
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic investigations under whole-cell conditions provide new insight into the metal hydride chemistry of [FeFe]-hydrogenase†
Lívia S.
Mészáros‡
 a,
Pierre
Ceccaldi‡
a,
Marco
Lorenzi
a,
Holly J.
Redman
a,
Emanuel
Pfitzner
b,
Joachim
Heberle
b,
Moritz
Senger
b,
Sven T.
Stripp
*b and
Gustav
Berggren
*a
a,
Pierre
Ceccaldi‡
a,
Marco
Lorenzi
a,
Holly J.
Redman
a,
Emanuel
Pfitzner
b,
Joachim
Heberle
b,
Moritz
Senger
b,
Sven T.
Stripp
*b and
Gustav
Berggren
*a
aMolecular Biomimetics, Dept. of Chemistry – Ångström Laboratory, Uppsala University, Lägerhyddsvägen 1, SE-75120, Uppsala, Sweden. E-mail: gustav.berggren@kemi.uu.se
bInstitute of Experimental Physics, Experimental Molecular Biophysics, Freie Universität Berlin, Arnimallee 14, Berlin, DE-14195, Germany. E-mail: sven.stripp@fu-berlin.de
First published on 14th April 2020
Abstract
Hydrogenases are among the fastest H2 evolving catalysts known to date and have been extensively studied under in vitro conditions. Here, we report the first mechanistic investigation of an [FeFe]-hydrogenase under whole-cell conditions. Functional [FeFe]-hydrogenase from the green alga Chlamydomonas reinhardtii is generated in genetically modified Escherichia coli cells by addition of a synthetic cofactor to the growth medium. The assembly and reactivity of the resulting semi-synthetic enzyme was monitored using whole-cell electron paramagnetic resonance and Fourier-transform Infrared difference spectroscopy as well as scattering scanning near-field optical microscopy. Through a combination of gas treatments, pH titrations, and isotope editing we were able to corroborate the formation of a number of proposed catalytic intermediates in living cells, supporting their physiological relevance. Moreover, a previously incompletely characterized catalytic intermediate is reported herein, attributed to the formation of a protonated metal hydride species.
Introduction
Hydrogenases are gas processing metalloenzymes that interconvert protons (H+) and molecular hydrogen (H2) with remarkable efficiency. The so-called [FeFe]-hydrogenases are considered the most efficient H2 producers in nature, with reported turnover frequencies up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 H2 per second.1,2 This reactivity makes [FeFe]-hydrogenases highly relevant for biotechnological H2 production as an alternative to platinum-based electrolysis3–6 and a biological blue-print for the design of synthetic catalysts.7–9 Consequently, intense efforts have been invested in elucidating the structure and catalytic mechanism of these enzymes.10,11
000 H2 per second.1,2 This reactivity makes [FeFe]-hydrogenases highly relevant for biotechnological H2 production as an alternative to platinum-based electrolysis3–6 and a biological blue-print for the design of synthetic catalysts.7–9 Consequently, intense efforts have been invested in elucidating the structure and catalytic mechanism of these enzymes.10,11
The reactivity of [FeFe]-hydrogenases is enabled by a hexanuclear iron complex, referred to as the hydrogen-forming cluster or simply the “H-cluster” (Fig. 1). This cofactor consists of a canonical iron–sulfur cluster ([4Fe–4S]H) coupled to an organometallic diiron subsite ([2Fe]H). The low-valent iron ions of the [2Fe]H subsite are bridged by an azadithiolate ligand (−SCH2NHCH2S−, adt) and further coordinated by strong-field ligands like carbon monoxide (CO) and cyanide (CN−).12–15 Due to its unique nature, the biosynthesis of the [2Fe]H subsite requires at least three hydrogenase specific maturation enzymes.16–20 Despite challenges in preparing the enzyme, extensive in vitro work has revealed a number of potential catalytic intermediates (Scheme 1). The oxidized resting state of the enzyme (Hox) exhibits a mixed-valence [2Fe]H subsite and an oxidized [4Fe–4S]H cluster ([4Fe–4S]H2+-[Fe(I)Fe(II)]H).21,22 Under acidic conditions HoxH is formed, attributed to a protonation at the [4Fe–4S]H cluster.23,24 Reduction of Hox by one electron results in either Hred′ or Hred (the latter also referred to as “HredH+”).23,25,26 The H-cluster shows a reduced and possibly protonated [4Fe–4S]H cluster in Hred′ whereas in Hred the [2Fe]H site is reduced and protonated.27 Further reduction of Hred generates the “super-reduced” state, Hsred, identified as a [4Fe–4S]H+-[Fe(I)Fe(I)]H species.28
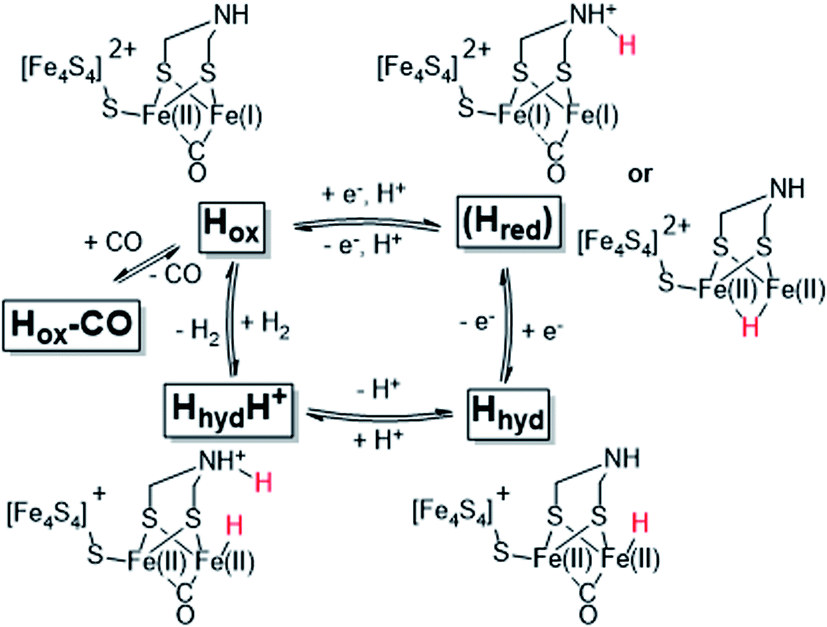 | ||
| Scheme 1 Schematic representation of the catalytic cycle of [FeFe]-hydrogenase, based on the states observed under whole-cell conditions including the protonated hydride species (HhydH+). The catalytic cycle is derived from ref. 10 and 29, and the debated Hred state is shown in parenthesis together with two proposed structures (protonated on the ligand or metal, respectively). | ||
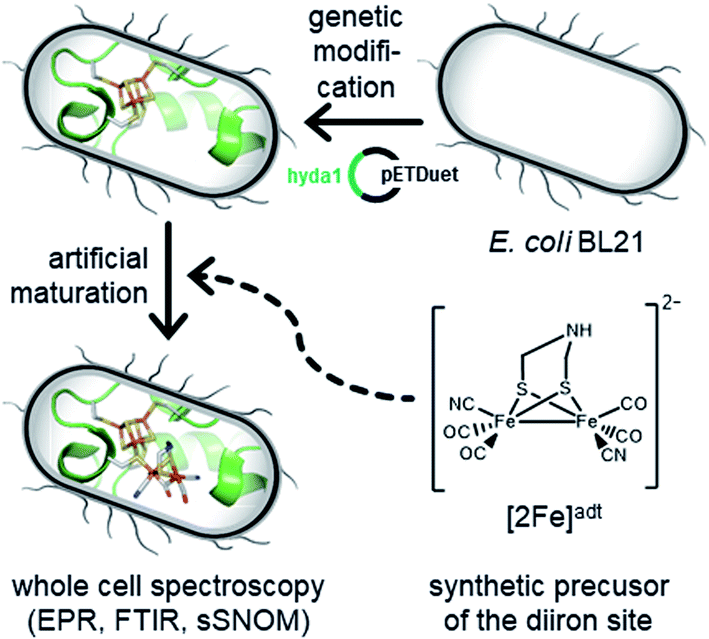 | ||
| Fig. 1 Schematic representation of the whole-cell assembly and spectroscopic investigation of the resulting semi-synthetic H-cluster. Genetic modification of E. coli for expression of [FeFe]-hydrogenase apo-protein, followed by synthetic maturation generates functional hydrogenase (the H-cluster is shown in the Hox state). The reactivity of this semi-synthetic enzyme is probed in whole cells by EPR and ATR FTIR spectroscopy. Proposed key intermediates were observed in cells for the first time, and conditions for accumulating a protonated hydride state (denoted HhydH+) are reported. In parallel, the integrity of the cells is verified by AFM imaging and near-field IR spectroscopy. | ||
