
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic enantioselective synthesis of tetrasubstituted chromanones via palladium-catalyzed asymmetric conjugate arylation using chiral pyridine-dihydroisoquinoline ligands†
Doohyun
Baek
 a,
Huijeong
Ryu
a,
Ji Yeon
Ryu
b,
Junseong
Lee
b,
Brian M.
Stoltz
*c and
Sukwon
Hong
*ad
a,
Huijeong
Ryu
a,
Ji Yeon
Ryu
b,
Junseong
Lee
b,
Brian M.
Stoltz
*c and
Sukwon
Hong
*ad
aDepartment of Chemistry, Gwangju Institute of Science and Technology, 123 Cheomdan-gwagiro, Buk-gu, Gwangju 61005, Republic of Korea. E-mail: shong@gist.ac.kr
bDepartment of Chemistry, Chonnam National University, 77 Yongbong-ro, Buk-gu, Gwangju 61186, Republic of Korea
cThe Warren and Katharine Schlinger Laboratory of Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
dSchool of Materials Science and Engineering, Gwangju Institute of Science and Technology, 123 Cheomdan-gwagiro, Buk-gu, Gwangju 61005, Republic of Korea
First published on 7th April 2020
Abstract
Highly enantioselective conjugate addition reactions of arylboronic acids to 2-substituted chromones catalyzed by palladium complexes with new chiral Pyridine-Dihydroisoquinoline (PyDHIQ) ligands have been developed. These reactions provide highly enantioselective access to chromanones containing tetrasubstituted stereocenters. Various arylboronic acids and 2-substituted chromones can be used in the catalytic reaction to afford the chiral tetrasubstituted chromanones in good yields and excellent enantioselectivities (25 examples, up to 98% yields, up to 99% ee).
Introduction
Optically active chromanone scaffolds having a stereocenter at the C2 position are prominent structural motifs in natural products, and possess numerous bioactivities (Scheme 1a).1 Chiral chromanones have been synthesized by various methods,2 such as intramolecular oxa-Michael additions,3 asymmetric conjugate additions,4 asymmetric reductions,5 and Mitsunobu cyclizations.6 Nevertheless, much of the previous research has focused on generation of trisubstituted rather than tetrasubstituted stereocenters. Direct asymmetric transformation to generate a tetrasubstituted oxygen-bearing stereocenter at the C2 position of chromanone has remained rather elusive. A sole exception was reported by the Kurth group, wherein the enantioselective synthesis of chiral tetrasubstituted spirocyclic chromanones was enabled via enamine-mediated aldol/oxa-Michael tandem reactions.7 To the best of our knowledge, there have been no other examples demonstrating the enantioselective construction of tetrasubstituted stereocenters at the C2 position of chromanones in a single step.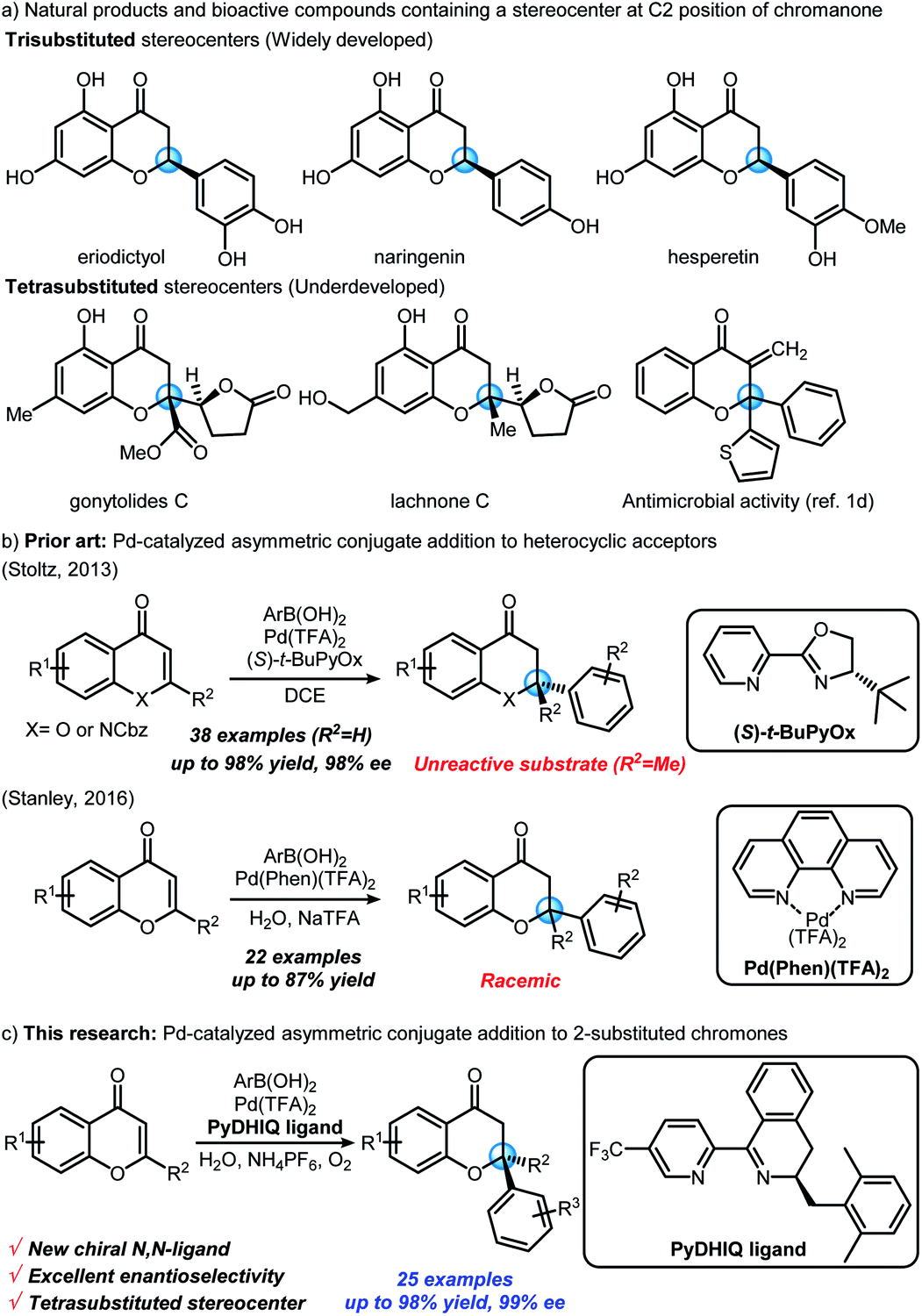 | ||
| Scheme 1 Synthesis of chromanones by palladium-catalyzed conjugate addition to chromones. | ||
Asymmetric conjugate addition reactions8 can provide a straightforward pathway to chiral C2-tetrasubstituted chromanones from relatively simple, achiral unsaturated acceptors (i.e., 2-substituted chromones). In 2013, the Stoltz group disclosed that 2-methylchromone is not a suitable electrophile for their well-established Pd(II)/PyOx4e,9 catalyzed asymmetric conjugate addition chemistry4e (Scheme 1b).
