
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free carbon monoxide-releasing micelles undergo tandem photochemical reactions for cutaneous wound healing†
Jian
Cheng
a,
Bin
Zheng
*b,
Sheng
Cheng
c,
Guoying
Zhang
a and
Jinming
Hu
 *a
*a
aCAS Key Laboratory of Soft Matter Chemistry, Hefei National Laboratory for Physical Science at the Microscale, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: jmhu@ustc.edu.cn
bSchool of Chemistry and Chemical Engineering, Hefei Normal University, Hefei, Anhui 230061, P. R. China. E-mail: paradise@mail.ustc.edu.cn
cInstrumental Analysis Center, Hefei University of Technology, Hefei, Anhui 230009, China
First published on 14th April 2020
Abstract
Carbon monoxide (CO) has shown broad biomedical applications. The site-specific delivery and controlled release of CO is of crucial importance to achieve maximum therapeutic benefits. The development of carbon monoxide (CO)-releasing polymers (CORPs) can increase the stability, optimize pharmacokinetic behavior, and reduce the side effects of small molecule precursors. However, almost all established CORPs were synthesized through a post functional approach, although the direct polymerization strategy is more powerful in controlling the chain compositions and architectures. Herein, a direct polymerization strategy is proposed toward metal-free CO-releasing polymers (CORPs) based on photoresponsive 3-hydroxyflavone (3-HF) derivatives. Such CO-releasing amphiphiles self-assemble into micelles, having excellent water-dispersity. Intriguingly, photo-triggered tandem photochemical reactions confer successive fluorescence transitions from blue-to-red-to-colorless, enabling self-reporting CO release in vitro and in vivo as a result of the incorporation of 3-HF derivatives. More importantly, the localized CO delivery of CORPs by taking advantage of the spatiotemporal control of light stimulus outperformed conventional metal carbonyls such as CORMs in terms of anti-inflammation and cutaneous wound healing. This work opens a novel avenue toward metal-free CORPs for potential biomedical applications.
Introduction
Carbon monoxide (CO), together with nitric oxide (NO) and hydrogen sulfide (H2S), has been recognized as an endogenous signal molecule. These signal molecules exert critical physiological functions, and can also be used as novel therapeutic agents. For example, CO is endogenously produced via the breakdown of heme catalyzed by heme oxygenase (HO), exhibiting pleiotropic functions such as anti-inflammation, anti-apoptosis, and cytoprotection.1–3 Because of the inconvenience of the direct inhalation of CO gas, it is of increasing interest to develop CO-releasing molecules (CORMs) for the controlled release of CO. Following the seminal work of Motterlini and coworkers,4–6 a variety of CORMs have been successfully exploited.7–10 These CORMs can be roughly categorized into two sorts, namely metal carbonyls and metal-free CORMs. Although metal carbonyls hold great promise in both laboratory and clinical investigations,9,11–26 the development of metal-free CORMs has recently been receiveing increasing attention.27 In this context, boranocarbonates,28 CO-releasing prodrugs operated in a “click and release” manner,29–31 and photoresponsive organic CORMs such as 3-hydroxyflavone (3-HF) derivatives,32–37 xanthene-9-carboxylic acid,38 BODIPY derivatives,39 and aromatic α-diketone derivatives40 have been developed.In addition to the fast development of small molecule-based CORMs, the fabrication of CO-releasing polymers (CORPs) represents a topic of intensive research.41 In comparison with small molecule precursors, the corresponding CORPs are characterized by increased half-lives and bioavailability, optimized pharmacokinetic performance, and decreased side effects. For example, Hubbell et al.42 fabricated CO-releasing micelles for immunotherapy using triblock copolymers bearing Ru(CO)3Cl(ornithinate) moieties in the middle blocks. The Boyer group43 reported the preparation of water-soluble CORPs based on the post-functionalization of poly(oligo(ethylene glycol)monoether acrylate)-b-poly(4-vinylbenzyl chloride) (POEGMA-b-PVBC) with metal carbonyls for antimicrobial applications. Notably, so far almost all the reported CORPs were synthesized through the post functional strategy either by physically loading CORMs into polymeric nanovectors or by covalently linking CORMs to the polymeric matrices.42–49 Very recently, Bazzi and coworkers50 reported the synthesis of green light-responsive CORPs through ring-opening metathesis polymerization (ROMP). To our knowledge, the preparation of metal-free CORPs through a direct polymerization technique has not been achieved, although the direct polymerization approach has the potential to more precisely control the spatial location of the CORMs and to more readily obtain well-defined chain architectures.
In this work, we propose a direct polymerization approach to fabricate photoresponsive CORPs based on 3-HF derivatives (Scheme 1a). A polymerizable monomer, 2-((((4-(3-((2-nitrobenzyl)oxy)-4-oxo-4H-benzo[g]chromen-2-yl)benzyl)oxy)carbonyl)amino)ethyl methacrylate (FNM) containing both photoresponsive o-nitrobenzyl ether and extended 3-HF derivatives was rationally designed and synthesized. The FNM monomer can be directly polymerized through reversible addition–fragmentation chain transfer (RAFT) polymerization with the formation of CO-releasing amphiphiles using a conventional poly(ethylene glycol) (PEG)-based macroRAFT agent. The resulting CO-releasing amphiphiles self-assembled into micellar nanoparticles in an aqueous solution, undergoing tandem photochemical reactions of the removal of o-nitrobenzyl moieties and photooxygenation of 3-HF residues under visible light irradiation. This process features a consecutive fluorescence transition from blue-to-red-to-colorless, allowing for the monitoring of the CO release by fluorescence readout (Scheme 1b).33
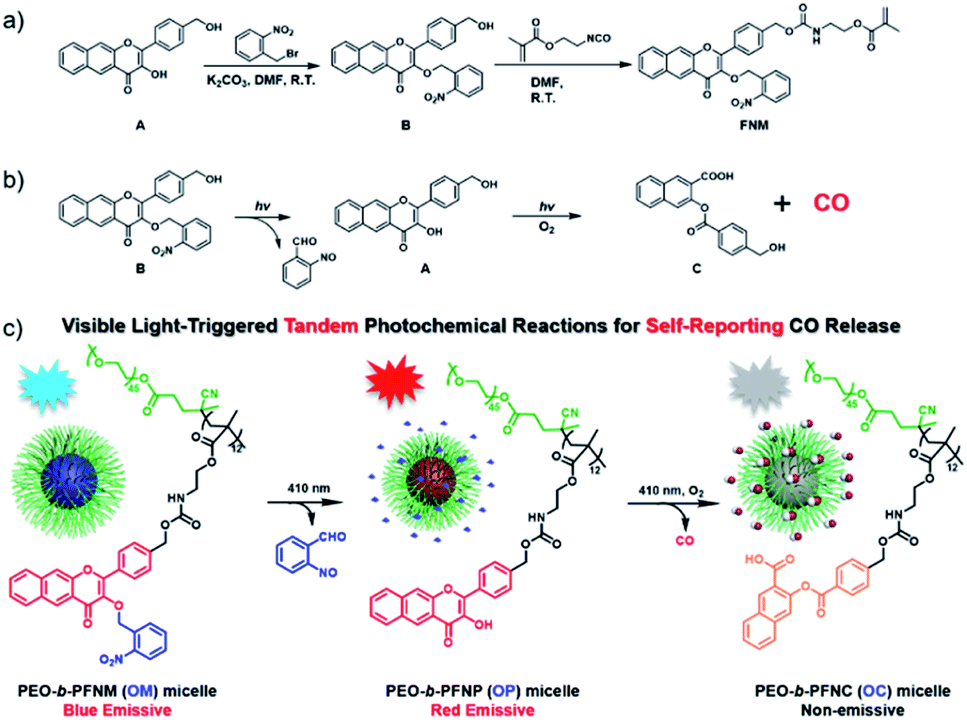 | ||
| Scheme 1 (a) Synthesis of the CO-releasing monomer (FNM) based on extended 3-HF derivatives. (b) CO release from compound B through tandem photochemical reactions. (c) Schematic illustration of the self-reporting CO release from OM micellar nanoparticles via visible light-triggered tandem photochemical reactions. | ||
Results and discussion
Rational design and synthesis of metal-free CORPs
3-HF derivatives are known for their unique excited-state intramolecular proton transfer (ESIPT) characteristic. The unmasking of the phenolic groups of 3-HF derivatives led to a large Stokes shift, which was previously used for the design of ratiometric fluorescent probes.51 Recently, Berreau and coworkers32–36 ingeniously found that extended 3-HF derivatives could release CO under visible light irradiation. In this case, the fluorescent extended 3-HF moieties were transformed into non-emissive 3-(benzoyloxy)-2-naphthalenecarboxylic acid (3-BNA) species, enabling self-reporting CO release. We surmised that 3-HF derivatives could be engineered as a polymerizable monomer and metal-free CORPs could be further synthesized through conventional radical polymerizations. However, the presence of a phenolic hydroxyl group at the 3-position was problematic for radical polymerization because a phenolic hydroxyl group is known as an inhibitor for radical polymerization.Notably, the hydroxyl groups of extended 3-HF have been successfully masked by thiol-,35 H2S-,36 and H2O2 (ref. 37)-reactive moieties and photo-triggered CO release could be manipulated in a logical manner. Given that 2-nitrobenzyl ethers underwent photo-degradation reactions with the recovery of the initial phenolic hydroxyl groups,52,53 we designed a hydroxyl-containing 3-HF derivative (i.e., compound A) and selectively protected the 3-OH group with 2-nitrobenzyl bromide with the formation of compound B (Scheme 1), containing two photoresponsive moieties (i.e., 2-nitrobenzyl ether and 3-HF derivative) within the same molecule. Interestingly, the different reactivities of the phenolic and alcoholic hydroxyl groups enabled selective masking of the 3-OH group without protecting the alcoholic hydroxyl group. Finally, the residual alcoholic hydroxyl group was converted to methacrylate (i.e., FNM) by reacting compound B with 2-isocyanatoethyl methacrylate (Scheme 1a). The chemical structures of the intermediates (A and B) and the named monomer (FNM) were characterized by nuclear magnetic resonance (NMR) spectroscopy, high-resolution mass spectroscopy (HR-MS), and high-performance liquid chromatography (HPLC) (Fig. S1–S4†).