The exact nature of these intermediate states and their relevance to the catalytic mechanism is under debate.10,27,30–33 However, all recent models agree that a terminal hydride is formed during catalysis. One such intermediate, denoted Hhyd, has been detected in vitro and shown to accumulate in amino acid variants with disrupted proton transfer networks, but also in the native enzyme under reducing conditions at low pH.29,34–39 The Hhyd state features a terminal hydride on the [2Fe]H subsite (Scheme 1) and the electronic structure of this state comprises a diamagnetic [Fe(II)Fe(II)]H subsite coupled to a paramagnetic [4Fe–4S]H+ cluster.29,35 Protonation of this latter species, presumably on the nitrogen bridgehead of the adt ligand, is expected to form the so-called HhydH+ state, as a potentially final metastable intermediate prior to H–H bond formation and H2 release. However, this latter state is still incompletely characterized and its electronic and geometric structure remains to be clarified.29,40
The biological maturation machinery required for H-cluster assembly can be circumvented by incubating [FeFe]-hydrogenase apo-protein with synthetic mimics of the [2Fe]H subsite. Under in vitro conditions it is well established that the incorporation of the [Fe2(adt)(CO)4(CN)2]2− complex ([2Fe]adt) results in the spontaneous assembly of semi-synthetic hydrogenases indistinguishable from the native enzyme ([2Fe]adt-HydA1).7,13,41–43 We recently adopted this strategy for in vivo applications, enabling the preparation of fully functional, semi-synthetic enzymes in both E. coli and cyanobacteria.6,44–46 Herein, we take advantage of this protocol to perform spectroscopic investigations of [FeFe]-hydrogenase in whole cells (Fig. 1). This approach provides new mechanistic insight as it allows us to probe the formation of proposed key intermediates under relatively mild conditions more similar to the native environment of the enzyme.
We combine whole-cell electron paramagnetic resonance (EPR) and Fourier-transform infrared (FTIR) spectroscopy to characterize the enzymatic activity and catalytic mechanism of an [FeFe]-hydrogenase from the photosynthetic green alga Chlamydomonas reinhardtii, HydA1.47 In parallel, individual cells were characterized by atomic force microscopy (AFM), scattering-type scanning near-field optical microscopy (sSNOM) and nano-FTIR spectroscopy, verifying cellular integrity and protein content. Through a combination of gas flushes and pH changes we detected a number of proposed catalytic intermediates, including Hox, HoxH, and Hred, supporting their catalytic relevance. The present study also provides the first demonstration of the formation of reactive metal hydride species in living cells, as the Hhyd state accumulates in whole cells under reducing conditions mimicking the native environment of the enzyme, i.e. the weakly alkaline stroma of the chloroplast in C. reinhardtii. Moreover, the formation of a protonated hydride state is observed under mildly acidic and strongly reducing conditions.
Results and discussion
In vivo assembly of the semi-synthetic H-cluster
To generate active hydrogenase in concentrations suitable for whole-cell spectroscopy, the HydA1 protein was heterologously expressed in BL21(DE3) E. coli cells, similar to previous reports.44–46 The absence of the [FeFe]-hydrogenase specific maturation machinery in E. coli results in the synthesis of an inactive form of the enzyme containing the [4Fe–4S]H cluster but lacking the [2Fe]H subsite (apo-HydA1). 50 mL cell cultures were concentrated to 2 mL and depleted of O2 at which point the synthetic cofactor mimic [2Fe]adt was added to the medium, and the H-cluster assembly monitored by EPR spectroscopy and H2 production assays. The apo-enzyme remained undetected in our experiments, due to the intensity of underlying signals attributed to other iron–sulfur proteins in E. coli (Fig. 2A and S1†). Conversely, the formation of the H-cluster was readily observable by whole-cell EPR spectroscopy (Fig. 2A). Incubation of the cell suspensions for 1 h in the presence of low concentrations (0.008–0.8 μM) of the synthetic [2Fe]adt cofactor resulted in the appearance of a rhombic EPR signal attributable to Hox (g = 2.100; 2.040; 1.998).48 This rhombic signal was clearly discernable in spectra, but of small intensity relative to the background signal from the cells (Fig. 2A compare the black spectrum, “control”, to blue and green, 0.08 μM–0.8 μM, spectra, see also Fig. S1† for non-background subtracted spectra and a comparison between background and H-cluster derived signals). For higher concentrations of [2Fe]adt in the cell medium (8–80 μM), the overall intensity of the EPR signal increased. Quantification of the signals versus a copper standard showed that a final total spin concentration of up to 10 μM was obtained following this protocol (Fig. 2B). However, the spectra became dominated by an axial signal originating from the CO inhibited state Hox–CO (g = 2.054; 2.007).48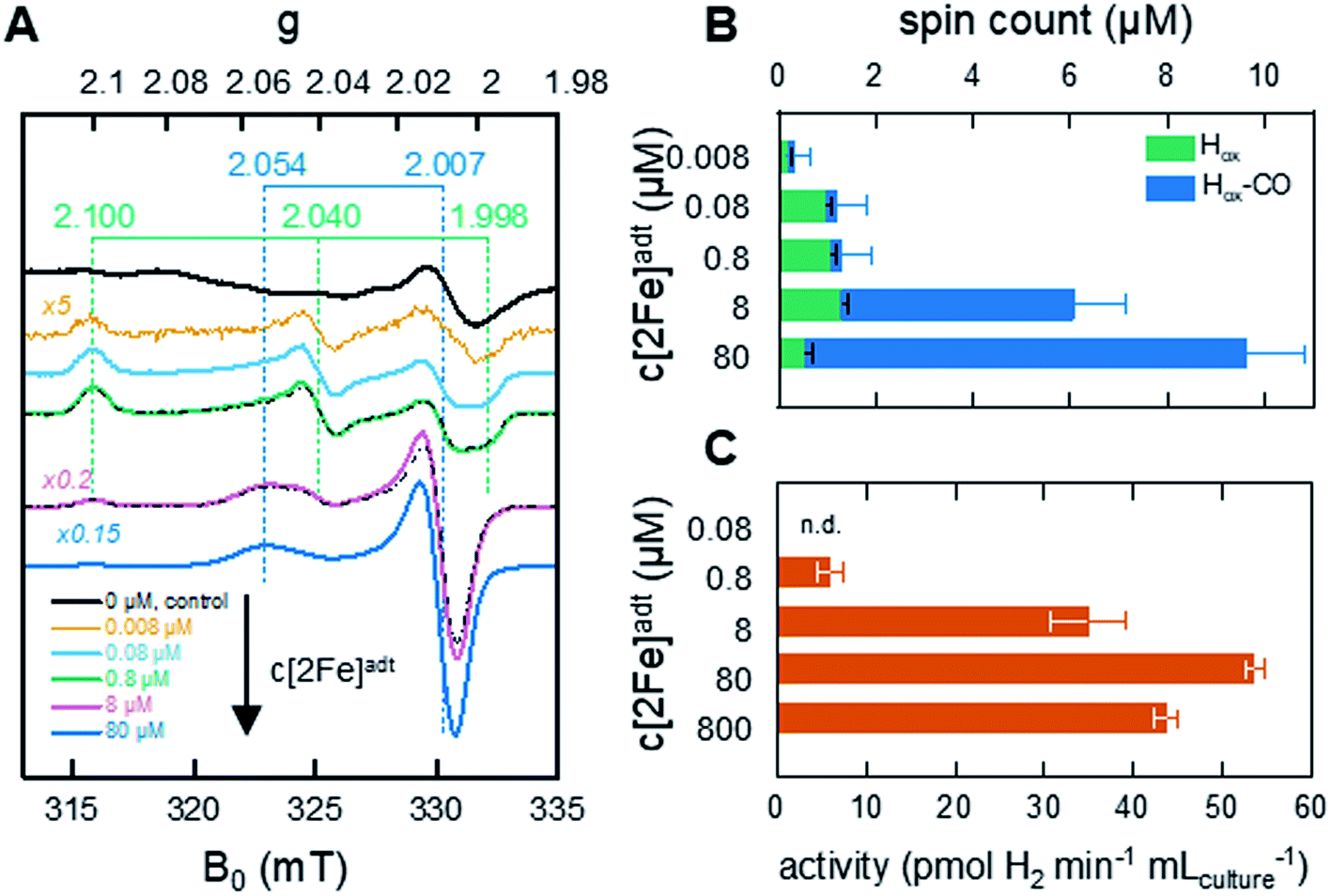 | ||