Interestingly, the Stanley group later reported the racemic version of such a conjugate addition of arylboronic acids to 2-substituted chromones catalyzed by a Pd(II)/phenanthroline complex in aqueous media10 (Scheme 1b). We envisioned that palladium-catalyzed asymmetric conjugate addition to 2-substituted chromones could be achieved by developing new chiral N,N-ligands, which can be effective in Stanley's aqueous conditions. We imagined that dihydroisoquinoline-based ligands, having chiral imine moieties, could be potential candidates. Herein, we report the first example of a palladium-catalyzed asymmetric conjugate addition of arylboronic acids to 2-substituted chromones with newly developed chiral N,N-ligands (Scheme 1c). This reaction provides highly enantioselective synthetic access to chiral chromanones bearing tetrasubstituted stereocenters in a single step.
Results and discussion
We have previously reported chiral dihydroisoquinoline-based N-heterocyclic carbene (NHC) ligands and (N,N) type diimine ligands, which were synthesized via Bischler–Napieralski cyclization.11 Following a similar synthetic protocol, a series of chiral Pyridine-Dihydroisoquinoline (PyDHIQ) ligands were successfully prepared (Scheme 2). Electronic and steric properties of PyDHIQ ligands can be easily modulated by varying substituents of the pyridine as well as the stereodifferentiating groups of the chiral imine moiety ((S)-3a–3e).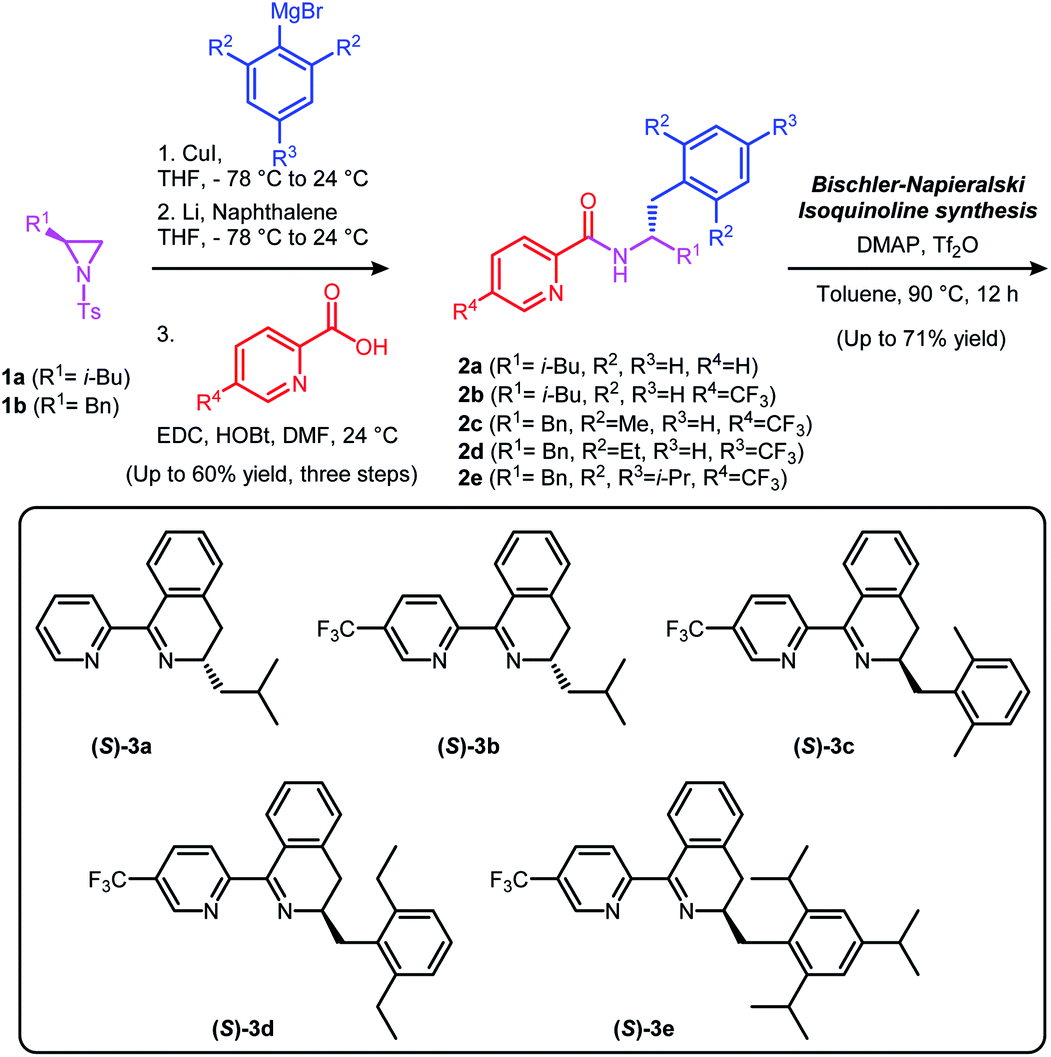 | ||
| Scheme 2 Chiral pyridine-dihydroisoquinoline(PyDHIQ) ligands. | ||
With the chiral PyDHIQ ligands in hand, the asymmetric conjugate addition reaction of phenylboronic acid to 2-methylchromone was investigated by employing the PyDHIQ ligands, Pd(TFA)2, H2O as a solvent, and NH4PF6 as an additive4e,10 (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T [°C] | ArB(OH)2 | Additive (30 mol%) | Ligand | Yieldc [%] | eed [%] |
| a All reactions were carried out with 4 (0.5 mmol, 1 equiv.), ArB(OH)2 (0.6 mmol, 1.2 equiv.), Pd(TFA)2 (0.025 mmol, 5 mol%), ligand (0.030 mmol, 6 mol%), additive (0.15 mmol, 30 mol%), H2O (0.35 mL) for 12 h. b (S)-5-CF3-t-BuPyOx also showed trace amount of products; see ESI for additional screening data. c Isolated yield of 6a or 6o. d Determined by HPLC with DAICEL chiralpak. | ||||||
| 1 | 60 | PhB(OH)2 | NH4PF6 | (S)-t-BuPyOx | Trace | — |
| 2 | 60 | PhB(OH)2 | NH4PF6 | (S)-3a | Trace | — |