Photo-triggered tandem photochemical reactions for CO release
Next, we examined the photoresponsive behavior of compound B under visible light irradiation. Notably, compound B exhibited a lower water solubility than that of the unmodified 3-HF precursor, which should be ascribed to the introduction of the hydrophobic 2-nitrobenzyl moiety. Due to the very limited water solubility of compound B, a mixed solvent of dimethylsulfoxide (DMSO) and water (v/v = 6/4) was applied for spectroscopic studies. Intriguingly, compound B was responsive to visible light (410 nm, 31.5 mW cm−2), as evidenced by the evolution of UV-Vis absorbance spectroscopy under illumination (Fig. S5†). Specifically, after irradiation for 10 min, all the absorbance intensities at 270 nm, 335 nm, and 385 nm gradually decreased. However, no changes were observed in UV-Vis spectroscopy without light irradiation under otherwise identical conditions (Fig. S6†), which is in good agreement with the previous results that 3-HF-based CO donors were relatively stable under dark conditions without premature CO leakage even in an aqueous medium.32–36From fluorescence spectroscopy, we can observe a remarkable fluorescence increase for compound B at 606 nm at the initial 40 s irradiation, corresponding to the deprotection of the 2-nitrobenzyl residue (Fig. S7a†). Upon increasing the irradiation time to 400 s, the fluorescence emission at 606 nm underwent a monotonous drop, in good agreement with the photo-mediated CO release with the formation of a non-emissive 3-BNA side product (Fig. S7b†). Notably, the fluorescence intensity of compound B after 40 s irradiation revealed a 60% decrease as compared to authentic compound A with the same concentration (Fig. S8†). Thus, the current design by integrating two photoresponsive moieties into one molecule enabled tandem photochemical reactions and rendered the CO release process visible by fluorescence changes by taking advantage of the unique property of 3-HF derivatives.32–36
To elucidate the underlying photoreaction mechanism of compound B under light irradiation, HPLC and HRMS were used to examine the degraded products. Upon irradiation, compound B was completely consumed within 10 min, while the formation of the intermediate compounds A and the final photolyzed product, compound C, was observed by HPLC (Fig. S9†). Notably, the coexistence of the intermediate compound A and compound C revealed that, once formed, compound A underwent a further photo-degradation. On the other hand, the generation of compound A, followed by the production of compound C, was unequivocally supported by HRMS analysis (Fig. S10†). Collectively, we proposed the following mechanism: under visible light irradiation, compound B was converted to the intermediate compound A through the removal of the photoresponsive 2-nitrobenzyl capping moiety; the resulting compound A was sensitive to the same light irradiation and subsequently transformed to compound C with the release of CO under an aerobic condition (Fig. S9a†). Notably, the 3-OH of extended 3-HF moieties has been previously functionalized with H2O2-37 or thiol-responsive36 groups. These designs allowed for the fabrication of logic gates for controlled CO release, while our design enabled the cascade photochemical reactions and regulated CO release by a single stimulus, namely light irradiation.
Although compound B with two photoresponsive moieties within one molecule underwent a progressive degradation with the release of CO under visible light irradiation, the insufficient water-solubility remarkably impeded the potential biomedical applications. Previously, the improved water-solubility of extended 3-HF moieties was achieved by the sulfonation of the 3-HF core structures;54 we surmised that it was possible to circumvent the poor water-solubility of compound B through the fabrication of amphiphilic CORPs which can self-assemble into diverse nanostructures in an aqueous solution. With the FNM monomer in hand, we first confirmed that the photodegradation was not affected by the introduction of the methacrylate residues. The HRMS results confirmed the formation of the 2-((((4-(3-hydroxy-4-oxo-4H-benzo[g]chromen-2-yl)benzyl)oxy)carbonyl)amino)ethyl methacrylate (FNP) monomer with the removal of the 2-nitrobenzyl moiety, followed by the generation of the 3-((4-((((2-(methacryloyloxy)ethyl)carbamoyl)oxy)methyl)benzoyl)oxy)-2-naphthoic acid (FNC) monomer (Fig. S11†), in line with the photo-mediated degradation mechanism of compound B (Fig. S9†).
The FNM monomer can be directly polymerized through RAFT polymerization using a PEO-based macroRAFT agent, with the formation of an amphiphilic PEO-b-PFNM (OM) diblock copolymer (Scheme 1b). For example, a representative block copolymer of PEO45-b-PFNM12 was synthesized and the degree of polymerization (DP) of the PFNM block was calculated to be 12 by 1H NMR analysis (Fig. S12a†). The as-synthesized diblock copolymer had a molecular weight (MW) of 7.1 kDa and an MW distribution of 1.08 (Fig. S12b†), revealing the feasibility to synthesize well-defined CORPs through a direct polymerization approach. By contrast, previously reported CORPs were dominantly prepared by the postmodification procedure and, to our knowledge, no metal-free CORPs have been exploited to date.43,55–57
The PEO45-b-PFNM12 diblock copolymer self-assembled into micellar nanoparticles in pure aqueous solution with an intensity-average hydrodynamic diameter of ∼52 nm, as confirmed by TEM observation and DLS analysis (Fig. 1a and c). The theoretical CO loading concentration was calculated to be ∼1.3 mM at a micelle concentration of 1.0 g L−1 if one mole of 3-HF moieties can release one mole of CO, which was much higher than that of the solubility of compound B in aqueous solution (<10 μM). Thus, the formation of CORPs provided a feasible solution to remarkably elevate the water dispersity of hydrophobic CORMs such as compound B. Notably, the water-solubility of 3-HF derivatives could be remarkably elevated by the introduction of the hydrophilic sulfonate group as well.54 Upon irradiation of the OM micelles with a hand-held LED lamp (410 nm, 31.5 mW cm−2), the micellar nanostructures were retained but there was a slight decrease in the diameter of micellar nanoparticles from 52 to 46 nm (Fig. 1b, c and S13†). This result was not surprising due to the transformation of FNM to FNC monomers under visible light irradiation (Fig. S11†). Despite the FNC monomer bearing carboxylic acid residues, the water solubility of the FNC monomer remained limited because of the presence of hydrophobic aromatic rings and aliphatic chains. However, there was a remarkable change of the polarity within the micellar nanoparticles—the fluorescence intensity of Nile red (NR)-loaded OM micelles at 638 nm gradually decreased under illumination (Fig. 1d), although NR itself was not sensitive to identical light irradiation (data not shown). Note that the fluorescence emission at 638 nm belonged to NR rather than the micellar nanoparticles because no emission was observed when the OM micelles were excited at 550 nm without NR loading (Fig. S14†).