| Fig. 2 In vivo H-cluster assembly monitored by EPR spectroscopy and H2 gas production. (A) Whole-cell EPR spectra of apo-HydA1 (control, 0 μM [2Fe]adt added) and [2Fe]adt-HydA1 containing cell suspensions, concentrated to 400 μL. H-cluster assembly occurs spontaneously upon addition of [2Fe]adt to the medium, with Hox being the favored state at low [2Fe]adt concentrations. As the concentration of the synthetic cofactor increases, the cell samples become dominated by the Hox–CO state. The g-values of Hox (green dashed lines) and Hox–CO (blue dashed lines) states are based on simulations of the two samples at 0.8 and 8 μM (black dash-dotted lines). All [2Fe]adt-HydA1 spectra were corrected for contribution from the cells by subtracting the signal of the apo-HydA1 control sample (black spectrum). EPR experimental conditions: T = 20 K, P = 1 mW, ν = 9.28 GHz. (B) Concentrations of the Hox (green bars) and Hox–CO (blue bars) signals in each sample, extrapolated from the simulations. The full length of the bar reflects total spin count. Error bars indicate estimated relative error from simulations, in addition an absolute error of approx. 0.5 μM is expected from the background subtraction (see Fig. S1† for details). (C) Average rate of hydrogen gas production during 1 h from anaerobic 2 mL E. coli cultures, concentrated from 100 mL batch cultures. | ||
The successful assembly of functional enzyme was further verified by monitoring H2 production from recombinant E. coli cells suspended in fresh M9 minimal medium. Hydrogen gas formation became clearly observable after the addition of 0.8 μM [2Fe]adt to the medium and increased up to 80 μM [2Fe]adt, concomitantly with the overall EPR signal intensity (Fig. 2C). A maximum rate of 50–55 pmol H2 per min per mL cell culture was observed, which corresponds to a specific activity of 10–15 nmol H2 per min per mg assuming that the entire H-cluster population is detectable by EPR spectroscopy. The same titration trend was observed in in vitro assays. Hydrogenase activity assays performed on lysed cells demonstrated that a major fraction (approx. 80%) of the total available apo-HydA1 pool had formed the active [2Fe]adt-HydA1 enzyme after 1 h incubation with 80 μM [2Fe]adt in the cell medium (Fig. S2†). Increasing the concentration of [2Fe]adt beyond this point resulted in a decline of in vivo H2 production. Due to the dominance of Hox–CO at higher [2Fe]adt concentration (Fig. 2B), the decreased H2 productivity is attributed to release of CO from excess cofactor in the cell medium resulting in inhibition of the enzyme. The effect of the hydrogenase activity on the cytoplasmic pH was found to be negligible, with a decrease from pH 7.6 (±0.2) to 7.5 (±0.3) after incubation of the cells with [2Fe]adt for 60 minutes observed in separate assays using pH dependent fluorescent dyes. Consequently, a concentration of 80 μM [2Fe]adt in the cell medium was determined as optimal for the following mechanistic studies.
Investigating the integrity and enzymatic activity of single cells
The integrity and viability of the cells following anaerobic incubation in the presence of 80 μM [2Fe]adt was verified on batch level through a combination of SDS-PAGE, in vitro enzymatic assays, and plating experiments (Fig. S3 and S4†). Moreover, the integrity of individual cells was monitored by atomic force microscopy (AFM) on diluted E. coli samples (factor 5 × 10−5). The topography suggested viable bacteria and only a limited number of cells were found to be morphologically altered (Fig. 3A and S5†). Subsequently, the local IR absorption of single cells was recorded using scattering-type scanning near-field optical microscopy (sSNOM). This label-free technique provides a chemical image of the cell at a spatial resolution of 30 nm.49 To visualize the protein content in a number of representative cells, the amide I band (v1 = 1660 cm−1) was corrected for a topography feature at v2 = 1710 cm−1 (Fig. 3A and Fig. S6† for additional examples). We found that the protein distribution was largely homogenous within the cells and no indication of secreted protein outside the cell was detected. Nano-FTIR near-field phase spectra showed typical amide I and amide II absorption within the cell, while such bands were not observed outside of the cell (Fig. 3B and S7† for additional examples). Moreover, only limited loss of H-cluster signal was observed upon washing E. coli cells in up to 108 time the volume of buffer (Fig. S8†). In combination with the aforementioned SDS-PAGE, in vitro enzymatic assays, and plating experiments (Fig. S3 and S4†), these findings underscore that our spectroscopy studies report on [FeFe]-hydrogenase located inside intact and viable cells.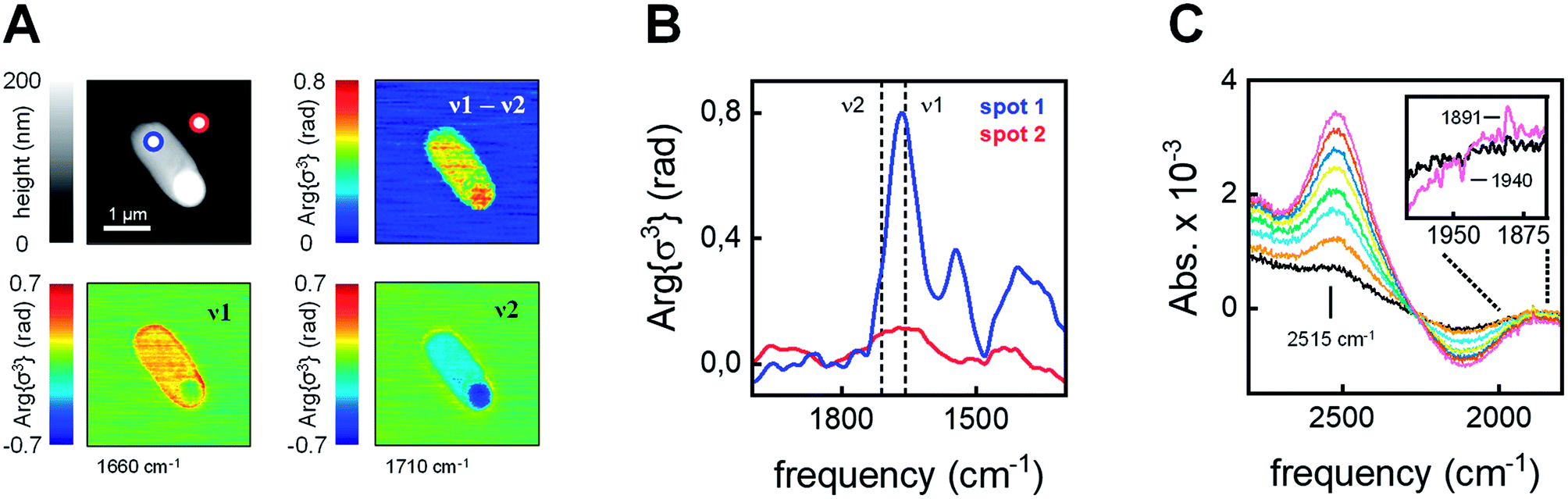 | ||
| Fig. 3 Analysis of individual cells. (A) Upper left: AFM topography of a [2Fe]adt–HydA1-containing E. coli cell. Lower left: sSNOM image mapped at v1 = 1660 cm−1 (amide I absorption). Lower right: sSNOM image mapped at v2 = 1710 cm−1 (topography artifact). Upper right: v1 − v2 sSNOM difference image. The protein is clearly localized to the cellular environment see Fig. S6 and S7† for additional examples. (B) Nano-FTIR near-field phase spectrum from 2000–1300 cm−1. Spectra were recorded on the cell (spot 1, blue) and next to the cell (spot 2, red) in the AFM topography (panel A). (C) ATR FTIR difference spectra showed an HDO band (2515 cm−1) increase in the presence of D2 (exposure time 0–90 s increasing from black to purple). Inset: a simultaneous enrichment of Hred (1891 cm−1) over Hox (1940 cm−1) was observed. | ||