| 3 | 60 | PhB(OH)2 | NH4PF6 | (S)-3b | 11 | −68 |
| 4 | 60 | PhB(OH)2 | NH4PF6 | (S)-3c | 97 | 95 |
| 5 | 60 | PhB(OH)2 | NH4PF6 | (S)-3d | 77 | 98 |
| 6 | 60 | PhB(OH)2 | NH4PF6 | (S)-3e | 70 | 98 |
| 7 | 60 | PhB(OH)2 | — | (S)-3c | 42 | 90 |
| 8 | 60 | PhB(OH)2 | NaBF4 | (S)-3c | 47 | 95 |
| 9 | 60 | PhB(OH)2 | NaPF6 | (S)-3c | 85 | 95 |
| 10 | 60 | PhB(OH)2 | NH4BF4 | (S)-3c | 60 | 90 |
| 11 | 60 | 3-CIPhB(OH)2 | NH4PF6 | (S)-3c | 17 | 90 |
| 12 | 70 | 3-CIPhB(OH)2 | NH4PF6 | (S)-3c | 33 | 90 |
| 13 | 80 | 3-CIPhB(OH)2 | NH4PF6 | (S)-3c | 35 | 89 |
| 14 | 70 | 3-CIPhB(OH)2 | NH4PF6, O2 balloon | (S)-3c | 55 | 92 |
It is important to note that (S)-t-BuPyOx ligand showed no reactivity in the current reaction conditions (Table 1, entry 1). While (S)-3a also showed no reactivity, electronically modified ligand (S)-3b showed low reactivity and moderate enantioselectivity (Table 1, entries 2 and 3). Surprisingly, a considerable increase of both yield and enantioselectivity was observed with ligand (S)-3c which has a (2,6-dimethylphenyl)methyl group (Table 1, entry 4). When the steric demands were increased in the ligands (S)-3d and (S)-3e, enantioselectivity was increased at the expense of the yield (Table 1, entries 5 and 6). Therefore, the ligand (S)-3c was chosen for further study as it showed a balanced performance in terms of reactivity and enantioselectivity. Encouraged by these results, we systematically altered the reaction conditions and searched for potential additives to further improve reactivity and enantioselectivity. Weakly coordinating counteranions such as BF4− and PF6− were tested, since these have previously been shown to beneficially impact the reactivity and selectivity in related conjugate addition reactions.9b All the additives tested resulted in better reactivity than without the additive (cf. entries 8–10 with entry 7). PF6− showed higher yields than BF4− (entries 8 vs. 9, and entries 4 vs. 10) and ammonium cation showed higher yields than the sodium cation (entries 4 vs. 9 and entries 8 vs. 10). As a result, NH4PF6 was selected because it showed excellent reactivity and high enantioselectivity (entry 4). In addition, the use of m-chlorophenylboronic acids resulted in very low yields (entry 11). By increasing the reaction temperature from 60 °C to 70 °C, the yield increased from 17% to 33% (entry 12). The ligand decomposition was observed at 80 °C by thin-layer chromatography monitoring (entry 13).
