Broecker, P. Stallforth, C. Anish and J. Hudon, J. Med. Chem., 2018, 61, 4918–4927 CrossRef PubMed
- B. Richichi, B. Thomas, M. Fiore, R. Bosco, H. Qureshi, C. Nativi, O. Renaudet and L. BenMohamed, Angew. Chem. Int. Ed., 2014, 53, 11917–11920 CrossRef CAS PubMed
- E. Kagan, G. Ragupathi, S. S. Yi, C. A. Reis, J. Gildersleeve, D. Kahne, H. Clausen, S. J. Danishefsky and P. O. Livingston, Cancer Immunol. Immunother., 2005, 54, 424–430 CrossRef CAS PubMed
- E. Kaltgrad, S. Sen Gupta, S. Punna, C.-Y. Huang, A. Chang, C.-H. Wong, M. G. Finn and O. Blixt, ChemBioChem, 2007, 8, 1455–1462 CrossRef CAS PubMed
- A. Miermont, H. Barnhill, E. Strable, X. Lu, K. A. Wall, Q. Wang, M. G. Finn and X. Huang, Chem.–Eur. J., 2008, 14, 4939–4947 CrossRef CAS PubMed
- A. L. Parry, N. A. Clemson, J. Ellis, S. S. R. Bernhard, B. G. Davis and N. R. Cameron, J. Am. Chem. Soc., 2013, 135, 9362–9365 CrossRef CAS PubMed
- J. Stagg, R. W. Johnstone and M. J. Smyth, Immunol. Rev., 2007, 220, 82–101 CrossRef CAS
- D. S. Vinay, E. P. Ryan, G. Pawelec, W. H. Talib, J. Stagg, E. Elkord, T. Lichtor, W. K. Decker, R. L. Whelan, H. M. C. S. Kumara, E. Signori, K. Honoki, A. G. Georgakilas, A. Amin, W. G. Helferich, C. S. Boosani, G. Guha, M. R. Ciriolo, S. Chen, S. I. Mohammed, A. S. Azmi, W. N. Keith, A. Bilsland, D. Bhakta, D. Halicka, H. Fujii, K. Aquilano, S. S. Ashraf, S. Nowsheen, X. Yang, B. K. Choi and B. S. Kwon, Semin. Canc. Biol., 2015, 35, S185–S198 CrossRef
- Z. Guo and Q. Wang, Curr. Opin. Chem. Biol., 2009, 13, 608–617 CrossRef CAS PubMed
- J. Heimburg-Molinaro, M. Lum, G. Vijay, M. Jain, A. Almogren and K. Rittenhouse-Olson, Vaccine, 2011, 29, 8802–8826 CrossRef CAS PubMed
- C.-C. Liu and X.-S. Ye, Glycoconj. J., 2012, 29, 259–271 CrossRef CAS PubMed
- F. Berti and R. Adamo, ACS Chem. Biol., 2013, 8, 1653–1663 CrossRef CAS PubMed
- C. Nativi and O. Renaudet, ACS Med. Chem. Lett., 2014, 5, 1176–1178 CrossRef CAS PubMed
- M.-M. Wei, Y.-S. Wang and X.-S. Ye, Med. Res. Rev., 2018, 38, 1003–1026 CrossRef PubMed
- Z. Yin, M. Comellas-Aragones, S. Chowdhury, P. Bentley, K. Kaczanowska, L. BenMohamed, J. C. Gildersleeve, M. G. Finn and X. Huang, ACS Chem. Biol., 2013, 8, 1253–1262 CrossRef CAS PubMed
- X. Wu, C. McKay, C. Pett, J. Yu, M. Schorlemer, S. Ramadan, S. Lang, S. Behren, U. Westerlind, M. G. Finn and X. Huang, ACS Chem. Biol., 2019, 14, 2176–2184 CAS
- R. Dagan, J. Poolman and C.-A. Siegrist, Vaccine, 2010, 28, 5513–5523 CrossRef CAS PubMed
- K. Pobre, M. Tashani, I. Ridda, H. Rashid, M. Wong and R. Booy, Vaccine, 2014, 32, 1423–1430 CrossRef CAS PubMed
- A. Jegerlehner, M. Wiesel, K. Dietmeier, F. Zabel, D. Gatto, P. Saudan and M. F. Bachmann, Vaccine, 2010, 28, 5503–5512 CrossRef CAS PubMed
- T. C. Shiao and R. Roy, New J. Chem., 2012, 36, 324–339 RSC
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC
- Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657–4708 RSC
- J. L. Jimenez Blanco, C. Ortiz Mellet and J. M. Garcia Fernandez, Chem. Soc. Rev., 2013, 42, 4518–4531 RSC
- S. Grigalevicius, S. Chierici, O. Renaudet, R. Lo-Man, E. Dériaud, C. Leclerc and P. Dumy, Bioconjugate Chem., 2005, 16, 1149–1159 CrossRef CAS PubMed
- P. Dumy, I. M. Eggleston, S. Cervigni, U. Sila, X. Sun and M. Mutter, Tetrahedron Lett., 1995, 36, 1255–1258 CrossRef CAS
- S. Peluso, T. Rückle, C. Lehmann, M. Mutter, C. Peggion and M. Crisma, Chembiochem, 2001, 2, 432–437 CrossRef CAS PubMed
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem.–Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed
- C. Pifferi, G. C. Daskhan, M. Fiore, T. C. Shiao, R. Roy and O. Renaudet, Chem. Rev., 2017, 117, 9839–9873 CrossRef CAS PubMed