In the next step, we used attenuated total reflection Fourier-transform infrared (ATR FTIR) spectroscopy to probe the catalytic competence of hydrated [2Fe]adt-HydA1-containing E. coli films. Following reported protocols,46E. coli cell suspensions were deposited on the silicon crystal of the ATR optic, dried under inert gas (i.e., N2 or Ar), and rehydrated by running the gas through a wash bottle, forming a bacterial film that can react to changes in atmospheric gas composition. When the [2Fe]adt-HydA1-containing E. coli film was kept under 100 mbar deuterium gas (D2) instead of N2, the appearance of a band at 2515 cm−1 suggests that deuterium ions (D+) were released into bulk H2O (Fig. 3C). This HDO band serves as direct verification of D2 oxidation activity,34 confirming that the [FeFe]-hydrogenase located inside the E. coli cells retained its activity in hydrated films. Additionally, H-cluster band shifts in the CO regime suggest reduction of Hox into Hred (Fig. 3C, inset). Bacterial films containing apo-HydA1 did not show HDO formation under D2 (Fig. S9†).
Mechanistic investigations on cell films using in situ ATR FTIR spectroscopy
The H-cluster-specific CO stretching vibrations in the frequency regime from 2050–1750 cm−1 were clearly resolved by ATR FTIR spectroscopy on [2Fe]adt-HydA1-containing E. coli films. Under N2, our data indicated predominantly oxidized enzyme with only minor contaminations of CO-inhibited cofactor (Fig. S9†). No trace of the free [2Fe]adt cofactor was detected. Exposing the film to 99% N2 and 1% H2 resulted in an enrichment of Hred over Hox and Hox–CO on a time-scale of seconds (Fig. 4A, spectrum a) whereas increasing the H2 content to 100% had no further effects on the composition of redox states (Fig. S9†). Notably, neither Hred′ nor Hsred were detected in [2Fe]adt-HydA1-containing E. coli films (main CO marker bands at 1933 cm−1 and 1882 cm−1, respectively). This is in contrast to purified [2Fe]adt-HydA1 exposed to 100% H2 and hints at specific differences (Fig. S10†). When the H2 atmosphere was changed to N2 again, the H-cluster converted quickly back into Hox (Fig. 4A, spectrum b). The fact that Hox–CO does not re-appear suggests a high degree of cofactor stability on the experimental time-scale (∼10 min). When exposed to 1% CO, the oxidized H-cluster converted to Hox–CO immediately (Fig. 4A, spectrum c).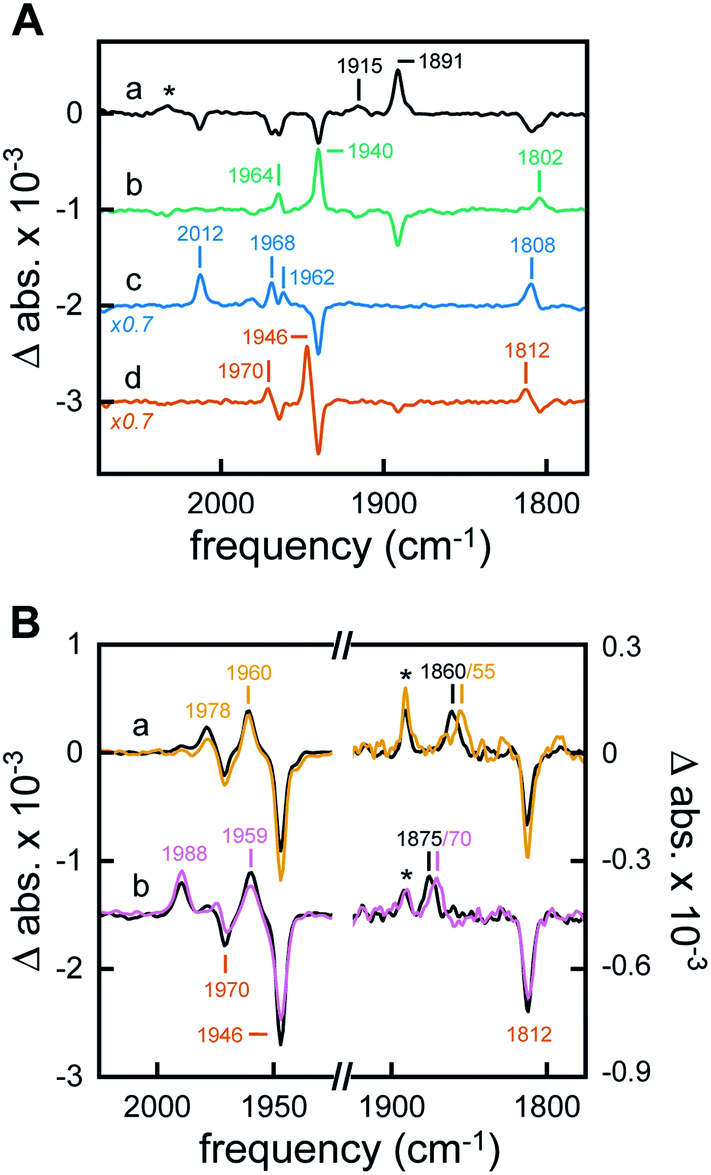 | ||
| Fig. 4 ATR FTIR analysis of [2Fe]adt-HydA1 containing E. coli cells. All difference spectra show the CO regime of the H-cluster. (A) In the presence of 1% H2 (pH 8, 2 mM NaDT) freshly prepared cells converted from Hox and Hox–CO to Hred (spectrum a). * The band at 2032 cm−1 was attributed to a CN− band of Hred. In the absence of H2 quantitative enrichment of Hox was observed (spectrum b). Exposure to 1% CO resulted in population of Hox–CO (spectrum c). Under non-reducing, acidic conditions (N2, pH 4, 2 mM NaDT) Hox converted into HoxH (spectrum d). (B) In the presence of 1% H2 (pH 4, 2 mM NaDT) formation of Hhyd over HoxH was observed (spectrum a, black line). Reduction with D2 gas reproduced the hydride-specific downshift of the μCO band from 1860 to 1855 cm−1 (spectrum a, dark yellow line). Increasing the NaDT in the aerosol to 100 mM (pH 4, 1% H2) facilitated accumulation of a new state, HhydH+ (spectrum b, black line). The downshift of the μCO band under D2 (1875 to 1870 cm−1) verified the presence of a hydride ligand (spectrum b, purple line). * Small fraction of Hred at low pH. | ||
Acidification of the [2Fe]adt-HydA1-containing E. coli film via the aerosol (mixed buffer set to pH 4) and addition of 2 mM sodium dithionite (NaDT) caused a complete conversion of Hox into HoxH within minutes (Fig. 4A, spectrum d and Fig. S10†).23 The effect of this treatment on the cytoplasmic pH of [2Fe]adt-HydA1 containing cells was determined in separate assays utilizing a commercial pH dependent fluorescent probe, as in the case of the H2 producing cell suspensions. Incubation in pH 4 media resulted in a decrease from pH 7.5 (±0.3) to 6.1 (±0.1), however, the presence of up to 100 mM NaDT had negligible effect on the cytoplasmic pH both in pH 7.5 and 4 media. In the presence of H2 or D2, a quantitative enrichment of Hhyd over HoxH was observed (Fig. 4B, both spectra a). Increasing the NaDT concentration in the buffer to 100 mM enabled detection of Hhyd at pH 8 (Fig. S12†), suggesting that strongly reducing conditions allow the accumulation of this reactive intermediate already at weakly alkaline pH. Similar trends have been observed for the in vitro enrichment of HoxH and Hred′H.23,26 Combining high NaDT concentrations and acidic pH resulted in the formation of a new species, shifted to higher frequencies by up to 15 cm−1 as compared to the previously reported Hhyd state (Fig. 4B, both spectra b). Reference experiments on purified [2Fe]adt-HydA1 in the presence of pH 4, 100 mM NaDT, and 1% H2 confirmed the possibility of generating this species in vitro. Additionally, it facilitated a precise assignment of the CO/CN− band positions (Fig. S12†). To verify that this new signal reflected a hydride-binding form of the H-cluster, the same species was generated using D2, which resulted in the expected downshift of the bridging carbonyl band40 from 1875 to 1870 cm−1 (Fig. 4B, purple spectrum b).