Remarkably, O2 atmosphere increased the isolated yield from 33% to 55% at 70 °C (entry 14).12 Therefore, the optimized reaction conditions including the use of NH4PF6 in O2 atmosphere at 70 °C showed the best combination of reactivity and enantioselectivity.
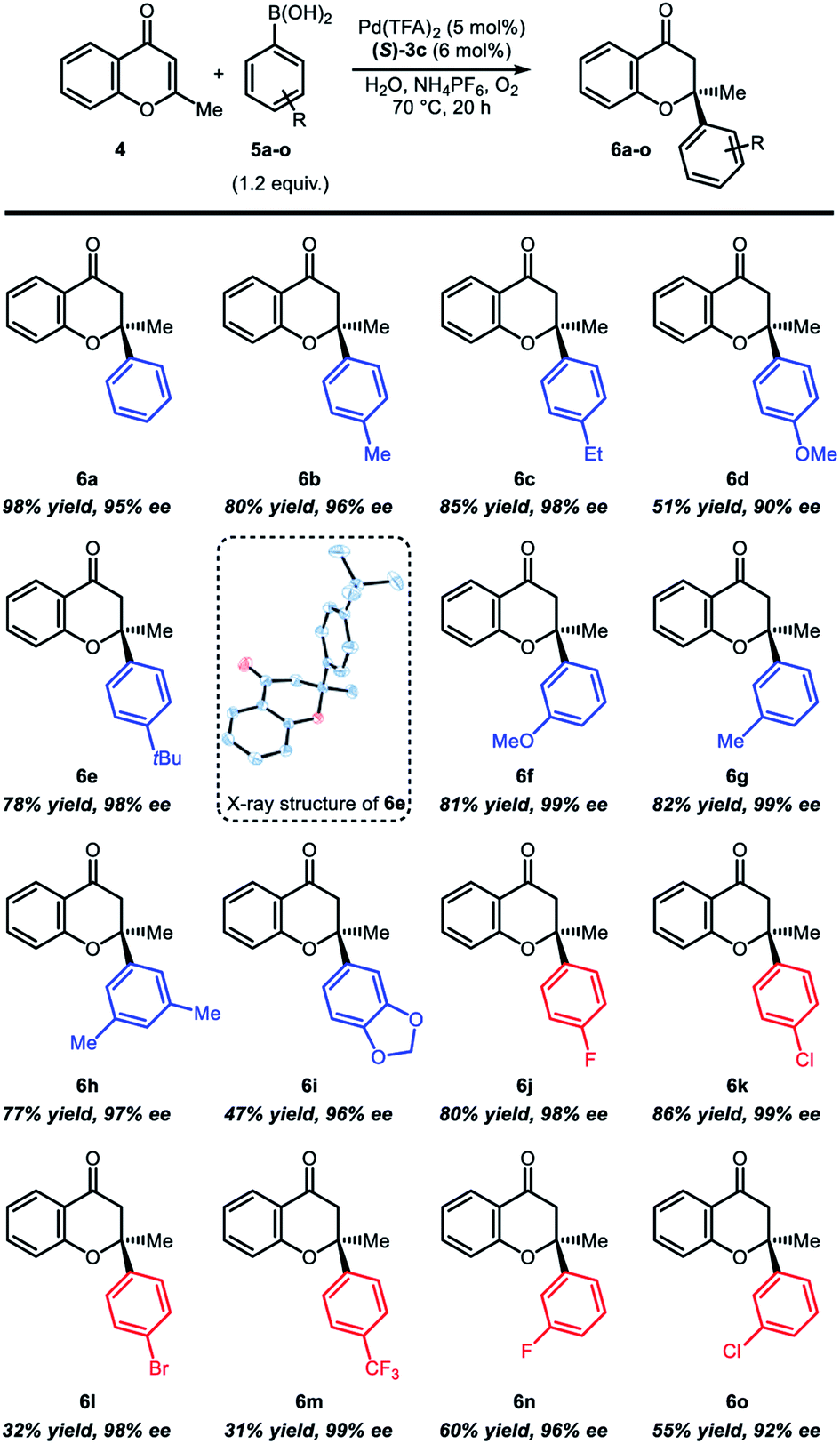
Under the optimized reaction conditions, the substrate scope was examined with various arylboronic acids (Table 2). In case of the reaction with para-substituted arylboronic acids, electron-rich boronic acids (Table 2, 6b–6e) resulted in mostly good yields (78–85%) and high enantioselectivity (96–99%), except for para-methoxyphenylboronic acid 6d (51% yields and 90% ee). Reactions with electron-deficient boronic acids such as para-fluoro- or para-chlorophenylboronic acid also showed good reactivity and high enantioselectivity (Table 2, 6j and 6k). However, reduced yields were observed with para-bromophenylboronic acid (6l, 32% yield and 98% ee) and para-trifluoromethylphenylboronic acid (6m, 31% yield and 99% ee). In case of the reaction with meta-substituted arylboronic acids, electron-rich boronic acids (Table 2, 6f–6i) showed good yield (77–82%) and high enantioselectivity (97–99%). An exceptional case is 3,4-(methylenedioxy)phenylboronic acid 6i, which was isolated in 47% yield and 96% ee. Reactions with electron-deficient arylboronic acids (6n and 6o) were also isolated in moderate yields (55–60%) and high enantioselectivity (92–96%). The absolute configuration of compound 6e was determined by X-ray crystallography, and all other products were assigned by analogy.