- O. Renaudet, L. BenMohamed, G. Dasgupta, I. Bettahi and P. Dumy, ChemMedChem, 2008, 3, 737–741 CrossRef CAS PubMed
- I. Bettahi, G. Dasgupta, O. Renaudet, A. A. Chentoufi, X. Zhang, D. Carpenter, S. Yoon, P. Dumy and L. BenMohamed, Cancer Immunol. Immunother., 2009, 58, 187–200 CrossRef CAS PubMed
- O. Renaudet, G. Dasgupta, I. Bettahi, A. Shi, A. B. Nesburn, P. Dumy and L. BenMohamed, PLoS One, 2010, 5, e11216 CrossRef PubMed
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef CAS PubMed
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed
- N. R. Bennett, D. B. Zwick, A. H. Courtney and L. L. Kiessling, ACS Chem. Biol., 2015, 10, 1817–1824 CrossRef CAS PubMed
- E. B. Puffer, J. K. Pontrello, J. J. Hollenbeck, J. A. Kink and L. L. Kiessling, ACS Chem. Biol., 2007, 2, 252–262 CrossRef CAS PubMed
- L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC
- A. Lanzavecchia, Nature, 1985, 314, 537–539 CrossRef CAS PubMed
- A. Lanzavecchia, Annu. Rev. Immunol., 1990, 8, 773–793 CrossRef CAS PubMed
- M.-I. Yuseff, P. Pierobon, A. Reversat and A.-M. Lennon-Dumenil, Nat. Rev. Immunol., 2013, 13, 475–486 CrossRef CAS PubMed
- M. Fiore, N. Berthet, A. Marra, E. Gillon, P. Dumy, A. Dondoni, A. Imberty and O. Renaudet, Org. Biomol. Chem., 2013, 11, 7113–7122 RSC
- C. Pifferi, B. Thomas, D. Goyard, N. Berthet and O. Renaudet, Chem. - Eur. J., 2017, 23, 16283–16296 CrossRef CAS PubMed
- D. Mazal, R. Lo-Man, S. Bay, O. Pritsch, E. Dériaud, C. Ganneau, A. Medeiros, L. Ubillos, G. Obal, N. Berois, M. Bollati-Fogolin, C. Leclerc and E. Osinaga, Cancer Immunol. Immunother., 2013, 62, 1107–1122 CrossRef CAS PubMed
- S. Foillard, M. O. Rasmussen, J. Razkin, D. Boturyn and P. Dumy, J. Org. Chem., 2008, 73, 983–991 CrossRef CAS PubMed
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed
- M. S. Bijker, S. J. F. van den Eeden, K. L. Franken, C. J. M. Melief, R. Offringa and S. H. van der Burg, J. Immunol., 2007, 179, 5033–5040 CrossRef CAS PubMed
- Y. Ikehara, N. Shiuchi, S. Kabata-Ikehara, H. Nakanishi, N. Yokoyama, H. Takagi, T. Nagata, Y. Koide, K. Kuzushima, T. Takahashi, K. Tsujimura and N. Kojima, Canc. Lett., 2008, 260, 137–145 CrossRef CAS PubMed
- M. Ito, K. Hayashi, E. Adachi, T. Minamisawa, S. Homma, S. Koido and K. Shiba, PLoS One, 2014, 9, e11042 Search PubMed
- M. Li, G. M. Davey, R. M. Sutherland, C. Kurts, A. M. Lew, C. Hirst, F. R. Carbone and W. R. Heath, J. Immunol., 2001, 166, 6099–6103 CrossRef CAS PubMed
- F. Albericio, D. Andreu, E. Giralt, C. Navalpotro, E. Pedroso, B. Ponsati and M. Rue-Gayo, Int. J. Pept. Protein Res., 1989, 34, 124–128 CrossRef CAS PubMed
- G. Mezö, N. Mihala, D. Andreu and F. Hudecz, Bioconjugate Chem., 2000, 11, 484–491 CrossRef PubMed
- B. Thomas, C. Pifferi, G. C. Daskhan, M. Fiore, N. Berthet and O. Renaudet, Org. Biomol. Chem., 2015, 13, 11529–11538 RSC
- A. Fernández-Tejada, D. S. Tan and D. Y. Gin, Acc. Chem. Res., 2016, 49, 1741–1756 CrossRef PubMed
-
C. Janeway, P. Travers, M. Walport and M. Shlomchik, Immunobiology: the immune system in health and disease, Garland Publishing, New York, 5th edn, 2001 Search PubMed
- S. L. Lambert, C. Y. Okada and R. Levy, J. Immunol., 2004, 172, 929–936 CrossRef CAS PubMed
- S. Huet, H. Groux, B. Caillou, H. Valentin, A. M. Prieur and A. Bernard, J. Immunol., 1989, 143, 798–801 CAS
- B. J. G. Baaten, C.-R. Li, M. F. Deiro, M. M. Lin, P. J. Linton and L. M. Bradley, Immunity, 2010, 32, 104–115 CrossRef CAS PubMed
- E. Aktas, U. C. Kucuksezer, S. Bilgic, G. Erten and G. Deniz, Cell. Immunol., 2009, 254, 149–154 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00544d |
| This journal is © The Royal Society of Chemistry 2020 |