Making use of in situ ATR FTIR spectroscopy on [2Fe]adt-HydA1-containing E. coli cells, we were able to observe several H-cluster states previously identified on purified hydrogenase. It is worth noting that Hsred and Hred′ were never observed under whole-cell conditions. The accumulation of Hred′ depends on intermolecular electron transfer and the concentration of enzyme,23 which explains the lack of signal in Fig. 4A. Fig. S11† demonstrates that no such correlation was observed for Hsred, providing indirect support for the notion that our data report exclusively on [2Fe]adt-HydA1 within E. coli cells. Conversely, Hhyd and the new hydride-like state readily accumulated under acidic conditions and could be selectively enriched as a function of NaDT concentration. A similar hydride-like state was reported earlier34,40 but has not been characterized in detail. Compared to Hhyd, the observed spectral up-shifts are larger and less consistent than those previously attributed to protonation changes at the [4Fe–4S]H cluster, e.g. in HoxH (Fig. S12†).23,26 DFT calculations carried out by Mulder et al. have attributed shifts of similar magnitude to protonation changes at the [2Fe]H subsite.40 Thus, we assign this species to a new hydride state featuring a nitrogen-protonated azadithiolate ligand, HhydH+.
Two different hydride-like states observed by EPR as a function of pH
The formation of Hhyd, as well as an alternative hydride-like state formed at low pH, was corroborated by EPR spectroscopy. A rhombic signal (gzyx = 2.078, 1.935, 1.878) in good agreement with the in vitro identified Hhyd state was observed for weakly alkaline [2Fe]adt-HydA1 containing E. coli suspensions, rapidly frozen after incubation with NaDT and H2 (Fig. 5, spectrum a and Fig. S13, Table S3†).35,39,40 The gy and gx positions of this Hhyd signal were readily apparent in spectra recorded at both 10 and 20 K (Fig. S14,† orange spectra), while the broad nature of the feature observed at g ≈ 2.08–2.07 complicated an exact assignment of the gz position. Spectra recorded at higher microwave power (P ≥ 8 mW) simplified the low-field region and were consequently used for the gz assignment (Fig. S15†). Small contributions from Hox were still visible in the spectrum, in combination with an additional minor rhombic signal (Fig. 5 and S13†). Conversely, including an Hox–CO component in the simulation did not improve the overall fit. Thus, the central g ≈ 2.0 feature is most likely attributable to EPR active species native to E. coli. Finally, it is noteworthy that the Hhyd signal was readily observable in cells incubated under an H2 atmosphere even in the absence of NaDT, supporting its physiological relevance.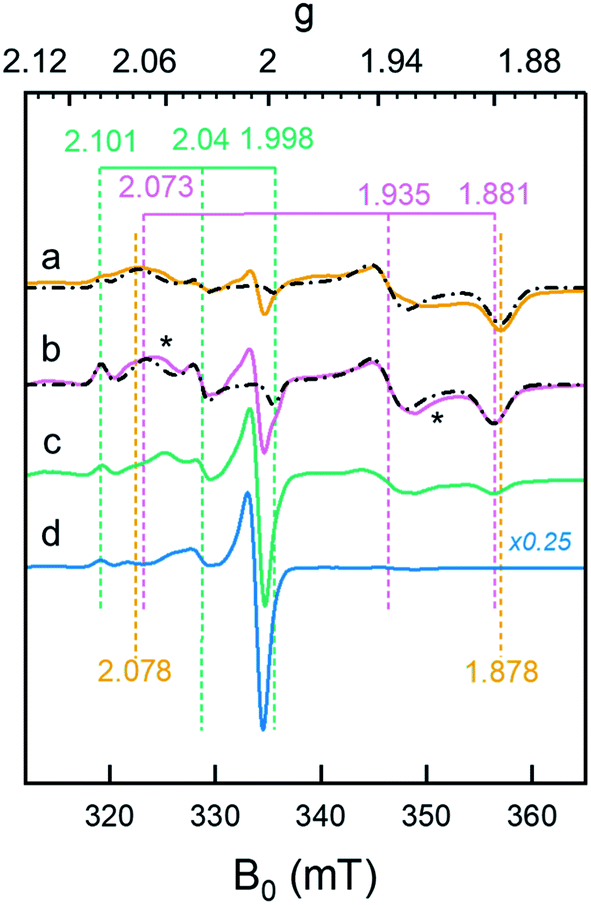 | ||
| Fig. 5 Whole-cell generation of the Hhyd and HhydH+ states under different conditions probed by EPR spectroscopy. (a) The Hhyd state was observable in EPR spectra recorded on [2Fe]adt-HydA1 containing cells at pH 7.5, complemented with 10 mM NaDT, flushed with H2 for 15 min and rapidly frozen (dark yellow spectrum); (b): A new signal attributed to HhydH+ was observed in samples prepared as lane a but at pH 4 (purple spectrum); (c) samples prepared as lane b but excluding NaDT generated a weaker HhydH+ signal (green spectrum); (d) samples prepared as lane b but excluding H2 flushing did not generate any discernable HhydH+ signal (blue spectrum). The g-values of Hox/HoxH (green dashed vertical lines), Hhyd (dark yellow dashed vertical lines) and HhydH+ (purple dashed vertical lines) states are based on the simulations of the spectra (black dash-dotted lines), for details see Fig. S13.† Unassigned weak signals potentially arising from an Htrans-like state indicated with asterisks.40 EPR experimental conditions: T = 20 K, P = 1 mW, ν = 9.38 GHz. | ||
A new rhombic Hhyd-like signal was observed when the analogous experiment was performed in media acidified to pH 4 (Fig. 5, spectrum b and S13–S15†). As the signal accumulates under conditions similar to those of HhydH+ in ATR FTIR, we assign this signal to the same protonated hydride state. Similar to the Hhyd signal, the low pH rhombic signal saturates by only 30% at 100 mW (T = 20 K), illustrating fast spin relaxation of the center (Fig. S15†). An exact assignment of the gz position was challenging also for HhydH+, due to the complex nature of the gz region at low microwave powers (Fig. S13–S15†). Still, in comparison to Hhyd the HhydH+ spectrum featured a clearly discernible decreased anisotropy (gzyx = 2.073, 1.935, 1.881, vertical purple dashed lines in Fig. 5), and the gz component became more distinct at higher microwave power (P ≥ 8mW, see Fig. S15†). We assign the observed shift of the Hoxgz position from 2.100 to 2.101 at low pH to the formation of HoxH. Attempts at simulating the high pH Hhyd spectrum as a combination of HhydH+ and Hsred did not yield reasonable results, further supporting that these three rhombic EPR signals arise from different states (Table S3†). Based on the similarity of the rhombic HhydH+ signal to that observed for the Hhyd state in particular,40 but also the Htrans state,21 it is attributed to a species featuring a reduced [4Fe–4S]H cluster coupled to a diamagnetic [Fe(II)Fe(II)]H subsite, closely resembling the Hhyd state.