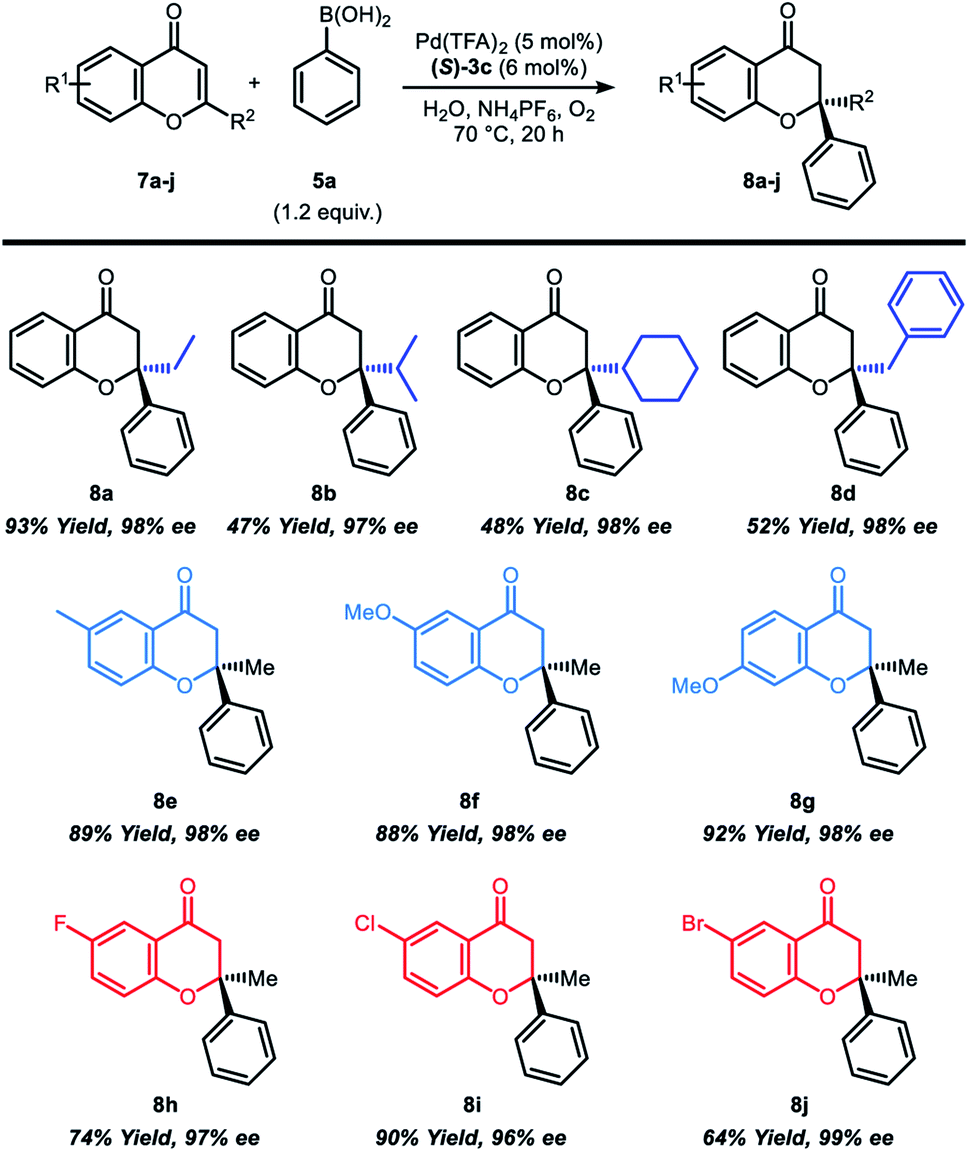
In sequence, the reaction scope was studied with various 2-substituted chromone derivatives (Table 3). 2-Ethyl-, 2-isopropyl-, 2-benzyl- and 2-cyclohexylchromones were synthesized according to previously reported literature5b to investigate the steric effect of 2-substituted chromones (Table 3, 8a–8d). Reactions with 2-ethylchromone, which has a linear β-alkyl substituent resulted in excellent yield and high enantioselectivity (8a, 93% yield and 98% ee). On the other hand, chromones having a branched β-alkyl substituent such as isopropyl or cyclohexyl group afforded the product 8b and 8c in moderate yield (47–48%), but still with high enantioselectivity (97–98%). 2-Benzylchromone also displayed moderate reactivity with outstanding enantioselectivity (8d, 52% yield and 98% ee). Notably, both electron-rich and electron-deficient chromones with substituents at the 6-position or 7-position were well tolerated in the reaction (8e–8j), furnishing good yields (64–92%)and high enantioselectivity (96–99%).
The [(S)-3d]–Pd(II) complex was prepared by the complexation of (S)-3d with palladium(II) chloride and characterized by X-ray crystallography (Fig. 1a). The X-ray structure of [(S)-3d]PdCl2 showed that Pd–Cl bond trans to the dihydroisoquinoline is slightly longer than Pd–Cl bond cis to the dihydroisoquinoline (2.287(2) Å vs. 2.280(2) Å).13 This suggests that the dihydroisoquinoline would exert a stronger trans influence than the pyridine. Based on the literature precedence, it might be reasonable to propose a stereochemistry-determining transition state where a Pd–phenyl bond is located trans to the dihydroisoquinoline moiety (Fig. 1b).9b,9g,12b,13Fig. 1b shows our simplified transition state model rationalizing the stereochemical pathways of asymmetric conjugate addition of phenylboronic acid 5a to 2-methylchromone 4. It has been known that the enantioselectivity-determining step involves a square-planar geometry transition state at the stage when the phenyl group inserts across the alkene.9b Among the two possible isomeric transition states TS-A and B (Fig. 1b), TS-A leading to the major product, (S)-6a, might be most favored owing to the minimized steric repulsions between the ligand and the substrate.