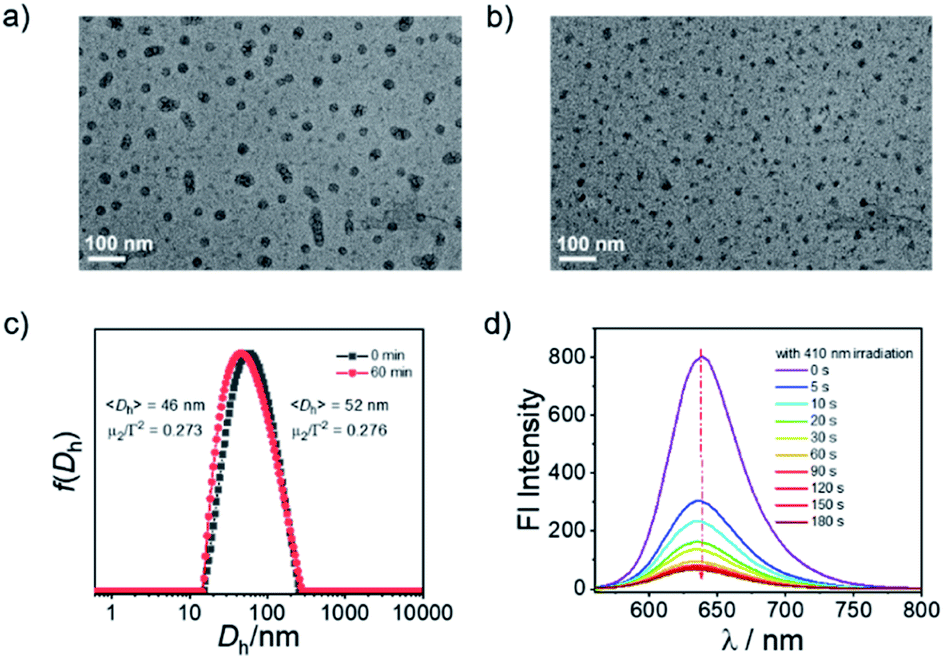 | ||
| Fig. 1 (a and b) TEM images of aqueous dispersions (0.1 g L−1) of OM micelles (a) before and (b) after irradiation for 60 min (scale bars: 100 nm). (c) Hydrodynamic distributions, f(Dh), of aqueous dispersions of OM micelles before and after irradiation. (d) Evolution of fluorescence emission spectra (λex = 550 nm; slit width: Ex. 5 nm, Em. 5 nm) of NR-loaded OM micelles under irradiation (410 nm, 31.5 mW cm−2). | ||
To investigate whether the OM micelles inherited the photoresponsive behavior of the B precursor, we recorded the UV-Vis and fluorescence spectroscopy spectra of the micelles in pure water under visible light irradiation. The absorption peaks centered at 270 nm, 336 nm, and 385 nm gradually declined under irradiation (Fig. 2a), akin to that of compound B (Fig. S5a†). Moreover, there was no detectable change in the UV-Vis spectra without light irradiation, indicating the good stability of the micelles in aqueous solution (Fig. S15†). The fluorescence changes of the OM micelle dispersion encompassed two distinct stages—the blue-to-red transition at the initial 20 s and the following red-to-colorless variation in the next 30 min (Fig. 2b and c). Notably, the photo-activated red emission of OM micelles in pure aqueous solution centered at 606 nm, whereas previous results regarding the extended 3-HF small molecules showed emissions at ∼575–585 nm depending on the concentrations and solvent conditions.35,37 The red-shift of the fluorescence emission was likely ascribed to the formation of 3-HF aggregates within the micellar cores, which was rather beneficial to eliminate background interferences. Moreover, the continuous and remarkable fluorescence transitions can be readily observed by the naked eye (insets in Fig. 2b and c). The photo-triggered fluorescence transition coincided well with the changes of B precursor as well (Fig. S7†), demonstrating that the formation of micelle nanoparticles in aqueous solution did not alter the photoreaction pathways. However, the fluorescence decay corresponding to the CO release rate within the micellar nanoparticles in pure aqueous solution appeared slower than that of B precursor in a mixed solvent. This result not only revealed that the photolysis process was highly relevant to the aggregated status of the 3-HF moieties but also provided a feasible strategy to modulate the CO release behavior.
 | ||
| Fig. 2 (a) UV-Vis spectra and (b and c) fluorescence emission spectra (λex = 405 nm; slit width: Ex. 5 nm, Em. 5 nm) of aqueous dispersions (0.1 g L−1) of OM micelles under irradiation. (d) CO release profiles from OM micelle solutions (0.1 g L−1) in pure water, PBS, and DMEM culture medium with or without irradiation. (e) CO release from OM micelle solutions (0.1 g L−1) measured by using a myoglobin assay. (f) Evolution of fluorescence emission spectra (λex = 490 nm; slit width: Ex. 5 nm, Em. 5 nm) of OM micelles in the presence of an allyl-Flu probe (5 μM) and PdCl2 (5 μM) under light irradiation (410 nm, 31.5 mW cm−2). | ||
To directly confirm the CO release under illumination, a portable CO detector was used to monitor the CO release into the vial headspace from the OM micelle solution. Importantly, there was no premature CO release detected without light irradiation (Fig. 2d). However, under irradiation, the released CO amounts in pure water gradually increased and stabilized after ∼180 min of irradiation (Fig. 2d). Notably, although metal carbonyls such as CORM-2 and CORM-3 have been widely used as CO donors, recent studies unexpectedly discovered that no CO release was detected in PBS buffer and cell culture media.20,21 It was, therefore, of particular interest to clarify whether CO could be released from the OM micelle dispersion in PBS and cell culture media. Intriguingly, we discovered that the CO release processes under visible light were not adversely affected in PBS or cell culture media (Fig. 2d) and the CO release contents were quite comparable to those in pure water. This result concurred well with the fact that the extended 3-HF small molecules could release CO both in aqueous solution and in the presence of bovine serum albumin under irradiation.34 Specifically, the CO release was calculated to be ∼0.45 μmol mg−1 for the OM diblock copolymer, which was equal to ∼33.6% of the loaded 3-HF residues. Notably, the quantum yields of CO release were calculated to be 0.003 ± 0.001 for the OM micelles in pure aqueous solution and 0.011 ± 0.005 for compound B (DMSO/H2O = 6/4, v/v).32 Although compound B could release CO in a quantitative manner (0.96 ± 0.02 equiv. relative to compound B), the decreased CO yield from the OM micelles was tentatively ascribed to the decreased quantum yield within the micelle cores. Besides, the photo-triggered CO release was also confirmed by a standard myoglobin (Mb) assay. However, the CO release content by the Mb assay was only determined to be ∼7.7% of the loaded 3-HF moieties. This result was understandable because the Mb assay was performed under a deoxygenated condition but the CO release of 3-HF moieties was more efficient under a normoxic condition.32,35
In addition to the CO detector and Mb assay, the release of CO from OM micelles under illumination was further confirmed using a previously reported fluorescein-based probe (i.e., allyl-Flu), which can be selectively activated by CO in the presence of Pd2+ ions with the formation of highly emissive fluorescein (Fig. 2f).58 In contrast, no fluorescence was detected in the absence of OM micelles under otherwise identical conditions (Fig. S16a†). Remarkably, the observed fluorescence did not originate from the OM micelles either since there was no emission when the OM micelles were excited at 490 nm (Fig. S16b†). Thus, the selective fluorescence increase at 515 nm should be solely ascribed to the photo-mediated CO release that, in turn, activated the allyl-Flu probe through the CO-mediated Tsuji–Trost reaction.58 Building on the above results, we can safely conclude that CO could be released from OM micelles under visible light irradiation.