Exposing apo-HydA1 containing cells to H2 in the presence or absence of NaDT did not result in any spectral features similar to HhydH+, neither did BL21(DE3) E. coli cells lacking the hyda1 gene. The transient nature of the HhydH+ state in cell suspensions was verified by exchanging the H2 atmosphere with Ar for 30 minutes, which resulted in a loss of HhydH+ and conversion back to a mixture of oxidized states (Hox and Hox–CO). Interestingly, the presence of NaDT in the cell medium during H2 treatment was not strictly required for the generation of the HhydH+ state (Fig. 5, compare spectra a and c). Increasing the NaDT concentration in the cell suspension up to 100 mM, i.e. conditions shown to favor accumulation of the HhydH+ state under ATR FTIR conditions (Fig. 4B), resulted in a more intense HhydH+ signal but no distinct new spectral features were observed. Conversely, no HhydH+ signal was discernible in the absence of H2, irrespective of NaDT concentration (Fig. 5, spectrum d).
Conclusions
The artificial maturation of apo-HydA1 in E. coli provides access to high concentrations of [FeFe]-hydrogenase in vivo. Furthermore, cellular H2 oxidation and production verify that the resulting semi-synthetic HydA1 enzyme is functional and connects to the cell metabolism, most likely via ferredoxins.6 This has facilitated the first mechanistic investigation of an [FeFe]-hydrogenase under whole-cell conditions, employing EPR and FTIR spectroscopy as complementary techniques. Despite differences in sample preparation and experimental conditions (i.e. cell suspension vs. cell film, cryogenic vs. ambient temperatures) both techniques reveal the same trends.The dominant oxidation states under weakly alkaline conditions are Hox and Hox–CO. In addition, FTIR spectroscopy on hydrated films showed that Hred was enriched in the presence of H2. This transition was rapid and quantitative but the enzyme converted back to Hox when H2 was removed from the gas phase. Similar reactivity (“auto-oxidation”) is generally observed for HydA1 in vitro and is clearly retained in vivo. It is noteworthy that diverging reactivity was recently reported for two putative sensory [FeFe]-hydrogenases of different origin, which appear to stabilize in an Hred-like state both in vitro and in whole cells.46,50 This arguably reflects the different physiological roles of sensory hydrogenases and the prototypical HydA1 enzyme.51
In addition to the relatively stable catalytic intermediates Hox and Hred, the study revealed complex metal-hydride chemistry occurring in living cells and two different hydride states were observed. The EPR spectra recorded on cell suspensions incubated under a neat H2 atmosphere in weakly alkaline buffer revealed the formation of the Hhyd state. This finding verifies that this proposed catalytic intermediate is generated under physiologically relevant conditions, i.e. in the absence of the artificial reductant dithionite and in weakly alkaline cell medium where the cytoplasmic pH is ≈7.5. The whole-cell formation of Hhyd was also confirmed by FTIR spectroscopy. Acidification of the cell medium resulted in decrease of cytoplasmic pH and rapid formation of HoxH. Exposing the acidified cells to reducing conditions resulted in accumulation of Hhyd, previously only observed in vitro. Moreover, the complementary nature of EPR and FTIR spectroscopy has allowed us to detect and characterize an additional hydride-like state, denoted HhydH+. The existence of alternative hydride-like states have been suggested earlier from work on HydA1, often in amino acid variants with impaired proton transfer capacity, but has so far been challenging to accumulate and consequently not characterized in detail.32–35,37,40 Herein, we show how such an alternative hydride-like state, HhydH+, can be selectively generated also in the fully functional enzyme, not only in vitro but also under whole-cell conditions. The assignment of HhydH+ as a hydride state is confirmed by the observed H/D isotopic shift of the μ-CO ligand. The pH dependence and spectral up-shift relative to Hhyd, as well as earlier DFT calculations, supports that this hydride state features a protonated nitrogen bridgehead atom.40 Additionally, the relatively small change observed in the corresponding EPR spectra supports a model in which the electronic configuration of Hhyd remains unchanged (i.e. [4Fe–4S]H+-[Fe(II)Fe(II)]H) upon protonation, as the latter technique reports primarily on the paramagnetic [4Fe–4S]H+ cluster. The relatively facile accumulation of this species is somewhat surprising, as it should readily proceed to produce H2. However, it is likely a result of the relatively flat energy landscape of H+/H2 interconversion necessary for the remarkable catalytic efficiency of the enzyme, which has already enabled the stabilization of a wide range of potential catalytic intermediates through gas, pH and redox potential manipulations. The possibility to accumulate the HhydH+ state also in vitro should enable in-depth studies, providing more detailed insight into its structure. Moreover, having elucidated its FTIR fingerprint will allow probing for it in transient spectroscopy to verify its catalytic competence.
In conclusion, the expansion of artificial maturation to in vivo conditions in combination with our capacity to manipulate and spectroscopically investigate [FeFe]-hydrogenases in whole cells has allowed us to support the physiological relevance of a number of proposed catalytic intermediates. In the context of mechanistic understanding, HhydH+ potentially represent the last isolatable intermediate in the transition from Hhyd to Hox with concomitant H2 formation. Deciphering and mapping out the reactivity of this hydride species represents an exciting future prospect, expected to provide critical insight into H2 formation and cleavage.