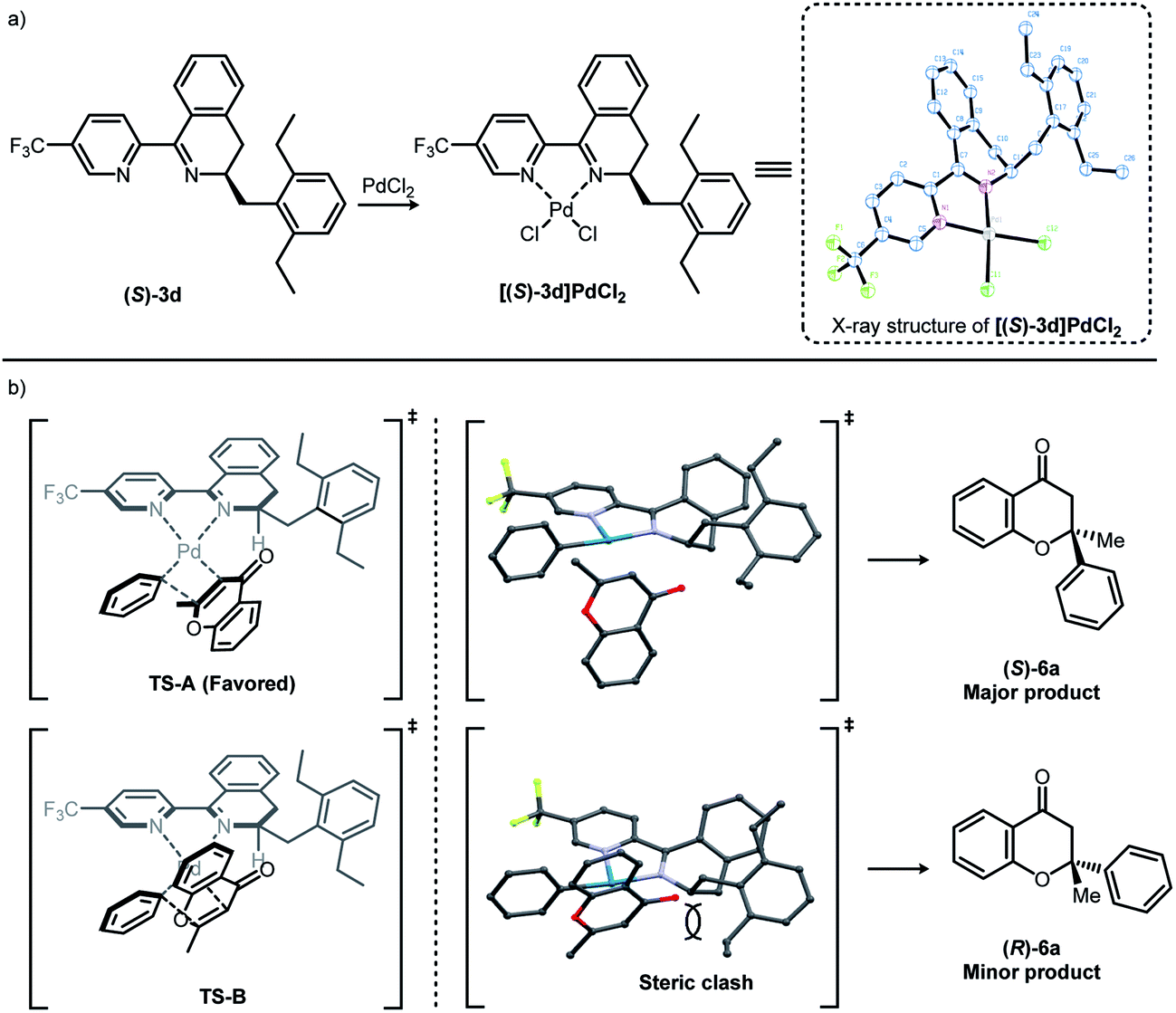 | ||
| Fig. 1 (a) Synthesis and X-ray structure of [(S)-3d]PdCl2 (thermal ellipsoids correspond to 50% probability) and (b) proposed stereochemical model for observed enantioselectivity. | ||
Conclusions
In conclusion, new chiral pyridine-dihydroisoquinoline (PyDHIQ) ligands have been developed for the palladium-catalyzed asymmetric conjugate addition reactions in aqueous media. This chemistry represents the first example of a highly enantioselective conjugate addition of arylboronic acids to 2-substituted chromones to afford hindered chromanone products containing tetrasubstituted stereocenters. Twenty-five examples of various arylboronic acids and 2-substituted chromones were demonstrated to provide good isolated yield (31–98%) and outstanding enantioselectivity (90–99%) under the optimized reaction conditions. The observed stereochemistry was rationalized by proposed transition state models. The application of this method to the synthesis of natural products is currently on-going in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the “GIST-Caltech Research Collaboration” grant funded by the GIST in 2017. This research was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science and ICT) (NRF-2020R1A2C1009123). The NIH-NIGMS (R01GM080269) and Caltech are also thanked for support of our research program.Notes and references
- (a) L. C. Chang and A. D. Kinghorn, Bioactive compounds from natural sources: isolation, characterization and biological properties, Taylor & Francis, London, 2001 Search PubMed; (b) R. J. Grayer and N. C. Veitch, Flavonoids: Chemistry, Biochemistry and Applications, CRC, Taylor & Francis, Boca Raton, FL, 2006 Search PubMed; (c) S. Emami and Z. Ghanbarimasir, Eur. J. Med. Chem., 2015, 93, 539–563 CrossRef CAS PubMed; (d) F. E. Ward, D. L. Garling and R. T. Buckler, J. Med. Chem., 1981, 24, 1073–1077 CrossRef CAS PubMed.