Photo-mediated CO release in vitro and in vivo
As stated above, the successive and remarkable fluorescence transitions endowed the CO release process with a self-reporting characteristic, which has been well investigated by Berreau and coworkers.32–36 We further evaluated the photo-controlled CO release behavior of OM micelles in living cells. Upon incubation of OM micelles with RAW264.7 cells, blue emission, originating from the OM micelles without light irradiation, was gradually intensified upon increasing the incubation time (Fig. S17†), suggesting that OM micelles could be easily taken up by cells. Quantitative analysis revealed that the fluorescence intensities after 4 h and 6 h were rather comparable (Fig. S17b†). As such, an incubation time of 4 h was applied for the subsequent cellular studies. Interestingly, the blue-to-red fluorescence transition could be observed within OM micelle-treated cells upon irradiation of the treated cell for 1 min (Fig. 3), which is in good agreement of the removal of 2-nitrobenzyl moieties in aqueous solution (Fig. 2b). Moreover, the red emission gradually faded after 15 min irradiation (Fig. 3), which should be ascribed to photo-triggered intracellular CO release with the formation of non-emissive derivatives.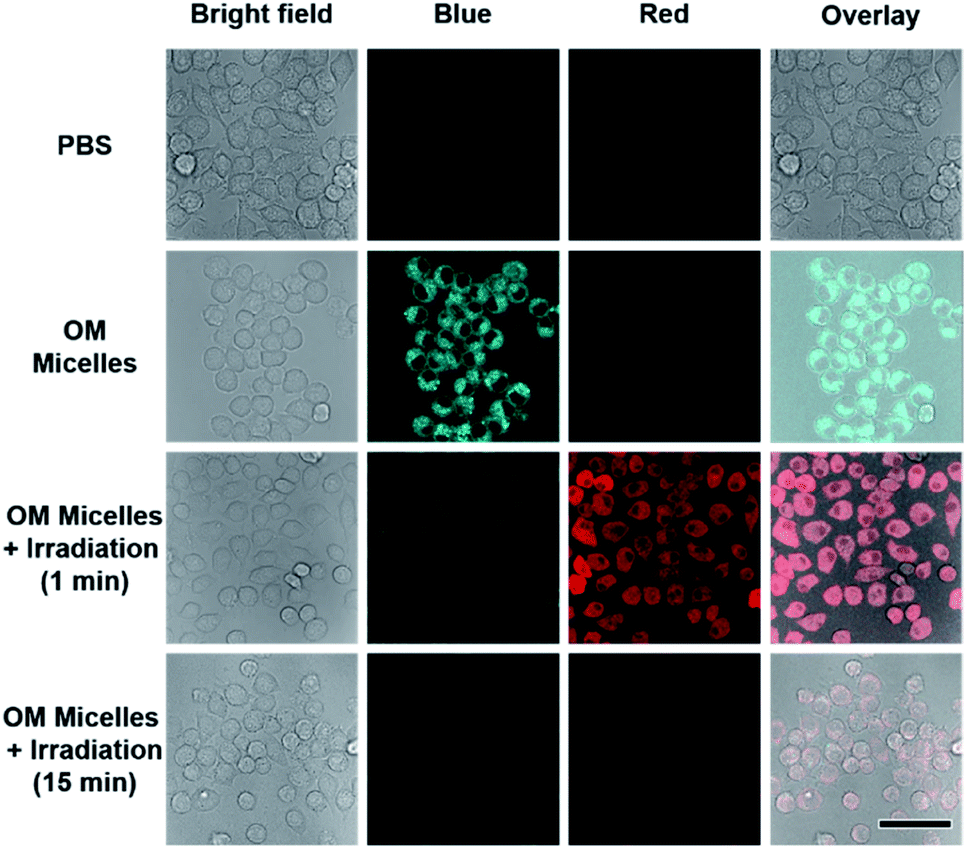 | ||
| Fig. 3 Confocal laser scanning microscopy (CLSM) images of RAW 264.7 cells after incubation with OM micelles without or with 410 nm irradiation. The OM micelle concentration was 0.1 g L−1. Scale bar is 50 μm. The blue and red channels were both excited at 405 nm and collected at 415–500 nm and 550–650 nm, respectively. | ||
Apart from self-reporting CO release, we further confirmed the photo-triggered CO release within cells using the allyl-Flu probe as a signal reporter. It should be pointed out that although COP-1 was previously widely used for monitoring CO release,59 we found that the COP-1 probe was photolabile and it was not suitable for the current study. Upon incubation of RAW264.7 cells with OM micelles, green emission of activated fluorescein can only be observed in the coexistence of OM micelles, allyl-Flu probe, Pd2+ ions, and light irradiation. The control experiments without OM micelles or without light irradiation did not give rise to the green emission, nor did OC micelles (OM micelles with pre-treated illumination) lead to green emission even in the presence of allyl-Flu, Pd2+ ions, and additional illumination (Fig. 4). Taken together, we concluded that the photo-triggered self-reporting CO release of OM micelles could operate not only under cell-free conditions but also in living cells.
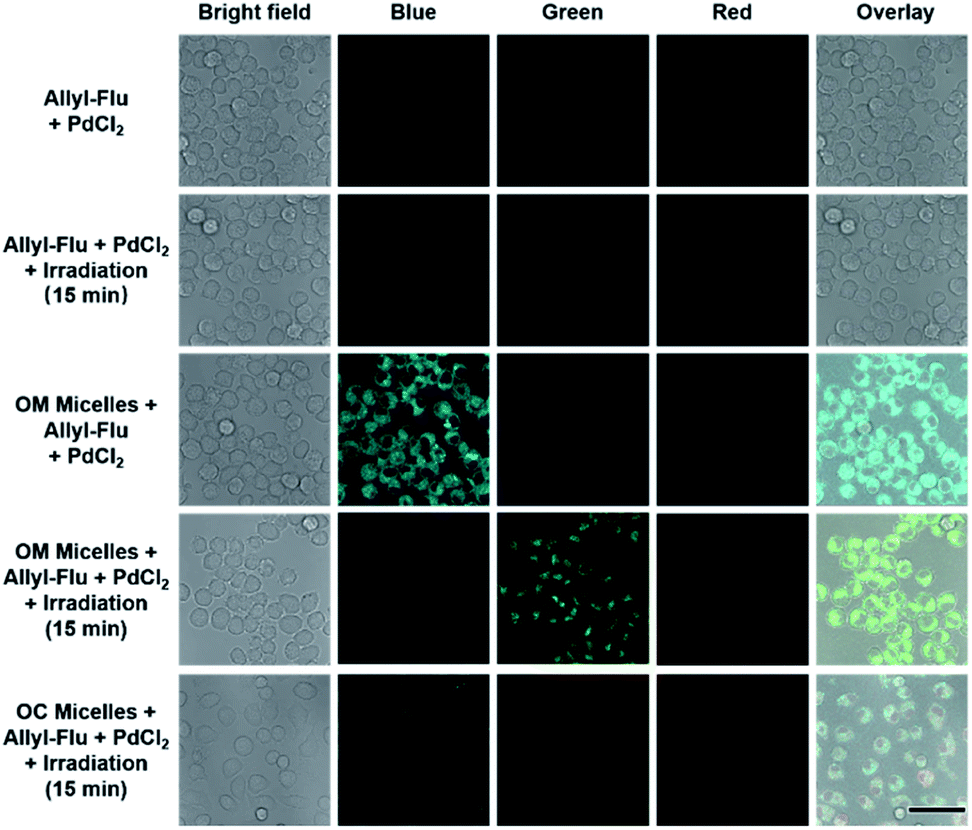 | ||
| Fig. 4 CLSM images of RAW 264.7 cells after incubation with OM micelles and allyl-Flu probe under various conditions. In all cases, the OM micelle concentration was 0.1 g L−1 and the concentrations of allyl-Flu and PdCl2 were 5 μM. Scale bar is 50 μm. The blue and red channels were both excited at 405 nm and collected at 415–500 nm and 550–650 nm, respectively. The green channel was excited at 488 nm and collected at 500–580 nm. | ||
Besides photo-triggered CO release in cells, we testified whether CO release could be modulated in living animals by taking advantage of the self-reporting fluorescence changes during CO release. Notably, the large Stokes shift of 3-HF derivatives was quite advantageous for minimizing the background interference. We monitored the red fluorescence changes in a mouse after a cutaneous injection, revealing an initial fluorescence increase followed by a fluorescence drop upon increasing irradiation time (Fig. 5a and b), in line with the sequential photochemical reactions of the deprotection of the 2-nitrobenzyl moiety and photooxygenation of 3-HF derivatives with CO release (Fig. 2b and c). However, no appreciable change of red fluorescence was observed without irradiation. This result demonstrated that 410 nm light could partially penetrate skin tissues and the current OM micelle-based CO donor could be potentially used for the treatment of cutaneous diseases. Moreover, both the OM micelles and OC micelles (OM micelles with pre-treated illumination) were nontoxic to RAW264.7 cells up to 0.2 g L−1 with the cell viability higher than 90% (Fig. 5c). Moreover, we did not observe significant cytotoxicity of the released small molecules either (Fig. S18†). The high CO loading contents, superior dispersity in biological fluids, photo-triggered sustainable CO release, and good biocompatibility may augur a promising future of these CORPs in biomedical applications.