Experimental
General
All chemicals were purchased from Sigma-Aldrich or VWR and used as received unless otherwise stated. All anaerobic work was performed in an MBRAUN glovebox ([O2] < 10 ppm). The expression vector encoding the hydA1 gene (pETDuet-CrHydA1-His) was kindly provided by Prof. Marc Fontecave (Collège de France, Paris/CEA, Grenoble). (Et4N)2[Fe2(adt)(CO)4(CN)2] ([2Fe]adt]) was synthesized in accordance to literature protocols with minor modifications, and verified by FTIR spectroscopy.52–55 The complex was dissolved in anaerobic potassium phosphate buffer (100 mM, pH 6.8) at 10 ng μL−1 to 10 μg μL−1 concentration and used directly. Protein content was analyzed by 10% SDS-PAGE minigels in a BioRad Mini-PROTEAN Tetra Cell system. The proteins were stained with Page Blue protein staining solution (Thermo Fisher Scientific) according to the supplier's instructions.Overexpression of the apo-HydA1 hydrogenase
Escherichia coli BL21(DE3) cells containing the HydA1 plasmid were grown in 50 mL M9 medium [22 mM Na2HPO4, 22 mM KH2PO4, 85 mM NaCl, 18 mM NH4Cl, 0.2 mM MgSO4, 0.1 mM CaCl2, 0.4% (v/v) glucose] under aerobic conditions until O.D.600 = 0.6–0.8 in the presence of ampicillin. The protein overproduction was induced with 1 mM IPTG and persisted at 20 °C for 16–18 h with continuous aeration. The media was supplemented with 100 μM FeSO4 at the time of the induction. Final O.D.600 of the cultures were 1.4 ± 0.2.In vivo formation of [2Fe]adt-HydA1
The preparation of the semi-synthetic hydrogenase was performed following a literature protocol with minor modifications.44,45 The apo-HydA1 protein was expressed in 50 mL E. coli cultures as described in the “Overexpression of the apo-HydA1 hydrogenase” section. After the 16–18 h expression period the cells were harvested, deaerated and transferred to the glove-box. The cells were re-suspended in fresh M9 medium (2 mL final volume), and formation of [2Fe]adt–HydA1 was achieved by treating the cell suspensions with 100 μg (156 nmol) [2Fe]adt complex (80 mM final conc.), unless otherwise stated, for 1 h at 37 °C under strictly anaerobic conditions.Whole-cell EPR and FTIR sample preparation
The 2 mL concentrated cell suspensions generated via the “In vivo formation of [2Fe]adt-HydA1” protocol were centrifuged and the cell pellet washed with 1 mL Tris–HCl buffer (100 mM Tris, 150 mM NaCl pH 7.5) three times under anaerobic conditions. For EPR samples the cells were diluted to a final volume of 400 μL with Tris–HCl buffer after the washing protocol and transferred into EPR tubes. The tubes were capped and directly frozen in liquid nitrogen. In case of FTIR samples four separate 2 mL sample preparations were combined, concentrated and diluted to 400 μL with Tris–HCl buffer and frozen in liquid nitrogen under anaerobic conditions.Generation of the Hhyd EPR samples
The apo-HydA1 containing cells were activated as described above. After activation the cells were washed with Tris–HCl buffer (100mM Tris, 150mM NaCl) and re-suspended 400 μL buffer under anaerobic conditions. When indicated, the Tris–HCl buffer was complemented with 10 or 100 mM Na-dithionite. The H2 treated samples were flushed with H2 gas for 15 min under anaerobic conditions. After flushing the cells were quickly transferred into EPR tubes. The tubes were capped and flash frozen in liquid nitrogen. The pH of the Tris–HCl buffer was either pH 7.5 or pH 4 according to the experiment, and verified before and after each flushing experiment. Plating the cells on ampicillin enriched LB-agar plates as well as SDS-PAGE gel analysis and in vitro enzymatic assays were used to ensure the viability and integrity of the cells also under low pH conditions (Fig. S3†).Hydrogenase activity measurements
In vivo and in vitro activity measurements were performed according to published protocols.44 Hydrogen production was determined by analyzing the head-space gas, using a gas chromatograph (GC; PerkinElmer LLC, MA, USA) equipped with a thermal conductivity detector (TCD) and a stainless-steel column packed with Molecular Sieve (60/80 mesh). A calibration curve was established by injecting known amounts of hydrogen. The operational temperatures of the injection port, the oven and the detector were 100 °C, 80 °C and 100 °C, respectively. Argon was used as the carrier gas at a flow rate of 35 mL min−1.EPR measurements
The EPR spectra shown are representative signals from at least two individual experiments. The individual experiments show some preparation dependent differences, but the amplitude of these background signals are negligible compared to the signal intensity of the [2Fe]adt activated HydA1. Measurements were performed on a Bruker ELEXYS E500 spectrometer using an ER049X SuperX microwave bridge in a Bruker SHQ0601 cavity (Fig. 2) or a Bruker EMX micro equipped with an EMX Premium bridge and an ER4119 HS resonator (Fig. 5 and S13–S15†), both equipped with an Oxford Instruments continuous flow cryostat and using an ITC 503 temperature controller (Oxford Instruments). Measurement temperatures ranged from 10 to 20 K, using liquid helium as coolant, with the following EPR settings unless otherwise stated: microwave power 1 mW, modulation amplitude 1 mT, modulation frequency 100 kHz. The spectrometer was controlled by the Xepr software package (Bruker).EPR spectra processing, simulations and spin quantification
The EPR spectra were processed using the softwares Matlab (Mathworks, Inc) and QSoas.56 Matlab served for converting the EPR files to ascii format, while QSoas was used to display the spectra as a function of g values, for visual inspection and subtraction of background signals emerging from the cells. The processed signals were used for Fig. 2, 5 and S13.† The simulations were performed using the easyspin toolbox (5.2.23) within Matlab.57 The positions and line-width of each signature were manually adjusted to fit the experimental features, and kept the same across all datasets. Other details of the procedure can be found in ref. 45.Infrared measurements
For whole-cell ATR FTIR spectroscopy, 1 μL E. coli suspension was deposited on the silicon crystal of an ATR cell in the beam path of a commercial FTIR spectrometer (Bruker). All experiments were performed at ambient temperature (∼24 °C) and pressure (∼1 atm), in the dark, and on hydrated films. The cell suspension was dried under 100% N2 gas and re-hydrated with buffer solution (100 mM Tris, MES, and PIPPS) in the humidified gas stream (aerosol), similar to what was reported for purified protein earlier.23 The utilized buffer mix allowed titrating the cell film between pH 9 and pH 4. Reduction of [FeFe]-hydrogenase in the cells was induced by adding 1–100% H2 to the N2 gas stream (flow volume 1.5 L min−1). In the absence of H2 (100% N2), the oxidized resting state (Hox or HoxH) recovered due to auto-oxidation. Plating experiments were performed to verify cell viability after the FTIR experiments (Fig. S4†) Transitions were followed with a spectral precision of 2 cm−1 and 1000 averages of interferometer scans per spectrum. Difference spectra were calculated by subtraction of single absorbance spectra recorded under varying conditions (e.g., H2 – N2, pH 4 – pH 8, etc.)Single cell analysis
For single-cell AFM, sSNOM and nano-FTIR cell suspensions were diluted by a factor of 5 × 105 (1:20000) and a 1 μL droplet was dried on template stripped gold substrate. Topographies, near-field maps and spectra were acquired under ambient conditions. For other technical details, the setup is described in the ESI Section, Fig. S16.†
Cytoplasmic pH quantification
The intracellular pH of the pH 4 and pH 7.5 incubated cells were quantified with pH Rhodo™ Green AM Intracellular pH Indicator in combination with the Intracellular pH Calibration Buffer Kit (Thermo Fisher Scientific). This method determines intracellular pH using the pHrodo™ Red fluorescent dye and is applicable to a range of cell types.58,59 Modification of pHrodo™ Red and Green dyes with acetomyxethyl (AM) ester groups results in an uncharged molecule that can permeate cell membranes. Once inside the cell, the lipophilic blocking groups are cleaved by nonspecific esterases, resulting in a compound that is retained within the intracellular space.More specifically, 50 mL cultures of E.coli cells containing the overproduced apo-HydA1 protein were concentrated down to 2 mL and parallel vials were either treated with 80 μM [2Fe]adt or left untreated for 1 h, before the cell suspensions were centrifuged and washed three times in 1 mL pH 7.5 or pH 4 Tris–HCl media respectively. After washing, the cells were further incubated in 1 mL pH 4 or pH 7.5 Tris -HCl media with or without 100 mM Na-dithionite for 30 minutes under anaerobic conditions. After removal of the incubation buffer through centrifugation, the cells were re-suspended in 500 uL Live Cell Imaging Solution (Thermo Scientific) containing 10 μL Rodo Green dye. 100 μL of the resulting solutions were pipetted into a 96-well microplate. Every experimental condition was measured in four parallels. The fluorescent signals were detected using a Hidex Chameleon plate reader with 495 nm excitation and 590 nm emission filters. The intercellular pH in the different samples were calculated using the calibration curve obtained using the Intracellular pH Calibration Buffer Kit (Thermo Sceintific). For the latter, 50 mL cultures of E.coli cells containing the overproduced apo-HydA1 were centrifuged and washed three times in 1 mL pH 7.5 Tris–HCl buffer. After washing, the cells were suspended in 500 μL Intracellular pH Calibration Buffer, component A, B, C or D (Thermo Scientific, covering a pH range of 4.5–7.5) containing 10 μM valinomicin/10 μM nigericin (Thermo Scientific).