- For general reviews, see; (a) A. E. Nibbs and K. A. Scheidt, Eur. J. Org. Chem., 2012, 449–462 Search PubMed; (b) L. Meng and J. Wang, Synlett, 2016, 27, 656–663 CASfor other reported researches to synthesize chiral chromaones, see; (c) Z. Zhang, C. Pan and Z. Wang, Chem. Commun., 2007, 4686–4688 RSC; (d) C. Wu, Y. Liu, H. Zeng, L. Liu, D. Wang and Y. Chen, Org. Biomol. Chem., 2011, 9, 253–256 RSC; (e) N.-J. Zhong, L. Liu, D. Wang and Y.-J. Chen, Chem. Commun., 2013, 49, 3697–3699 RSC; (f) G. Wen, Y. Su, G. Zhang, Q. Lin, Y. Zhu, Q. Zhang and X. Fang, Org. Lett., 2016, 18, 3980–3983 CrossRef CAS PubMed; (g) A. M. Hardman-Baldwin, M. D. Visco, J. M. Wieting, C. Stern, S.-I. Kondo and A. E. Mattson, Org. Lett., 2016, 18, 3766–3769 CrossRef CAS PubMed; (h) L. G. DeRatt, M. Pappoppula and A. Aponick, Angew. Chem., Int. Ed., 2019, 58, 8416–8420 CrossRef CAS PubMedfor kinetic resolution approaches, see; (i) E. J. Corey and R. B. Mitra, J. Am. Chem. Soc., 1962, 84, 2938–2941 CrossRef CAS; (j) T. L. No, P. Press and G. Britain, Tetrahedron Lett., 1970, 2305–2308 Search PubMed; (k) A. Keßberg and P. Metz, Angew. Chem., Int. Ed., 2016, 55, 1160–1163 CrossRef PubMed; (l) E. R. Ashley, E. C. Sherer, B. Pio, R. K. Orr and R. T. Ruck, ACS Catal., 2017, 7, 1446–1451 CrossRef CAS.
- (a) M. M. Biddle, M. Lin and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 3830–3831 CrossRef CAS PubMed; (b) C. Dittmer, G. Raabe and L. Hintermann, Eur. J. Org. Chem., 2007, 5886–5898 CrossRef CAS; (c) L. Wang, X. Liu, Z. Dong, X. Fu and X. Feng, Angew. Chem., Int. Ed., 2008, 47, 8670–8673 CrossRef CAS PubMed; (d) H.-F. Wang, H.-F. Cui, Z. Chai, P. Li, C.-W. Zheng, Y.-Q. Yang and G. Zhao, Chem.–Eur. J., 2009, 15, 13299–13303 CrossRef CAS PubMed; (e) H. F. Wang, H. Xiao, X. W. Wang and G. Zhao, Tetrahedron, 2011, 67, 5389–5394 CrossRef CAS; (f) L. Hintermann and C. Dittmer, Eur. J. Org. Chem., 2012, 5573–5584 CrossRef CAS; (g) Y.-L. Zhang and Y.-Q. Wang, Tetrahedron Lett., 2014, 55, 3255–3258 CrossRef CAS; (h) B. R. McDonald, A. E. Nibbs and K. A. Scheidt, Org. Lett., 2015, 17, 98–101 CrossRef CAS PubMed; (i) M. Shaikh, K. K. Atyam, M. Sahu and K. V. S. Ranganath, Chem. Commun., 2017, 53, 6029–6032 RSC.
- (a) M. K. Brown, S. J. Degrado and A. H. Hoveyda, Angew. Chem., Int. Ed., 2005, 44, 5306–5310 CrossRef CAS PubMed; (b) J. Chen, J. Chen, F. Lang, X. Zhang, L. Cun, J. Zhu, J. Deng and J. Liao, J. Am. Chem. Soc., 2010, 132, 4552–4553 CrossRef CAS PubMed; (c) F. Han, G. Chen, X. Zhang and J. Liao, Eur. J. Org. Chem., 2011, 2928–2931 CrossRef CAS; (d) T. Korenaga, K. Hayashi, Y. Akaki, R. Maenishi and T. Sakai, Org. Lett., 2011, 13, 2022–2025 CrossRef CAS PubMed; (e) J. C. Holder, A. N. Marziale, M. Gatti, B. Mao and B. M. Stoltz, Chem.–Eur. J., 2013, 19, 74–77 CrossRef CAS PubMed; (f) C. Vila, V. Hornillos, M. Fañanás-Mastral and B. L. Feringa, Chem. Commun., 2013, 49, 5933–5935 RSC; (g) Q. He, C. M. So, Z. Bian, T. Hayashi and J. Wang, Chem.–Asian J., 2015, 10, 540–543 CrossRef CAS PubMed; (h) B. M. Trost, E. Gnanamani, C. A. Kalnmals, C.-I. Hung and J. S. Tracy, J. Am. Chem. Soc., 2019, 141, 1489–1493 CrossRef CAS PubMed.
- (a) M.-K. Lemke, P. Schwab, P. Fischer, S. Tischer, M. Witt, L. Noehringer, V. Rogachev, A. Jäger, O. Kataeva, R. Fröhlich and P. Metz, Angew. Chem., Int. Ed., 2013, 52, 11651–11655 CrossRef CAS PubMed; (b) D. Zhao, B. Beiring and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 8454–8458 CrossRef CAS PubMed; (c) D. Xiong, W. Zhou, Z. Lu, S. Zeng and J. Wang, Chem. Commun., 2017, 53, 6844–6847 RSC; (d) Y. Ma, J. Li, J. Ye, D. Liu and W. Zhang, Chem. Commun., 2018, 54, 13571–13574 RSC.
- (a) K. J. Hodgetts, Tetrahedron Lett., 2001, 42, 3763–3766 CrossRef CAS; (b) Y. Noda and M. Watanabe, Helv. Chim. Acta, 2002, 85, 3473–3477 CrossRef CAS; (c) K. J. Hodgetts, Tetrahedron, 2005, 61, 6860–6870 CrossRef CAS.
- R. D. Carpenter, J. C. Fettinger, K. S. Lam and M. J. Kurth, Angew. Chem., Int. Ed., 2008, 47, 6407–6410 CrossRef CAS PubMed.
- For general reviews, see; (a) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 9, 1279–1300 CrossRef; (b) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC; (c) K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586–7656 CrossRef CAS PubMed.
- For reported examples on asymmetric conjugate addition reactions by Stoltz group with Pd(II)/PyOx system; (a) K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, J. Am. Chem. Soc., 2011, 133, 6902–6905 CrossRef CAS PubMed; (b) J. C. Holder, L. Zou, A. N. Marziale, P. Liu, Y. Lan, M. Gatti, K. Kikushima, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 14996–15007 CrossRef CAS PubMed; (c) C. L. Boeser, J. C. Holder, B. L. H. Taylor, K. N. Houk, B. M. Stoltz and R. N. Zare, Chem. Sci., 2015, 6, 1917–1922 RSC; (d) J. C. Holder, E. D. Goodman, K. Kikushima, M. Gatti, A. N. Marziale and B. M. Stoltz, Tetrahedron, 2015, 71, 5781–5792 CrossRef CAS PubMed; (e) S. E. Shockley, J. C. Holder and B. M. Stoltz, Org. Process Res. Dev., 2015, 19, 974–981 CrossRef CAS PubMedsee also selected examples on redox-relay oxidative Heck reactions by Sigman using similar Pd(II)/PyOx system; (f) M. S. Sigman, E. W. Werner, T.-s. Mei and A. J. Burckle, J. Am. Chem. Soc., 2013, 135, 6830–6833 CrossRef PubMed; (g) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960–1967 CrossRef CAS PubMed; (h) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS PubMed; (i) M. J. Hilton, L.-P. Xu, P.-O. Norrby, Y.-D. Wu, O. Wiest and M. S. Sigman, J. Org. Chem., 2014, 79, 11841–11850 CrossRef CAS PubMed; (j) C. Zhang, C. B. Santiago, L. Kou and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 7290–7293 CrossRef CAS PubMed; (k) Z.-M. Chen, M. J. Hilton and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 11461–11464 CrossRef CAS PubMedsee also a general review of dinitrogen type ligands; (l) G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
- A. L. Gerten and L. M. Stanley, Tetrahedron Lett., 2016, 57, 5460–5463 CrossRef CAS.
- (a) H. Seo, D. Hirsch-Weil, K. A. Abboud and S. Hong, J. Org. Chem., 2008, 73, 1983–1986 CrossRef CAS PubMed; (b) D. Hirsch-Weil, K. A. Abboud and S. Hong, Chem. Commun., 2010, 46, 7525–7527 RSC; (c) M. J. Rodig, H. Seo, D. Hirsch-Weil, K. A. Abboud and S. Hong, Tetrahedron: Asymmetry, 2011, 22, 1097–1102 CrossRef CAS.
- During the reaction optimization, an undesired homocoupling reaction of arylboronic acids was observed. Generation of Pd-black was also observed. Thus, we envisioned that Pd(0) could be reoxidized to Pd(II) in oxygen atmosphere. There are reported examples on oxidation of Pd(0) to Pd(II) using molecular oxygen. A general review for Pd(II)-catalyzed enantioselective oxidative Heck-coupling; (a) A.-L. Lee, Org. Biomol. Chem., 2016, 14, 5357–5366 RSCa reported example of enantioselective conjugate addition reaction enhanced by introducing molecular oxygen; (b) C. J. C. Lamb, F. Vilela and A.-L. Lee, Org. Lett., 2019, 21, 8689–8694 CrossRef CAS PubMed.
- The bond length difference between two Pd–Cl bonds is 0.007 Å (see X-ray crystallographic data for [(S)-3d]PdCl2 in ESI;† Pd–Cl bond length trans to dihydroisoquinoline group: 2.287(2) Å, Pd–Cl bond length cis to dihydroisoquinoline group: 2.280(2) Å). This value is similar to the bond length difference between two Pd–Cl bonds of [(S)-t-BuPyOx]PdCl2 complex (0.009 Å) reported by Jung group (Pd–Cl bond length trans to oxazoline group: 2.283(2) Å, Pd–Cl bond length cis to oxazoline group: 2.274(2) Å); K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O'Neill and K. W. Jung, Org. Lett., 2007, 9, 3933–3935 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1843946 and 1954885. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00412j |
| This journal is © The Royal Society of Chemistry 2020 |