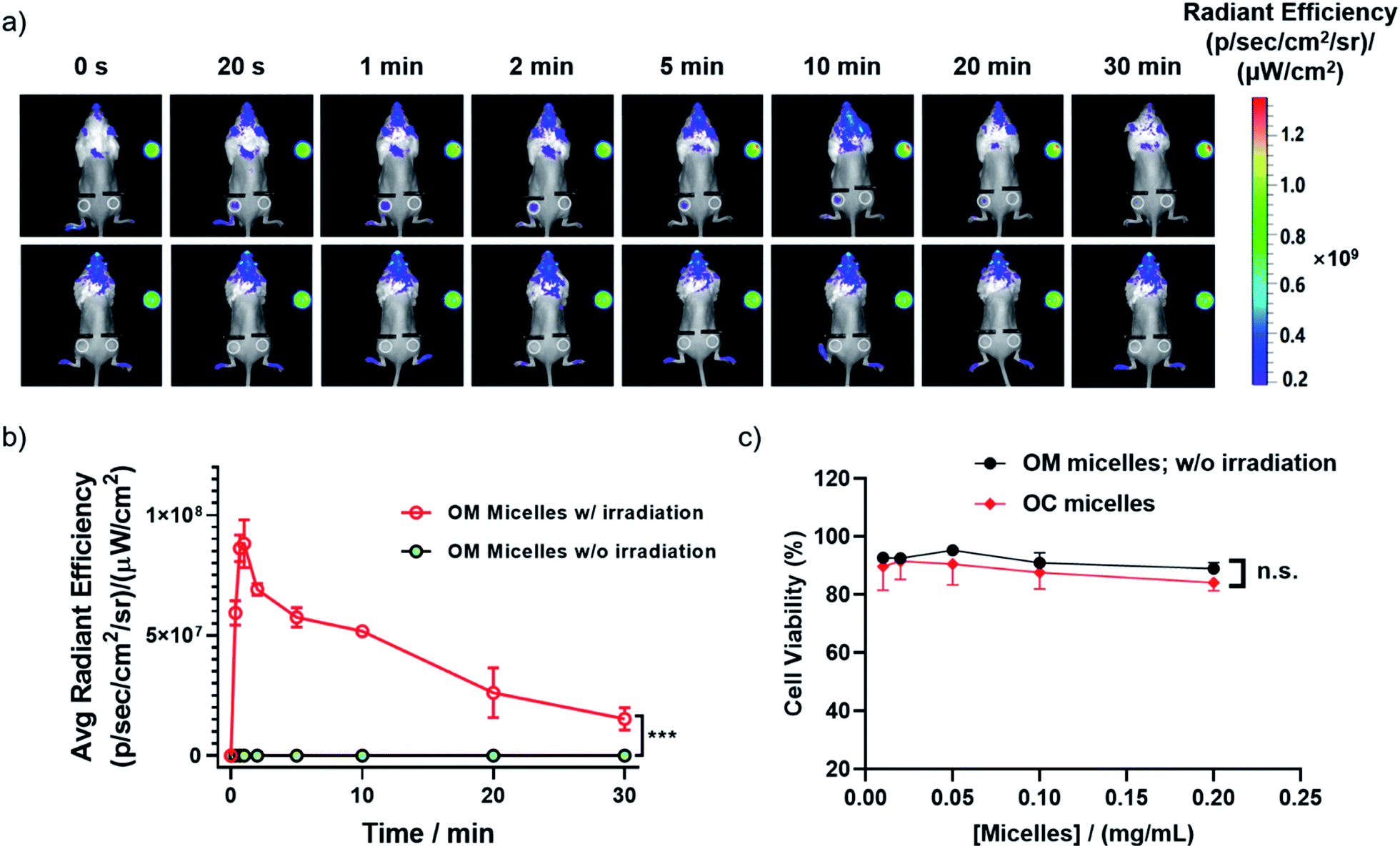 | ||
| Fig. 5 (a) In vivo fluorescence images of an individual BALB/c mouse after a cutaneous injection of 100 μL of OM micelle solution (0.1 g L−1) with (upper panel) or without (bottom panel) light irradiation. PBS buffer was used as the control. The injection sites are marked by white circles. (b) Analysis of the fluorescence changes of the mice (mean ± s.d.; ***p < 0.001 vs. the non-irradiated group). (c) Cell viability determined by using the MTT assay of OM micelles without irradiation and OC micelles. Data represented as mean ± s.d.; n.s. denote not significant. | ||
Anti-inflammatory performance and cutaneous wound healing of CORPs
It has been well-documented that CO exhibits pleiotropic physiological functions such as vasoactive response, cytoprotection, redox control, and immunological modulation.8 Next, we evaluated the anti-inflammatory performance of the CO-releasing micelles by attenuating lipopolysaccharide (LPS)-induced nitrite and tumor necrosis factor alpha (TNF-α) production.54,60 After incubating RAW264.7 cells with LPS (1 μg mL−1), there were remarkable increases in nitrite and TNF-α levels. However, photo-triggered CO release from OM micelles can efficiently alleviate LPS-induced nitrite and TNF-α production, exhibiting a good anti-inflammatory performance (Fig. 6a and b). Notably, the OC micelles with pre-treated illumination without CO release were unable to attenuate LPS-induced nitrite and TNF-α production, suggesting that photo-mediated CO release from OM micelles played a crucial role in the anti-inflammatory capacity. Notably, there were no remarkable changes in cell viability, indicating that the variations of nitrite and TNF-α concentrations were not correlated to the potential cytotoxicity of the micellar nanoparticles (Fig. 6c).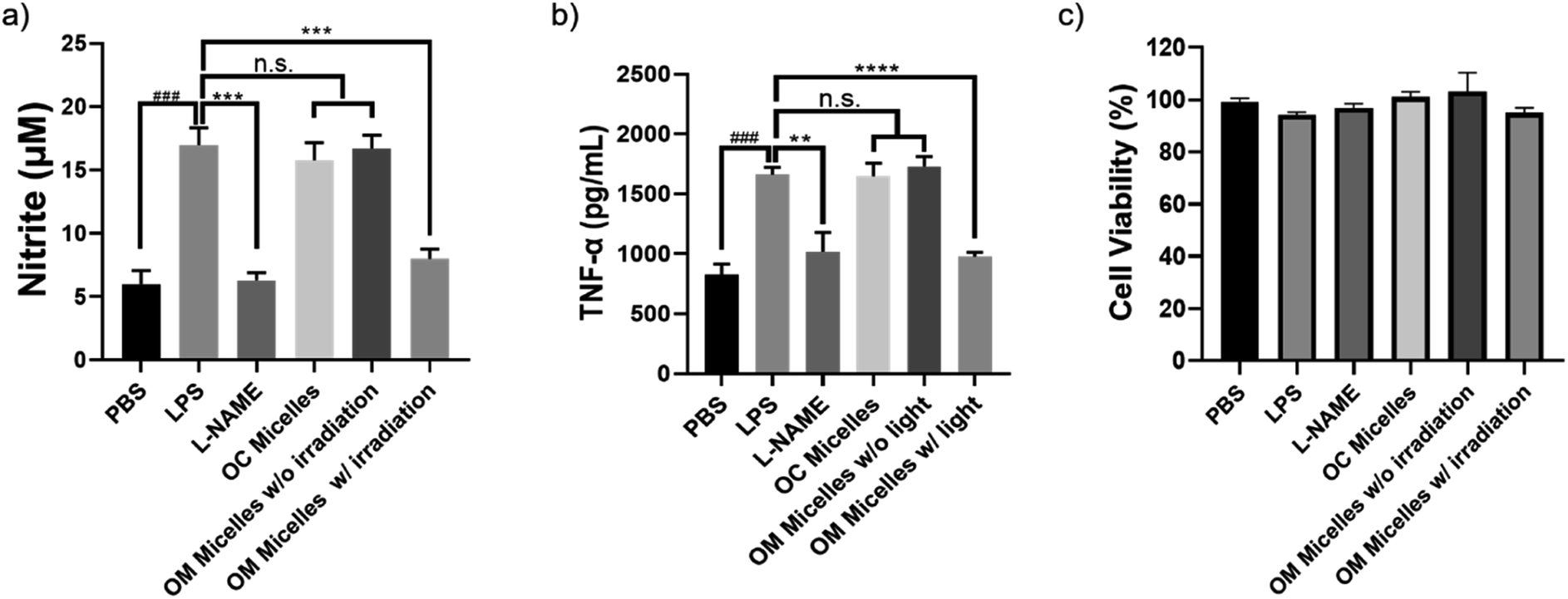 | ||
| Fig. 6 Quantitative analysis of intracellular (a) nitrite and (b) TNF-α concentrations by the Griess assay after treatment of RAW264.7 cells under various conditions. Data represent mean ± s.d. (n = 5), ### indicates p < 0.001 vs. PBS control; ** indicates p < 0.01, *** indicates p < 0.001 and **** indicates p < 0.0001 vs. LPS-treated group, and n.s. denotes not significant. (c) Cell viability determined by using the MTT assay after incubation of RAW264.7 under varying conditions. | ||
Finally, given the unique therapeutic effects of CO, we evaluated the performance of the CO-releasing micelles in a murine full-thickness cutaneous wound model. In comparison with the control groups treated with PBS buffer and OM micelles without light irradiation, the group treated with OM micelles with light radiation exhibited a faster wound healing rate (Fig. 7), as exemplified by the smaller wound areas especially after 7 day treatments (Fig. 7b). However, OC micelles with pre-treated light irradiation performed similar to PBS buffer, suggesting that the faster healing performance was attributed to photo-triggered CO release.
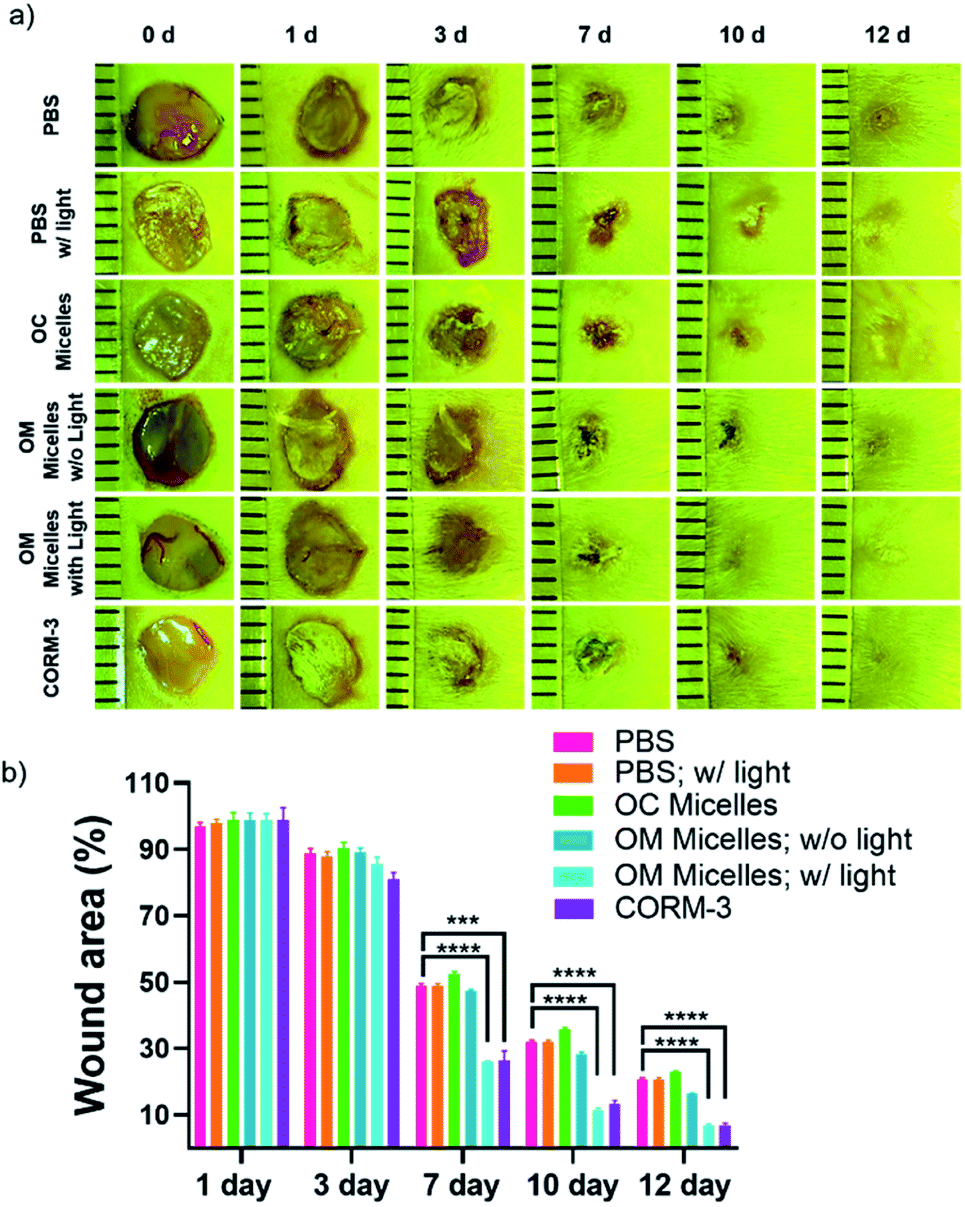 | ||
| Fig. 7 (a) Representative images of cutaneous wounds treated with PBS buffer, OC micelles, OM micelles (0.1 g L−1, 50 μL each day) without or with light irradiation for 15 min, and CORM-3, respectively. (b) Quantitative analysis of the residual wounded areas treated with PBS buffer and OM micelles (0.1 g L−1) without or with light irradiation using ImageJ software (mean ± s.d.; ***p < 0.001, ****p < 0.0001 vs. the PBS-treated group). | ||
H&E staining analyses revealed that, on day 3, the CORM-3 and OM micelle-treated group with irradiation exhibited less inflammatory cell infiltration, presumably due to the anti-inflammatory effect of CO (Fig. 6).61 Meanwhile, Sirius red staining demonstrated increased collagen deposition and CD31 immunostaining revealed enhanced neovascularization as reflected by the green emission of abundant CD31-expressed endothelial cells (Fig. 8). Moreover, OM micelles exhibited higher green emission than that of the CORM-3-treated group, indicating better neovascularization for OM micelles with sustained CO release under illumination. On the other hand, on day 12, the OM micelle-treated group with irradiation showed complete re-epithelialization in the wound area, and the histological results were quite comparable to those of healthy skin. By contrast, the PBS-, PBS with light irradiation-, OC micelle-, and OM micelle without light irradiation-treated groups exhibited inflammatory cell infiltration and lacked collagen deposition and angiogenesis (Fig. 8).62
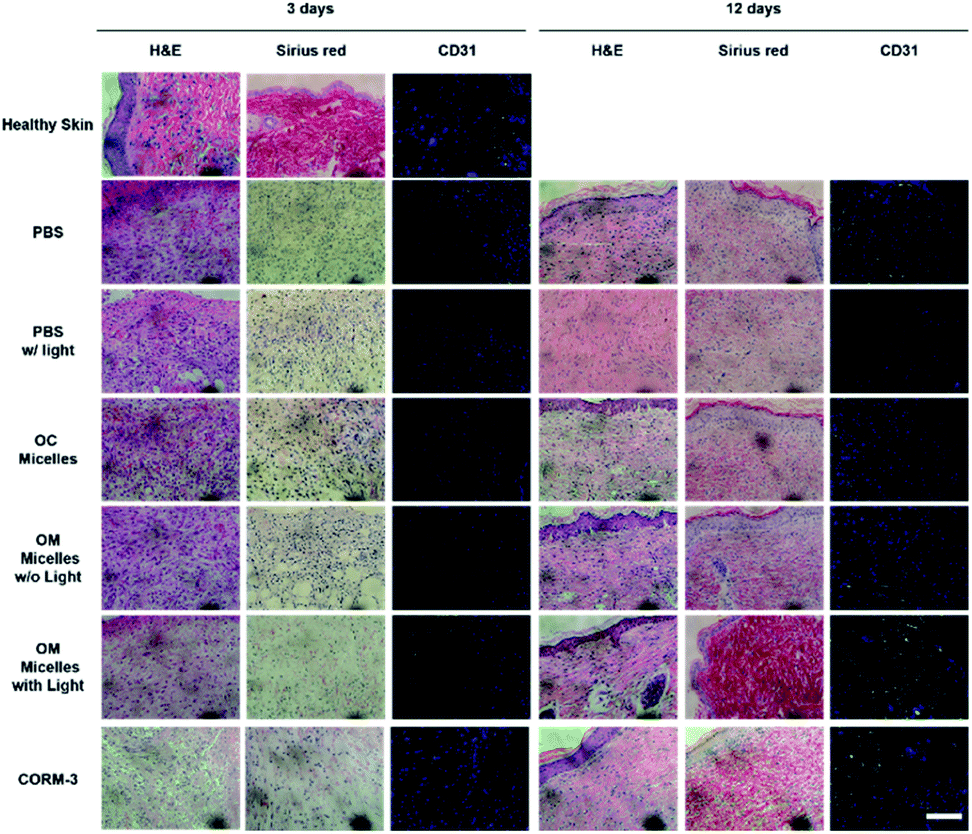 | ||
| Fig. 8 Hematoxylin and eosin (H&E), Sirius red, and immunofluorescent CD31-stained sections of control (healthy skin) and wound tissues on days 3 and 12 treated with PBS buffer, OC micelles, OM micelles (0.1 g L−1) without or with light irradiation (410 nm, 31.5 mW cm−2), and CORM-3, respectively. Scale bars are 100 μm. | ||
Notably, in comparison with CORM-3, OM micelles exerted a comparable effect on anti-inflammation (Fig. 6) but better performance on cutaneous wound healing (Fig. 7 and 8). Moreover, the very limited half-life of CORM-3 in water (∼1 min; pH 7.4, 37 °C)1,63 entailed a fresh stock solution to be prepared just prior to the treatment, and the premature leakage of CO from CORM-3 was difficult to eliminate. However, OM micellar nanoparticles were relatively stable in the dark even after 60 days of incubation in PBS buffer (Fig. S19†), which appeared more convenient for controlled CO delivery and exhibited sustainable CO release under illumination.
Conclusions
In summary, in contrast to conventional CORPs fabricated by the post-modification procedure, we herein demonstrate a new strategy to construct CORPs through a direct polymerization approach by engineering metal-free photoCORMs as CO-releasing monomers. The rational design allowed for the preparation of well-defined CO-releasing block copolymers, which can self-assemble into micellar nanoparticles, exhibiting photo-triggered CO release not only in pure water but also in PBS buffer and cell culture media. More importantly, the photo-mediated CO release process was characterized by a sequential fluorescence transition from blue-to-red-to-colorless, enabling self-reporting CO release both in living cells and animals. Besides, the polymeric CO donor has potential applications in anti-inflammation and accelerating wound healing. This work provides a novel strategy to fabricate CORPs with self-reporting release behavior, which may open an avenue toward CO-based potent therapeutic agents.Ethical statement
Handling and care of all animals were performed in strict accordance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals by the Laboratory Animal Center in the University of Science and Technology of China (USTC) and all procedures in this study were approved by the USTC Animal Care and Use Committee.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Natural Science Foundation of China (NNSFC) project (51690150, 51690154, 51722307, 51673179, and 51773190) and the Fundamental Research Funds for the Central Universities (WK3450000003) is gratefully acknowledged.References
- X. Y. Ji, K. Damera, Y. Q. Zheng, B. C. Yu, L. E. Otterbein and B. H. Wang, J. Pharm. Sci., 2016, 105, 406–416 CrossRef CAS PubMed.
- C. de la Torre, A. Toscani, C. Marin-Hernandez, J. A. Robson, M. C. Terencio, A. J. P. White, M. J. Alcaraz, J. D. E. T. Wilton-Ely, R. Martinez-Manez and F. Sancenon, J. Am. Chem. Soc., 2017, 139, 18484–18487 CrossRef PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- R. Motterlini, Biomed. Pharmacother., 2002, 56, 349–350 Search PubMed.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, E17–E24 CrossRef CAS PubMed.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, E2–E8 CrossRef CAS PubMed.
- K. Ling, F. Men, W. C. Wang, Y. Q. Zhou, H. W. Zhang and D. W. Ye, J. Med. Chem., 2018, 61, 2611–2635 CrossRef CAS PubMed.
- S. Garcia-Gallego and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 53, 9712–9721 CrossRef CAS PubMed.
- A. E. Pierri, P. J. Huang, J. V. Garcia, J. G. Stanfill, M. Chui, G. Wu, N. Zheng and P. C. Ford, Chem. Commun., 2015, 51, 2072–2075 RSC.
- H. Meyer, F. Winkler, P. Kunz, A. M. Schmidt, A. Hamacher, M. U. Kassack and C. Janiak, Inorg. Chem., 2015, 54, 11236–11246 CrossRef CAS PubMed.
- S. H. C. Askes, G. U. Reddy, R. Wyrwa, S. Bonnet and A. Schiller, J. Am. Chem. Soc., 2017, 139, 15292–15295 CrossRef CAS PubMed.
- S. Romanski, B. Kraus, U. Schatzschneider, J. M. Neudorfl, S. Amslinger and H. G. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 2392–2396 CrossRef CAS PubMed.
- I. Kim, E. H. Han, W. Y. Bang, J. Ryu, J. Y. Min, H. C. Nam, W. H. Park, Y. H. Chung and E. Lee, Adv. Funct. Mater., 2018, 28, 1803051 CrossRef.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896–4898 RSC.
- R. Sakla and D. A. Jose, ACS Appl. Mater. Interfaces, 2018, 10, 14214–14220 CrossRef CAS PubMed.
- I. Chakraborty, S. J. Carrington, J. Hauser, S. R. J. Oliver and P. K. Mascharak, Chem. Mater., 2015, 27, 8387–8397 CrossRef CAS.
- S. Diring, A. Carne-Sanchez, J. C. Zhang, S. Ikemura, C. Kim, H. Inaba, S. Kitagawa and S. Furukawa, Chem. Sci., 2017, 8, 2381–2386 RSC.
- I. Chakraborty, S. J. Carrington and P. K. Mascharak, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056–1060 CrossRef CAS PubMed.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romao and M. J. Romao, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- M. Chaves-Ferreira, I. S. Albuquerque, D. Matak-Vinkovic, A. C. Coelho, S. M. Carvalho, L. M. Saraiva, C. C. Romao and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2015, 54, 1172–1175 CrossRef CAS PubMed.
- V. Ramu, G. U. Reddy, J. J. Liu, P. Hoffmann, R. Sollapur, R. Wyrwa, S. Kupfer, C. Spielmann, S. Bonnet, U. Neugebauer and A. Schiller, Chem.–Eur. J., 2019, 25, 8453–8458 CrossRef CAS PubMed.
- I. Kim, W. Y. Bang, W. H. Park, E. H. Han and E. Lee, Nanoscale, 2019, 11, 17327–17333 RSC.
- K. Fujita, Y. Tanaka, T. Sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, S. Kizaka-Kondoh and T. Ueno, J. Am. Chem. Soc., 2014, 136, 16902–16908 CrossRef CAS PubMed.
- U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS PubMed.
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS.
- N. Abeyrathna, K. Washington, C. Bashur and Y. Liao, Org. Biomol. Chem., 2017, 15, 8692–8699 RSC.
- T. I. Ayudhya, C. C. Raymond and N. N. Dingra, Dalton Trans., 2017, 46, 882–889 RSC.
- Y. Q. Zheng, X. Y. Ji, B. C. Yu, K. L. Ji, D. Gallo, E. Csizmadia, M. Y. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. X. Pan, Z. N. Yuan, L. E. Otterbein and B. H. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed.
- X. Y. Ji, K. L. Ji, V. Chittavong, B. C. Yu, Z. X. Pan and B. H. Wang, Chem. Commun., 2017, 53, 8296–8299 RSC.
- X. Y. Ji, C. Zhou, K. L. Ji, R. E. Aghoghovbia, Z. X. Pan, V. Chittavong, B. W. Ke and B. H. Wang, Angew. Chem., Int. Ed., 2016, 55, 15846–15851 CrossRef CAS PubMed.
- S. N. Anderson, J. M. Richards, H. J. Esquer, A. D. Benninghoff, A. M. Arif and L. M. Berreau, ChemistryOpen, 2015, 4, 590–594 CrossRef CAS PubMed.
- T. Soboleva and L. M. Berreau, Isr. J. Chem., 2019, 59, 339–350 CrossRef CAS PubMed.
- M. Popova, T. Soboleva, S. Ayad, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2018, 140, 9721–9729 CrossRef CAS PubMed.
- T. Soboleva, H. J. Esquer, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2017, 139, 9435–9438 CrossRef CAS PubMed.
- T. Soboleva, A. D. Benninghoff and L. M. Berreau, ChemPlusChem, 2017, 82, 1408–1412 CrossRef CAS PubMed.
- Y. Li, Y. Z. Shu, M. W. Liang, X. L. Xie, X. Y. Jiao, X. Wang and B. Tang, Angew. Chem., Int. Ed., 2018, 57, 12415–12419 CrossRef CAS PubMed.
- L. A. P. Antony, T. Slanina, P. Sebej, T. Solomek and P. Klan, Org. Lett., 2013, 15, 4552–4555 CrossRef CAS PubMed.
- E. Palao, T. Slanina, L. Muchova, T. Solomek, L. Vitek and P. Klan, J. Am. Chem. Soc., 2016, 138, 126–133 CrossRef CAS PubMed.
- P. Peng, C. M. Wang, Z. Shi, V. K. Johns, L. Y. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Org. Biomol. Chem., 2013, 11, 6671–6674 RSC.
- D. Nguyen and C. Boyer, ACS Biomater. Sci. Eng., 2015, 1, 895–913 CrossRef CAS.
- U. Hasegawa, A. J. van der Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS PubMed.
- D. Nguyen, T. K. Nguyen, S. A. Rice and C. Boyer, Biomacromolecules, 2015, 16, 2776–2786 CrossRef CAS PubMed.
- A. C. Kautz, P. C. Kunz and C. Janiak, Dalton Trans., 2016, 45, 18045–18063 RSC.
- A. Elgattar, K. S. Washington, S. Talebzadeh, A. Alwagdani, T. Khalil, O. Alghazwat, S. Alshammri, H. Pal, C. Bashur and Y. Liao, Photochem. Photobiol. Sci., 2019, 18, 2666–2672 RSC.
- H. Z. Yin, J. Fang, L. Liao, H. Nakamura and H. Maeda, J. Controlled Release, 2014, 187, 14–21 CrossRef CAS PubMed.
- M. N. Pinto, I. Chakraborty, C. Sandoval and P. K. Mascharak, J. Controlled Release, 2017, 264, 192–202 CrossRef CAS PubMed.
- D. Nguyen, S. Oliver, N. N. M. Adnan, C. Herbert and C. Boyer, RSC Adv., 2016, 6, 92975–92980 RSC.
- L. H. Wu, X. J. Cai, H. F. Zhu, J. H. Li, D. X. Shi, D. F. Su, D. Yue and Z. W. Gu, Adv. Funct. Mater., 2018, 28, 1804324 CrossRef.
- U. R. Gandra, A. Sinopoli, S. Moncho, M. NandaKumar, D. B. Ninkovic, S. D. Zaric, M. Sohail, S. Al-Meer, E. N. Brothers, N. A. Mazloum, M. Al-Hashimi and H. S. Bazzi, ACS Appl. Mater. Interfaces, 2019, 11, 34376–34384 CrossRef CAS PubMed.
- A. C. Sedgwick, L. L. Wu, H. H. Han, S. D. Bull, X. P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef CAS PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- T. Soboleva, C. R. Simons, A. Arcidiacono, A. D. Benninghoff and L. M. Berreau, J. Med. Chem., 2019, 62, 9990–9995 CrossRef CAS PubMed.
- P. Govender, S. Pai, U. Schatzschneider and G. S. Smith, Inorg. Chem., 2013, 52, 5470–5478 CrossRef CAS PubMed.
- A. J. van der Vlies, R. Inubushi, H. Uyama and U. Hasegawa, Bioconjugate Chem., 2016, 27, 1500–1508 CrossRef CAS PubMed.
- N. E. Bruckmann, M. Wahl, G. J. Reiss, M. Kohns, W. Watjen and P. C. Kunz, Eur. J. Inorg. Chem., 2011, 29, 4571–4577 CrossRef.
- S. M. Feng, D. D. Liu, W. Y. Feng and G. Q. Feng, Anal. Chem., 2017, 89, 3754–3760 CrossRef CAS PubMed.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- P. Sawle, R. Foresti, B. E. Mann, T. R. Johnson, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2005, 145, 800–810 CrossRef CAS PubMed.
- L. E. Otterbein, F. H. Bach, J. Alam, M. Soares, H. T. Lu, M. Wysk, R. J. Davis, R. A. Flavell and A. M. K. Choi, Nat. Med., 2000, 6, 422–428 CrossRef CAS PubMed.
- Y. Kang, J. Kim, Y. M. Lee, S. Im, H. Park and W. J. Kim, J. Controlled Release, 2015, 220, 624–630 CrossRef CAS PubMed.
- R. Foresti, J. Hammad, J. E. Clark, T. R. Johnson, B. E. Mann, A. Friebe, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2004, 142, 453–460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00135j |
| This journal is © The Royal Society of Chemistry 2020 |