Conflicts of interest
There are no conflicting interests to declare.Acknowledgements
We thank Dr. Basem Soboh for support with the plating experiments and Dr. Ping Huang for discussions concerning EPR spectra analysis. The European Research Council (GB, StG contract no. 714102) and the Olle Engkvist Byggmästare foundation (GB and LM) are gratefully acknowledged for funding. The Deutsche Forschungsgemeinschaft (DFG) is acknowledged for financial support to STS (STR 1554/5-1) and JH (HE 2063/5-1). Part of the project is funded by the DFG under Germany's Excellence Strategy – EXC 2008/1-390540038 (UniSysCat) to JH. The European Union’s Horizon 2020 research and innovation programme is gratefully acknowledged for funding to MS (under the Marie Skłodowska-Curie grant agreement No. 897555).Notes and references
- C. Madden, M. D. Vaughn, I. Díez-Pérez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- A. Wegelius, N. Khanna, C. Esmieu, G. D. Barone, F. Pinto, P. Tamagnini, G. Berggren and P. Lindblad, Energy Environ. Sci., 2018, 11, 3163–3167 RSC.
- C. Esmieu, P. Raleiras and G. Berggren, Sustainable Energy Fuels, 2018, 2, 724–750 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- M. Haumann and S. T. Stripp, Acc. Chem. Res., 2018, 51, 1755–1763 CrossRef CAS PubMed.
- B. L. Greene, G. J. Schut, M. W. W. Adams and R. B. Dyer, ACS Catal., 2017, 7, 2145–2150 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- M. C. Posewitz, P. W. King, S. L. Smolinski, L. Zhang, M. Seibert and M. L. Ghirardi, J. Biol. Chem., 2004, 279, 25711–25720 CrossRef CAS PubMed.
- P. W. King, M. C. Posewitz, M. L. Ghirardi and M. Seibert, J. Bacteriol., 2006, 188, 2163–2172 CrossRef CAS PubMed.
- E. M. Shepard, F. Mus, J. N. Betz, A. S. Byer, B. R. Duffus, J. W. Peters and J. B. Broderick, Biochemistry, 2014, 53, 4090–4104 CrossRef CAS PubMed.
- G. Caserta, L. Pecqueur, A. Adamska-Venkatesh, C. Papini, S. Roy, V. Artero, M. Atta, E. Reijerse, W. Lubitz and M. Fontecave, Nat. Chem. Biol., 2017, 13, 779–784 CrossRef CAS PubMed.
- G. Rao, L. Tao, D. L. M. Suess and R. D. Britt, Nat. Chem., 2018, 10, 555–560 CrossRef CAS PubMed.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- A. Silakov, E. J. Reijerse, S. P. J. Albracht, E. C. Hatchikian and W. Lubitz, J. Am. Chem. Soc., 2007, 129, 11447–11458 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- B. Bennett, B. J. Lemon and J. W. Peters, Biochemistry, 2000, 39, 7455–7460 CrossRef CAS PubMed.
- S. Katz, J. Noth, M. Horch, H. S. Shafaat, T. Happe, P. Hildebrandt and I. Zebger, Chem. Sci., 2016, 7, 6746–6752 RSC.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, J. Am. Chem. Soc., 2018, 140, 7623–7628 CrossRef CAS PubMed.
- M. L. K. Sanchez, C.-H. Wu, M. W. W. Adams and R. B. Dyer, Chem. Commun., 2019, 55, 5579–5582 RSC.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS PubMed.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Comm., 2017, 8, 16115 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- S. Rumpel, C. Sommer, E. Reijerse, C. Farès and W. Lubitz, J. Am. Chem. Soc., 2018, 140, 3863–3866 CrossRef CAS PubMed.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 16894–16902 CrossRef CAS PubMed.
- C. C. Pham, D. W. Mulder, V. Pelmenschikov, P. W. King, M. W. Ratzloff, H. Wang, N. Mishra, E. E. Alp, J. Zhao, M. Y. Hu, K. Tamasaku, Y. Yoda and S. P. Cramer, Angew. Chem., Int. Ed., 2018, 57, 10605–10609 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, E. M. Shepard, A. S. Byer, S. M. Noone, J. W. Peters, J. B. Broderick and P. W. King, J. Am. Chem. Soc., 2013, 135, 6921–6929 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, Joule, 2017, 1, 61–76 CrossRef CAS.
- N. Khanna, C. Esmieu, L. S. Meszaros, P. Lindblad and G. Berggren, Energy Environ. Sci., 2017, 10, 1563–1567 RSC.
- L. S. Mészáros, B. Németh, C. Esmieu, P. Ceccaldi and G. Berggren, Angew. Chem., Int. Ed., 2018, 57, 2596–2599 CrossRef PubMed.
- H. Land, P. Ceccaldi, L. S. Mészáros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2019, 10, 9941–9948 RSC.
- S. T. Stripp and T. Happe, Dalton Trans., 2009, 9960–9969, 10.1039/b916246a.
- C. Kamp, A. Silakov, M. Winkler, E. J. Reijerse, W. Lubitz and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 410–416 CrossRef CAS PubMed.
- I. Amenabar, S. Poly, W. Nuansing, E. H. Hubrich, A. A. Govyadinov, F. Huth, R. Krutokhvostov, L. Zhang, M. Knez, J. Heberle, A. M. Bittner and R. Hillenbrand, Nat. Comm., 2013, 4, 2890 CrossRef PubMed.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rüdiger, W. Lubitz and H. Ogata, J. Am. Chem. Soc., 2018, 140, 1057–1068 CrossRef CAS PubMed.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, ISME J., 2015, 10, 761 CrossRef PubMed.
- A. Le Cloirec, S. C. Davies, D. J. Evans, D. L. Hughes, C. J. Pickett, S. P. Best and S. Borg, Chem. Commun., 1999, 2285–2286 RSC.
- E. J. Lyon, I. P. Georgakaki, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed., 1999, 38, 3178–3180 CrossRef CAS PubMed.
- M. Schmidt, S. M. Contakes and T. B. Rauchfuss, J. Am. Chem. Soc., 1999, 121, 9736–9737 CrossRef CAS.
- H. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS PubMed.
- V. Fourmond, Anal. Chem., 2016, 88, 5050–5052 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. Ogawa, N. Kosaka, C. A. Regino, M. Mitsunaga, P. L. Choyke and H. Kobayashi, Mol. BioSyst., 2010, 6, 888–893 RSC.
- L. V. Deriy, E. A. Gomez, G. Zhang, D. W. Beacham, J. A. Hopson, A. J. Gallan, P. D. Shevchenko, V. P. Bindokas and D. J. Nelson, J. Biol. Chem., 2009, 284, 35926–35938 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Including additional EPR and FTIR data, AFM images of E. coli cells, cell viability data, in vitro enzymatic assay data, as well as an overview of the sSNOM and nanoFTIR setup. See DOI: 10.1039/d0sc00512f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |